

Abstract
Alcohol withdrawal syndrome (AWS) symptoms include hyperexcitability, anxiety, and sleep disorders. Chronic intermittent ethanol (CIE) treatment of rats with subsequent withdrawal of ethanol (EtOH) reproduced AWS symptoms in behavioral assays, which included tolerance to the sleep-inducing effect of acute EtOH and its maintained anxiolytic effect. Electrophysiological assays demonstrated a CIE-induced long-term loss of extrasynaptic GABAA receptor (GABAAR) responsiveness and a gain of synaptic GABAAR responsiveness of CA1 pyramidal and dentate granule neurons to EtOH that we were able to relate to behavioral effects. After CIE treatment, the α4 subunit-preferring GABAAR ligands 4,5,6,7 tetrahydroisoxazolo[5,4-c]pyridin-3-ol, La3+, and Ro15-4513 (ethyl-8-azido-5,6-dihydro-5-methyl-6-oxo-4H-imidazo[1,5α][1,4]benzodiazepine-3-carboxylate) exerted decreased effects on extrasynaptic currents but had increased effects on synaptic currents. Electron microscopy revealed an increase in central synaptic localization of α4 but not δ subunits within GABAergic synapses on the dentate granule cells of CIE rats. Recordings in dentate granule cells from δ subunit-deficient mice revealed that this subunit is not required for synaptic GABAAR sensitivity to low [EtOH]. The profound alterations in EtOH sensitivity and α4 subunit localization at hippocampal GABAARs of CIE rats suggest that such changes in these and other relevant brain circuits may contribute to the development of tolerance to the sleep-inducing effects and long-term dependence on alcohol.
Keywords: subunit composition; anxiety; sleep; electron microscopy; alcoholism; inhibitory neurotransmission, GABAA receptor
Introduction
Alcohol abuse represents a significant problem in our society with an estimated 14 million people in the United States being dependent on alcohol (McGinnis and Foege, 1999). The alcohol withdrawal syndrome (AWS) is a particularly severe manifestation of alcohol abuse, presenting with a variety of symptoms such as anxiety, insomnia, agitation, and seizures. Clinical literature abounds with evidence that alcohol dependence is a chronic relapsing disorder in which alcoholics go through cycles of intense ethanol (EtOH) intoxication alternating with periods of abstinence; this results in a withdrawal syndrome, the severity of which is positively correlated to the number of intoxication and withdrawal cycles (Brown et al., 1988; Booth and Blow, 1993). Laboratory studies in rodents fully support these clinical findings (McCown and Breese, 1990; Becker and Hale, 1993; Kokka et al., 1993; Veatch and Gonzalez, 1996). Such studies have demonstrated the importance of intermittent EtOH administration in producing long-term alterations in the function of both GABAA receptors (GABAARs) and NMDA receptors (NMDARs) (Hu and Ticku, 1997; Becker et al., 1998). EtOH withdrawal-induced increases in the extracellular glutamate levels are also positively correlated to the number of withdrawals (Dahchour and De Witte, 2003). Other studies showed that repeated EtOH treatment and withdrawal leads to higher alcohol intake and preference than continuous EtOH treatment (Rimondini et al., 2003; O’Dell et al., 2004).
GABAARs represent one of the several important pharmacological targets of EtOH (Allan and Harris, 1986; Franks and Lieb, 1987; Mehta and Ticku, 1988; Weiner et al., 1994; Lovinger, 1997; Harris, 1999; Ariwodola et al., 2003; Aroor and Shukla, 2004). A family of heteropentameric GABAAR isoforms of different subunit composition accounts for variable sensitivity to other modulatory drugs such as benzodiazepines, barbiturates, neurosteroids, and general anesthetics (Olsen and Homanics, 2000; Whiting et al., 2000). GABAAR function and expression are altered after chronic administration of EtOH (Morrow et al., 1988; Mhatre et al., 1993; Kumar et al., 2004). EtOH tolerance and dependence also appear to be attributable, at least in part, to changes in the function of GABAARs, possibly involving alterations in native GABAAR subunit composition and trafficking (Kumar et al., 2004). In the chronic intermittent ethanol (CIE) model of alcohol withdrawal and dependence, rats are exposed to intermittent episodes (≥60 doses) of EtOH intoxication and withdrawal (approximating binge drinking episodes in humans), leading to behavioral hyperexcitability that includes decreased seizure thresholds and increased anxiety (Kokka et al., 1993). This is accompanied by presumably causal changes in GABAAR expression and physiology including: (1) persistent decreases in hippocampal GABAAR-mediated paired-pulse inhibition (Kang et al., 1996), (2) changes in levels of several GABAAR subunits (Mahmoudi et al., 1997; Cagetti et al., 2003), and (3) remarkable alterations in the effectiveness of several clinically important drug classes that act through allosteric modulation of GABAAR function (Kang et al., 1998; Cagetti et al., 2003; Liang et al., 2004). For example, there is a loss of sleep-inducing actions of the benzodiazepine flurazepam and the neuroactive steroid anesthetic alphaxalone. These changes are paralleled by the loss of synaptic and extrasynaptic GABAAR-mediated responses to such compounds in hippocampal CA1 neurons (Cagetti et al., 2003; Liang et al., 2004).
Several recent studies have suggested that GABAARs containing α4βδ or α6βδ subunit combinations, which are normally found outside or at the edges of GABAergic synapses (Nusser et al., 1998; Wei et al., 2003), are particularly sensitive to low millimolar concentrations of EtOH (Sundstrom-Poromaa et al., 2002; Wallner et al., 2003; Wei et al., 2004). We hypothesized that the demonstrated alterations in the hippocampal levels of α1, α4, and δ subunits after CIE treatment are responsible for the altered function and pharmacological sensitivity of hippocampal GABAARs (Cagetti et al., 2003; Liang et al., 2004). Therefore, we evaluated the effects of CIE treatment and withdrawal on re-exposure to EtOH in anxiety and sleep assays and related these to changes in the effects of acute EtOH application on hippocampal GABAAR-mediated synaptic and extrasynaptic currents. Then, we used electron microscopic immunogold labeling techniques to examine possible alterations in the localization of α4 and δ subunits after CIE treatment. Our results are consistent with a net loss of extrasynaptic and a gain of synaptic GABAAR responsiveness to EtOH, which are concomitant with increased α4 but not δ subunit localization within hippocampal GABAergic synapses of CIE rats.
Materials and Methods
Production of CIE rats.
The Institutional Animal Care and Use Committee approved all animal experiments. Male Sprague Dawley rats (170–190 g) were housed in the vivarium under a 12 h light/dark cycle and had ad libitum access to food and water. Intoxicating doses of EtOH (Pharmco Products, Brookfield, CT) were administered by oral intubation on a chronic regimen: for the first five doses, rats received 5 g/kg of body weight as a 25% (w/v) solution in saline once every other day, and for the following 55 doses, they received 6 g/kg of 30% (w/v) EtOH once every day. The control group received saline (20 ml/kg of body weight). With this EtOH regimen, rats experience multiple cycles of intoxication and withdrawal phases leading to a kindling-like state with a persistent decrease in pentylenetetrazol seizure threshold (Kokka et al., 1993) and reduced hippocampal GABAAR-mediated inhibition (Kang et al., 1996). After the treatment and 2 or 40 d of withdrawal, rats were subjected to behavioral experiments, or they were killed and tissues prepared for experiments.
Sleep-time assay.
The hypnotic effect of EtOH (3 g/kg, i.p.) was tested on saline- and CIE-treated rats. EtOH (DSP-CT-18; Pharmco Products) was diluted to 17.8% in 0.9% saline. Injection volume was 2 ml/kg. Sleep time was determined as follows: after drug injection and loss of righting reflex, rats were placed on their backs in a V-shaped trough, and a timer was started. The sleep time period ended when animals were able to flip over three times in 30 s after being repeatedly placed on their backs.
Elevated plus maze assay.
CIE- and saline-treated rats were tested for the anxiolytic effect of EtOH (0.5 g/kg) on the elevated plus maze (Cagetti et al., 2003). Rats were randomly divided into four groups: saline controls treated with vehicle (n = 7) or with EtOH (n = 7) and CIE rats treated with vehicle (n = 6) or EtOH (n = 7). EtOH was diluted in saline (0.9%) and administered to rats via gavage 30 min before testing. Rats were placed on the central area of the maze, tested for 5 min, and videotaped. The following measures were scored: number of entries into open arms, closed arms, or center platform and time spent in each of these areas. To measure the locomotor activity, the number of total entries was measured for each rat. Data are reported as percentage of number of entries in arms, percentage of time spent in different entries, and number of total entries. Statistical differences were determined using ANOVA.
Production of δ −/− mice.
Mice with a targeted disruption of the δ subunit gene were produced and genotyped as described previously (Mihalek et al., 1999). All mice were of a mixed C57BL/6J × 129Sv/SvJ genetic background and were derived from heterozygote matings. Experiments were performed on male mice at 6 months of age.
Electrophysiological recordings.
Transverse slices (400 μm thick) of rat or mouse dorsal hippocampus were obtained using standard techniques (Kang et al., 1996; Spigelman et al., 2003). Whole-cell patch-clamp recordings were obtained from cells located in the CA1 pyramidal or dentate granule (DG) cell layers at 34 ± 0.5°C during perfusion with artificial CSF (ACSF) composed of the following (in mm): 125 NaCl, 2.5 KCl, 2 CaCl2, 2 MgCl2, 26 NaHCO3, and 10 d-glucose. The ACSF was bubbled continuously with a 95/5% mixture of O2/CO2 to ensure adequate oxygenation of slices and a pH of 7.4. Patch pipettes contained the following (in mm): 135 cesium gluconate, 2 MgCl2, 1 CaCl2, 11 ethylene glycol-bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid, 10 N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid, 2 K2ATP, and 0.2 Na2GTP, pH adjusted to 7.25 with CsOH. GABAAR-mediated miniature IPSCs (mIPSCs) were pharmacologically isolated by adding tetrodotoxin (TTX; 0.5 μm), d(−)-2-amino-5-phosphonopentanoate (40 μm), 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX; 10 μm), and CGP 54626 (5,7,8,9-tetrahydro-5-hydroxy-6H-benzocyclohepten-6-ylideneacetic acid) (1 μm) to the ACSF from stock solutions. Stock solutions of CNQX were made with pure dimethyl sulfoxide (DMSO). Final concentration of DMSO did not exceed 42 μm in the recording chamber. Signals were recorded in voltage-clamp mode with an amplifier (Axoclamp 2B; Molecular Devices, Union City, CA). Whole-cell access resistances were in the range of 8–20 MΩ before electrical compensation by ∼90%. During voltage-clamp recordings, access resistance was monitored by measuring the size of the capacitative transient in response to a 5 mV step command, and experiments were abandoned if changes >20% were encountered. At least 10 min was allowed for equilibration of the pipette solution with the intracellular milieu before commencing recordings. Data were acquired with pClamp 8 software (Molecular Devices), digitized at 20 kHz (Digidata 1200B; Molecular Devices), and analyzed using Clampfit software (Molecular Devices) and the Mini Analysis Program (versions 5.2.2 and 5.4.8; Synaptosoft, Decatur, GA).
Detection and analysis of mIPSCs and tonic currents.
The recordings were low-pass filtered off-line (Clampfit software) at 2 kHz. The mIPSCs were detected (Mini Analysis Program) with threshold criteria of: amplitude, 5 pA and area, 20 pA·ms. Frequency of mIPSCs was determined from all automatically detected events in a given 100 s recording period. For kinetic analysis, only single-event mIPSCs with a stable baseline, sharp rising phase, and exponential decay were chosen during visual inspection of the recording trace. Double and multiple peak mIPSCs were excluded. The mIPSC kinetics was obtained from analysis of the averaged chosen single events (>120 events per 100 s recording period) aligned with half rise time in each cell. Decay time constants were obtained by fitting a double exponential to the falling phase of the averaged mIPSCs. The tonic current magnitudes were obtained from the mean baseline current during the 100 s recording periods. The investigator performing the recordings and mIPSC analysis was blind to the treatment (saline or CIE) that the rats received.
Tissue preparation for electron microscopy.
In preparation for postembedding immunogold labeling for the α4 and δ subunits of GABAA receptors, four CIE- and four saline-treated rats were anesthetized deeply with sodium pentobarbital (90 mg/kg, i.p.) and perfused through the ascending aorta with a fixative solution of 4% paraformaldehyde and 0.1% glutaraldehyde in 0.1 m phosphate buffer (PB), pH 7.3. The brains were removed, postfixed in the same fixative for 2 h, and then rinsed thoroughly in PB. Coronal brain sections were cut at 0.5–1 mm with a razor blade, and small blocks of the dentate gyrus were trimmed from these sections. Specimens were immersed in 5% sucrose in PB, pH 7.4, and then cryoprotected in 10, 20, and 30% glycerol in PB for 2 h each. Cryoprotected sections were frozen at −190°C in a cryofixation unit (EM CPC; Leica, Wien, Austria) and then transferred to a cryosubstitution unit (EM AFS; Leica), which was programmed for all subsequent steps (Wei et al., 2003). Specimens were immersed in 4% uranyl acetate (Electron Microscopy Sciences, Fort Washington, PA), dissolved in anhydrous methanol for 24 h at −90°C, rinsed in methanol at −45°C, and infiltrated with Lowicryl HM20 resin (Electron Microscopy Sciences) for 48 h at −45°C. The resin was polymerized with ultraviolet light (360 nm) for 24 h at −45°C and then warmed in 4°C steps to 0°C.
Immunogold labeling for electron microscopy.
Ultrathin sections were cut on a microtome (Reichert-Jung, Vienna, Austria), picked up on nickel mesh grids, and processed for immunogold labeling with previously described methods (Wei et al., 2003). Briefly, ultrathin sections were treated with 0.2% NaOH in distilled water for 5 min and then with 0.1% NaBH4 in 0.01 m Tris-buffered saline (TBS), pH 7.4, for 10 min and incubated in 2% human serum albumin (HSA) (Sigma, St. Louis, MO) and 0.05 m glycine in TBS containing 0.1% Triton X-100 for 10 min and 7 min, respectively. Sections were blocked in 2% HSA in TBS for 1.5 h and then incubated in the primary antiserum, rabbit anti-α4 subunit (1:300; AB5457; Chemicon, Temecula, CA), or rabbit anti-δ subunit (1:100; gift from Dr. Werner Sieghart, Medical University of Vienna, Vienna, Austria) in TBS containing 2% HSA for 18–24 h at room temperature. After a rinse with 0.05 m Tris-HCl buffer (THB) containing polyethylene glycol (50 mg/ml), sections were incubated for 2.5 h in an appropriate secondary antisera conjugated to 10 nm colloidal gold particles, diluted 1:30 in 0.05 m THB, pH 8.0, containing 2% HSA. The secondary antisera were either goat anti-rabbit IgG (Amersham Biosciences, Piscataway, NJ) or goat anti-rabbit IgG (Aurion; distributed by Electron Microscopy Sciences). After labeling, sections were stained with uranyl acetate for 40 min and lead citrate for 4 min.
Quantitative analysis.
Randomly selected series of α4 or δ subunit-labeled synaptic profiles within the molecular layer of the dentate gyrus were studied and photographed with a Jeol (Peabody, MA) 100CX II electron microscope at a primary magnification of 19,000×. The localization of colloidal gold particles was determined for each symmetric synapse that exhibited immunogold labeling in these photomicrographs. Symmetric synaptic contacts were operationally defined as regions with close apposition between an axon terminal and putative granule cell dendrite at which the presynaptic and postsynaptic membranes were precisely parallel. Such contacts generally included a thin postsynaptic density and some electron-dense material in the cleft between the membranes. The quantitative analyses included 126 and 140 α4 subunit-labeled synapses from saline- (n = 3) and CIE-treated (n = 3) rats, respectively.
Gold particle positioning along the synaptic membranes was operationally defined as either (1) perisynaptic or (2) synaptic. Labeling was classified as perisynaptic if the gold particles were located either directly at the ends of the synaptic contact, within 30 nm of the ends of the synaptic contact, or along the extrasynaptic membranes that extended up to 100 nm beyond the end of the synapse. Gold particles that were located farther than 100 nm from the ends of a synaptic contact were not included in this analysis. Labeling was classified as synaptic if gold particles were located directly at synaptic contacts, excluding the perisynaptic sites indicated above. Such labeling was frequently concentrated near the center of the synapse.
Statistical analysis.
In sleep-time assays, statistical significance was calculated by Student’s t test. In the elevated plus maze assay, statistical differences were determined using ANOVA. In electron microscopy (EM) analysis, statistical differences were determined with ANOVA. In all electrophysiological recordings, comparisons of group differences in mIPSC kinetics and drug effects were made with ANOVA (Sigmastat; SPSS, Chicago, IL).
Results
CIE rats show tolerance to the hypnotic effect of EtOH
Using a sleep-time assay, we compared the hypnotic effect of EtOH (3 g/kg, i.p.) in CIE- and saline-treated controls. CIE rats had a profoundly shorter sleep duration (3.5 ± 2.9 min; n = 14) compared with controls (37.2 ± 5.4 min; n = 12) after administration of EtOH, representing a significant reduction (89%; p < 0.001) in sleep time. These results indicated that CIE rats become tolerant to the sleep-inducing effect of EtOH.
CIE rats retain the anxiolytic effect of EtOH
The anxiolytic effect of EtOH (0.5 g/kg, p.o.) was tested in CIE- and saline-treated rats on the elevated plus maze. As shown previously using the same test (Cagetti et al., 2003), vehicle-treated CIE rats showed increased anxiety, and they did not enter the open arms (Fig. 1A). EtOH significantly increased the number of open entries in control (p = 0.03) and CIE (p = 0.02) rats. EtOH also increased the time CIE rats spent in the open arms (p = 0.027). Thus, despite showing tolerance to the sleep-inducing effect of EtOH, CIE rats are still sensitive to the anxiolytic effect of EtOH.
Figure 1.
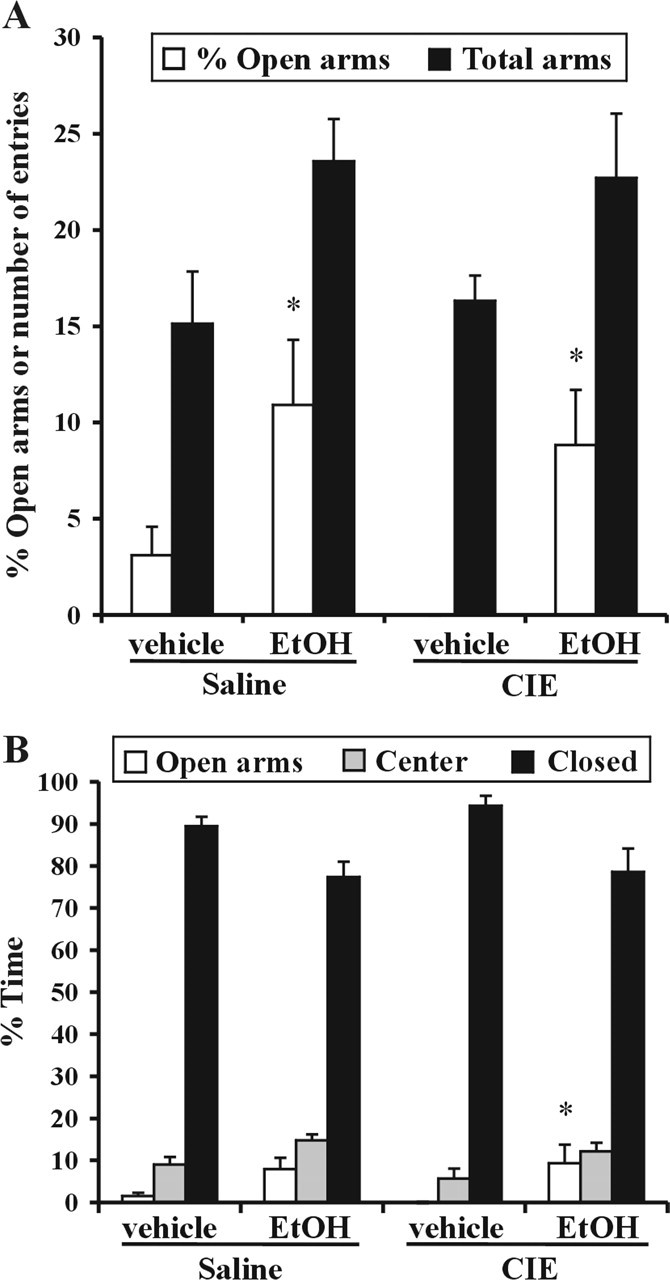
Increased anxiety of CIE rats is alleviated by EtOH. The anxiolytic effect of EtOH (0.5 g/kg, p.o.) was tested on the elevated plus maze. Rats were divided in four groups: vehicle- and EtOH-treated controls and vehicle- and EtOH-treated CIE rats. Data are reported as mean ± SEM of percentage of entries in open arms and number of total entries (A) and percentage of time in different arms (B). EtOH significantly increased the number of open arms entries in CIE (*p = 0.02) and in controls (*p = 0.03) and for CIE rats, the time spent in open arms (*p = 0.027).
EtOH potentiates extrasynaptic but not synaptic GABAAR currents in CA1 neurons from normal rats
To explore the possible mechanisms of EtOH actions, we recorded pharmacologically isolated GABAAR currents in CA1 pyramidal and dentate granule cells in hippocampal slices from saline- and CIE-treated rats. In these and all subsequent whole-cell patch recordings, membrane voltage was clamped at 0 mV, and the initial extracellular solution containing TTX, ionotropic glutamate, and GABAB receptor blockers was applied for at least 10 min. Bath application of EtOH produced increases in the holding current (Ihold) in CA1 neurons from saline-treated rats. We and others previously showed Ihold to be mediated by extrasynaptic GABAARs, because during selective blockade of synaptic GABAAR currents by 1 μm gabazine, this tonic current is enhanced by various GABAAR agonists and blocked by 50 μm picrotoxin (Bai et al., 2001; Liang et al., 2004). In CA1 neurons, increases in Ihold were observed at 50 mm ETOH but became statistically significant only at 100 mm (Fig. 2). In contrast, the kinetics and frequency of mIPSCs were unaffected even by 100 mm EtOH (Fig. 2A,B).
Figure 2.

EtOH effects of on GABAAR-mediated currents in CA1 neurons are altered after CIE treatment. A, EtOH potentiates the tonic current (Ihold) in a CA1 neuron from a saline-treated rat. The kinetics of mIPSCs (top traces) averaged over the indicated 100 s periods during continuous recordings (bottom traces) are unaffected even by 100 mm EtOH. In a CA1 neuron from a CIE-treated rat (bottom traces), mIPSCs are sensitive to 10 mm EtOH. B, Summary graphs of total charge transfer of averaged mIPSCs (top graph) and tonic current (bottom graph) before and after EtOH application in saline- and CIE-treated rats. Each point represents a mean ± SEM value from six to eight neurons (2–3 rats per group). *p < 0.05 between saline and CIE groups; †p < 0.05 from pre-EtOH value (two-way repeated-measures ANOVA). Note that CIE treatment results in novel sensitivity of mIPSCs to EtOH and loss of tonic current sensitivity to EtOH.
CIE-induced switch of EtOH actions from extrasynaptic to synaptic GABAARs in CA neurons
The profile of EtOH action was quite different in CA1 neurons from CIE-treated rats. In these recordings, EtOH (10–100 mm) no longer had an effect on the tonic GABAAR current, whereas the mIPSCs became very sensitive to EtOH, such that the total charge transfer (area of averaged mIPSCs) was significantly increased from predrug condition by 10 mm EtOH. After application of 50 and 100 mm EtOH, the total charge transfer of mIPSCs surpassed that of mIPSCs from saline-treated rats (Fig. 2B). It is noteworthy that the total charge transfer of mIPSCs was significantly smaller in CIE rats without acute EtOH application, compared with saline controls. This was because of faster mIPSC τ1 and τ2 decay time constants, as described previously (Cagetti et al., 2003; Liang et al., 2004). The baseline mIPSC frequency was also significantly reduced from 12.1 ± 0.4 to 9.7 ± 0.4 Hz after CIE treatment and remained unaffected by acute EtOH application. In contrast, no differences were seen in the magnitude of Ihold between CA1 neurons from saline- and CIE-treated rats (Fig. 2B). The above results were both qualitatively and quantitatively similar to those obtained in slices from saline- and CIE-treated rats at 40 d of withdrawal (Fig. 2B), indicating long-term alterations.
Both synaptic and extrasynaptic GABAARs of dentate granule cells from control rats are sensitive to EtOH
Differences in subunit combinations of synaptic and extrasynaptic GABAARs are known to profoundly affect both the kinetics of GABAAR activation and their response to various GABAergic drugs (Olsen and Homanics, 2000; Whiting et al., 2000). Recent studies have suggested that certain subunit combinations, namely those GABAARs containing α4βδ subunits, are highly sensitive to EtOH and the partial GABA agonist 4,5,6,7 tetrahydroisoxazolo[5,4-c]pyridin-3-ol (THIP) (Brown et al., 2002; Sundstrom-Poromaa et al., 2002; Wallner et al., 2003; Hanchar et al., 2005). Increases in hippocampal α4 subunit levels were reported after chronic ethanol treatment (Mahmoudi et al., 1997; Matthews et al., 1998), and we recently used preferential benzodiazepine ligands [bretazenil and ethyl-8-azido-5,6-dihydro-5-methyl-6-oxo-4H-imidazo[1,5α][1,4]benzodiazepine-3-carboxylate (Ro15-4513)] at α4 subunit-containing GABAARs to provide electrophysiological support for these data in CA1 neurons (Cagetti et al., 2003; Liang et al., 2004). Because the dentate gyrus is particularly enriched in α4 and δ subunits (Pirker et al., 2000; Peng et al., 2002), we hypothesized that GABAARs of DG cells would exhibit greater sensitivity to EtOH than GABAARs of CA1 neurons. Recordings in slices from saline-treated rats revealed this to be the case. Thus, potentiation of both mIPSCs and Ihold was detectable at 10 mm EtOH and significantly potentiated at 50 mm EtOH in DG cells (Fig. 3A,B).
Figure 3.

Altered EtOH effects on synaptic and extrasynaptic GABAARs of DG cells after CIE treatment. A, Tonic GABAAR-mediated current (Ihold) is visibly potentiated by 50 mm EtOH in a DG cell from a saline-treated rat. Small potentiation of mIPSCs (increased decay time) is also observed (top traces). After CIE treatment, Ihold is no longer potentiated (bottom traces), whereas mIPSC potentiation by EtOH is more prominent. Note the faster decay of mIPSCs before EtOH application after CIE treatment. B, Summary graphs of total charge transfer of averaged mIPSCs (top graph) and tonic current (bottom graph) before and after EtOH application in saline- and CIE-treated rats. Each point represents a mean ± SEM value from 6–11 neurons (2–4 rats per group). *p < 0.05 between saline and CIE groups; †p < 0.05 from pre-EtOH value (two-way repeated-measures ANOVA). Note the proportionately greater mIPSC potentiation by EtOH and loss of the tonic current sensitivity to EtOH after CIE treatment.
CIE-induced alterations in EtOH sensitivity of extrasynaptic and synaptic GABAARs in DG cells
Analogous to the effects in CA1 neurons, EtOH (10–100 mm) no longer significantly potentiated the tonic GABA current in DG cells after CIE treatment (Fig. 3). Furthermore, the sensitivity of mIPSCs in DG cells to EtOH was increased, such that 10 mm EtOH application produced significant increases in total charge transfer of averaged mIPSCs (Fig. 3B). Under baseline conditions (without acute EtOH application), faster decay accounted for the significantly smaller total charge transfer of averaged mIPSCs in DG neurons from CIE rats, compared with saline controls. The baseline frequency of DG neuron mIPSCs appeared to be slightly reduced after CIE treatment; inclusion of additional baseline recordings revealed significantly smaller mIPSC frequency of CIE rats (11.0 ± 0.3 Hz; n = 23 neurons, 8 rats) compared with saline controls (12.4 ± 0.4 Hz; n = 19 neurons, 5 rats).
CIE treatment leads to enhanced mIPSC responsiveness to THIP, La3+, and Ro15-4513
To obtain further insight into the GABAAR subunit composition changes that may be present after CIE treatment, we first compared the effects of a partial GABAAR agonist THIP and the antagonist LaCl3 on mIPSCs and tonic currents in DG cells from saline- and CIE-treated rats. In recombinant receptor studies, these compounds were shown to have a particularly high affinity for the α4β3δ GABAAR subunit combination (Brown et al., 2002). Based on this, we hypothesized that similar to EtOH, both THIP and La3+, should have reduced effectiveness at extrasynaptic and increased effectiveness at synaptic GABAARs after CIE treatment. Application of THIP (1 μm) produced large increases in Ihold and a small but significant prolongation of the mIPSC decay time in DG cells from saline-treated rats. Subsequent addition of LaCl3 (100 μm) produced a significant reduction in the THIP-induced tonic current, without affecting mIPSC decay (Fig. 4A,B). In DG cells from CIE rats, THIP produced smaller increases in Ihold but proportionately greater potentiation of mIPSCs. However, subsequent addition of LaCl3 (100 μm) had no effect on Ihold but decreased the THIP-induced mIPSC potentiation.
Figure 4.

Altered synaptic and extrasynaptic sensitivity to THIP and LaCl3 after CIE treatment. A, α4β3δ-preferring agonist (THIP) and antagonist (La3+) preferentially affect DG cell tonic current in saline-treated rats and synaptic currents in CIE-treated rats, respectively. Note the loss of La3+ effect on Ihold and increased THIP effect on mIPSCs in CIE rats. B, Summary graphs of total charge transfer of averaged mIPSCs (top graph) and Ihold (bottom graph) before and after THIP and La3+ application in saline- and CIE-treated rats. Each point is a mean ± SEM value from six neurons (1 rat per group). *p < 0.05 between saline and CIE groups; †p < 0.05 from pre-THIP value (two-way repeated-measures ANOVA).
Next, we examined the effect of CIE treatment on responses to Ro15-4513, a partial inverse agonist at the benzodiazepine site of α1- and α2-containing GABAARs, which was also shown to bind with high affinity at α4-containing GABAARs (Knoflach et al., 1996). Importantly, Ro15-4513 has agonist activity at α4β3γ2 GABAARs but does not modulate α4β3δ GABAARs (Brown et al., 2002). In DG cells from saline-treated rats, Ro15-4513 (0.3 μm) slightly but significantly potentiated both mIPSCs and Ihold (Fig. 5A,B). After CIE treatment, mIPSC potentiation by Ro15-4513 was markedly increased, whereas Ihold was now inhibited by the drug.
Figure 5.

Altered synaptic and extrasynaptic sensitivity to Ro15-4513 after CIE treatment. A, The α4β3γ2-prefering partial inverse agonist Ro15-4513 potentiates both mIPSCs and tonic current in DG neurons from saline-treated rats. Note the greater potentiation of mIPSCs and a reduction in Ihold by Ro15-4513 after CIE treatment. B, Summary graphs of total charge transfer of averaged mIPSCs (top graph) and Ihold (bottom graph) before and after Ro15-4513 application in saline- and CIE-treated rats (points are mean ± SEM values from 5–6 neurons; 2–3 rats per group). *p < 0.05 between saline and CIE groups; †p < 0.05 from pre-Ro15-4513 value (two-way repeated-measures ANOVA).
α4 but not δ subunits assume a more central synaptic location after CIE treatment
To provide additional evidence in support of the hypothesis that GABAAR subunit alterations mediate the switch in responsiveness of synaptic and extrasynaptic GABAARs to EtOH after CIE treatment, we examined the subcellular location of α4 and δ subunits with electron microscopic immunogold labeling. In the dendrites of DG cells from saline-treated rats, the labeling for both α4 and δ subunits was found predominantly in regions immediately adjacent to or outside symmetric GABAergic synapses, as demonstrated previously for the δ subunit in mice (Wei et al., 2003). Quantitative analysis demonstrated a perisynaptic location of the α4 subunit in 93% of α4-labeled synapses (117 of 126 synaptic profiles; n = 3 rats) (Fig. 6A,C). However, in EM sections from CIE-treated rats, immunogold labeling of α4 subunits was detected predominantly near the center of postsynaptic densities of symmetric synapses (78% of α4-labeled synapses; 109 of 140 synaptic profiles; n = 3 rats) (Fig. 6B,D). Statistical analysis revealed significant (p < 0.001; ANOVA) increases in α4 subunit labeling at the center of symmetric synapses (Fig. 6E). Although the incidence of δ subunit immunogold labeling in CIE rats was quite low [consistent with decreased levels of the δ subunit in these animals (Cagetti et al., 2003)], labeling was detected predominantly at perisynaptic locations on DG cell dendrites from CIE rats (91% of δ-labeled synapses; 32 of 35 labeled profiles; n = 3) (Fig. 6F).
Figure 6.
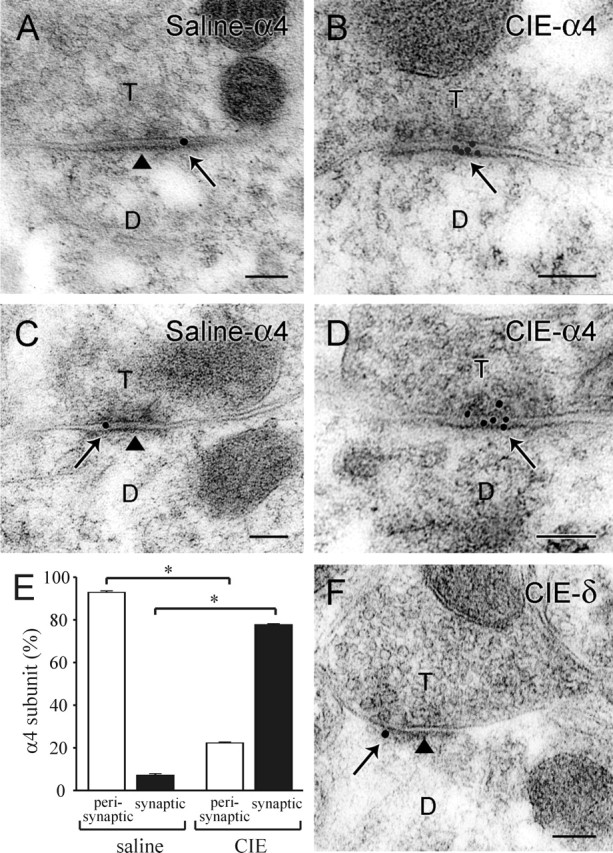
Postembedding immunogold labeling reveals a change in α4 but not in δ subunit location from perisynaptic to synaptic sites in the molecular layer of the dentate gyrus after CIE treatment. A, C, In saline-treated rats, colloidal gold labeling of the α4 subunit (arrows) was present on or near the plasma membrane of dendrites (D) that contacted axon terminals (T). Gold particles were found predominantly at the outer edges of symmetric synapses (arrows) but not at the center of these synapses (arrowheads). B, D, After CIE treatment, labeling for α4 subunits was found mainly in the center of symmetric synapses (arrows). E, Quantitative analysis showed that perisynaptic labeling was found at 93% of α4 subunit-labeled synapses (open bar) in saline-treated rats (n = 3 rats). In CIE-treated rats (n = 3), perisynaptic labeling was observed at 22% (open bar) of labeled synapses, but synaptic labeling was evident at 78% of labeled synapses (black bar). *p < 0.001 between saline and CIE groups. Error bars represent SEM. F, In contrast to the α4 subunit labeling (B, D), δ subunit labeling (arrow) in CIE-treated rats was present at perisynaptic locations but not within the synaptic contact (arrowhead). Scale bars, 0.1 μm.
δ Subunit is not required for synaptic GABAAR sensitivity to low [EtOH]
One implication of these findings is that sensitivity of synaptic GABAARs to low [EtOH] may not require the presence of the δ subunit. To test this, we compared EtOH sensitivity of synaptic and extrasynaptic GABAAR-mediated currents in DG cells from δ subunit null mice and their wild-type counterparts. Although the magnitude of the tonic current was similar between the two groups, its potentiation by EtOH was greatly reduced in δ −/− mice (Fig. 7). Consistent with our previous report (Spigelman et al., 2003), the total charge transfer of averaged mIPSCs from δ −/− mice was smaller compared with that of δ +/+ mice (Fig. 7). However, the EtOH potentiation of mIPSCs from δ −/− mice was actually increased compared with δ +/+ mice (Fig. 7).
Figure 7.

Decreased tonic current potentiation, but enhanced mIPSCs potentiation, by EtOH in δ subunit-deficient mice. Summary graphs of changes in total charge transfer of averaged mIPSCs (right) and tonic current Ihold (left) before and after EtOH application in δ +/+ and δ −/− DG cells are shown. Each point represents a mean ± SEM value from 6–12 neurons, two mice per group. *p < 0.05 between +/+ and −/− groups; †p < 0.05 from pre-EtOH value (two-way repeated-measures ANOVA). Note the smaller total charge transfer of averaged mIPSCs in DG cells from δ −/− mice. Also note that 10 mm EtOH significantly potentiates mIPSCs from δ −/− but not δ +/+ mice.
Discussion
Relationship between sleep-inducing effects of EtOH and tonic inhibitory currents
In this study, we demonstrate profound tolerance to the sleep-inducing but not anxiolytic effects of EtOH in rats after withdrawal from CIE treatment. Alcoholics also exhibit tolerance to the sleep-inducing effects of EtOH (Allen et al., 1977; Brower, 2001; Roehrs and Roth, 2001) as well as cross-tolerance to barbiturate and benzodiazepine sedative hypnotic drugs (Miller, 1995). Tolerance to the actions of EtOH and other GABAergic drugs in CIE rats has previously been related to alterations in the pharmacological sensitivity and subunit composition of GABAARs (Mahmoudi et al., 1997; Kang et al., 1998; Cagetti et al., 2003; Liang et al., 2004). In hippocampal homogenates from CIE rats, these changes include increases in α4 and γ2 and reductions in α1 and δ subunit protein levels (Cagetti et al., 2003). The mechanisms by which the relative abundance and localization of specific GABAAR subunits are altered by CIE treatment are not known. However, selective endocytosis and recycling of receptors based on subunit composition, distinct assembly signals, and sensitivity to protein kinase C phosphorylation may be involved (for review, see Brandon et al., 2000; Kumar et al., 2004). Tolerance to the sleep-inducing effects of EtOH in CIE rats coincides with tolerance to acute EtOH potentiation of tonic GABAAR currents. A similar relationship between decreased sleep time and decreased tonic current enhancement has been observed for alphaxalone and THIP in CIE rats (Cagetti et al., 2003; Liang et al., 2004). Together, these data suggest that the sleep-inducing effects of GABAergic drugs may be mediated primarily by potentiation of extrasynaptic GABAARs whose sensitivity for these drugs is decreased after CIE treatment. It has also been shown that the general anesthetics propofol and thiopental exert most of their inhibitory effects on intrinsic excitability of hippocampal neurons by potentiation of the tonic, and not phasic, GABAAR-mediated currents (Bieda and MacIver, 2004). Similarly, low concentrations of the volatile anesthetic isoflurane selectively enhance tonic GABAergic currents in CA1 neurons (Caraiscos et al., 2004), whereas the sedative hypnotic THIP selectively enhances tonic currents but not mIPSCs in CA1 neurons (Liang et al., 2004) and in ventrobasal thalamocortical neurons (Belelli et al., 2005; Cope et al., 2005), at concentrations that enhance slow-wave sleep activity (Belelli et al., 2005).
Relationship between altered mIPSC kinetics and hyperexcitability after CIE treatment
Consistent with our previous reports (Cagetti et al., 2003; Liang et al., 2004), CIE treatment results in decreased total charge transfer of mIPSCs in both CA1 and DG neurons, in the absence of significant changes in baseline tonic currents (Figs. 2–5). In addition, small but significant decreases in the frequency of mIPSCs are observed. Such decreases in synaptic inhibition may account for the decreased paired-pulse inhibition (Kang et al., 1996) and abnormal discharges (Veatch and Gonzalez, 1996) recorded in the CA1 region of the hippocampus after CIE treatment. Analogous decreases in synaptic inhibition of other affected circuits (e.g., amygdala) are likely to contribute to the hyperexcitability and anxiety exhibited by CIE rats.
Synaptic GABAAR subunit composition and EtOH sensitivity
This report provides pharmacological and electron microscopic evidence for increases in the α4- but not δ-containing GABAARs at synaptic locations. Because of the relatively low sensitivity of immunogold labeling methods (Somogyi et al., 1996), the current labeling presumably identified sites with the highest densities of each subunit. Therefore, our findings do not preclude the presence of low densities of either subunit at additional synaptic and extrasynaptic locations. With this caveat in mind, our results suggest that after CIE treatment there is a net “shift” of α4 subunits from perisynaptic to synaptic locations at GABAergic synapses, without concomitant changes in δ subunit localization. The α4βδ GABAARs are particularly sensitive to low [EtOH] (Sundstrom-Poromaa et al., 2002; Wallner et al., 2003; Hanchar et al., 2005), which under normal conditions, preferentially potentiates extrasynaptic receptors that mediate tonic inhibition (Wei et al., 2003, 2004). Recently, changes in the levels of δ subunit protein were linked to cyclic changes in tonic inhibition during the ovarian cycle of mice (Maguire et al., 2005). Analogous increases in α4β2δ subunit combinations were proposed to underlie the behavioral and physiological changes induced by withdrawal from progesterone treatment, a model of premenstrual syndrome (Smith et al., 1998; Sundstrom-Poromaa et al., 2002). In the CIE model of alcohol tolerance and dependence, there is a loss of tonic current potentiation by EtOH but a large increase in synaptic GABAAR responsiveness to EtOH. This is concomitant with a decrease in δ subunit protein levels (Cagetti et al., 2003), without electron microscopic evidence of a δ subunit shift to synaptic locations (Fig. 6). Therefore, our data suggest that CIE treatment and withdrawal lead to the formation of α4 subunit-containing synaptic GABAARs that do not possess the δ subunit. The enhanced EtOH sensitivity of mIPSCs in δ subunit knock-out mice provides further support to the notion that δ subunit presence is not an absolute requirement for sensitivity of GABAARs to low [EtOH]. The discriminative stimulus effects of EtOH are also preserved in δ −/− mice (Shannon et al., 2004).
Relationship of altered EtOH sensitivity of GABAARs to alcoholism
The development of alcohol dependence is thought to involve an incremental neuroadaptation to the presence of alcohol (Robinson and Berridge, 1993; Koob and Le Moal, 1997). In CIE rats, the development of alcohol dependence, as measured by the decrease in seizure threshold, is gradual, requiring multiple cycles of EtOH intoxication and withdrawal to produce long-lasting changes (Kokka et al., 1993). Recent studies have confirmed the existence of a temporal threshold of a persistent alcoholic state, measured as a long-lasting increase in voluntary EtOH consumption after intermittent intoxication and withdrawal (Rimondini et al., 2003). It was also shown that intermittent exposure to EtOH is much more effective than continuous EtOH exposure to produce enhanced EtOH self-administration after withdrawal (Rimondini et al., 2003; Spanagel, 2003; O’Dell et al., 2004).
On the basis of many studies in rodents, nonhuman primates, and humans, it has been proposed that the negative affective state produced by alcohol deprivation can contribute to craving and subsequent relapse behavior after re-exposure to alcohol (Higley et al., 1991; Anton, 1999; Breese et al., 2005). Considerable evidence implicates an imbalance between excitatory and inhibitory neurotransmission among mechanisms that lead to the development of the alcohol withdrawal-induced negative affective state (De Witte, 2004). This includes the demonstrated increases in release of glutamate in the hippocampus (Dahchour and De Witte, 2003), amygdala (Roberto et al., 2004), and nucleus accumbens (Dahchour and De Witte, 2000) after chronic EtOH treatment and withdrawal. Chronic continuous EtOH and/or CIE treatment with subsequent withdrawal also increase the binding levels and activation of NMDARs (Nie et al., 1994; Hu and Ticku, 1995; Hu and Ticku, 1997), presumably as a result of increased expression of various NMDAR subunits (Trevisan et al., 1994; Follesa and Ticku, 1995; Nelson et al., 2005; Roberto et al., 2006). The resultant increases in glutamatergic transmission are exacerbated by the decrements in synaptic GABAAR function (Cagetti et al., 2003), together contributing to the early withdrawal symptoms, as well as the long-term dependence and craving for alcohol.
Our demonstration of the persistent switch in EtOH actions from hippocampal extrasynaptic to synaptic GABAARs in CIE rats suggests a possible mechanism by which alcohol dependence may be maintained. In CIE rats, hyperexcitability and anxiety symptoms likely brought on by the demonstrated decreases in the function of synaptic GABAARs are effectively relieved by low doses of EtOH, despite tolerance to its sleep-inducing actions. We suggest that the CIE-induced switch of EtOH actions to synaptic GABAARs in the appropriate circuits may represent the physiological substrate of its anxiolytic effects, which in vivo leads to enhanced preference for alcohol consumption. The enhanced EtOH action at inhibitory synapses of CIE rats is likely to be further accentuated by the increased EtOH responsiveness at excitatory synapses. Acute EtOH is well known to inhibit NMDARs (Hoffman et al., 1989; Lovinger et al., 1989). After chronic EtOH treatment and withdrawal, the responsiveness of NMDAR-mediated synaptic currents to acute EtOH challenges is increased (Nelson et al., 2005; Roberto et al., 2006). This dual increase in the effectiveness of EtOH at excitatory and inhibitory synapses should have potent physiological effects.
Although the hippocampus plays a role in anxiolysis (Gray, 1982; Ferreira et al., 1999), we view it as a model circuit for studying alcohol-induced alterations. Clearly, future studies will need to determine whether mechanisms analogous to those described here for hippocampal synapses occur in other brain areas, such as amygdala, nucleus accumbens, and prefrontal cortex, which are thought to play key roles in the modulation of stress and drug-seeking behavior (Koob and Le Moal, 1997; Nestler, 2001; Stevenson and Gratton, 2003).
In summary, our data provide an explanation for the apparent discrepancies between studies that demonstrated an absence of tolerance to acute EtOH potentiation of evoked inhibitory synaptic potentials in brain slice recordings (Kang et al., 1998; Signore and Yeh, 2000) and the marked tolerance to the sedative–hypnotic (Khanna et al., 1991), but not anxiolytic (Fig. 1), effects of acute EtOH after chronic EtOH treatment. It also underscores the importance of subunit composition of native receptors in determining the contribution of synaptic or extrasynaptic GABAARs to EtOH actions.
Footnotes
This work was supported by National Institutes of Health Grants AA07680, NS35985, and NS051311. We thank Drs. Gregg Homanics and Istvan Mody for providing the δ subunit −/− mice, Werner Sieghart for the δ subunit antibody, and Thomas Otis for helpful discussions and comments on this manuscript.
References
- Allan AM, Harris RA (1986). Gamma-aminobutyric acid and alcohol actions: neurochemical studies of long sleep and short sleep mice. Life Sci 39:2005–2015. [DOI] [PubMed] [Google Scholar]
- Allen RP, Wagman AM, Funderburk FR (1977). Slow wave sleep changes: alcohol tolerance and treatment implications. Adv Exp Med Biol 85A:629–640. [DOI] [PubMed] [Google Scholar]
- Anton RF (1999). What is craving? Models and implications for treatment. Alcohol Res Health 23:165–173. [PMC free article] [PubMed] [Google Scholar]
- Ariwodola OJ, Crowder TL, Grant KA, Daunais JB, Friedman DP, Weiner JL (2003). Ethanol modulation of excitatory and inhibitory synaptic transmission in rat and monkey dentate granule neurons. Alcohol Clin Exp Res 27:1632–1639. [DOI] [PubMed] [Google Scholar]
- Aroor AR, Shukla SD (2004). MAP kinase signaling in diverse effects of ethanol. Life Sci 74:2339–2364. [DOI] [PubMed] [Google Scholar]
- Bai D, Zhu G, Pennefather P, Jackson MF, MacDonald JF, Orser BA (2001). Distinct functional and pharmacological properties of tonic and quantal inhibitory postsynaptic currents mediated by γ-aminobutyric acidA receptors in hippocampal neurons. Mol Pharmacol 59:814–824. [DOI] [PubMed] [Google Scholar]
- Becker HC, Hale RL (1993). Repeated episodes of ethanol withdrawal potentiate the severity of subsequent withdrawal seizures: an animal model of alcohol withdrawal “kindling”. Alcohol Clin Exp Res 17:94–98. [DOI] [PubMed] [Google Scholar]
- Becker HC, Veatch LM, Diaz-Granados JL (1998). Repeated ethanol withdrawal experience selectively alters sensitivity to different chemoconvulsant drugs in mice. Psychopharmacology (Berl) 139:145–153. [DOI] [PubMed] [Google Scholar]
- Belelli D, Peden DR, Rosahl TW, Wafford KA, Lambert JJ (2005). Extrasynaptic GABAA receptors of thalamocortical neurons: a molecular target for hypnotics. J Neurosci 25:11513–11520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bieda MC, MacIver MB (2004). Major role for tonic GABAA conductances in anesthetic suppression of intrinsic neuronal excitability. J Neurophysiol 92:1658–1667. [DOI] [PubMed] [Google Scholar]
- Booth BM, Blow FC (1993). The kindling hypothesis: further evidence from a U.S. national study of alcoholic men. Alcohol Alcohol 28:593–598. [PubMed] [Google Scholar]
- Brandon NJ, Smart TG, Moss SJ (2000). Regulation of GABAA receptors by protein phosphorylation. In: GABA in the nervous system: the view at fifty years (Martin DL, Olsen RW, eds) , pp. 191–206. Philadelphia: Lippincott Williams and Wilkins.
- Breese GR, Overstreet DH, Knapp DJ (2005). Conceptual framework for the etiology of alcoholism: a “kindling”/stress hypothesis. Psychopharmacology (Berl) 178:367–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brower KJ (2001). Alcohol’s effects on sleep in alcoholics. Alcohol Res Health 25:110–125. [PMC free article] [PubMed] [Google Scholar]
- Brown ME, Anton RF, Malcolm R, Ballenger JC (1988). Alcohol detoxification and withdrawal seizures: clinical support for a kindling hypothesis. Biol Psychiatry 23:507–514. [DOI] [PubMed] [Google Scholar]
- Brown N, Kerby J, Bonnert TP, Whiting PJ, Wafford KA (2002). Pharmacological characterization of a novel cell line expressing human α4β3δ GABAA receptors. Br J Pharmacol 136:965–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cagetti E, Liang J, Spigelman I, Olsen RW (2003). Withdrawal from chronic intermittent ethanol treatment changes subunit composition, reduces synaptic function, and decreases behavioral responses to positive allosteric modulators of GABAA receptors. Mol Pharmacol 63:53–64. [DOI] [PubMed] [Google Scholar]
- Caraiscos VB, Newell JG, You T, Elliott EM, Rosahl TW, Wafford KA, MacDonald JF, Orser BA (2004). Selective enhancement of tonic GABAergic inhibition in murine hippocampal neurons by low concentrations of the volatile anesthetic isoflurane. J Neurosci 24:8454–8458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cope DW, Hughes SW, Crunelli V (2005). GABAA receptor-mediated tonic inhibition in thalamic neurons. J Neurosci 25:11553–11563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahchour A, De Witte P (2000). Taurine blocks the glutamate increase in the nucleus accumbens microdialysate of ethanol-dependent rats. Pharmacol Biochem Behav 65:345–350. [DOI] [PubMed] [Google Scholar]
- Dahchour A, De Witte P (2003). Excitatory and inhibitory amino acid changes during repeated episodes of ethanol withdrawal: an in vivo microdialysis study. Eur J Pharmacol 459:171–178. [DOI] [PubMed] [Google Scholar]
- De Witte P (2004). Imbalance between neuroexcitatory and neuroinhibitory amino acids causes craving for alcohol. Addict Behav 29:1325–1339. [DOI] [PubMed] [Google Scholar]
- Ferreira VM, Valenzuela CF, Morato GS (1999). Role of nitric oxide-dependent pathways in ethanol-induced anxiolytic effects in rats. Alcohol Clin Exp Res 23:1898–1904. [PubMed] [Google Scholar]
- Follesa P, Ticku MK (1995). Chronic ethanol treatment differentially regulates NMDA receptor subunit mRNA expression in rat brain. Brain Res Mol Brain Res 29:99–106. [DOI] [PubMed] [Google Scholar]
- Franks NP, Lieb WR (1987). Are the biological effects of ethanol due to primary interactions with lipids or with proteins? Alcohol Alcohol Suppl 1:139–145. [PubMed] [Google Scholar]
- Gray JA (1982). In: The neuropsychology of anxiety Oxford: Oxford UP.
- Hanchar HJ, Dodson PD, Olsen RW, Otis TS, Wallner M (2005). Alcohol-induced motor impairment caused by increased extrasynaptic GABAA receptor activity. Nat Neurosci 8:339–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris RA (1999). Ethanol actions on multiple ion channels: which are important? Alcohol Clin Exp Res 23:1563–1570. [PubMed] [Google Scholar]
- Higley JD, Hasert MF, Suomi SJ, Linnoila M (1991). Nonhuman primate model of alcohol abuse: effects of early experience, personality, and stress on alcohol consumption. Proc Natl Acad Sci USA 88:7261–7265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman PL, Rabe CS, Moses F, Tabakoff B (1989). N-methyl-d-aspartate receptors and ethanol: inhibition of calcium flux and cyclic GMP production. J Neurochem 52:1937–1940. [DOI] [PubMed] [Google Scholar]
- Hu XJ, Ticku MK (1995). Chronic ethanol treatment upregulates the NMDA receptor function and binding in mammalian cortical neurons. Brain Res Mol Brain Res 30:347–356. [DOI] [PubMed] [Google Scholar]
- Hu XJ, Ticku MK (1997). Functional characterization of a kindling-like model of ethanol withdrawal in cortical cultured neurons after chronic intermittent ethanol exposure. Brain Res 767:228–234. [DOI] [PubMed] [Google Scholar]
- Kang MH, Spigelman I, Sapp DW, Olsen RW (1996). Persistent reduction of GABAA receptor-mediated inhibition in rat hippocampus after chronic intermittent ethanol treatment. Brain Res 709:221–228. [DOI] [PubMed] [Google Scholar]
- Kang MH, Spigelman I, Olsen RW (1998). Alterations in the sensitivity of GABAA receptors to allosteric modulatory drugs in rat hippocampus following chronic intermittent ethanol treatment. Alcohol Clin Exp Res 22:2165–2173. [PubMed] [Google Scholar]
- Khanna JM, Kalant H, Shah G, Chau A (1991). Tolerance to ethanol and cross-tolerance to pentobarbital and barbital in four rat strains. Pharmacol Biochem Behav 39:705–709. [DOI] [PubMed] [Google Scholar]
- Knoflach F, Benke D, Wang Y, Scheurer L, Luddens H, Hamilton BJ, Carter DB, Mohler H, Benson JA (1996). Pharmacological modulation of the diazepam-insensitive recombinant γ-aminobutyric acidA receptors α4β2γ2 and α6β2γ2. Mol Pharmacol 50:1253–1261. [PubMed] [Google Scholar]
- Kokka N, Sapp DW, Taylor AM, Olsen RW (1993). The kindling model of alcohol dependence: similar persistent reduction in seizure threshold to pentylenetetrazol in animals receiving chronic ethanol or chronic pentylenetetrazol. Alcohol Clin Exp Res 17:525–531. [DOI] [PubMed] [Google Scholar]
- Koob GF, Le Moal M (1997). Drug abuse: hedonic homeostatic dysregulation. Science 278:52–58. [DOI] [PubMed] [Google Scholar]
- Kumar S, Fleming RL, Morrow AL (2004). Ethanol regulation of γ-aminobutyric acidA receptors: genomic and nongenomic mechanisms. Pharmacol Ther 101:211–226. [DOI] [PubMed] [Google Scholar]
- Liang J, Cagetti E, Olsen RW, Spigelman I (2004). Altered pharmacology of synaptic and extrasynaptic GABAA receptors on hippocampal CA1 pyramidal neurons is consistent with subunit changes in a model of alcohol withdrawal and dependence. J Pharmacol Exp Ther 310:1234–1245. [DOI] [PubMed] [Google Scholar]
- Lovinger DM (1997). Alcohols and neurotransmitter gated ion channels: past, present and future. Naunyn Schmiedebergs Arch Pharmacol 356:267–282. [DOI] [PubMed] [Google Scholar]
- Lovinger DM, White G, Weight FF (1989). Ethanol inhibits NMDA-activated ion current in hippocampal neurons. Science 243:1721–1724. [DOI] [PubMed] [Google Scholar]
- Maguire JL, Stell BM, Rafizadeh M, Mody I (2005). Ovarian cycle-linked changes in GABAA receptors mediating tonic inhibition alter seizure susceptibility and anxiety. Nat Neurosci 8:797–804. [DOI] [PubMed] [Google Scholar]
- Mahmoudi M, Kang MH, Tillakaratne N, Tobin AJ, Olsen RW (1997). Chronic intermittent ethanol treatment in rats increases GABAA receptor α4-subunit expression: possible relevance to alcohol dependence. J Neurochem 68:2485–2492. [DOI] [PubMed] [Google Scholar]
- Matthews DB, Devaud LL, Fritschy JM, Sieghart W, Morrow AL (1998). Differential regulation of GABAA receptor gene expression by ethanol in the rat hippocampus versus cerebral cortex. J Neurochem 70:1160–1166. [DOI] [PubMed] [Google Scholar]
- McCown TJ, Breese GR (1990). Multiple withdrawals from chronic ethanol “kindles” inferior collicular seizure activity: evidence for kindling of seizures associated with alcoholism. Alcohol Clin Exp Res 14:394–399. [DOI] [PubMed] [Google Scholar]
- McGinnis JM, Foege WH (1999). Mortal and morbidity attributable to use of addictive substances in the United States. Proc Assoc Am Physicians 111:109–118. [DOI] [PubMed] [Google Scholar]
- Mehta AK, Ticku MK (1988). Ethanol potentiation of GABAergic transmission in cultured spinal cord neurons involves γ-aminobutyric acidA-gated chloride channels. J Pharmacol Exp Ther 246:558–564. [PubMed] [Google Scholar]
- Mhatre MC, Pena G, Sieghart W, Ticku MK (1993). Antibodies specific for GABAA receptor α subunits reveal that chronic alcohol treatment down-regulates α-subunit expression in rat brain regions. J Neurochem 61:1620–1625. [DOI] [PubMed] [Google Scholar]
- Mihalek RM, Banerjee PK, Korpi ER, Quinlan JJ, Firestone LL, Mi ZP, Lagenaur C, Tretter V, Sieghart W, Anagnostaras SG, Sage JR, Fanselow MS, Guidotti A, Spigelman I, Li Z, DeLorey TM, Olsen RW, Homanics GE (1999). Attenuated sensitivity to neuroactive steroids in γ-aminobutyrate type A receptor delta subunit knockout mice. Proc Natl Acad Sci USA 96:12905–12910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller NS (1995). Pharmacotherapy in alcoholism. J Addict Dis 14:23–46. [DOI] [PubMed] [Google Scholar]
- Morrow AL, Suzdak PD, Karanian JW, Paul SM (1988). Chronic ethanol administration alters γ-aminobutyric acid, pentobarbital and ethanol-mediated 36Cl− uptake in cerebral cortical synaptoneurosomes. J Pharmacol Exp Ther 246:158–164. [PubMed] [Google Scholar]
- Nelson TE, Ur CL, Gruol DL (2005). Chronic intermittent ethanol exposure enhances NMDA-receptor-mediated synaptic responses and NMDA receptor expression in hippocampal CA1 region. Brain Res 1048:69–79. [DOI] [PubMed] [Google Scholar]
- Nestler EJ (2001). Molecular basis of long-term plasticity underlying addiction. Nat Rev Neurosci 2:119–128. [DOI] [PubMed] [Google Scholar]
- Nie Z, Madamba SG, Siggins GR (1994). Ethanol inhibits glutamatergic neurotransmission in nucleus accumbens neurons by multiple mechanisms. J Pharmacol Exp Ther 271:1566–1573. [PubMed] [Google Scholar]
- Nusser Z, Sieghart W, Somogyi P (1998). Segregation of different GABAA receptors to synaptic and extrasynaptic membranes of cerebellar granule cells. J Neurosci 18:1693–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Dell LE, Roberts AJ, Smith RT, Koob GF (2004). Enhanced alcohol self-administration after intermittent versus continuous alcohol vapor exposure. Alcohol Clin Exp Res 28:1676–1682. [DOI] [PubMed] [Google Scholar]
- Olsen RW, Homanics GE (2000). Function of GABAA receptors. In: GABA in the nervous system: the view at fifty years (Martin DL, Olsen RW, eds) , pp. 81–96. Philadelphia: Lippincott Williams and Wilkins.
- Peng Z, Hauer B, Mihalek RM, Homanics GE, Sieghart W, Olsen RW, Houser CR (2002). GABAA receptor subunit changes in δ subunit-deficient mice: altered expression of α4 and γ2 subunits in the forebrain. J Comp Neurol 446:179–197. [DOI] [PubMed] [Google Scholar]
- Pirker S, Schwarzer C, Wieselthaler A, Sieghart W, Sperk G (2000). GABAA receptors: immunocytochemical distribution of 13 subunits in the adult rat brain. Neuroscience 101:815–850. [DOI] [PubMed] [Google Scholar]
- Rimondini R, Sommer W, Heilig M (2003). A temporal threshold for induction of persistent alcohol preference: behavioral evidence in a rat model of intermittent intoxication. J Stud Alcohol 64:445–449. [DOI] [PubMed] [Google Scholar]
- Roberto M, Schweitzer P, Madamba SG, Stouffer DG, Parsons LH, Siggins GR (2004). Acute and chronic ethanol alter glutamatergic transmission in rat central amygdala: an in vitro and in vivo analysis. J Neurosci 24:1594–1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberto M, Bajo M, Crawford E, Madamba SG, Siggins GR (2006). Chronic ethanol exposure and protracted abstinence alter NMDA receptors in central amygdala. Neuropsychopharmacology in press. [DOI] [PubMed]
- Robinson TE, Berridge KC (1993). The neural basis of drug craving: an incentive-sensitization theory of addiction. Brain Res Brain Res Rev 18:247–291. [DOI] [PubMed] [Google Scholar]
- Roehrs T, Roth T (2001). Sleep, sleepiness, sleep disorders and alcohol use and abuse. Sleep Med Rev 5:287–297. [DOI] [PubMed] [Google Scholar]
- Shannon EE, Shelton KL, Vivian JA, Yount I, Morgan AR, Homanics GE, Grant KA (2004). Discriminative stimulus effects of ethanol in mice lacking the γ-aminobutyric acid type A receptor δ subunit. Alcohol Clin Exp Res 28:906–913. [DOI] [PubMed] [Google Scholar]
- Signore AP, Yeh HH (2000). Chronic exposure to ethanol alters GABAA receptor-mediated responses of layer II pyramidal cells in adult rat piriform cortex. J Neurophysiol 84:247–254. [DOI] [PubMed] [Google Scholar]
- Smith SS, Gong QH, Hsu FC, Markowitz RS, ffrench-Mullen JM, Li X (1998). GABAA receptor α4 subunit suppression prevents withdrawal properties of an endogenous steroid. Nature 392:926–930. [DOI] [PubMed] [Google Scholar]
- Somogyi P, Fritschy JM, Benke D, Roberts JD, Sieghart W (1996). The γ2 subunit of the GABAA receptor is concentrated in synaptic junctions containing the α 1 and β2/3 subunits in hippocampus, cerebellum and globus pallidus. Neuropharmacology 35:1425–1444. [DOI] [PubMed] [Google Scholar]
- Spanagel R (2003). Alcohol addiction research: from animal models to clinics. Best Pract Res Clin Gastroenterol 17:507–518. [DOI] [PubMed] [Google Scholar]
- Spigelman I, Li Z, Liang J, Cagetti E, Samzadeh S, Mihalek RM, Homanics GE, Olsen RW (2003). Reduced inhibition and sensitivity to neurosteroids in hippocampus of mice lacking the GABAA receptor δ subunit. J Neurophysiol 90:903–910. [DOI] [PubMed] [Google Scholar]
- Stevenson CW, Gratton A (2003). Basolateral amygdala modulation of the nucleus accumbens dopamine response to stress: role of the medial prefrontal cortex. Eur J Neurosci 17:1287–1295. [DOI] [PubMed] [Google Scholar]
- Sundstrom-Poromaa I, Smith DH, Gong QH, Sabado TN, Li X, Light A, Wiedmann M, Williams K, Smith SS (2002). Hormonally regulated α4β2δ GABAA receptors are a target for alcohol. Nat Neurosci 5:721–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trevisan L, Fitzgerald LW, Brose N, Gasic GP, Heinemann SF, Duman RS, Nestler EJ (1994). Chronic ingestion of ethanol up-regulates NMDAR1 receptor subunit immunoreactivity in rat hippocampus. J Neurochem 62:1635–1638. [DOI] [PubMed] [Google Scholar]
- Veatch LM, Gonzalez LP (1996). Repeated ethanol withdrawal produces site-dependent increases in EEG spiking. Alcohol Clin Exp Res 20:262–267. [DOI] [PubMed] [Google Scholar]
- Wallner M, Hanchar HJ, Olsen RW (2003). Ethanol enhances α4β3δ and α6β3δ γ-aminobutyric acid type A receptors at low concentrations known to affect humans. Proc Natl Acad Sci USA 100:15218–15223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei W, Zhang N, Peng Z, Houser CR, Mody I (2003). Perisynaptic localization of δ subunit-containing GABAA receptors and their activation by GABA spillover in the mouse dentate gyrus. J Neurosci 23:10650–10661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei W, Faria LC, Mody I (2004). Low ethanol concentrations selectively augment the tonic inhibition mediated by δ subunit-containing GABAA receptors in hippocampal neurons. J Neurosci 24:8379–8382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner JL, Zhang L, Carlen PL (1994). Potentiation of GABAA-mediated synaptic current by ethanol in hippocampal CA1 neurons: possible role of protein kinase C. J Pharmacol Exp Ther 268:1388–1395. [PubMed] [Google Scholar]
- Whiting PJ, Wafford KA, McKernan RM (2000). Pharmacologic subtypes of GABAA receptors based on subunit composition. In: GABA in the nervous system: the view at fifty years (Martin DL, Olsen RW, eds) , pp. 113–126. Philadelphia: Lippincott Williams and Wilkins.