

Abstract
The formation of dendritic spines during development and their structural plasticity in the adult brain are critical aspects of synaptogenesis and synaptic plasticity. Many different factors and proteins have been shown to control dendritic spine development and remodeling (Ethell and Pasquale, 2005). The extracellular matrix (ECM) components and their cell surface receptors, integrins, have been found in the vicinity of synapses and shown to regulate synaptic efficacy and play an important role in long-term potentiation (Bahr et al., 1997; Chavis and Westbrook, 2001; Chan et al., 2003; Lin et al., 2003; Bernard-Trifilo et al., 2005). Although molecular mechanisms by which integrins affect synaptic efficacy have begun to emerge, their role in structural plasticity is poorly understood. Here, we show that integrins are involved in spine remodeling in cultured hippocampal neurons. The treatment of 14 d in vitro hippocampal neurons with arginine–glycine–aspartate (RGD)-containing peptide, an established integrin ligand, induced elongation of existing dendritic spines and promoted formation of new filopodia. These effects were also accompanied by integrin-dependent actin reorganization and synapse remodeling, which were partially inhibited by function-blocking antibodies against β1 and β3 integrins. This actin reorganization was blocked with the NMDA receptor (NMDAR) antagonist MK801 [(+)-5-methyl-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5,10-imine hydrogen maleate]. The Ca2+/calmodulin-dependent protein kinase II (CaMKII) inhibitor KN93 (N-[2-[N-(4-chlorocinnamyl)-N-methylaminomethyl]phenyl]-N-(2-hydroxyethyl)-4-methoxybenzenesulfonamide) also suppressed RGD-induced actin reorganization and synapse remodeling. Our findings show that integrins control ECM-mediated spine remodeling in hippocampal neurons through NMDAR/CaMKII-dependent actin reorganization.
Keywords: actin, dendritic spines, hippocampal neurons, integrin, NMDAR, CaMKII
Introduction
Dendritic spines are the postsynaptic sites of most excitatory synapses in the CNS (Harris, 1999; Hering and Sheng, 2001; Sheng, 2001). Dendritic spines develop from long, thin, finger-like dendritic extensions called filopodia (Ziv and Smith, 1996; Fiala et al., 1998). During the early stages of synaptogenesis, dendrites appear bristling with many filopodia that rapidly extend and retract within minutes (Dailey and Smith, 1996; Lendvai et al., 2000; Dunaevsky et al., 2001). As synapses form, the number of filopodia declines and more spines become established. Dendritic spines remain dynamic in the adult brain and can change in response to certain forms of long-term potentiation (LTP)-inducing stimuli (Yuste and Bonhoeffer, 2001; Carlisle and Kennedy, 2005; B. Lin et al., 2005). Actin filaments are thought to be the basis for both the formation of dendritic spines during development and their structural plasticity at mature synapses (Fischer et al., 2000; Matus et al., 2000).
Many cell surface molecules have been shown to influence spine properties in response to external signals by mediating cell adhesion, affecting Rho GTPases or altering Ca2+ fluxes that ultimately control actin dynamics (Ethell and Pasquale, 2005). Several of these molecules provide linkages between the presynaptic and postsynaptic membranes, whereas others connect these membranes to the extracellular matrix (ECM). Although the role of the ECM in synapses is not completely understood, many ECM components and their cell surface receptors have been shown to regulate synaptic plasticity (Dityatev and Schachner, 2003).
Integrins form a large family of heterodimeric (α/β subunits) membrane-spanning ECM receptors (Humphries, 2000; Van der Flier and Sonnenberg, 2001) that bind to an arginine–glycine–aspartate (RGD) motif found in many ECM proteins. This interaction activates a diverse array of intracellular signaling cascades, leading to reorganization of actin cytoskeleton (Ruoslahti, 1996; Aplin et al., 1998; Plow et al., 2000). Currently, 18 α subunits, 8 β subunits, and >20 different α/β heterodimers have been described, with several subunits localized to synapses (Einheber et al., 1996; Nishimura et al., 1998; Chan et al., 2003; Gall and Lynch, 2004). Recent studies have revealed that integrins can modulate NMDA receptor (NMDAR)-mediated synaptic currents and play an important role in LTP (Bahr et al., 1997; Chavis and Westbrook, 2001; Chun et al., 2001; Kramar et al., 2002; Chan et al., 2003; Lin et al., 2003; Bernard-Trifilo et al., 2005). The molecular mechanisms by which integrins affect synaptic efficacy have begun to emerge, but their role in structural plasticity is poorly understood.
Here, we show that integrins are involved in spine turnover in cultured hippocampal neurons. Treatment of 14 d in vitro (DIV) hippocampal neurons with RGD-containing peptide induced elongation of existing dendritic spines and promoted formation of new filopodia. These effects were accompanied by actin reorganization and synapse remodeling, which were partially inhibited by function-blocking antibodies against β1 and β3 integrins. RGD-induced actin reorganization was also blocked by an NMDAR antagonist (+)-5-methyl-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5,10-imine hydrogen maleate (MK801) and a Ca2+/calmodulin-dependent protein kinase II (CaMKII) inhibitor (N-[2-[N-(4-chlorocinnamyl)-N-methylaminomethyl]phenyl]-N-(2-hydroxyethyl)-4-methoxybenzenesulfonamide) (KN93), suggesting that ECM–integrin interactions can regulate spine remodeling through NMDAR/CaMKII-mediated actin reorganization.
Materials and Methods
Hippocampal neuron culture, transfection.
Cultures of hippocampal neurons were prepared from embryonic day 15 (E15) to E16 mouse embryos as previously described with modifications (Ethell et al., 2001). Briefly, after treatment with papain (0.5 mg/ml) and DNase (0.6 μg/ml) for 20 min at 37°C and mechanical dissociation, hippocampal cells were plated on coverslips coated with poly-dl-ornithine (0.5 mg/ml in borate buffer) and laminin (5 μg/ml in PBS). The cells were cultured in Neurobasal medium with 25 μm glutamine, 1% penicillin–streptomycin, and B27 supplement (Invitrogen, Carlsbad, CA) under 5% CO2/10% O2 atmosphere at 37°C. The hippocampal cultures were transiently transfected with green fluorescent protein (GFP) at 1–5 DIV, using the calcium phosphate method as previously described (Ethell et al., 2001).
Treatments.
For RGD treatments, 7 or 14 DIV mouse hippocampal neurons were treated with 500 μm RGD-containing peptide [Gly-Arg-Gly-Asp-Thr-Pro (GRGDTP)] (Calbiochem, La Jolla, CA; EMD Biosciences, San Diego, CA) or the control peptide RAD-containing peptide [Gly-Arg-Ala-Asp-Ser-Pro (GRADSP)] (Calbiochem; EMD Biosciences) for 1 h before analysis. For NMDA application, cultures were treated with 50 μm NMDA (M-102; Research Biochemicals International, Natick, MA) for 10 min in Mg2+-free HBSS containing 1.8 mm CaCl2 (Invitrogen), followed by removal of NMDA and the incubation of cultures in complete medium for 1 h before analysis. For MK801 studies, the cultures were pretreated with 10 μm MK801 (M-107; Research Biochemicals International) for 2 h before RGD/RAD treatment. For KN93 studies, 10 μm KN93 (K1385; Sigma, St. Louis, MO) or its control analog 2-[N-(4′-methoxybenzenesulfonyl)]amino-N-(4′-chlorophenyl)-2-propenyl-N-methylbenzylamine) (KN92) (K112; Sigma) was applied 30 min before RGD/RAD treatment. The function-blocking anti-β1 (10 μg/ml; Chemicon, Temecula, CA) and/or anti-β3 (10 μg/ml; Chemicon) integrin antibodies were applied to 14 DIV cultured hippocampal neurons 30 min before RGD treatment.
Immunostaining.
Cultures of hippocampal neurons at 7 or 14 DIV were fixed in 2% paraformaldehyde, permeabilized in 0.1% Triton X-100, and blocked in PBS containing 5% normal goat serum and 1% BSA. Dendritic spines and filopodia were visualized by GFP fluorescence. To detect polymerized filamentous actin (F-actin), the cultures were incubated with rhodamine-coupled phalloidin (1:40; R-415; Invitrogen). The primary antibodies used were the following: mouse anti-PSD-95 (33 μg/ml; clone 6G6; Affinity BioReagents, Golden, CO), rabbit anti-NMDAR2A/B (1 μg/ml; AB1548; Chemicon), rabbit anti-β3 integrin and rabbit anti-β5 integrin (20 μg/ml; AB1932 and AB1926; Chemicon), mouse anti-β1 integrin (20 μg/ml; MAB1987Z; Chemicon), mouse anti-MAP2 (5 μg/ml; clone HM-2; Sigma), rabbit anti-GAD (1 μg/ml; GAD-65; Chemicon), and mouse anti-synaptophysin (61 μg/ml; SVP-38; Sigma). The secondary antibodies used were the following: 4 μg/ml Alexa Fluor 488-conjugated chicken anti-mouse IgG, 4 μg/ml Alexa Fluor 594-conjugated donkey anti-rabbit IgG, 4 μg/ml Alexa Fluor 594-conjugated chicken anti-mouse IgG, 4 μg/ml Alexa Fluor 488-conjugated chicken anti-rabbit IgG, and 4 μg/ml Alexa Fluor 350-conjugated goat anti-rabbit IgG (all Invitrogen). Immunostaining was analyzed under a confocal laser-scanning microscope (model LSM 510; Carl Zeiss MicroImaging, Oberkochen, Germany) or an inverted fluorescent microscope (model TE2000; Nikon, Melville, NY).
Live imaging.
We monitored the morphology of dendritic spines and filopodia in 7 or 14 DIV GFP-expressing neurons before and after the application of RGD or control RAD peptides. Time-lapse imaging was performed under an inverted fluorescent microscope (model TE2000; Nikon) with a 40× air Fluor objective and monitored by a 12-bit CCD camera (model ORCA-AG; Hamamatsu, Hamamatsu City, Japan) using Image-Pro software (Media Cybernetics, Silver Spring, MD). During imaging, the cultures were maintained in Hank’s solution supplemented with 1.8 mm CaCl2, 0.45% glucose, and 0.1% BSA at 37°C and 5% CO2, and images were captured at 3 min intervals for 1 h. Two baseline images were captured before RGD or RAD application.
Image analysis.
The effects of RGD on dendritic morphology were examined in 7 or 14 DIV GFP-expressing hippocampal neurons as previously described (Ethell and Yamaguchi, 1999). Briefly, experimental and control samples were encoded for blind analysis, and GFP-expressing pyramidal neurons were randomly selected and imaged with a confocal laser-scanning microscope (model LSM 510; Carl Zeiss MicroImaging). Note that nonspiny neurons were excluded from the analysis. The proximal dendrites (identified as MAP2-positive processes extending from the neuronal cell body and at least 1 μm in diameter) were selected for analysis of the length and number of dendritic protrusions. Hidden protrusions that protruded toward the back or front of the viewing plane were not counted. Fifteen GFP-expressing neurons were randomly selected for each experimental group, and three to five proximal dendrites per each neuron were analyzed (∼2000 μm of total dendritic length per group). The length of protrusion was determined by measuring the distance between its tip and the base using Image-Pro Plus software. Experimental values represent mean ± SEM. Three independent experiments were performed for each condition. Statistical analysis was performed using Microsoft (Redmond, WA) Excel. Statistical differences between RAD-treated control and RGD-treated experimental groups of dendritic protrusions were compared by Student’s t test. Statistical differences for multiple groups were assessed by one-way ANOVA followed by Newman–Keuls post hoc tests.
For analysis of F-actin organization, experimental and control samples were encoded for blind analysis; spiny pyramidal hippocampal neurons labeled with rhodamine-coupled phalloidin were randomly selected; and images were taken under a confocal laser-scanning microscope (model LSM 510; Carl Zeiss MicroImaging). Note that nonspiny GABAergic neurons, identified by immunostaining for glutamic acid decarboxylase with anti-GAD65 antibody, were excluded from the analysis. For quantification of F-actin staining, inverted fluorescent images (single channel) were analyzed using Image-Pro Plus software. F-actin clusters (branched F-actin) were defined operationally as 0.5–2.0 μm F-actin-enriched puncta along dendrites (with an average pixel intensity at least 50% above that in the adjacent dendritic region) and were counted manually (see Fig. 4). Ten to 15 neurons were randomly selected for each experimental group, and three to four proximal dendrites per each neuron were analyzed (∼2000 μm of total dendritic length per group). Experimental values represent the mean ± SEM. Three independent experiments were performed for each condition. Statistical differences for multiple groups were assessed by one-way ANOVA followed by Newman–Keuls post hoc tests.
Figure 4.

RGD induced actin reorganization and synapse remodeling in 14 DIV hippocampal neurons. A–C, Confocal images of 14 DIV mouse hippocampal neurons untreated (A), or treated with 500 μm RAD-containing peptide (B) or with 500 μm RGD-containing peptide (C). Detection of polymerized F-actin with rhodamine-coupled phalloidin (red) and presynaptic boutons by synaptophysin immunostaining (green) is shown. Bottom panels show inverted fluorescent images of dendritic fragments with characteristic F-actin labeling for each group. Control untreated (A) or control RAD-treated (B) 14 DIV neurons demonstrated F-actin clusters in close proximity to synaptophysin-positive presynaptic boutons. RGD-treated (C) neurons showed a significant reduction in the number of F-actin puncta (circled), the appearance of hair-like extensions (arrowheads), and a decreased number of synaptophysin-positive terminals. Moreover, rope-like actin bundles were seen in the dendritic shaft of RGD-treated neurons (arrows). Scale bar: top panels, 10 μm; bottom panels, 5 μm. D, Quantitative analysis of the number of F-actin, synaptophysin, and actin/synaptophysin double-positive clusters per 10 μm of dendrite. RGD-treated neurons showed reduction in the number of F-actin clusters, synaptophysin-positive presynaptic boutons, and spiny synapses. Data represent the average number of clusters per 10 μm of dendrite. Error bars indicate SEM (n = 15 neurons per group). ***p < 0.001 with one-way ANOVA.
Biochemical assays.
For immunoprecipitation, 14 DIV hippocampal neurons were treated with 500 μm RGD or RAD and lysed in ice-cold TBS (25 mm Tris, pH 7.4; 0.15 mm NaCl) containing 1% Triton X-100, 5 mm EDTA, 0.5 mm pervanadate, and protease inhibitor mixture (Sigma). Cell lysates were cleared by centrifugation at 12,000 × g. Protein lysates were incubated with 2 μg of anti-NR2A/B antibody and protein A-Sepharose beads (Sigma) for 4 h at 4°C. The beads were washed three times with ice-cold lysis buffer. Bound materials were eluted with SDS-PAGE sample buffer, resolved on 8–16% Tris-glycine gels, and transferred onto nitrocellulose membranes. NMDAR phosphorylation on tyrosine was detected by HRP-conjugated anti-phosphotyrosine antibody (PY20; BD Transduction Laboratories, Bedford, MA) with Super Signal West Pico chemiluminescent substrate (Pierce Chemical, Rockford, IL). Equal loading was confirmed by immunodetection of NR2A/B.
Subcellular fractionation of adult mouse hippocampi was performed as previously described with modification (Ethell et al., 2000; Kramar et al., 2002). Briefly, hippocampi were homogenized with cold 4 mm HEPES, pH 7.4, containing 0.32 m sucrose and protease inhibitor mixture (Sigma). Homogenates (H) were cleared at 1000 × g for 10 min. The supernatant was collected and centrifuged at 16,000 × g for 15 min, yielding the pellet, a crude synaptosome fraction (P2). The P2 was resuspended in homogenization buffer to yield a protein concentration of 1.5 mg/ml and represented crude synaptosome fraction. The crude synaptosome fraction was lysed by osmotic shock by adding 10 vol of ice-cold water containing protease inhibitors. The synaptic membrane fraction (LP1) was pelletted from this homogenate by centrifugation at 48,000 × g for 30 min and resuspended in homogenization buffer to yield a protein concentration of 1.5 mg/ml. Protein concentration was determined using the BCA Protein Assay kit (Pierce Chemical). Equal amounts of protein from each fraction (12 μg) were resolved by SDS-PAGE on an 8–16% Tris-glycine gel (Invitrogen), transferred to a nitrocellulose membrane, and analyzed by immunoblotting. The proteins of interest were detected using specific primary antibody and corresponding secondary HRP-conjugated antibody with Super Signal West Pico chemiluminescent substrate (Pierce Chemical). The primary antibodies used were the following: rabbit anti-β3 integrin and rabbit anti-β5 integrin (2 μg/ml; AB1932, AB1926; Chemicon), rabbit anti-NMDAR2A/B (1 μg/ml; AB1548; Chemicon), and mouse anti-PSD-95 (33 μg/ml; clone 6G6; Affinity BioReagents). The secondary antibodies used were the following: HRP-conjugated goat anti-rabbit (0.08 μg/ml; Jackson ImmunoResearch, West Grove, PA) and HRP-conjugated donkey anti-mouse (0.08 μg/ml; Jackson ImmunoResearch).
Results
Synaptic localization of β1, β3, and β5 integrin subunits in hippocampalneurons
We first addressed the cellular distribution of integrins in 14 DIV hippocampal neurons by immunostaining cultures for several β integrin subunits. β3 and β5 integrin subunits were found in close proximity to synaptophysin-positive presynaptic boutons (Fig. 1A,B). More specifically, β3 integrin-immunopositive puncta colocalized with postsynaptic density protein of 95 kDa (PSD-95) (Fig. 1C) and GFP-labeled dendritic spines (Fig. 1D). The localization of β1 and β3 subunits in dendritic spines was also confirmed by labeling polymerized F-actin with rhodamine-coupled phalloidin (Fig. 1E,F). Furthermore, β3 and β5 integrin subunits were detected in crude synaptosome and synaptic membrane fractions of adult mouse hippocampus with a fractionation pattern that was similar to NMDAR and PSD-95 (Fig. 1G). Thus, our data show that integrins are accumulated in dendritic spines/synapses of hippocampal neurons in vitro and in vivo.
Figure 1.

Integrin subunits β1, β3, and β5 are localized in synapses of hippocampal neurons. A, B, Immunofluorescent labeling of 14 DIV hippocampal neurons showed localization of β3 (red; A) and β5 (red; B) in close proximity to synaptophysin-positive presynaptic boutons (green). Arrows indicate colocalization of integrins (red) and synaptophysin (green). Scale bars: top panel, 10 μm; bottom panel, 5 μm. C, Coimmunolabeling of β3 integrin clusters (red) and postsynaptic scaffold protein PSD-95 (green) in 14 DIV hippocampal neurons. β3-Immunopositive puncta (red) colocalized with PSD-95 (green). Arrows point to examples of synaptic integrin clusters. Scale bars: top panel, 10 μm; bottom panel, 5 μm. D, Immunofluorescent labeling of β3 integrin (red) in GFP-expressing 14 DIV hippocampal neurons. Hippocampal neurons were transfected with GFP at 5 DIV. At 14 DIV, hippocampal neurons were immunolabeled with anti-β3 integrin antibody. Arrows point to examples of β3 integrin clusters (red) that are localized in dendritic spines (green). Scale bars: top panel, 10 μm; bottom panel, 5 μm. E, F, Immunofluorescent labeling of 14 DIV hippocampal neurons shows localization of β3 (green; E) and β1 (green; F) in close proximity to polymerized actin (F-actin) that is labeled with rhodamine-coupled phalloidin (red). Arrows indicate colocalization of integrins (green) and F-actin (red). Scale bars: top panel, 10 μm; bottom panel, 5 μm. G, Western blot analysis of the subcellular distribution of β3 and β5 integrins in adult mouse hippocampus. Subcellular fractions were prepared from adult mouse hippocampus as described in Materials and Methods. Equal amounts (12 μg) of protein from each fraction were resolved on an 8–16% Tris-glycine gels and immunoblotted with specific antibodies against β3, β5, NR2A/B, and PSD-95. H, Homogenate; P2, crude synaptosome fraction; LP1, presynaptic and postsynaptic membrane fraction.
RGD induces dendritic spine elongation and promotes new filopodia formation in 14 DIV hippocampalneurons
To determine whether integrins contribute to synaptic plasticity by mediating ECM-induced dendritic spine remodeling, we mimicked the effects of ECM on integrin signaling in cultured hippocampal neurons by applying an RGD-containing peptide and then examined its effects on dendritic spine morphology. During the first week in vitro, hippocampal neurons extend many dendritic filopodia that subsequently transform into dendritic spines between 7 and 14 DIV. As dendritic spines form, the number of long, thin dendritic filopodia declines. At 14 DIV, most dendritic protrusions are spines and exhibit a characteristic mushroom-like morphology (Fig. 2A). Treatment of 14 DIV hippocampal neurons with 500 μm RGD-containing peptide induced elongation of existing dendritic spines, altered dendritic spine morphology, and increased the number of filopodia-like protrusions (Fig. 2C,F), whereas treatment with control RAD-containing peptide resulted in no changes in dendritic spine morphology or length (Fig. 2B,E). Quantitative analysis revealed that the changes in overall dendritic protrusion length were statistically significant, with an average protrusion length of 1.88 ± 0.03 μm in control untreated neurons, 1.85 ± 0.04 μm in control RAD-treated neurons, and 2.46 ± 0.05 μm in RGD-treated neurons (mean ± SEM; p < 0.001). A significant increase in mean protrusion length was first seen ∼30 min after RGD treatment (p < 0.05) (Fig. 2H). There were also more protrusions per unit length of dendrite in RGD-treated neurons compared with the control RAD-treated or control untreated neurons. RGD-treated neurons showed 3.69 ± 0.27 protrusions per 10 μm of dendrite, whereas RAD-treated neurons had 2.32 ± 0.19 protrusions and untreated neurons 2.24 ± 0.11 protrusions per 10 μm of dendrite (mean ± SEM; p < 0.001) (Fig. 2I). Time-lapse imaging also revealed a significant increase in the number of dendritic protrusion ∼30 min after RGD application (p < 0.05) (Fig. 2J).
Figure 2.
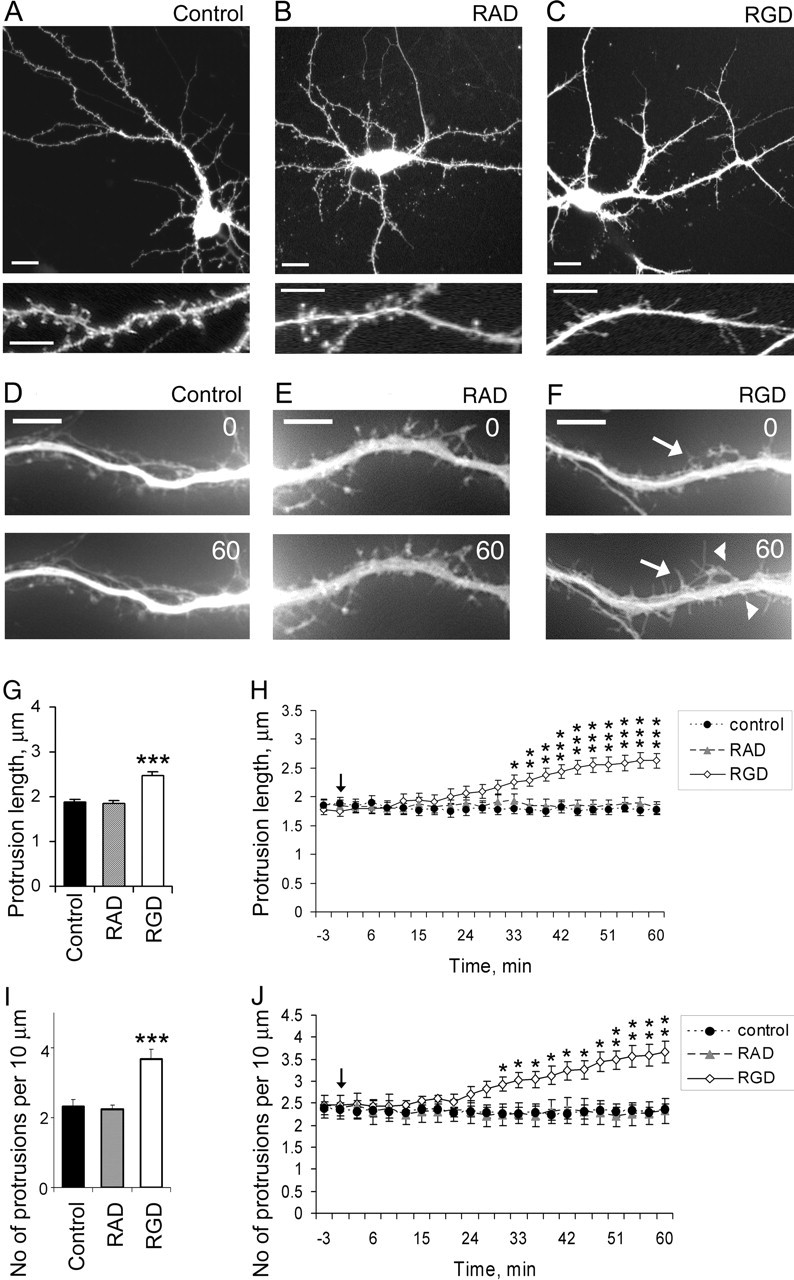
Dendritic spine elongation and new filopodia formation are induced by RGD-containing peptide in 14 DIV hippocampal neurons. A–C, Confocal images of GFP-labeled 14 DIV mouse hippocampal neurons untreated (A), treated with 500 μm control RAD-containing peptide (B), or treated with 500 μm RGD-containing peptide (C). GFP fluorescence was used to visualize dendritic morphology. RGD-treated neurons showed increased number of filopodia-like protrusions and increased length of dendritic spines, whereas neurons treated with control RAD-containing peptide showed no changes in dendritic spine morphology or length. Scale bars: top panel, 10 μm; bottom panel, 5 μm. D–F, Live images of GFP-labeled dendritic spines in 14 DIV hippocampal neurons before (0 min) and after (60 min) treatment with blank (D), control RAD (E), or RGD (F). RGD-treated neurons showed formation of new filopodia (arrowheads) and elongation of existing dendritic spines (arrows). G, H, Quantification of dendritic protrusion length in 14 DIV GFP-labeled hippocampal neurons before and after treatment with blank (control), control RAD, or RGD. G, Dendritic protrusions were significantly longer in RGD-treated neurons. Data represent the mean protrusion length. Error bars indicate SEM (n = 15 neurons per group). H, The time course of the elongation of dendritic protrusions showed a significant increase in mean protrusion length starting ∼30 min after RGD treatment. Live images were taken at 3 min intervals for 1 h. The arrow indicates RGD/RAD application. Data represent the mean protrusion length. Error bars indicate SEM (n = 5 neurons per group). *p < 0.05; **p < 0.01; ***p < 0.001 with one-way ANOVA. I, J, Quantification of dendritic protrusion density in 14 DIV GFP-labeled hippocampal neurons before and after treatment with blank (control), control RAD, or RGD. I, There were more dendritic protrusions in RGD-treated neurons. Data represent the average number of protrusions per 10 μm of dendrite. Error bars indicate SEM (n = 15 neurons per group). J, Time-lapse imaging has revealed a significant increase in the number of dendritic protrusion ∼30 min after RGD application. Live images were taken at 3 min interval for 1 h. The arrow indicates RGD/RAD application. Data represent the average number of protrusions per 10 μm of dendrite. Error bars indicate SEM (n = 5 neurons per group). *p < 0.05; **p < 0.01; ***p < 0.001 with one-way ANOVA.
The ability of RGD-containing peptide, an established integrin ligand, to induce the elongation of existing dendritic spines and promote new filopodia formation suggests that integrins play a role in dendritic spine remodeling.
RGD promotes new filopodia formation and elongation in 7 DIV hippocampalneurons
In addition to activating integrins, RGD-containing peptide may also interfere with endogenous ECM–integrin interactions at already established synapses in 14 DIV hippocampal neurons (Ruoslahti, 1996; Blystone, 2002; Bernard-Trifilo et al., 2005). To address this issue, we also examined RGD effects on hippocampal neurons at 7 DIV, before the appearance of established synapses. The 7 DIV hippocampal neurons usually show many dendritic spine precursors, filopodia, that rapidly extend and retract searching for axonal partners. RGD treatment of 7 DIV hippocampal neurons induced filopodia elongation and promoted new filopodia formation similar to 14 DIV cultures (Fig. 3C,F). The filopodia were longer in RGD-treated neurons with a mean length of 3.94 ± 0.14 μm compared with 2.75 ± 0.09 μm in control untreated neurons and 2.90 ± 0.11 μm in control RAD-treated neurons (mean ± SEM; p < 0.001) (Fig. 3G). A significant elongation of dendritic filopodia in 7 DIV hippocampal neurons was first seen ∼30 min after RGD treatment (p < 0.05) (Fig. 3H). There were also more filopodia per unit length of dendrite in RGD-treated neurons compared with RAD-treated or untreated neurons. The number of filopodia was 3.28 ± 0.19 per 10 μm of dendrite in control untreated neurons, 3.18 ± 0.22 in control RAD-treated neurons, and 5.48 ± 0.46 in RGD-treated neurons (mean ± SEM; p < 0.001) (Fig. 3I). Time-lapse imaging also revealed an extension of new filopodia as soon as ∼15 min after RGD application (p < 0.05) (Fig. 3J).
Figure 3.
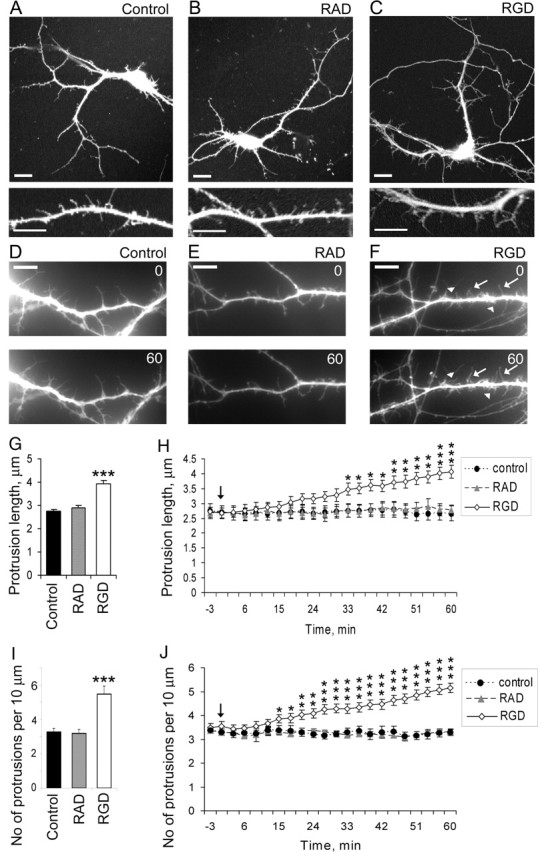
RGD induced filopodia elongation and promoted new filopodia formation in 7 DIV hippocampal neurons. A–C, Confocal images of GFP-labeled 7 DIV mouse hippocampal neurons untreated (A), treated with 500 μm control RAD-containing peptide (B), or treated with 500 μm RGD-containing peptide (C). GFP fluorescence was used to visualize dendritic morphology. RGD-treated neurons showed increased length and number of filopodia-like protrusions, whereas neurons treated with control RAD-containing peptide showed no changes in dendritic filopodia number or length. Scale bars: top panel, 10 μm; bottom panel, 5 μm. D–F, Live images of GFP-labeled dendritic spines in 7 DIV hippocampal neurons before (0 min) and after (60 min) treatment with blank (D), control RAD (E), or RGD (F). RGD-treated neurons showed formation of new (arrowheads) and elongation of existing filopodia (arrows). G, H, Quantification of dendritic protrusion length in 7 DIV GFP-labeled hippocampal neurons before and after treatment with blank (control), control RAD, or RGD. G, Dendritic protrusions were significantly longer in RGD-treated neurons. Data represent the mean protrusion length. Error bars indicate SEM (n = 15 neurons per group). H, Time course of the elongation of dendritic protrusions showed a significant increase in mean protrusion length starting ∼20 min after RGD treatment. Live images were taken at 3 min intervals for 1 h. The arrow indicates RGD/RAD application. Data represent the mean protrusion length. Error bars indicate SEM (n = 5 neurons per group). *p < 0.05; **p < 0.01; ***p < 0.001 with one-way ANOVA. I, J, Quantification of dendritic protrusion density in 7 DIV GFP-labeled hippocampal neurons before and after treatment with blank (control), control RAD, or RGD. I, There were more dendritic protrusions in RGD-treated neurons. Data represent the average number of protrusions per 10 μm of dendrite. Error bars indicate SEM (n = 15 neurons per group). J, Time-lapse imaging revealed a significant increase in the number of dendritic protrusion ∼20 min after RGD application. Live images were taken at 3 min intervals for 1 h. The arrow indicates RGD/RAD application. Data represent the average number of protrusions per 10 μm of dendrite. Error bars indicate SEM (n = 5 neurons per group). *p < 0.05; **p < 0.01; ***p < 0.001 with one-way ANOVA.
These results show that RGD-containing peptide induced the elongation of filopodia and promoted new filopodia formation in 7 DIV hippocampal neurons.
RGD induces actin reorganization and synapse remodeling in 14 DIV hippocampalneurons
The actin cytoskeleton plays a major role in the morphological development and plasticity of dendritic spines and filopodia (Matus et al., 1982; Cohen et al., 1985; Smart and Halpain, 2000). Therefore, we examined whether changes in actin cytoskeleton underlie RGD-induced remodeling of existing dendritic spines or formation of new filopodia. To observe RGD effects on actin organization in dendritic spines, we visualized polymerized F-actin with rhodamine-coupled phalloidin and adjacent presynaptic terminals with immunostaining for synaptophysin. In the early stages of spine development, 7 DIV hippocampal neurons extend many motile thin filopodia, which are driven by linear organized F-actin polymers appearing as hair-like extensions along the dendrite, whereas most F-actin polymers are found within the dendritic shaft (supplemental Fig. 1A, available at www.jneurosci.org as supplemental material). As hippocampal neurons mature, F-actin becomes concentrated in spine heads forming highly branched, stable structures, which appear as intense puncta along the dendrite (supplemental Fig. 1B, available at www.jneurosci.org as supplemental material) (Fig. 4A). At 14 DIV, most pyramidal hippocampal neurons show dendritic spines with spine heads intensively stained with rhodamine-coupled phalloidin, many of which are adjacent to presynaptic boutons, and a small proportion of F-actin staining in the dendritic shaft (Fig. 4A). We found that RGD treatment of 14 DIV cultures induced striking rearrangements of F-actin, including decreased F-actin puncta in dendritic spines, as well as the appearance of hair-like linear organized actin in filopodia and the formation of rope-like actin bundles within the dendritic shaft (Fig. 4C,D). Moreover, immunolabeling of synaptophysin-positive presynaptic boutons revealed a decrease in the number of synapses on dendritic spines (spiny synapses) and an increase in the proportion of synapses located directly on the dendritic shaft after RGD treatment, suggesting that RGD induced a redistribution of presynaptic sites from dendritic spines to the dendritic shaft (Fig. 4C,D).
Thus, RGD treatment induced the reorganization of F-actin in dendritic spines, concurrent with their transformation into motile filopodia-like protrusions and the formation of bundle-like actin polymers in the dendritic shaft.
NMDAR blockade with MK801 prevents RGD-induced actin reorganization at synapses and dendritic spineremodeling
Previous studies have established that RGD enhances Ca2+ influx through NMDAR in hippocampal slices (Lin et al., 2003; Bernard-Trifilo et al., 2005). Our studies suggested that RGD treatment of 14 DIV hippocampal neurons might also induce NMDAR activation given increased tyrosine phosphorylation of NR2A/B subunit of NMDAR after RGD treatment (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). Moreover, NMDAR activation with a low concentration of NMDA resulted in actin changes that were similar to actin reorganizations seen in RGD-treated cultures, such as the formation of rope-like actin bundles within the dendritic shaft (Fig. 5F) (Halpain et al., 1998; Hering and Sheng, 2003).
Figure 5.
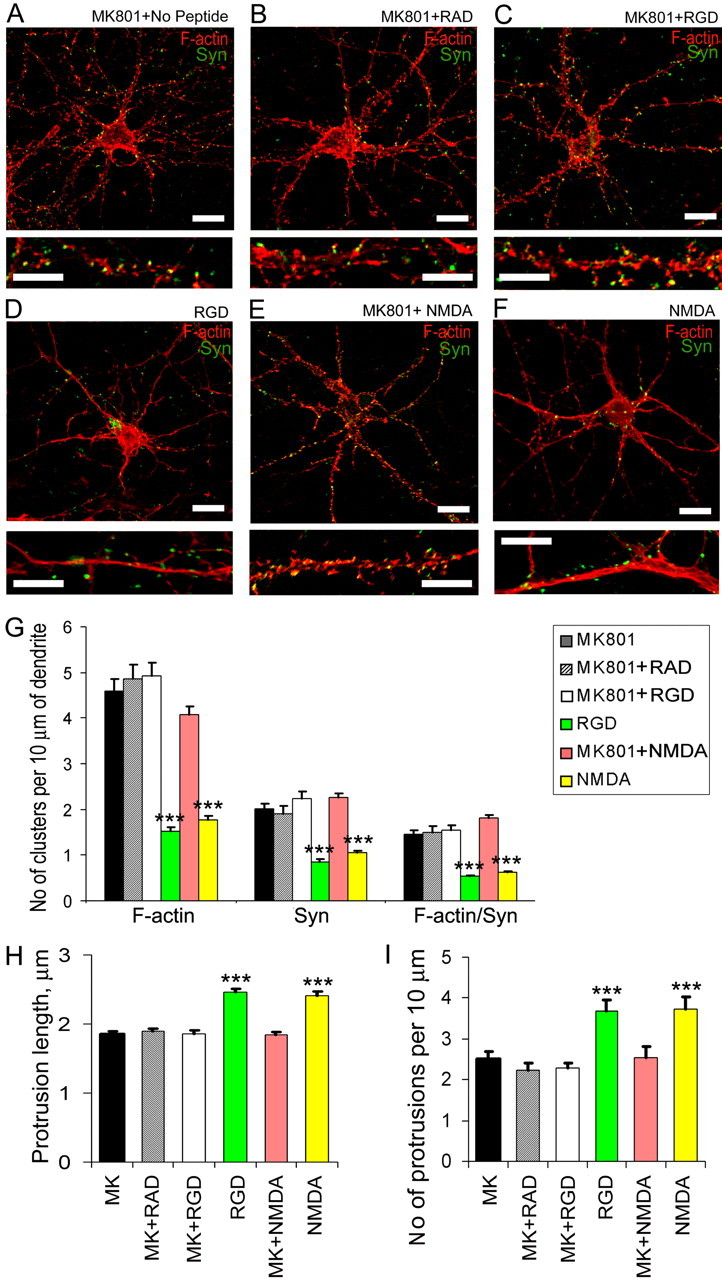
Blockade of NMDAR with MK801 prevented RGD-induced actin reorganization at synapses and dendritic spine remodeling. A–F, Confocal images of 14 DIV hippocampal neurons from cultures treated with MK801 (A), MK801 plus RAD (B), MK801 plus RGD (C), RGD (D), MK801 plus NMDA (E), or NMDA (F). Detection of polymerized F-actin with rhodamine-coupled phalloidin (red) and presynaptic boutons by synaptophysin immunostaining (green) is shown. NMDAR blockade with its antagonist MK-801 (10 μm) prevented both RGD-induced and NMDA-mediated actin rearrangements. Scale bar: top panels, 10 μm; bottom panels, 5 μm. G, Quantitative analysis of the number of F-actin, synaptophysin, and actin/synaptophysin double-positive clusters per 10 μm of dendrite. Treatment with MK801 blocked RGD-induced and NMDA-mediated reduction in the number of F-actin clusters, synaptophysin-positive presynaptic boutons, and spiny synapses. Data represent the average number of clusters per 10 μm of dendrite. Error bars indicate SEM (n = 10 neurons per group). ***p < 0.001 with one-way ANOVA. H, I, Quantification of dendritic protrusion length (H) and number (I) in 14 DIV GFP-labeled hippocampal neurons after treatment with MK801, MK801 plus RAD, MK801 plus RGD, RGD, MK801 plus NMDA, or NMDA. Treatment with MK801 blocked RGD-induced and NMDA-mediated dendritic spine elongation and new filopodia extension. Data represent the mean protrusion length (H) or average number of protrusions per 10 μm of dendrite (I). Error bars indicate SEM (n = 10 neurons per group). ***p < 0.001 by one-way ANOVA.
To test whether the activation of NMDAR is responsible for RGD-induced actin reorganization, we examined the effects of RGD-containing peptide on actin organization and synapses in the presence of specific NMDAR antagonist MK-801. NMDAR blockade with MK-801 (10 μm) prevented both RGD-induced actin re-arrangements and synapse remodeling (Fig. 5C). In the presence of MK-801, RGD-treated hippocampal neurons showed no changes in the number of actin clusters or synaptophysin-positive puncta compared with untreated or control RAD-treated neurons (Fig. 5G). Moreover, MK-801 also suppressed RGD-induced dendritic spine remodeling and the extension of new filopodia (Fig. 5H) (supplemental Figs. 3 and 4, available at www.jneurosci.org as supplemental material). Our findings reveal that RGD effects on actin organization and dendritic spine morphology in 14 DIV hippocampal neurons were mediated by NMDAR.
CaMKII inhibitor KN93 prevents RGD-induced actin reorganization atsynapses
The elevation of intracellular Ca2+ levels through NMDAR can activate CaMKII, which is highly expressed in neurons, and can bind to F-actin and regulate its polymerization (Shen et al., 1998; Lisman et al., 2002; Petersen et al., 2003). To investigate whether RGD-induced actin reorganization depends on NMDAR-mediated activation of CaMKII, we examined the effects of RGD on actin organization in the presence of the CaMKII inhibitor KN93, which specifically targets the calmodulin binding site of CaMKII. Treatment of 14 DIV hippocampal neurons with KN93, but not its inactive analog KN92, prevented actin reorganization and synapse remodeling induced by RGD application (Fig. 6).
Figure 6.
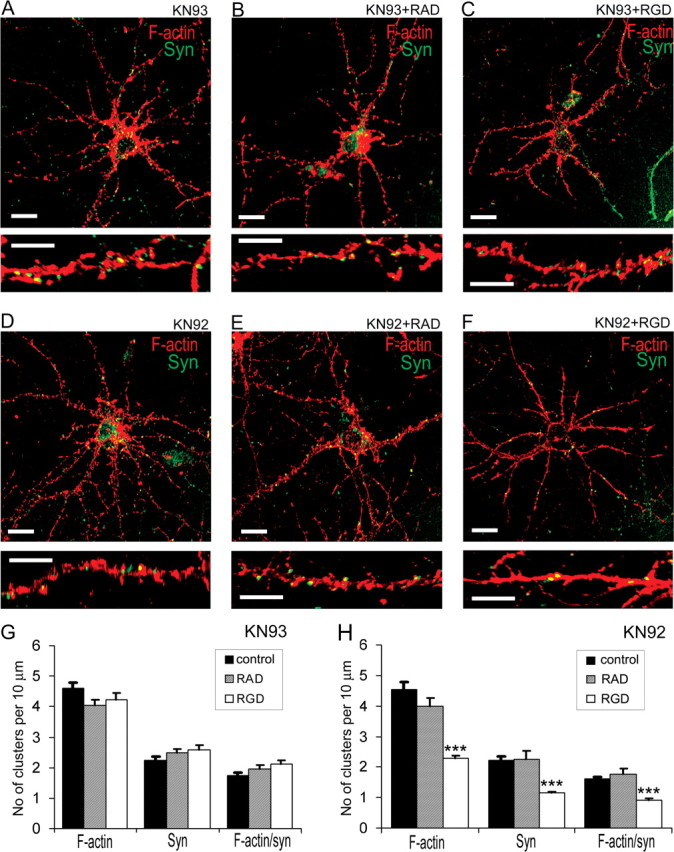
CaMKII inhibitor KN93 prevented the actin reorganization induced by RGD in 14 DIV hippocampal neurons. A–F, Confocal images of 14 DIV hippocampal neurons from cultures treated with KN93 (A), KN93 plus RAD (B), KN93 plus RGD (C), KN92 (D), KN92 plus RAD (E), or KN92 plus RGD (F). Detection of polymerized F-actin with rhodamine-coupled phalloidin (red) and presynaptic boutons by synaptophysin immunostaining (green) is shown. Pretreatment with CaMKII inhibitor KN93, but not its inactive homolog KN92, blocked RGD-induced actin reorganization. G, H, Quantitative analysis of the number of F-actin, synaptophysin, and actin/synaptophysin double-positive clusters per 10 μm of dendrite. Treatments with KN93 (G), but not KN92 (H), blocked RGD-induced reduction in the number of F-actin clusters, synaptophysin-positive presynaptic boutons, and spiny synapses. Data represent the average number of clusters per 10 μm of dendrite. Error bars indicate SEM (n = 10 neurons per group). ***p < 0.001 with one-way ANOVA.
These findings show that RGD induced changes in actin cytoskeleton through NMDAR-mediated activation of CaMKII, suggesting that ECM–integrin interactions can regulate spine remodeling through NMDAR/CaMKII-mediated actin reorganization.
Function-blocking antibodies against β1 and β3 integrin partially block RGD-induced actin reorganization and dendritic spine remodeling
To verify whether RGD-induced actin reorganization and spine remodeling are integrin mediated and to gain information on the specific integrins involved, we selectively inhibited RGD-induced activation of β1 and/or β3 subunits of integrins with function-blocking antibodies. As illustrated in Figure 7, function-blocking anti-β1 and anti-β3 antibodies partially inhibited RGD-induced actin reorganization, dendritic spine elongation, and new filo-podia extension. In the presence of both function-blocking antibodies, neurons showed no changes in the number of synapses or dendritic spines and only a minor decrease in F-actin puncta along the dendrites of RGD-treated neurons (Fig. 7). These results suggest that β1 and β3 integrin subunits play important role in RGD-mediated actin reorganization and synapse remodeling.
Figure 7.
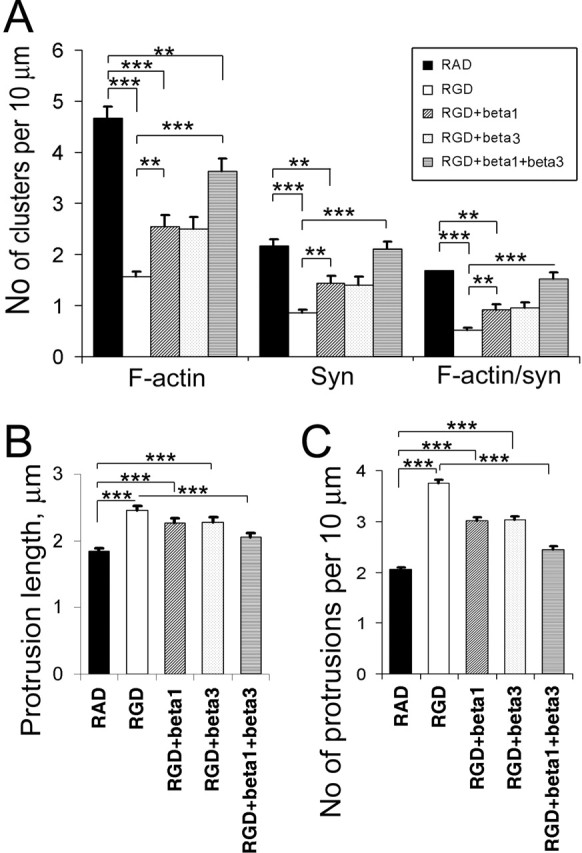
Function-blocking antibodies against β1 and β3 integrin partially blocked RGD-induced actin reorganization and dendritic spine elongation. A, Quantitative analysis of the number of F-actin, synaptophysin, and actin/synaptophysin double-positive clusters per 10 μm of dendrite. Data represent the average number of clusters per 10 μm of dendrite. Error bars indicate SEM (n = 10 neurons per group). **p < 0.01; ***p < 0.001 by one-way ANOVA. B, C, Quantification of dendritic protrusion length (B) and number (C) in 14 DIV GFP-labeled hippocampal neurons after treatment with RAD, RGD, RGD plus anti-β1 antibody, RGD plus anti-β3 antibody, or RGD plus anti-β1 and anti-β3 antibodies. Data represent the mean protrusion length (B) or average number of protrusions per 10 μm of dendrite (C). Error bars indicate SEM (n = 10 neurons per group). ***p < 0.001 by one-way ANOVA.
Discussion
This study revealed that integrins, which are the major ECM receptors, are involved in dendritic spine turnover and actin reorganization in cultured hippocampal neurons. Treatment of 14 DIV hippocampal neurons with RGD-containing peptide induced the elongation of existing dendritic spines and promoted the formation of new filopodia. These effects were also accompanied by actin reorganization and synapse remodeling, which were partially blocked by function-blocking antibodies against β1 and β3 integrins. Moreover, NMDAR antagonist MK801 and CaMKII inhibitor KN93 suppressed RGD-induced actin reorganization, suggesting that integrins control spine remodeling through NMDAR/CaMKII-dependent actin reorganization.
Extracellular space in the CNS is thought to be occupied by a prominent perineuronal net that is enriched in ECM molecules (Bruckner et al., 1993). ECM surrounds neuronal cell bodies and dendrites and extends into the synaptic cleft (Tian et al., 1997; Pappas et al., 2001). Although the role of ECM in synapses is not completely understood, many ECM components have been shown to regulate synaptic plasticity, including laminin, proteoglycans (Tenascin-R, Tenascin-C, Brevican, and Neurocan) and glycoproteins (for review, see Dityatev and Schachner, 2003). The effects of ECM molecules on synaptic efficacy depend, at least in part, on their interactions with integrins, which are localized on the synaptic membrane and activate intracellular signaling events.
Different combinations of α and β integrin heterodimers recognize several ECM molecules (fibronectin, laminin, vitronectin, and tenascin) in most cases by binding to the small peptide sequence RGD motif common to these molecules. Such interactions induce integrin-mediated reorganization of the submembrane actin cytoskeleton and initiate an array of intracellular signaling cascades modulating diverse cellular responses, including cell proliferation, motility, migration, spreading, survival, and differentiation (Ruoslahti, 1996; Aplin et al., 1998; Plow et al., 2000). With the exception of α9 and αE, all of the integrin subunits are expressed in the brain, although differentially across subdivisions (Pinkstaff et al., 1999; Bi et al., 2001; Kramar et al., 2002; Chan et al., 2003; Gall and Lynch, 2004; C. Y. Lin et al., 2005). Several types of integrin subunits have been found in synapses, including α3, α5, αv, α8, β1, β3, and β8. For example, α8 and β8 were localized in dendritic spines of pyramidal neurons, where they are associated with the postsynaptic density (Einheber et al., 1996; Nishimura et al., 1998), and α3, α5, and β1 were identified in synaptosomes (Pinkstaff et al., 1999; Kramar et al., 2002; Chan et al., 2003).
Our studies show that integrin subunits β1, β3, and β5 are present in synapses/dendritic spines of 14 DIV hippocampal neurons and that at least the first two of these exert potent effects on cytoskeletal organization and morphology at these sites. Specifically, treatment of hippocampal neurons with the integrin ligand RGD caused rapid (30 min) and pronounced changes in dendritic spine morphology as well as in the organization and distribution of F-actin. These effects were largely suppressed by neutralizing antibodies directed at the β1 or β3 subunits, with strongest effects obtained using both antibodies.
RGD-containing peptide is known to mimic integrin activation (Blystone, 2002; Bernard-Trifilo et al., 2005), although some studies suggest that it could interfere with endogenous binding of integrins to elements of ECM (Ruoslahti, 1996). Therefore, RGD treatment of 14 DIV hippocampal neurons could induce integrin signaling and at the same time inhibit endogenous integrin binding to ECM at already established synapses, which may result in synaptic remodeling. Interestingly, RGD treatment of young 7 DIV hippocampal neurons, which lack established synapses and spines, also induced new filopodia formation similar to established 14 DIV hippocampal cultures, suggesting that RGD effects on filopodia extension resulted from integrin activation. The ability of RGD to induce filopodia extension in both 7 and 14 DIV hippocampal neurons, together with the ability of function-blocking anti-integrin antibodies to inhibit RGD-mediated F-actin reorganization and synapse remodeling, suggests that these effects are mediated by RGD-induced activation of integrins in our cultures.
Based on previous reports showing that a rapid extension of new filopodia at the site of synaptic stimulation required activation of NMDA receptors (Engert and Bonhoeffer, 1999; Maletic-Savatic et al., 1999), we tested whether RGD effects on actin reorganization and dendritic spine remodeling in 14 DIV hippocampal neurons also depended on NMDAR activation. Our results showed that blocking the NMDAR channels with MK-801 (10 μm) prevented both RGD-induced actin rearrangements and dendritic spine remodeling. The application of RGD-containing peptide to hippocampal slices has been reported to produce a twofold increase in the amplitude and duration of NMDAR-gated synaptic currents, causing a transient influx of Ca2+ into dendritic spines (Lin et al., 2003; Bernard-Trifilo et al., 2005). In dendritic spines, Ca2+ functions both as a charge carrier and as a signaling molecule that influences the activities of many proteins, including several actin regulatory proteins (Yuste et al., 2000; Nimchinsky et al., 2002). Increases in intracellular Ca2+ can have opposing effects on spine morphology depending on magnitude and duration (Segal et al., 2000). Moderate and transient elevations in intraspine Ca2+ through NMDAR has been shown to induce spine elongation (Korkotian and Segal, 1999). These increases can also activate CaMKII, which is bound to F-actin and enriched in dendritic spines (Shen et al., 1998). Our findings show that a CaMKII inhibitor can prevent RGD-induced actin reorganization and synapse remodeling in 14 DIV hippocampal cultures, suggesting that integrin-mediated actin reorganization at synapses was dependent on CaMKII.
CaMKII is a highly prevalent serine/threonine kinase in the postsynaptic density of glutamatergic synapses (Kennedy, 2000; Colbran and Brown, 2004). Activity-dependent activation of CaMKII is triggered by Ca2+ entry and is critical for LTP induction and experience-dependent plasticity (Pettit et al., 1994; Lledo et al., 1995; Soderling and Derkach, 2000; Lisman et al., 2002). A number of CaMKII substrates have been identified in the postsynaptic density, including AMPA and NMDA receptors, scaffolding proteins of the PSD-95 family, Ras-GTPase activating protein synGAP, and RhoGEF Tiam 1 (Shen et al., 1998; Fleming et al., 1999; Yoshimura et al., 2000; Colbran and Brown, 2004; Mauceri et al., 2004; Oh et al., 2004). Most recently, the calcium-dependent phosphorylation of Tiam 1 in response to NMDAR stimulation has been implicated in the effects of the NMDAR on dendritic development through Rac1-dependent actin remodeling (Tolias et al., 2005). Aside from its kinase activity, CaMKII is also known to directly associate with actin in dendritic spines and regulate actin reorganization. CaMKII has been shown to control the extension and branching of filopodia as well as the number of synapses in dissociated hippocampal neuron cultures (Fink et al., 2003). The role of CaMKII in actin reorganization was also supported by another observation that the activation of CaMKII in hippocampal slice cultures was sufficient to trigger filopodia growth and new spine formation, whereas the blockade of CaMKII prevented structural remodeling (Jourdain et al., 2003). Our studies showed that RGD-induced extension of new filopodia and restructuring of existing spines in established hippocampal neuron cultures also depended on CaMKII activity.
Whereas it is clear that the RGD effects on actin reorganization in spines and extension of new filopodia are mediated through the NMDAR/CaMKII pathway, mechanisms by which integrins regulate NMDAR activity in cultured hippocampal neurons are unclear. However, recent work showed that RGD-containing peptide and fibronectin can rapidly activate integrin signaling in synaptosomes purified from adult rat brain through phosphorylation/activation of focal adhesion kinase (FAK), proline-rich tyrosine kinase 2 (PYK2), and Src nonreceptor tyrosine kinase (Bernard-Trifilo et al., 2005). Moreover, RGD-mediated Src activation increased tyrosine phosphorylation of NMDA receptors (subunits NR2A and NR2B) in both synaptosomes and acute hippocampal slices (Bernard-Trifilo et al., 2005). Therefore, the RGD effects in cultured hippocampal neurons might also be mediated by FAK activation and Src-dependent phosphorylation of NMDAR. In conclusion, our studies provide the first evidence that integrins can regulate actin reorganization in dendritic spines through NMDAR and that these effects are accompanied by marked changes in spine morphology.
Footnotes
This work was supported by National Institute of Mental Health Grant MH67121 (I.M.E.). We thank Drs. Peter Hickmott and Douglas Ethell for helpful discussions and critical reading of this manuscript. We also thank members of the Ethell laboratory for advice and Laura Heraty for editing this manuscript.
References
- Aplin AE, Howe A, Alahari SK, Juliano RL (1998). Signal transduction and signal modulation by cell adhesion receptors: the role of integrins, cadherins, immunoglobulin-cell adhesion molecules, and selectins. Pharmacol Rev 50:197–263. [PubMed] [Google Scholar]
- Bahr BA, Staubli U, Xiao P, Chun D, Ji ZX, Esteban ET, Lynch G (1997). Arg-Gly-Asp-Ser-selective adhesion and the stabilization of long-term potentiation: pharmacological studies and the characterization of a candidate matrix receptor. J Neurosci 17:1320–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernard-Trifilo JA, Kramar EA, Torp R, Lin CY, Pineda EA, Lynch G, Gall CM (2005). Integrin signaling cascades are operational in adult hippocampal synapses and modulate NMDA receptor physiology. J Neurochem 93:834–849. [DOI] [PubMed] [Google Scholar]
- Bi X, Lynch G, Zhou J, Gall CM (2001). Polarized distribution of alpha5 integrin in dendrites of hippocampal and cortical neurons. J Comp Neurol 435:184–193. [DOI] [PubMed] [Google Scholar]
- Blystone SD (2002). Kinetic regulation of beta 3 integrin tyrosine phosphorylation. J Biol Chem 277:46886–46890. [DOI] [PubMed] [Google Scholar]
- Bruckner G, Brauer K, Hartig W, Wolff JR, Rickmann MJ, Derouiche A, Delpech B, Girard N, Oertel WH, Reichenbach A (1993). Perineuronal nets provide a polyanionic, glia-associated form of microenvironment around certain neurons in many parts of the rat brain. Glia 8:183–200. [DOI] [PubMed] [Google Scholar]
- Carlisle HJ, Kennedy MB (2005). Spine architecture and synaptic plasticity. Trends Neurosci 28:182–187. [DOI] [PubMed] [Google Scholar]
- Chan CS, Weeber EJ, Kurup S, Sweatt JD, Davis RL (2003). Integrin requirement for hippocampal synaptic plasticity and spatial memory. J Neurosci 23:7107–7116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavis P, Westbrook G (2001). Integrins mediate functional pre- and postsynaptic maturation at a hippocampal synapse. Nature 411:317–321. [DOI] [PubMed] [Google Scholar]
- Chun D, Gall CM, Bi X, Lynch G (2001). Evidence that integrins contribute to multiple stages in the consolidation of long term potentiation in rat hippocampus. Neuroscience 105:815–829. [DOI] [PubMed] [Google Scholar]
- Cohen RS, Chung SK, Pfaff DW (1985). Immunocytochemical localization of actin in dendritic spines of the cerebral cortex using colloidal gold as a probe. Cell Mol Neurobiol 5:271–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colbran RJ, Brown AM (2004). Calcium/calmodulin-dependent protein kinase II and synaptic plasticity. Curr Opin Neurobiol 14:318–327. [DOI] [PubMed] [Google Scholar]
- Dailey ME, Smith SJ (1996). The dynamics of dendritic structure in developing hippocampal slices. J Neurosci 16:2983–2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dityatev A, Schachner M (2003). Extracellular matrix molecules and synaptic plasticity. Nat Rev Neurosci 4:456–468. [DOI] [PubMed] [Google Scholar]
- Dunaevsky A, Blazeski R, Yuste R, Mason C (2001). Spine motility with synaptic contact. Nat Neurosci 4:685–686. [DOI] [PubMed] [Google Scholar]
- Einheber S, Schnapp LM, Salzer JL, Cappiello ZB, Milner TA (1996). Regional and ultrastructural distribution of the alpha 8 integrin subunit in developing and adult rat brain suggests a role in synaptic function. J Comp Neurol 370:105–134. [DOI] [PubMed] [Google Scholar]
- Engert F, Bonhoeffer T (1999). Dendritic spine changes associated with hippocampal long-term synaptic plasticity. Nature 399:66–70. [DOI] [PubMed] [Google Scholar]
- Ethell IM, Pasquale EB (2005). Molecular mechanisms of dendritic spine development and remodeling. Prog Neurobiol 75:161–205. [DOI] [PubMed] [Google Scholar]
- Ethell IM, Yamaguchi Y (1999). Cell surface heparan sulfate proteoglycan syndecan-2 induces the maturation of dendritic spines in rat hippocampal neurons. J Cell Biol 144:575–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ethell IM, Hagihara K, Miura Y, Irie F, Yamaguchi Y (2000). Synbindin, a novel syndecan-2-binding protein in neuronal dendritic spines. J Cell Biol 151:53–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ethell IM, Irie F, Kalo MS, Couchman JR, Pasquale EB, Yamaguchi Y (2001). EphB/syndecan-2 signaling in dendritic spine morphogenesis. Neuron 31:1001–1013. [DOI] [PubMed] [Google Scholar]
- Fiala JC, Feinberg M, Popov V, Harris KM (1998). Synaptogenesis via dendritic filopodia in developing hippocampal area CA1. J Neurosci 18:8900–8911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fink CC, Bayer KU, Myers JW, Ferrell JE Jr, Schulman H, Meyer T (2003). Selective regulation of neurite extension and synapse formation by the beta but not the alpha isoform of CaMKII. Neuron 39:283–297. [DOI] [PubMed] [Google Scholar]
- Fischer M, Kaech S, Wagner U, Brinkhaus H, Matus A (2000). Glutamate receptors regulate actin-based plasticity in dendritic spines. Nat Neurosci 3:887–894. [DOI] [PubMed] [Google Scholar]
- Fleming IN, Elliott CM, Buchanan FG, Downes CP, Exton JH (1999). Ca2+/calmodulin-dependent protein kinase II regulates Tiam1 by reversible protein phosphorylation. J Biol Chem 274:12753–12758. [DOI] [PubMed] [Google Scholar]
- Gall CM, Lynch G (2004). Integrins, synaptic plasticity and epileptogenesis. Adv Exp Med Biol 548:12–33. [DOI] [PubMed] [Google Scholar]
- Halpain S, Hipolito A, Saffer L (1998). Regulation of F-actin stability in dendritic spines by glutamate receptors and calcineurin. J Neurosci 18:9835–9844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris KM (1999). Structure, development, and plasticity of dendritic spines. Curr Opin Neurobiol 9:343–348. [DOI] [PubMed] [Google Scholar]
- Hering H, Sheng M (2001). Dendritic spines: structure, dynamics and regulation. Nat Rev Neurosci 2:880–888. [DOI] [PubMed] [Google Scholar]
- Hering H, Sheng M (2003). Activity-dependent redistribution and essential role of cortactin in dendritic spine morphogenesis. J Neurosci 23:11759–11769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphries MJ (2000). Integrin structure. Biochem Soc Trans 28:311–339. [PubMed] [Google Scholar]
- Jourdain P, Fukunaga K, Muller D (2003). Calcium/calmodulin-dependent protein kinase II contributes to activity-dependent filopodia growth and spine formation. J Neurosci 23:10645–10649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy MB (2000). Signal-processing machines at the postsynaptic density. Science 290:750–754. [DOI] [PubMed] [Google Scholar]
- Korkotian E, Segal M (1999). Bidirectional regulation of dendritic spine dimensions by glutamate receptors. NeuroReport 10:2875–2877. [DOI] [PubMed] [Google Scholar]
- Kramar EA, Bernard JA, Gall CM, Lynch G (2002). Alpha3 integrin receptors contribute to the consolidation of long-term potentiation. Neuroscience 110:29–39. [DOI] [PubMed] [Google Scholar]
- Lendvai B, Stern EA, Chen B, Svoboda K (2000). Experience-dependent plasticity of dendritic spines in the developing rat barrel cortex in vivo. Nature 404:876–881. [DOI] [PubMed] [Google Scholar]
- Lin B, Arai AC, Lynch G, Gall CM (2003). Integrins regulate NMDA receptor-mediated synaptic currents. J Neurophysiol 89:2874–2878. [DOI] [PubMed] [Google Scholar]
- Lin B, Kramar EA, Bi X, Brucher FA, Gall CM, Lynch G (2005). Theta stimulation polymerizes actin in dendritic spines of hippocampus. J Neurosci 25:2062–2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CY, Lynch G, Gall CM (2005). AMPA receptor stimulation increases alpha5beta1 integrin surface expression, adhesive function and signaling. J Neurochem 94:531–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisman J, Schulman H, Cline H (2002). The molecular basis of CaMKII function in synaptic and behavioural memory. Nat Rev Neurosci 3:175–190. [DOI] [PubMed] [Google Scholar]
- Lledo PM, Hjelmstad GO, Mukherji S, Soderling TR, Malenka RC, Nicoll RA (1995). Calcium/calmodulin-dependent kinase II and long-term potentiation enhance synaptic transmission by the same mechanism. Proc Natl Acad Sci USA 92:11175–11179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maletic-Savatic M, Malinow R, Svoboda K (1999). Rapid dendritic morphogenesis in CA1 hippocampal dendrites induced by synaptic activity. Science 283:1923–1927. [DOI] [PubMed] [Google Scholar]
- Matus A, Ackermann M, Pehling G, Byers HR, Fujiwara K (1982). High actin concentrations in brain dendritic spines and postsynaptic densities. Proc Natl Acad Sci USA 79:7590–7594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matus A, Brinkhaus H, Wagner U (2000). Actin dynamics in dendritic spines: a form of regulated plasticity at excitatory synapses. Hippocampus 10:555–560. [DOI] [PubMed] [Google Scholar]
- Mauceri D, Cattabeni F, Di Luca M, Gardoni F (2004). Calcium/calmodulin-dependent protein kinase II phosphorylation drives synapse-associated protein 97 into spines. J Biol Chem 279:23813–23821. [DOI] [PubMed] [Google Scholar]
- Nimchinsky EA, Sabatini BL, Svoboda K (2002). Structure and function of dendritic spines. Annu Rev Physiol 64:313–353. [DOI] [PubMed] [Google Scholar]
- Nishimura SL, Boylen KP, Einheber S, Milner TA, Ramos DM, Pytela R (1998). Synaptic and glial localization of the integrin alphavbeta8 in mouse and rat brain. Brain Res 791:271–282. [DOI] [PubMed] [Google Scholar]
- Oh JS, Manzerra P, Kennedy MB (2004). Regulation of the neuron-specific Ras GTPase-activating protein, synGAP, by Ca2+/calmodulin-dependent protein kinase II. J Biol Chem 279:17980–17988. [DOI] [PubMed] [Google Scholar]
- Pappas GD, Kriho V, Pesold C (2001). Reelin in the extracellular matrix and dendritic spines of the cortex and hippocampus: a comparison between wild type and heterozygous reeler mice by immunoelectron microscopy. J Neurocytol 30:413–425. [DOI] [PubMed] [Google Scholar]
- Petersen JD, Chen X, Vinade L, Dosemeci A, Lisman JE, Reese TS (2003). Distribution of postsynaptic density (PSD)-95 and Ca2+/calmodulin-dependent protein kinase II at the PSD. J Neurosci 23:11270–11278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettit DL, Perlman S, Malinow R (1994). Potentiated transmission and prevention of further LTP by increased CaMKII activity in postsynaptic hippocampal slice neurons. Science 266:1881–1885. [DOI] [PubMed] [Google Scholar]
- Pinkstaff JK, Detterich J, Lynch G, Gall C (1999). Integrin subunit gene expression is regionally differentiated in adult brain. J Neurosci 19:1541–1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plow EF, Haas TA, Zhang L, Loftus J, Smith JW (2000). Ligand binding to integrins. J Biol Chem 275:21785–21788. [DOI] [PubMed] [Google Scholar]
- Ruoslahti E (1996). RGD and other recognition sequences for integrins. Annu Rev Cell Dev Biol 12:697–715. [DOI] [PubMed] [Google Scholar]
- Segal I, Korkotian I, Murphy DD (2000). Dendritic spine formation and pruning: common cellular mechanisms? Trends Neurosci 23:53–57. [DOI] [PubMed] [Google Scholar]
- Shen K, Teruel MN, Subramanian K, Meyer T (1998). CaMKIIbeta functions as an F-actin targeting module that localizes CaMKIIalpha/beta heterooligomers to dendritic spines. Neuron 21:593–606. [DOI] [PubMed] [Google Scholar]
- Sheng M (2001). Molecular organization of the postsynaptic specialization. Proc Natl Acad Sci USA 98:7058–7061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smart FM, Halpain S (2000). Regulation of dendritic spine stability. Hippocampus 10:542–554. [DOI] [PubMed] [Google Scholar]
- Soderling TR, Derkach VA (2000). Postsynaptic protein phosphorylation and LTP. Trends Neurosci 23:75–80. [DOI] [PubMed] [Google Scholar]
- Tian M, Hagg T, Denisova N, Knusel B, Engvall E, Jucker M (1997). Laminin-alpha2 chain-like antigens in CNS dendritic spines. Brain Res 764:28–38. [DOI] [PubMed] [Google Scholar]
- Tolias KF, Bikoff JB, Burette A, Paradis S, Harrar D, Tavazoie S, Weinberg RJ, Greenberg ME (2005). The Rac1-GEF Tiam1 couples the NMDA receptor to the activity-dependent development of dendritic arbors and spines. Neuron 45:525–538. [DOI] [PubMed] [Google Scholar]
- Van der Flier A, Sonnenberg A (2001). Function and interactions of integrins. Cell Tissue Res 305:285–298. [DOI] [PubMed] [Google Scholar]
- Yoshimura Y, Aoi C, Yamauchi T (2000). Investigation of protein substrates of Ca2+/calmodulin-dependent protein kinase II translocated to the postsynaptic density. Brain Res Mol Brain Res 81:118–128. [DOI] [PubMed] [Google Scholar]
- Yuste R, Bonhoeffer T (2001). Morphological changes in dendritic spines associated with long-term synaptic plasticity. Annu Rev Neurosci 24:1071–1089. [DOI] [PubMed] [Google Scholar]
- Yuste R, Majewska A, Holthoff K (2000). From form to function: calcium compartmentalization in dendritic spines. Nat Neurosci 3:653–659. [DOI] [PubMed] [Google Scholar]
- Ziv NE, Smith SJ (1996). Evidence for a role of dendritic filopodia in synaptogenesis and spine formation. Neuron 17:91–102. [DOI] [PubMed] [Google Scholar]