

Abstract
NMDA-type glutamate receptors (NMDARs) contribute to many forms of long-term potentiation (LTP) and long-term depression (LTD). NMDARs are heteromers containing calcium-permeating neuronal receptor 1 (NR1) subunits and a variety of NR2 subunits. Evidence suggests that, in the CA1 region of the hippocampus, NR2A-containing NMDARs promote LTP whereas NR2B-containing receptors promote LTD. However, the calcium sensors that distinguish between these signals to promote the appropriate form of synaptic plasticity are not known. Ras-guanine nucleotide-releasing factor 1 (Ras-GRF1) and Ras-GRF2 are highly similar calcium-stimulated exchange factors that activate Ras and Rac GTPases. Here, using a set of Ras-GRF knock-out mice, we show that Ras-GRF2 contributes predominantly to the induction of NMDAR-dependent LTP, whereas Ras-GRF1 contributes predominantly to the induction of NMDAR-dependent LTD in the CA1 region of the hippocampus of postpubescent mice (postnatal days 25–36). In contrast, neither Ras-GRF protein influences synaptic plasticity in prepubescent mice (postnatal days 14–18). Ras-GRF2 mediates signaling from (R)-[(S)-1-(4-bromo-phenyl)-ethylamino]-(2,3-dioxo-1,2,3,4-tetrahydroquinoxalin-5-yl)-methyl-phosphonic acid-sensitive (NVP-AAM077-sensitive) (NR2A-containing) NMDARs to the Ras effector extracellular signal-related protein kinase 1/2 (Erk1/2) mitogen-activated protein (MAP) kinase, a promoter of NMDAR-induced LTP at this site. In contrast, Ras-GRF1 mediates signaling from ifenprodil-sensitive (NR2B-containing) NMDARs to the Rac effector p38 MAP kinase, a promoter of LTD. These findings show that, despite their similar functional domain organization, Ras-GRF1 and Ras-GRF2 mediate opposing forms of synaptic plasticity by coupling different classes of NMDARs to distinct MAP kinase pathways. Moreover, the postnatal appearance of Ras-GRF-dependent LTP and LTD coincides with the emergence of hippocampal-dependent behavior, implying that Ras-GRF proteins contribute to forms of synaptic plasticity that are required specifically for mature hippocampal function.
Keywords: Ras-GRF1, Ras-GRF2, LTP, LTD, synaptic plasticity, Ras, NMDA receptors
Introduction
Long-term potentiation (LTP) and long-term depression (LTD) are forms of synaptic plasticity that are thought to contribute to information storage in the brain. Recent data indicate that the mechanisms underlying these processes vary, depending on the synapse and circuit in which they function, the stage of brain development, and the nature of the inducing stimulus. Nevertheless, many forms of both LTP and LTD involve calcium influx via NMDA-type glutamate receptors (NMDARs) (Malenka and Bear, 2004).
NMDAR-dependent LTP and LTD have been studied most intensively at the CA1 region of the hippocampus. At this site NMDARs promote both LTP and LTD in part via a signaling cascades that involves activation of Ras family GTPases, which then activates mitogen-activated protein (MAP) kinase family members. For example, NMDAR activation of the Ras GTPase/extracellular signal-related protein kinase (Erk) MAP signaling cascade can promote both early and late forms of NMDAR-dependent LTP (Thomas and Huganir, 2004), whereas NMDAR activation of the Rap GTPase/p38 MAP kinase signaling cascade can promote some forms of NMDAR-dependent LTD (Bolshakov et al., 2000; Zhu et al., 2002).
A missing link in our understanding of NMDAR regulation of synaptic plasticity is the identification of specific calcium sensors that transmit signals from NMDARs to the induction of either LTP or LTD. Also missing is the identification of specific guanine nucleotide exchange factors (GEFs) that couple calcium influx via NMDARs to activation of the appropriate GTPases. Ras-guanine nucleotide-releasing factor (Ras-GRF) proteins are candidate mediators of both of these functions because both p140 Ras-GRF1 (Shou et al., 1992) and the closely related p130 Ras-GRF2 (Fam et al., 1997) are activated by calcium via calcium/calmodulin binding to their N-terminal IQ motifs (Shou et al., 1992; Farnsworth et al., 1995; Fam et al., 1997). They are also present predominantly in the CNS, where they are expressed in many neuronal cell types, but not in glial cells (Zippel et al., 1997). Both Ras-GRF1 and Ras-GRF2 contain two GEF domains, a C-terminal cell division cycle 25 (CDC25) domain that activates the Ras/Erk signaling cascade (Shou et al., 1992; Fam et al., 1997) and a more N-terminal dbl homology (DH) domain that activates a Rac/p38 cascade (Fan et al., 1998; Innocenti et al., 1999; Kiyono et al., 1999; Buchsbaum et al., 2002).
Using Ras-GRF knock-out mice, we previously revealed that both Ras-GRF proteins contribute to NMDAR activation of Ras/Erk and cAMP response element-binding protein (CREB) signaling in the cortex of mature mice, and both play a protective role in stroke-induced neurotoxicity (Tian et al., 2004). Moreover, Ras-GRF1 binds directly to the neuronal receptor 2B (NR2B) subunit of NMDARs (Krapivinsky et al., 2003). Interestingly, the contribution of Ras-GRFs to NMDAR function is regulated developmentally, because the Sos-GEFs, not Ras-GRFs, mediate this NMDAR activation of Erk in immature animals (Tian et al., 2004).
Here we show that Ras-GRF1 and Ras-GRF2 play opposing roles in the generation of synaptic plasticity specifically in postpubescent mice. Ras-GRF2 contributes primarily to the induction of LTP, whereas Ras-GRF1 contributes predominantly to LTD, consistent with the observation that Ras-GRF2 mediates NMDAR activation of Erk MAP kinase via NR2A subunit-containing NMDARs and Ras-GRF1 mediates p38 MAP kinase activation via NR2B-containing NMDARs.
Materials and Methods
Hippocampal slice preparation.
The generation of ras-grf1(−/−), ras-grf2(−/−), and double ras-grf1/ras-grf2(−/−) mice was described previously (Tian et al., 2004). ras-grf1 mice were bred in the C57BL/6J genetic background while ras-grf2(−/−) and double ras-grf1/ras-grf2(−/−) mice were maintained on a mixed C57BL/6x129 genetic background. Genetically matched wild-type mice (B6129 F2/J; Charles River Laboratories, Wilmington, MA) were bred for controls for each experiment. Mutant and control mice (14–18 or 26–35 d old) were killed after asphyxiation with CO2. The brain was removed quickly and submerged in ice-cold oxygenated sucrose-replaced artificial CSF (ACSF) cutting solution [containing the following (in mm): 206 sucrose, 2 KCl, 2 MgSO4, 1.25 NaH2PO4, 1 CaCl2, 1 MgCl2, 26 NaHCO3, 10 d-glucose, pH 7.4, 315 mOsm]. Transverse slices (350 μm thickness) from the middle portion of each hippocampus were cut with a vibroslicer in a 2% agar block. After dissection the slices were incubated in ACSF that contained the following (in mm): 124 NaCl, 2 KCl, 2 MgSO4, 1.25 NaH2PO4, 2 CaCl2, 10 d-glucose, and 26 NaHCO3, pH 7.4, 310 mOsm, in which they were allowed to recover for 60–90 min before recording. Then a single slice was transferred to the recording chamber and submerged beneath a continuously perfusing ACSF that had been saturated with 95% O2/5% CO2. Slices were incubated in the recording chamber for 20 min before stimulation. All animal procedures were performed according to protocols approved by the Tufts–New England Medical Center Institutional Animal Care and Use Committee, with approval on May 10, 2005, protocol 85-04.
Electrophysiological recordings.
To record field EPSPs (fEPSPs) in the CA1 region of the hippocampus, we used standard procedures. A unipolar stimulating electrode (World Precision Instruments, Sarasota, FL) was placed in the Schaffer collaterals to deliver test and conditioning stimuli. A borosilicate glass recording electrode filled with ACSF was positioned in stratum radiatum of CA1, 250–350 μm from the stimulating electrode. Test stimuli were applied at low frequency (0.05 Hz) at a stimulus intensity that elicited a fEPSP amplitude that was 40–50% of maximum, and the test responses were recorded for 20–30 min before the experiment was begun to ensure stability of the response. To induce LTP, we used three protocols in this study: (1) two consecutive trains (1 s) of stimuli at 100 Hz separated by 20 s; (2) theta burst stimulation (TBS), which consisted of 15 bursts of four pulses at 100 Hz delivered at an interburst interval of 200 ms; (3) four trains of stimuli (100 Hz for 1 s), delivered 5 min apart, which induced late-phase LTP. To induce LTD, we delivered 900 pulses at 1 Hz. The field potentials were amplified 100×, using an Axoprobe 1A. The data were sampled at 10 kHz and filtered at 2 kHz. Traces were analyzed by using the LTP Program (Anderson and Collingridge, 2001).
Data analysis.
The fEPSP magnitude was measured by using the initial fEPSP slope; three consecutive responses (1 min) were averaged and normalized to the mean value recorded between 20 and ∼30 min before the conditioning stimulus. Data were pooled across animals of the same age and genotype and are presented as the mean ± SEM. Values expressed here represent 60 min time points after the conditioning stimulus unless stated otherwise. Paired pulse responses were monitored at baseline stimulation strengths at 20, 40, 60, 100, 200, and 400 ms interstimulus intervals. The facilitation ratio was calculated as EPSP2 slope/EPSP1 slope. The following statistical analysis was performed: the same time window samples of the control and mutant mice or drug administrations were compared by using paired two-tailed Student’s t test. The difference between the effects of different groups was tested by one-way ANOVA. An effect was considered significant if p < 0.05.
Hippocampal brain slice preparation for biochemical studies and immunoblotting.
Brains were removed within 1 min of decapitation and submerged immediately in ice-cold Krebs–Ringer solution [containing the following (in mm): 11.1 glucose, 1.1 MgCl2, 1 Na2HPO4, 1.3 CaCl2, 25 NaHCO3, 120 NaCl, 4.7 KCl] presaturated with 95% O2/5% CO2 at 25°C for hippocampal dissection. Slices (300 μm, transverse) were prepared by using a vibratome and an agar backing (2%), and each slice then was transferred into six-well plates (two to four slices per well). The brain slices were incubated 60 min before pharmacological treatment to prevent initial neuronal firing caused by the slicing procedure. For the protein activation experiments 100 μm NMDA (Sigma–Aldrich, St. Louis, MO) was applied in each experiment at different times, as indicated. At the end of the experiment the slices were removed rapidly from the wells and lysed immediately in ice-cold buffer containing the following (in mm): 10 Tris-HCl, pH 7.5, 1 EDTA, 150 NaCl, 1 PMSF, and 1 NaVO3 plus 1% NP-40, 0.1% SDS, 10 mg/ml aprotinin, and 10 mg/ml leupeptin. Insoluble material was removed by centrifugation (13,000 × g for 20 min at 4°C). Nucleus-free supernatants were subjected to SDS-PAGE and immunoblotting with activation-specific phospho-Erk or p38 antibodies (Cell Signaling Technology, Beverly, MA). Then the samples were normalized by total Erk or total p38 antibodies (Cell Signaling Technology), as indicated. Digital images of blots were produced by densitometric scans of autoradiographs and quantified by using NIH Image 1.63 software working within the linear range of the readouts.
Results
Ras-GRF proteins are calcium-regulated GEFs for Ras and Rac GTPases. Because we showed that both p140 Ras-GRF1 and p130 Ras-GRF2 contribute to the ability of NMDA glutamate receptors to activate Ras and Erk MAP kinase in mature mice (Tian et al., 2004), an investigation into the role of the Ras-GRF family of GEFs in NMDAR-dependent synaptic plasticity was performed first on double knock-out mice [grf1/grf2(−/−)] and then on single grf1(−/−) and single grf2(−/−) knock-out mice to search for isoform-specific functions.
Normal basal synaptic function in double Ras-GRF1/Ras-GRF2 knock-out mice
Basal synaptic transmission was measured in the CA1 region of the hippocampus from double ras-grf1/ras-grf2 knock-out mice and strain-matched wild-type control mice. Brain slices from postnatal days 26–35 (P26–P35) were used, because we previously showed that NMDARs use Ras-GRF proteins to activate Erk MAP kinase only after ∼P20. A stimulus–response curve was generated by measuring the extracellular field potential in response to varying stimulus intensities. Stimulation intensity (mA) was plotted against the slope of the fEPSP (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). In addition, the size of the presynaptic fiber volley (PSFV), which is proportional to the number of presynaptic neurons recruited by stimulation, was measured (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Both of these methods showed similar input–output curves in hippocampal slices from strain-matched wild-type and double knock-out mice. Similar results were obtained in ras-grf1(−/−) or ras-grf2(−/−) single knock-out mice (data not shown). Thus Ras-GRF proteins do not contribute to basal synaptic transmission.
NMDAR-dependent LTP is reduced in mature, but not immature, Ras-GRF double knock-out mice
To study hippocampal long-term synaptic plasticity at the Schaffer→CA1 synapse, we used a well studied NMDAR-dependent paradigm. In particular, two trains of high frequency stimulation (HFS; 100 Hz) tetanus were used to induce LTP (Fig. 1 A). In hippocampal slices from P26–P35 mice the LTP was reduced significantly, but not completely, in samples from double knock-out mice when compared with those from wild-type mice [fEPSP slopes were 149.48 ± 7.35% (n = 15 slices from eight wild-type mice) vs 115.58 ± 3.32% (n = 13 slices from nine grf1/grf2(−/−) mice); p < 0.001].
Figure 1.

LTP is impaired in the Schaffer collateral→CA1 pathway of hippocampal slices from postpubescent (P26–P35), but not in prepubescent (P14–P18) double Ras-GRF knock-out mice. HFS (two 100 Hz trains) was used to induce LTP in hippocampal slices from P26–P35 (A) and P14–P18 (B) wild-type and double Ras-GRF [grf1/grf2(−/−)] mice. Both groups received two trains of 100 Hz (1 s, 20 s delay) indicated by the arrow. The recorded fEPSP slope is expressed as a percentage of baseline ± SEM, and data were pooled from each group [closed circles, wild-type slices (A, n = 15; B, n = 12); open circles, grf1/grf2(−/−) slices (A, n = 13; B, n = 10)]. Insets, Representative fEPSPs recorded before (1) and after (2) HFS in wild-type and grf1/grf2(−/−) slices. Calibration: horizontal, 10 ms; vertical, 1 mV. C, Hippocampal brain slices were pretreated with buffer containing DMSO (closed boxes; n = 7), the MEK inhibitor U0126 (20 μm; upward triangle; n = 7), or the NMDAR inhibitor d-AP-5 (100 μm; downward triangle; n = 4) before HFS stimulation. Data are expressed as in A, B. D, TBS was used in mature wild-type (closed circles; n = 8) and ras-grf1/grf2(−/−) slices (open circles; n = 7); the recorded data are expressed as in A–C. E, Late-phase LTP was induced by four trains of 1 s, 100 Hz tetani (5 min delay) in mature wild-type (closed circles; n = 8) and grf1/grf2(−/−) slices (open circles; n = 8); the data are displayed as in A–C.
Then, these experiments were repeated on P14–P18 mice, because we showed previously that NMDARs activate Erk MAP kinase in a Ras-GRF-independent mechanism in mice this age and younger (Fig. 1B). Consistent with these earlier findings, the loss of both Ras-GRF proteins in double knock-out mice had no statistically significant effect on the induction of LTP [wild type, 137.87 ± 5.34% (n = 12 slices from eight mice); grf1/grf2(−/−), 133.46 ± 4.78% (n = 10 slices from six mice); p > 0.05], showing that the mechanism of HFS-mediated LTP induction changes from P14–P18 to P26–P35 from a Ras-GRF-independent to a Ras-GRF-dependent process.
We then compared the data generated from double knock-out mice with those obtained after exposure of wild-type mice to inhibitors of either NMDARs or the Erk MAP kinase activator MEK. Consistent with previous reports, HFS LTP was blocked completely by the selective NMDA receptor antagonist d-aminophosphonopentanoic acid (d-AP-5; 100 μm; 99.29 ± 3.88%; n = 4) (see Fig. 2C), In contrast, the MEK inhibitor 1,4-diamino-2,3-dicyano-1,4-bis(2-aminophenylthio)-butadiene (U0126; 20 μm; dissolved in DMSO) (fEPSP slope was 114.12 ± 4.87%; n = 8), only partially blocked LTP induced by HFS to a level comparable to that found in mice lacking both Ras-GRF proteins (Fig. 1C). Moreover, the addition of U0126 to the double knock-out slices failed to inhibit LTP further [with U0126, 116.28 ± 2.97% (n = 9 slices from five grf1/grf2(−/−) mice) vs 115.58 ± 3.32% (n = 13 slices from nine grf1/grf2(−/−) mice); p = 0.24]. Finally, the p38 inhibitor 4-(4-fluorophenyl)-2-[4-(methylsulfonylphenyl)-5-(4-pyridyl)-1H-imidazole (SB203580; 10 μm) had no effect (data not shown). These findings imply that NMDA receptors promote HFS LTP via multiple pathways, one of which is Erk activation via Ras-GRF proteins.
Figure 2.
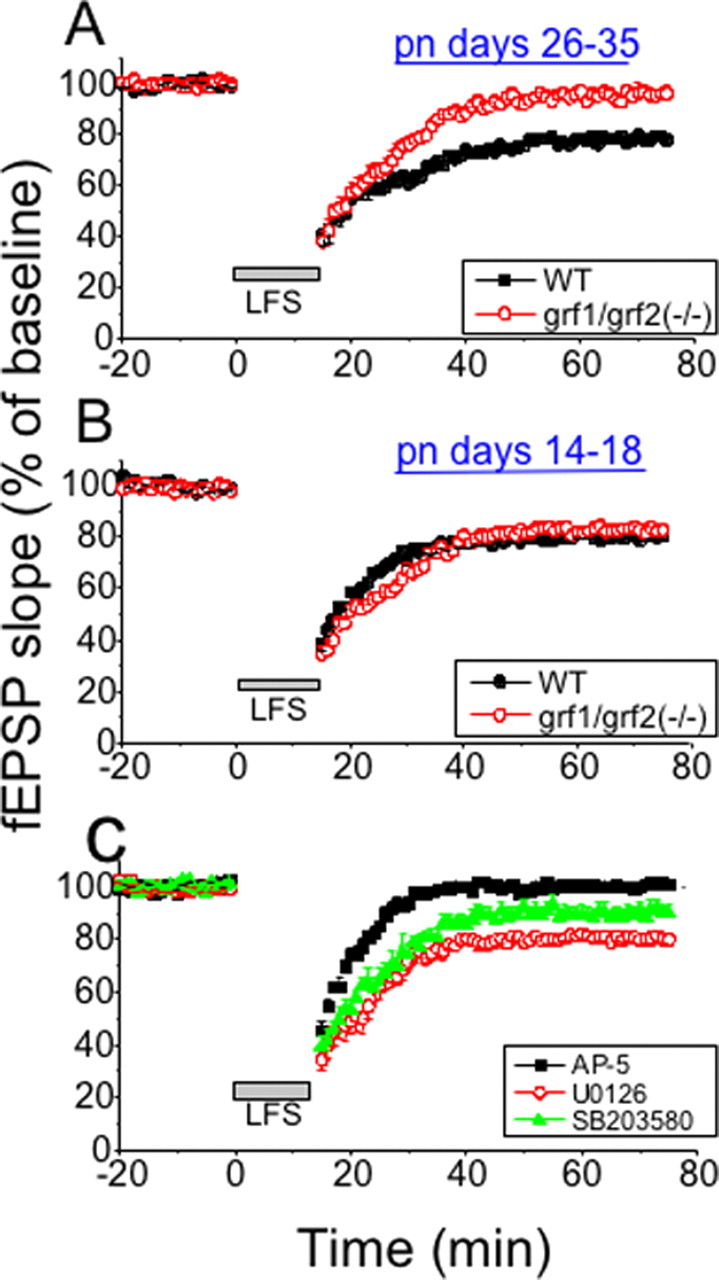
LTD is impaired in the Schaffer collateral→CA1 pathway of hippocampal slices from mature (P26–P35), but not in immature (P14–P18), double Ras-GRF knock-out mice. LFS (900 pulses at 1 Hz) was used to induce LTD in mature (A) and immature (B) hippocampal brain slices from wild-type (WT) and double Ras-GRF knock-out mice [grf1/grf2(−/−)]. The recorded fEPSP slope is expressed as a percentage of baseline ± SEM; data were pooled from each group [closed boxes, WT slices (n = 8 in A, B); open circles, grf1/grf2(−/−) slices (n = 9 in A, B)]. C, Hippocampal brain slices were pretreated with buffer containing the p38 inhibitor SB203580 (10 μm; filled triangle; n = 6), the MEK inhibitor U0126 (20 μm; open circle; n = 7), or the NMDAR inhibitor d-AP-5 (100 μm; filled square; n = 7).
A second LTP stimulation protocol, TBS, was used, which is thought to mimic more closely the physiological firing properties of hippocampal neurons. LTP induced by TBS also was reduced in samples from mature double Ras-GRF knock-out mice [wild type, 138.13 ± 8.18% (n = 8 slices from five mice); grf1/grf2(−/−), 115.41 ± 5.08% (n = 7 slices from five mice); p < 0.001] (Fig. 1D). TBS-induced LTP also was reduced partially by exposure to a MEK inhibitor (U0126; 109.10 ± 2.83%; n = 7) and totally inhibited by d-AP-5 (103.37 ± 2.56%; n = 4).
The protocols described above were recorded for 1 h, reflecting early LTP. To test the role of Ras-GRF proteins in late LTP (L-LTP), we used four 100 Hz tetani (each 1 s long, spaced by 5 min), which generates L-LTP (>3 h). Figure 1E shows that double Ras-GRF mice are also defective in L-LTP [3 h after fourth tetanus: wild type, 155.97 ± 4.51% (n = 8 slices from five mice); grf1/grf2(−/−), 123.16 ± 3.83% (n = 8 slices from five mice); p < 0.001].
NMDAR-dependent LTD generation is also defective in postpubescent, but not prepubescent, Ras-GRF double knock-out mice
NMDARs also can be involved in the generation of LTD, so this form of synaptic plasticity also was studied in mature (P26–P35) mice lacking Ras-GRF proteins. Induction of LTD by LFS (900 pulses at 1 Hz) was reduced significantly in hippocampal slices from double knock-out mice as compared with those from strain-matched controls [grf1/grf2(−/−), 96.29 ± 3.01% (n = 9 slices from six mice); wild type, 78.12 ± 1.34% (n = 8 slices from six mice); p < 0.001] (Fig. 2 A). Consistent with LTP experiments, no statistically significant defect in the induction of LTD was observed in P14–P18 double knock-out mice [wild type, 80.77 ± 2.43% (n = 8 slices from five mice) vs grf1/grf2(−/−), 82.91 ± 2.07% (n = 9 slices from six mice)] (Fig. 2B).
LFS induction of LTD at CA1 has been shown to be dependent on NMDAR activation. Addition of the NMDAR inhibitor d-AP-5 to mature brain slices (P26–P35) blocked LTD more effectively than that of Ras-GRF double knock-out mice (p < 0.05 ANOVA), indicating that LTD is induced via multiple signaling pathways, one of which is mediated by Ras-GRF proteins (Fig. 2C). LFS-induced LTD at CA1 also has been shown to be mediated, at least in part, by p38, but not Erk, MAP kinase. The p38 inhibitor SB203580 partially suppressed LTD (91.93 ± 3.75%; n = 6) to a level not significantly different from that found for Ras-GRF knock-out mice (p > 0.05), whereas the MEK inhibitor U0126 had no effect (83.13 ± 2.31%; n = 8).
Thus the findings described so far show that Ras-GRF proteins are involved in the induction of both NMDA-mediated LTP and NMDA-mediated LTD in the CA1 region of the hippocampus of 26- to 35-d-old mice, possibly via the regulation of Erk and p38 MAP kinases. Moreover, the mechanisms by which LTP and LTD are generated change with postnatal differentiation between ∼P18 and ∼P25 from Ras-GRF-independent to Ras-GRF-dependent processes.
Ras-GRF1 contributes predominantly to LTD, whereas Ras-GRF2 contributes predominantly to LTP in mature mice
To assess the contribution of individual Ras-GRF family members to the induction of LTP and LTD, we investigated single Ras-GRF knock-out mice (P26–P35) as described above for double knock-out mice. Figure 3 A shows that hippocampal slices from Ras-GRF1 knock-out mice displayed a small but statistically significant defect in HFS-induced LTP as compared with LTP induction observed in control mice [ras-grf1(−/−), 126.0 ± 5.13% (n = 10 slices from eight mice); wild type, 139.38 ± 3.16% (n = 7 slices from five mice); p < 0.05] (Fig. 3A). However, no significant defect was observed in response to theta burst induction of LTP (Fig. 3B), [ras-grf1(−/−), 134.3 ± 3.4% (n = 12 slices from five mice); wild type, 140.81 ± 3.32% (n = 10 slices from six mice); p > 0.05]. Thus Ras-GRF1 contributes little, if any, to LTP induction at this synapse.
Figure 3.

Ras-GRF1 predominantly affects LTD, whereas Ras-GRF2 predominantly affects LTP in P26–P35 mice. A, Two trains of 100 Hz (1 s duration, 20 s delay) were used to induce LTP in P26–P35 Ras-GRF1 knock-out mice [grf1(−/−), open squares; n = 10] and same genetic background wild-type slices (filled squares; n = 7). B, TBS was used in mature wild-type (filled squares; n = 10) and ras-grf1(−/−) slices (open squares; n = 12); the recorded data are expressed as in A. C, LFS (900 pulses at 1 Hz) was used to induce LTD in wild-type (closed squares; n = 6) and Ras-GRF1 knock-out mice [grf1(−/−), open squares; n = 10]. D, Two trains of 100 Hz were used to induce LTP in wild-type (closed circles; n = 15) and Ras-GRF2 knock-out mice [grf2(−/−), open triangles; n = 11]. E, TBS was used in mature wild-type (filled circles; n = 6) and ras-grf2(−/−) slices (open triangles; n = 8); the recorded data are expressed as in A. F, LFS (900 pulses at 1 Hz) was used to induce LTD in wild-type (closed boxes; n = 8) and single Ras-GRF2 knock-out mice [grf2(−/−), closed triangles; n = 11]. The recorded fEPSP slope is expressed as a percentage of the baseline ± SEM; data were pooled from each group.
In contrast, LFS-induced LTD was impaired severely in Ras-GRF1 knock-out mice [ras-grf1(−/−), 95.42 ± 2.73% (n = 10 slices from seven mice); wild type, 72.69 ± 4.48% (n = 6 slices from four mice); p < 0.001] (Fig. 3C) to a degree comparable that observed in double knock-out mice. Thus Ras-GRF1 contributes modestly to LTP but much more significantly to LTD at the CA1 region of the hippocampus.
Strikingly, the results for Ras-GRF2 knock-out mice were the inverse. Both HFS and theta burst-induced LTP clearly were impaired in mutant mice (Fig. 3D,E) [HFS ras-grf2(−/−), 112.87 ± 3.46% (n = 11 slices from six mice); wild type, 149.48 ± 7.35% (n = 15 slices from eight mice); p < 0.001; theta burst ras-grf2(−/−), 115.6 ± 1.46% (n = 11 slices from six mice); wild type, 142.7 ± 8.7% (n = 8 slices from six mice); p < 0.001], defects that were comparable to those found in double knock-out mice. In contrast, Ras-GRF2 knock-out mice showed no statistically significant defect in LFS-induced LTD [ras-grf2(−/−), 82.78 ± 2.99% (n = 11 slices from six mice); wild type, 78.12 ± 1.34% (n = 8 slices from six mice); p < 0.05] (Fig. 3F). As expected, no defect in either LTP or LTD was observed when single knock-out animals 14–18 d old were used (data not shown).
Overall, the study of single and double knock-out mice shows that Ras-GRF proteins are involved in the generation of both LTP and LTD at the CA1 region of the hippocampus, with the role of Ras-GRF2 restricted to the induction of LTP and the role of Ras-GRF1 predominantly in the induction of LTD.
Ras-GRF2 is a major regulator of Erk activation via NR2A subunit-containing NMDARs, whereas Ras-GRF1 is a minor regulator of Erk activation via NR2B-containing NMDARs in mature mice
To begin to understand why Ras-GRF2 plays a more dominant role in the generation of LTP than does Ras-GRF1, we investigated individual Ras-GRF family members for their contribution to NMDAR activation of Erk in hippocampal slices, because Erk has been shown to contribute to many forms of LTP generation. We previously showed that both Ras-GRF proteins contribute to NMDAR activation of Ras and its effector Erk in cortical slices from mature mice, because NMDAR activation of Erk was inhibited totally in cortical slices when double, but not single, Ras-GRF knock-out mice were studied (Tian et al., 2004).
Here, we investigated NMDAR activation of Erk in hippocampal slices from mature (P26–P35) wild-type and Ras-GRF knock-out mice and found a similar result. In particular, NMDA activation of wild-type hippocampal slices led to ∼4.8-fold increase in activated Erk. As we showed previously, NMDA stimulated Erk transiently in brain slices from control mice such that maximum activation occurred at 5 min, which receded to baseline by ∼20 min. In slices from double Ras-GRF knock-out mice NMDA activation was inhibited completely (Fig. 4 A) (the small increase in basal Erk activity seen in the sample from double knock-out mice when compared with control samples was not found routinely) (Fig. 4C). As expected, no defect was observed in hippocampal slices from P14–P18 double or single knock-out mice (Fig. 4B).
Figure 4.

Ras-GRF1 and Ras-GRF2 make different contributions to NMDA-induced Erk activation via different classes of NMDARs in the hippocampus of postpubescent mice. A, Hippocampal brain slices from mature (> P25) double Ras-GRF knock-out mice, as well as genetically matched control mice, were treated with NMDA (100 μm) for various times. Then the slices were lysed in detergent, and immunoblots of lysates were performed by using activation-specific phospho-Erk1/2 antibodies or antibodies to total Erk1/2. Quantification of the fold increase in Erk1/2 activation after NMDA stimulation also is shown (n > 3; ± SD, **p < 0.01). B, Similar analysis was performed on hippocampal brain slices from P14–P18 control and double Ras-GRF knock-out mice except that only the 5 min time point is shown. The data are representative of at least two independent experiments. C, Control, grf1(−/−), and grf2(−/−) knock-out mice (> P25) were analyzed along with double grf1/grf2(−/−) mice as described in B. No significant difference in NMDA-induced Erk activation was observed among control strains used for grf1(−/−), grf2(−/−), or double knock-out mice, so a single value of pooled controls was used to represent control samples from wild-type (WT) mice. Band intensities of phospho-Erk1/2 were normalized to the band intensity of total Erk (n ≥ 4; ± SD; **p < 0.01). D, Hippocampal brain slices from mature (> P25) ras-grf1(−/−) mice were treated with NMDA for 5 min as described in A. In some samples the brain slices were pretreated with ifenprodil (IF) (5 μm) for 30 min or NVP-AAM077 (NVP; 0.6 μm) for 50 min before NMDA stimulation. Erk activation was detected, and the fold increase was quantified as above (n ≥ 4; ± SD; **p < 0.01). E, Similar experiments were performed as in A except that the hippocampal brain slices were prepared from adult ras-grf2(−/−) mice (n ≥ 4; ± SD; **p < 0.01).
Then, hippocampal slices from single ras-grf1(−/−) and ras-grf2(−/−) knock-out mice were investigated and found to have partial effects. Importantly, the inhibition of Erk was more dramatic in ras-grf2(−/−) samples than in ras-grf1(−/−) samples (Fig. 4C). In fact, the defect in ras-grf1(−/−) mice was so small that we could not detect a statistically significant difference when compared with wild-type mice. However, residual NMDA activation of Erk was observed in Ras-GRF2 knock-out mice (Fig. 4C), and this activity was lost in double knock-out mice (Fig. 4A). Thus Ras-GRF1 may make a small but significant contribution to NMDAR activation of Erk that is difficult to detect with the use of these techniques. Nevertheless, it is clear that the contribution of Ras-GRF2 to NMDAR activation of Erk is greater than that of Ras-GRF1, consistent with our finding that Ras-GRF2 makes a greater contribution to the induction of LTP.
Next, we determined whether Ras-GRF1 and Ras-GRF2 mediate the action of distinct NMDAR subfamilies to activate Erk in the hippocampus. To this end we used inhibitors of either NR2A subunit-containing NMDARs or NR2B subunit-containing NMDARs in hippocampal slices from mature single knock-out mice stimulated in vitro with NMDA. In hippocampal slices from single Ras-GRF1 knock-out mice in which NMDAR signaling to Ras and Erk is mediated solely by Ras-GRF2, the observed ∼4.8-fold increase in Erk activity was blocked completely by pretreatment with (R)-[(S)-1-(4-bromo-phenyl)-ethylamino]-(2,3-dioxo-1,2,3,4-tetrahydroquinoxalin-5-yl)-methyl-phosphonic acid (NVP-AAM077), which preferentially suppresses NMDARs containing NR2A subunits (Fig. 4D). In contrast, pretreatment with ifenprodil, which preferentially blocks NMDARs containing NR2B subunits, had no effect. These results imply that Ras-GRF2 specifically couples NR2A-containing receptors to Erk activation in the hippocampus of mature mice. This finding is also consistent with the observation that NR2A subunits have been associated with Erk activation (Kim et al., 2005) and induction of LTP in the CA1 region of the hippocampus (Liu et al., 2004; Massey et al., 2004).
An analogous study of mature single Ras-GRF2 knock-out mice, in which signaling from NMDARs to Erk is via Ras-GRF1 only, showed the opposite result. As described above, Ras-GRF1 makes a smaller contribution to NMDAR activation of Erk than does Ras-GRF2, so in these samples NMDA activates Erk only approximately twofold (Fig. 4E). However, this activation was blocked completely by the NR2B inhibitor ifenprodil, but not blocked at all by the NR2A inhibitor NVP-AAM077. Thus these results imply that Ras-GRF1 specifically couples NR2B-containing NMDARs to Erk in the hippocampus of mature mice. This finding is consistent with previous studies showing that Ras-GRF1 binds directly to NR2B subunits (Krapivinsky et al., 2003). Overall, these findings show that Ras-GRF1 and Ras-GRF2 contribute to NMDAR Erk regulation to different degrees via different subclasses of NMDARs.
Ras-GRF1, but not Ras-GRF2, mediates the activation of p38 MAP kinase induced by NR2B-containing NMDARs in mature mice
To begin to understand why Ras-GRF1 makes a major contribution to LTD but Ras-GRF2 does not, we assessed the contribution of each Ras-GRF family member to NMDAR activation of p38 MAP kinase, because NMDAR activation of p38 activity has been implicated in the induction of LTD (Bolshakov et al., 2000; Zhu et al., 2002). We previously showed that Ras-GRF1 can regulate p38 via its ability to activate the Rac GTPase and via its interaction with IB2/JIP2, a scaffold for the kinase cascade that couples Rac specifically to p38 (Buchsbaum et al., 2002). Thus we investigated the role of individual Ras-GRF family members in the regulation of p38 MAP kinase as we did for Erk MAP kinase described above, except that activation-specific p38 antibodies were used. Figure 5 A shows that NMDA stimulation of hippocampal brain slices with NMDA led to ∼3.5-fold increase in active p38 after 5 min in control mice. Like NMDAR activation of Erk, this activation was transient and fell back to control levels by 20 min. In contrast, p38 activation was blocked completely in hippocampal brain slices from P26–P35 double grf1/grf2(−/−) knock-out mice (Fig. 5A). As expected, no defect in NMDAR activation of p38 was observed in hippocampal slices from P14–P18 double knock-out mice (Fig. 5B).
Figure 5.

Ras-GRF1, but not Ras-GRF2, couples NMDAR to p38 activation in hippocampal brain slices from postpubescent mice. A, Hippocampal brain slices from mature (> P25) or wild-type (WT) and double Ras-GRF knock-out mice were treated with NMDA (100 μm) for 5 min. p38 activation was measured by immunoblots that used activation-specific phospho-p38 antibodies, and the density of the bands was determined as shown in the graphs, normalized for total p38, and quantified for fold stimulation relative to the control in the absence of NMDA (n = 4; ± SD; **p < 0.01). B, Experiments were performed as in A except that P14–P18 mice were used and only the peak 5 min time point is shown. The data are representative of at least two independent experiments. C, Hippocampal brain slices from mature > P25 control, ras-grf1(−/−), or ras-grf2(−/−) mice were treated as above (n = 3; ± SD; **p < 0.01). D, Hippocampal brain slices from mature > P25 ras-grf2(−/−) mice were treated with NMDA as described above. In some samples the brain slices were pretreated with ifenprodil (IF; 5 μm) for 30 min or NVP-AAM077 (NVP; 0.6 μm) for 50 min before NMDA stimulation. The p38 activation was detected, and the fold increase was quantified as above (n ≥ 4; ± SD; **p < 0.01).
In hippocampal slices of mature (P26–P35) single Ras-GRF1 knock-out mice, in which only Ras-GRF2 remains coupled to NMDA receptor signaling, NMDA activation of p38 also was blocked completely when compared with brain slices from control mice (Fig. 5C). In contrast, in hippocampal slices of mature Ras-GRF2 knock-out mice, in which only Ras-GRF1 remains coupled to NMDA receptor signaling, NMDA activation of p38 was unaltered when compared with control mice (Fig. 5C). Thus Ras-GRF1, not Ras-GRF2, couples NMDARs to p38 in this tissue.
Furthermore, in Ras-GRF2 knock-out mice the NR2B inhibitor ifenprodil, but not the NR2A inhibitor NVP-AAM077, blocked NMDAR activation of p38, indicating that Ras-GRF1 mediates p38 regulation by NR2B-containing NMDARs (Fig. 5D). These results are consistent with our finding described above that Ras-GRF1 makes a much more substantial contribution to NMDAR-mediated LTD induction than does Ras-GRF2. Finally, these findings were not attributable to changes in NMDAR subunit expression levels, because immunoblotting detected no significant difference in NR2A or NR2B levels in either single or double Ras-GRF knock-out mice (data not shown).
Ras-GRF2 also contributes to short-term synaptic plasticity in postpubescent mice
Paired pulse facilitation (PPF) is a measure of short-term plasticity in which fEPSP responses to two stimuli delivered at short intervals are measured. It is observed as an increased synaptic response to a second stimulus. Compared with genetically matched controlled wild-type mice, PPF in Ras-GRF double knock-out mice (P26–P35) was significantly lower, especially in 20 through ∼100 ms interpulse intervals, as compared with that of control mice (p < 0.05; n = 6) (Fig. 6 A,B). Thus Ras-GRF proteins make a significant contribution to this form of short-term plasticity.
Figure 6.

Ras-GRF2, but not Ras-GRF1, contributes to a form of short-term plasticity. PPF was generated at the CA1 region in mature wild-type mice, double grf1/grf2(−/−) knock-out mice, and single grf1(−/−) or grf2(−/−) knock-out mice. A, Traces show typical fEPSPs evoked with a 40 ms interpulse interval in different genotype mice. Calibration: horizontal, 10 ms; vertical, 1 mV. B, The ratio of the slope of the fEPSPs evoked by the second stimulus and that evoked by the first stimulus is plotted at various interpulse intervals [closed squares, wild-type mice (n = 20); closed circles, ras-grf1/grf2 double knock-out mice (n = 21); closed triangles, ras-grf2(−/−) mice (n = 22)]. Data points represent the mean ± SEM. C, Separate control mice (n = 12) were used for grf1(−/−) mice (n = 25 slices), because we found that the magnitude of PPF was smaller in the mouse strain used to propagate ras-grf1(−/−) mice (see Materials and Methods). Data points represent the mean ± SEM.
It is possible that the reduction of PPF observed in mice lacking Ras-GRF proteins contributes to defects in LTP observed in these mice. However, among brain slices from control and mutant mice that have similar PPF values [wild type, 1.71 ± 0.09 and 1.84 ± 0.09 (n = 10); ras-grf1/grf2(−/−), 1.73 ± 0.07 and 1.66 ± 0.04 at interstimulus intervals for 40 and 60 ms, respectively (n = 9); p > 0.05], LTP in this ras-grf1/grf2(−/−) group still was reduced significantly as compared with control slices [grf1/grf2(−/−), 118.28 ± 2.49% (n = 9); wild type, 137.04 ± 3.61% (n = 10); p < 0.01], suggesting that the two defects caused by the loss of Ras-GRF2 were independent phenomena. Supporting this view are other knock-out mouse models in which PPF was inhibited, but no effect on LTP was observed (Moresco et al., 2003).
Interestingly, samples from ras-grf2(−/−) mice showed a defect comparable to that found in samples from double Ras-GRF knock-out mice (Fig. 6B), implying that Ras-GRF2, not Ras-GRF1, contributes to PPF. This conclusion was confirmed when single ras-grf1(−/−) mice were compared with their genetically matched controls (which displayed a smaller PPF response), and, as predicted, no significant defect was observed (Fig. 6C).
Discussion
This work establishes Ras-GRF1 and Ras-GRF2 as important regulators of multiple forms of hippocampal synaptic plasticity, specifically in postpubescent mice. This conclusion is based on the observation that three forms of LTP, LTD, and PPF were found to be defective in the CA1 region of the hippocampus from 25- to 36-d-old double Ras-GRF knock-out mice, but not from 14- to 18-d-old knock-out animals.
Surprisingly, analysis of single Ras-GRF knock-out mice revealed that, despite their similar overall structural organization, Ras-GRF1 and Ras-GRF2 function as calcium sensors for different classes of NMDARs to drive the activation of distinct MAP kinase family members that affect opposing forms of synaptic plasticity (Fig. 7). In particular, we found that Ras-GRF2 makes a greater contribution to HFS and theta burst-induced LTP than Ras-GRF1 does. These differences are likely attributable to the fact that Ras-GRF2 has a greater role in NMDAR activation of Erk MAP kinase, because Erk activation has been shown to contribute to HFS and theta burst-induced LTP in the rodent hippocampus (Selcher et al., 2003; Thomas and Huganir, 2004). Additional support derives from our observations that an Erk activation inhibitor suppresses HFS and theta burst LTP induction to levels comparable to those found in Ras-GRF2 knock-out mice and that the inhibitor could not additionally suppress LTP in samples from knock-out mice. Although the contribution of Erk to theta burst LTP generally is accepted, the role of Erk in HFS-induced LTP remains unsettled because, unlike our findings, some labs have found that HFS-induced LTP in the hippocampus of mice (but not rats) is Erk-independent (Selcher et al., 2003; Kelleher et al., 2004). Differences between mouse strains or HFS induction protocols used in these studies may be involved.
Figure 7.

Model for Ras-GRF family participation in LTP and LTD and in the regulation of specific MAP kinase family members at the CA1 region of the hippocampus of mature mice (P26 and older). Based on a combination of electrophysiology and biochemistry experiments described in this study, we propose that Ras-GRF proteins contribute to both NMDAR-dependent LTP and LTD at the CA1 region of the hippocampus. Ras-GRF2 contributes almost exclusively to LTP, based on its ability to be the major regulator of NMDAR activation of Erk via its Ras-activating CDC25 domain in response to NR2A subunit-containing receptors. In contrast, Ras-GRF1 exclusively contributes to the generation of LTD, based on its ability to activate p38 via its Rac-activating DH domain in response to NR2B subunit-containing receptors. Importantly, Ras-GRF proteins play a significant role in synaptic plasticity only after ∼P20. The reason that, despite their similar signaling potential, Ras-GRF2 is more effective at activating Erk in response to NMDAR activation, whereas Ras-GRF1 is more effective at activating p38 in the hippocampus, remains to be determined.
Ras-GRF2 and Ras-GRF1 also mediate Erk activation by different subclasses of NMDARs; Ras-GRF1 responds to receptors containing NR2B subunits, whereas Ras-GRF2 responds to receptors containing NR2A subunits. This conclusion is based on studies that used ifenprodil and NVP-AAM077, inhibitors that display preference for NR2B and NR2A subunits, respectively. Ifenprodil blocked NMDA activation of Erk in Ras-GRF2 knock-out slices in which NMDARs signal to Erk only via Ras-GRF1. However, the drug had no effect on Erk regulation in Ras-GRF1 knock-out slices in which NMDARs signal only via Ras-GRF2. In contrast, NVP-AAM077 had the opposite effect, blocking NMDAR activation of Erk in brain slices expressing only Ras-GRF2, but not in slices expressing only Ras-GRF1. Recent studies have revealed that NVP-AAM077 is not totally specific for NR2A subunits, because it has some inhibitory activity against NR2B subunits (Berberich et al., 2005). This cross-reactivity issue does not seem to be significant in the experimental system used here, because NVP-AAM077 had no detectable effect on signaling to Erk or p38 via Ras-GRF1, although the NR2B inhibitor ifenprodil did.
The dominant role of Ras-GRF2 in NMDAR-mediated induction of LTP and Erk activation is consistent with previous data showing that NR2A-containing NMDARs promote LTP and Erk in hippocampal neurons (Liu et al., 2004; Massey et al., 2004; Kim et al., 2005). However, the contribution of Ras-GRF1 to HFS LTP, albeit a small one, would not have been predicted from some previous studies that implicated NR2B-containing receptors in LTD rather than in LTP (Liu et al., 2004; Massey et al., 2004). However, other studies have shown that both subclasses of receptors can induce LTP (Tang et al., 1999; Wong et al., 2002; Berberich et al., 2005; Zhao et al., 2005). Thus our findings are consistent with the latter studies as well.
The mechanism by which Ras-GRF1 contributes to HFS LTP is not clear, because we could detect only a small, if any, defect in NMDAR activation of Erk in hippocampal brain slices from Ras-GRF1 knock-out mice, yet these mice displayed a significant defect in HFS LTP induction. Thus Ras-GRF1 may contribute to HFS LTP via Ras effector pathways other than Erk or via Rac effector pathways other than p38 MAP kinase. Why does Ras-GRF1 lead to such a minor activation of Erk at NR2B-containing receptors (Fig. 7)? A recent finding showed that a negative regulator of Ras activity, Syn-GAP, is associated with NMDARs containing NR2B subunits, but not NR2A subunits (Kim et al., 2005). Thus Ras/Erk activation may be downregulated preferentially when mediated by NR2B-NMDAR/Ras-GRF1 signaling.
Analysis of single Ras-GRF knock-out mice revealed that Ras-GRF1 makes a much more substantial contribution to LTD than Ras-GRF2 does. This finding is consistent with the fact that Ras-GRF1 binds directly to the NR2B subunit of NMDARs (Krapivinsky et al., 2003), because receptors containing this subunit have been implicated in the induction of LTD (Liu et al., 2004; Massey et al., 2004). However, NMDAR-mediated LTD in mature neurons has been shown both here and in previous studies to require p38 MAP kinase activation, whereas Ras-GRF proteins are best known for their ability to activate Ras and Erk via their CDC25-GEF domains, which are participants in NMDAR-induced LTP, but not LTD, at this brain region (Zhu et al., 2002). However, we showed previously that Ras-GRF1 also has the ability to activate Rac and its target p38 MAP kinase via its DH-GEF domain. In fact, we have shown that Ras-GRF1 binds to the scaffold IB2/JIP2, which specifically promotes p38 activation among multiple Rac effectors by targeting activated Rac to components of the p38 kinase signaling cascade (Buchsbaum et al., 2002). In the present study we documented that Ras-GRF1, but not Ras-GRF2, mediates p38 activation by NMDARs, consistent with its ability to influence LTD. We also found that NMDAR activation of p38 via Ras-GRF1 is blocked by the NR2B inhibitor ifenprodil, not the NR2A inhibitor NVP-AAM077, consistent with the demonstrated binding of Ras-GRF1 directly to NR2B, but not NR2A, NMDAR subunits.
We also detected a role for the Ras-GRF family of GEFs in short-term plasticity, as evidenced by a defect in PPF in postpubescent Ras-GRF2 knock-out mice. Strikingly, PPF also is regulated in an isoform-specific manner, because no defect was observed in Ras-GRF1 knock-out mice. There is substantial evidence that PPF regulation occurs via changes in presynaptic rather than postsynaptic events (Nicoll, 2003). Thus it is possible that Ras-GRF2 mediates the action of presynaptic NMDARs or other neuronal regulators that elevate calcium in cells. Alternatively, postsynaptic Ras-GRF2 may influence PPF by a messenger transmitted to the presynaptic cell.
Another important conclusion of this work is the striking age dependence of the contribution Ras-GRF proteins make to synaptic plasticity. Thus we could detect no change in LTP, LTD, or short-term plasticity in hippocampal brain slices from mice 14–18 d of age in double Ras-GRF knock-out or single Ras-GRF knock-out mice, nor could we detect any defects in NMDAR activation of Erk or p38 MAP kinases. However, ∼10 d later striking defects in all three forms of synaptic plasticity, as well as NMDA signaling to cytoplasmic MAP kinases, were clearly detectable. The expression levels of both Ras-GRF proteins are regulated developmentally (Zippel et al., 1997; Tian et al., 2004), which could contribute to this phenomenon, but the precise mechanisms responsible for the age-dependent coupling of Ras-GRFs to NMDARs remain to be determined.
Appreciation for developmental changes in the mechanisms underlying the generation of synaptic plasticity is important for many reasons. As pointed out previously (Malenka and Bear, 2004), many studies are performed on neurons prepared from neonatal animals and cultured for various amounts of time in vitro, such that the precise developmental stage is not clear. In addition, many studies are performed on brain slices from pubescent mice. This issue could, in fact, explain inconsistencies that have arisen in the literature regarding signaling pathways involved in synaptic plasticity regulation. For example, we found that NMDAR regulation of p38 is via Ras-GRF1, whereas a previous study implicated the Rap1 GTPase (Zhu et al., 2002). This difference may be attributable to the fact that the earlier study used brain slices isolated from animals at an age when Ras-GRFs have not yet become physiologically important. We previously demonstrated that NMDARs switch from Sos to Ras-GRF GEFs to activate Erk during late postnatal development of the brain (Tian et al., 2004). An analogous change in the mechanism of p38 activation by NMDARs may take place, causing a switch from a Rap-GEF/Rap/p38 pathway to a Ras-GRF/Rac/p38 pathway between P18 and P26 d in mice.
Moreover, the present study reinforces the growing appreciation that the mechanisms of synaptic plasticity are regulated developmentally (Malenka and Bear, 2004). Because Ras-GRF proteins contribute to synaptic plasticity only after ∼P20, they likely contribute to the regulation of mature synapses, not developing ones. Strikingly, the appearance of a Ras-GRF contribution to synaptic plasticity after birth coincides with the emergence of a role for the hippocampus in context and spatial memories in rodents (Rudy, 1993; Dumas, 2005). This implicates both Ras-GRF1 and Ras-GRF2 as significant components of signaling pathways that may be specific for mature hippocampal-dependent learning and memory. In fact, we previously have documented some defects in hippocampal-dependent behavior in Ras-GRF1 knock-out mice (Giese et al., 2001) and presently are comparing those findings with Ras-GRF2 knock-out mice.
Footnotes
*S.L. and X.T. contributed equally to this work.
This research was supported by National Institutes of Health Grant CA 047391 to L.A.F.; by the Tufts Center for Neuroscience Research (P30 NS047243), Electrophysiology and Biophysics Core Facility; and the Gastrointestinal Research on Absorptive and Secretory Process (GRASP) Digestive Disease Center (P30-DK34928 GRASP). We thank John Lisman for helpful comments on this manuscript.
References
- Anderson WW, Collingridge GL (2001). The LTP program: a data acquisition program for on-line analysis of long-term potentiation and other synaptic events. J Neurosci Methods 108:71–83. [DOI] [PubMed] [Google Scholar]
- Berberich S, Punnakkal P, Jensen V, Pawlak V, Seeburg PH, Hvalby O, Kohr G (2005). Lack of NMDA receptor subtype selectivity for hippocampal long-term potentiation. J Neurosci 25:6907–6910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolshakov VY, Carboni L, Cobb MH, Siegelbaum SA, Belardetti F (2000). Dual MAP kinase pathways mediate opposing forms of long-term plasticity at CA3–CA1 synapses. Nat Neurosci 3:1107–1112. [DOI] [PubMed] [Google Scholar]
- Buchsbaum RJ, Connolly BA, Feig LA (2002). Interaction of Rac exchange factors Tiam1 and Ras-GRF1 with a scaffold for the p38 mitogen-activated protein kinase cascade. Mol Cell Biol 22:4073–4085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumas TC (2005). Late postnatal maturation of excitatory synaptic transmission permits adult-like expression of hippocampal-dependent behaviors. Hippocampus 15:562–578. [DOI] [PubMed] [Google Scholar]
- Fam NP, Fan W-T, Wang Z, Zhang L-J, Chen Z, Moran MF (1997). Cloning and characterization of Ras-GRF2, a novel guanine nucleotide exchange factor for Ras. Mol Cell Biol 17:1396–1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan WT, Koch CA, de Hoog CL, Fam NP, Moran MF (1998). The exchange factor Ras-GRF2 activates Ras-dependent and Rac-dependent mitogen-activated protein kinase pathways. Curr Biol 8:935–938. [DOI] [PubMed] [Google Scholar]
- Farnsworth CL, Freshney NW, Rosen LB, Ghosh A, Greenberg ME, Feig LA (1995). Calcium activation of Ras mediated by the neuronal exchange factor Ras-GRF. Nature 376:524–527. [DOI] [PubMed] [Google Scholar]
- Giese KP, Friedman E, Telliez JB, Fedorov NB, Wines M, Feig LA, Silva AJ (2001). Hippocampus-dependent learning and memory is impaired in mice lacking the Ras-guanine nucleotide-releasing factor 1 (Ras-GRF1). Neuropharmacology 41:791–800. [DOI] [PubMed] [Google Scholar]
- Innocenti M, Zippel R, Brambilla R, Sturani E (1999). CDC25Mm/Ras-GRF1 regulates both Ras and Rac signaling pathways. FEBS Lett 460:357–362. [DOI] [PubMed] [Google Scholar]
- Kelleher RJ III, Govindarajan A, Jung HY, Kang H, Tonegawa S (2004). Translational control by MAPK signaling in long-term synaptic plasticity and memory. Cell 116:467–479. [DOI] [PubMed] [Google Scholar]
- Kim MJ, Dunah AW, Wang YT, Sheng M (2005). Differential roles of NR2A- and NR2B-containing NMDA receptors in Ras-ERK signaling and AMPA receptor trafficking. Neuron 46:745–760. [DOI] [PubMed] [Google Scholar]
- Kiyono M, Satoh T, Kaziro Y (1999). G-protein βγ subunit-dependent Rac-guanine nucleotide exchange activity of Ras-GRF1/CDC25Mm Proc Natl Acad Sci USA 96:4826–4831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krapivinsky G, Krapivinsky L, Manasian Y, Ivanov A, Tyzio R, Pellegrino C, Ben-Ari Y, Clapham DE, Medina I (2003). The NMDA receptor is coupled to the ERK pathway by a direct interaction between NR2B and RasGRF1. Neuron 40:775–784. [DOI] [PubMed] [Google Scholar]
- Liu L, Wong TP, Pozza MF, Lingenhoehl K, Wang Y, Sheng M, Auberson YP, Wang YT (2004). Role of NMDA receptor subtypes in governing the direction of hippocampal synaptic plasticity. Science 304:1021–1024. [DOI] [PubMed] [Google Scholar]
- Malenka RC, Bear MF (2004). LTP and LTD: an embarrassment of riches. Neuron 44:5–21. [DOI] [PubMed] [Google Scholar]
- Massey PV, Johnson BE, Moult PR, Auberson YP, Brown MW, Molnar E, Collingridge GL, Bashir ZI (2004). Differential roles of NR2A- and NR2B-containing NMDA receptors in cortical long-term potentiation and long-term depression. J Neurosci 24:7821–7828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moresco EM, Scheetz AJ, Bornmann WG, Koleske AJ, Fitzsimonds RM (2003). Abl family nonreceptor tyrosine kinases modulate short-term synaptic plasticity. J Neurophysiol 89:1678–1687. [DOI] [PubMed] [Google Scholar]
- Nicoll RA (2003). Expression mechanisms underlying long-term potentiation: a postsynaptic view. Philos Trans R Soc Lond B Biol Sci 358:721–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudy JW (1993). Contextual conditioning and auditory cue conditioning dissociate during development. Behav Neurosci 107:887–891. [DOI] [PubMed] [Google Scholar]
- Selcher JC, Weeber EJ, Christian J, Nekrasova T, Landreth GE, Sweatt JD (2003). A role for ERK MAP kinase in physiologic temporal integration in hippocampal area CA1. Learn Mem 10:26–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shou C, Farnsworth CL, Neel BG, Feig LA (1992). Molecular cloning of cDNAs encoding a guanine nucleotide-releasing factor for Ras p21. Nature 358:351–354. [DOI] [PubMed] [Google Scholar]
- Tang YP, Shimizu E, Dube GR, Rampon C, Kerchner GA, Zhuo M, Liu G, Tsien JZ (1999). Genetic enhancement of learning and memory in mice. Nature 401:63–69. [DOI] [PubMed] [Google Scholar]
- Thomas GM, Huganir RL (2004). MAPK cascade signaling and synaptic plasticity. Nat Rev Neurosci 5:173–183. [DOI] [PubMed] [Google Scholar]
- Tian X, Gotoh T, Tsuji K, Lo EH, Huang S, Feig LA (2004). Developmentally regulated role for Ras-GRFs in coupling NMDA glutamate receptors to Ras, Erk, and CREB. EMBO J 23:1567–1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong RW, Setou M, Teng J, Takei Y, Hirokawa N (2002). Overexpression of motor protein KIF17 enhances spatial and working memory in transgenic mice. Proc Natl Acad Sci USA 99:14500–14505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao MG, Toyoda H, Lee YS, Wu LJ, Ko SW, Zhang XH, Jia Y, Shum F, Xu H, Li BM, Kaang BK, Zhuo M (2005). Roles of NMDA NR2B subtype receptor in prefrontal long-term potentiation and contextual fear memory. Neuron 47:859–872. [DOI] [PubMed] [Google Scholar]
- Zhu JJ, Qin Y, Zhao M, Van Aelst L, Malinow R (2002). Ras and Rap control AMPA receptor trafficking during synaptic plasticity. Cell 110:443–455. [DOI] [PubMed] [Google Scholar]
- Zippel R, Gnesutta N, Matus-Leibovitch N, Mancinelli E, Saya D, Vogel Z, Sturani E (1997). Ras-GRF, the activator of Ras, is expressed preferentially in mature neurons of the central nervous system. Brain Res Mol Brain Res 48:140–144. [DOI] [PubMed] [Google Scholar]