

Abstract
Dendritic filopodia are highly dynamic structures, and morphological maturation from dendritic filopodia to spines is intimately associated with the stabilization and strengthening of synapses during development. Here, we report that telencephalin (TLCN), a cell adhesion molecule belonging to the Ig superfamily, is a negative regulator of spine maturation. Using cultured hippocampal neurons, we examined detailed localization and functions of TLCN in spine development and synaptogenesis. At early stages of synaptogenesis, TLCN immunoreactivity gradually increased and was present in dendritic shafts and filopodia. At later stages, TLCN tended to be excluded from mature spine synapses in which PSD-95 (postsynaptic density-95) clusters were apposed to presynaptic synaptophysin clusters. To elucidate the function of TLCN in spine maturation, we analyzed the dendrite morphology of TLCN-overexpressing and TLCN-deficient neurons. Overexpression of TLCN caused a dramatic increase in the density of dendritic filopodia and a concomitant decrease in the density of spines. Conversely, TLCN-deficient mice showed a decreased density of filopodia and an acceleration of spine maturation in vitro as well as in vivo. These results demonstrate that TLCN normally slows spine maturation by promoting the filopodia formation and negatively regulating the filopodia-to-spine transition. In addition, we found that spine heads of mature neurons were wider in TLCN-deficient mice compared with wild-type mice. Thus, the preservation of immature synapses by TLCN may be an essential step for refinement of functional neural circuits in the telencephalon, that take charge of higher brain functions such as learning, memory, and emotion.
Keywords: telencephalin, cell adhesion molecule, synaptogenesis, dendritic filopodia, spine, hippocampus
Introduction
In the mammalian brain, >90% of excitatory synapses occur on dendritic spines, tiny bulbous protrusions on dendrites (Harris and Kater, 1994). Spines are shaped depending on animal’s experiences in developing as well as adult brains (Lendvai et al., 2000; Trachtenberg et al., 2002; Zuo et al., 2005). For instance, the long-term potentiation (LTP) in the hippocampus is accompanied by alteration of spine morphology and formation of new spines (Engert and Bonhoeffer, 1999; Maletic-Savatic et al., 1999; Toni et al., 1999; Matsuzaki et al., 2004). Additionally, structural abnormalities of dendritic protrusions are associated with various neurological disorders including Fragile-X and Down syndromes (Irwin et al., 2000; Kaufmann and Moser, 2000).
During development, spines emerge after the appearance of dendritic filopodia, which are thin, long, and headless protrusions (Saito et al., 1992; Fiala et al., 1998; Lendvai et al., 2000). It has been proposed that dendritic filopodia are a precursor of the spine, although the precise cellular mechanisms of spine formation are still controversial (Yuste and Bonhoeffer, 2004). Filopodia are highly motile structures that may play important roles in the initial formation of axon–dendrite contacts. Thereafter, filopodia appear to be morphologically and functionally transformed into mature spines (Ziv and Smith, 1996).
In the past decade, a wealth of knowledge has accumulated on functional molecules involved in the formation, maintenance, and remodeling of spines (Hering and Sheng, 2001). Particularly, several cell recognition/adhesion molecules have been identified as regulators of spine morphogenesis, including cadherin/catenins, Ephs/ephrins, and syndecan-2 (Ethell and Yamaguchi, 1999; Murase et al., 2002; Togashi et al., 2002; Murai et al., 2003; Abe et al., 2004). In contrast to spines, however, little is known about molecular and cellular mechanisms underlying the formation and maintenance of dendritic filopodia.
Telencephalin (TLCN) [intercellular adhesion molecule 5 (ICAM-5)] is a cell adhesion molecule belonging to the Ig superfamily (Yoshihara and Mori, 1994; Yoshihara et al., 1994) and shows the highest homology to ICAMs that have crucial functions in the immune system (Hayflick et al., 1998). TLCN is expressed in neurons within the telencephalon (Mori et al., 1987; Oka et al., 1990), where it is specifically localized to dendrites but not to axons (Benson et al., 1998; Mitsui et al., 2005). Many of the TLCN-expressing neurons bear spines such as olfactory bulb granule cells, hippocampal and neocortical pyramidal cells, and striatal medium spiny neurons (Murakami et al., 1991; Sakurai et al., 1998) (data not shown). The expression of TLCN is developmentally regulated in that its appearance parallels dendritic elongation, spine formation, and synaptogenesis in individual telencephalic regions postnatally (Mori et al., 1987; Imamura et al., 1990; Yoshihara et al., 1994).
These accumulating lines of evidence imply a likely function of TLCN in dendritic morphogenesis. Here, we examined detailed localization of TLCN in dendrites of hippocampal neurons and effects of gain-of-function and loss-of-function of TLCN on morphology of dendritic protrusions. The results suggest that TLCN is a critical molecule for the formation and maintenance of dendritic filopodia and the first example of a cell-surface molecule that slows spine maturation.
Materials and Methods
Animals.
Production of TLCN-deficient mice was reported previously (Nakamura et al., 2001). Heterozygous F1 animals were backcrossed to C57BL/6J mice through >10 generations. C57BL/6J mice were used as a control strain. Genotyping was performed by tail DNA PCR using two pairs of primers specific to the wild-type and TLCN-deficient alleles. A forward primer (TLCN-550, 5′-CATCCTCACATTCTCCTCTTAGAACCACAT-3′; a sequence located upstream of the TLCN gene) was used for detection of both wild-type and mutant alleles. Two reverse primers were used for detection of either the wild-type allele (TLCN-8, 5′-GTGGTGGCAGAAGCGTGGGAAGCTAGAAGA-3′; a sequence in the first intron) or the mutant allele (CreR-3, 5′-GTTCGAACGCTAGAGCCTGTTTTGCACGTT-3′; a sequence in Cre recombinase).
DiI labeling.
Mice (n = 3 for each genotype) were deeply anesthetized with pentobarbital and perfused with 4% paraformaldehyde (PFA) in PBS. Brains were removed from the skull and postfixed. The fixed brains were sectioned at 100 μm using a vibratome (Dosaka, Kyoto, Japan), and small crystals of DiI (Molecular Probes, Eugene, OR) were placed on the CA1 pyramidal cell layer of the hippocampus using glass micropipettes. After DiI injection, slices were incubated in a fixative for 12–48 h at 4°C, and confocal images were captured as described below. The regions with a small number of labeled neurons were used for analysis of dendritic morphology. Five 50 μm segments of apical dendrites from the middle portion of the stratum radiatum were collected from individual animals and used for morphological analysis.
Antibodies.
The antibodies used were as follows: rabbit polyclonal anti-TLCN recognizing the cytoplasmic tail peptide of TLCN (1:5000) (Sakurai et al., 1998); mouse monoclonal anti-TLCN recognizing the extracellular epitope of only rabbit TLCN (clone 271A6; 1:3000) (Mori et al., 1987); guinea pig polyclonal anti-TLCN/Fc recognizing the extracellular region of TLCN (1:3000) (S. Mitsui, M. Saito, K. Mori, and Y. Yoshihara, unpublished observations); rat monoclonal anti-αN-catenin (1:1; a gift from M. Takeichi, RIKEN Center for Developmental Biology, Kobe, Japan); mouse monoclonal anti-neurofilament (clone 2H3; 1:3000; American Research Products, Belmont, MA); mouse monoclonal anti-microtubule-associated protein 2 (MAP-2) (clone AP20; 1:500; Chemicon, Temecula, CA); rat monoclonal anti-green fluorescent protein (GFP) (clone GF090R; 1:500; Nacalai Tesque, Kyoto, Japan); mouse monoclonal anti-Bassoon (clone SAP7F407; 1:100; StressGen Biotechnologies, San Diego, CA); mouse monoclonal anti-glutamate receptor 2 (GluR2) (MAB397; 1:500; Chemicon); mouse monoclonal anti-GABAA receptor β-chain (MAB341; 1:100; Chemicon); mouse monoclonal anti-postsynaptic density-95 (PSD-95) (clone 7E3–1B8; 1:100; Affinity BioReagents, Golden, CO); rabbit polyclonal anti-synaptophysin (1:10; Zymed, San Francisco, CA); mouse monoclonal anti-CD8 (clone HIT8a; 1:100; BD PharMingen, San Diego, CA); and mouse monoclonal anti-calmodulin-dependent protein kinase IIα (CaMKIIα) [clone 6G9(2); 1:200; Chemicon]. Alexa Fluor dye (Alexa488, Alexa568, and Alexa647)-conjugated anti-mouse, anti-rabbit, anti-guinea pig, and anti-rat secondary antibodies (Molecular Probes) were used at a concentration of 6.7 μg/ml.
For triple immunofluorescence labeling, rabbit anti-TLCN and anti-synaptophysin polyclonal antibodies were directly labeled with fluorescent dyes by using Alexa Fluor Labeling kits (Molecular Probes) according to the manufacturer’s instructions and were used in combination with other antibodies.
Cell cultures.
Mouse hippocampal neuronal cultures were prepared essentially as described previously (Okabe et al., 1999, 2001). Hippocampi were dissected from embryonic day 17 mice and dissociated with 0.25% trypsin (Invitrogen, San Diego, CA). The cells were plated at a density of 5000–7000 cells/cm2 on poly-l-lysine-coated cover glasses. They were maintained in MEM containing B27 supplements, 5% fetal bovine serum, and 0.5 mm glutamine (Invitrogen).
DNA constructs and transfection.
Rabbit TLCN deletion mutants and CD8/TLCN chimeric cDNA were constructed by standard molecular biological techniques. All constructs were subcloned into a mammalian expression vector (pCAGGS) that harbors a powerful and ubiquitous CAG promoter (Niwa et al., 1991). For detailed analysis of dendritic morphology, especially dendritic filopodia and spines, super-enhanced GFP and Venus (a gift from A. Miyawaki, RIKEN Brain Science Institute, Wako, Japan) with a palmitoylation signal of GAP-43 were used as a membrane-targeted fluorescent reporter.
Neurons were microinjected with 0.5–1 μg/μl plasmid DNAs by using a microinjector (FemtoJet; Eppendorf, Hamburg, Germany). After the transfection, neurons were cultured for an additional 24–48 h and immunostained.
Immunocytochemistry.
In most cases, cultured hippocampal neurons were fixed with 1% PFA for 3 min and with −30°C methanol for 15 min. For Bassoon and GABAA receptor immunostaining and for GFP fluorescence, the neurons were fixed with 4% PFA for 15 min. After permeabilization in 0.25% Triton X-100 and blocking with 5% normal goat serum, neurons were incubated in primary antibodies overnight at 4°C and visualized with Alexa Fluor dye-conjugated secondary antibodies.
Microscopy.
Fluorescence images were acquired using TCS SP2 (Leica, Nussloch, Germany) and Fluoview FV1000 (Olympus, Tokyo, Japan) confocal laser scanning microscopes. For visualizing fluorescent clusters containing TLCN, αN-catenin, PSD-95, and synaptophysin immunoreactivities, single optical sections were captured. For quantitative analysis of dendritic protrusions (filopodia and spines), Z-stacked images were acquired with 0.6 μm intervals, and maximal images were projected to XY planes. All dendritic images for quantitation were taken with a 100× objective lens, 2× digital zoom, and the same acquisition settings.
Image analysis and quantitation.
Quantitation was performed as described previously (Ethell et al., 2001; Pak et al., 2001; Sala et al., 2001) with some modification. More than 10 neurons were collected randomly from at least three independent experiments for each construct or developmental time point. The synaptic clusters and protrusions in proximal two to three dendritic segments (∼50 μm for each segment; total length of >100 μm) were analyzed for individual neurons, because the speed of synapse maturation differs between the proximal and distal segments of dendrites (Rao et al., 1998). When the PSD-95 cluster showed at least 0.15 μm overlap with the synaptophysin cluster, this structure was considered as a “synaptic cluster.” If TLCN or αN-catenin clusters showed overlap with more than a one-half area of the synaptic PSD-95 cluster, it was considered as a TLCN or αN-catenin positive synaptic cluster. Dendritic spines and filopodia were classified using the following criteria: spines were defined as protrusions of 0.5–1.5 μm length that had distinct mushroom-shaped heads, and filopodia were defined as headless filamentous protrusions longer than 1.5 μm. When larger than one-half areas of the protrusions contained TLCN immunoreactivity, they were defined as TLCN-positive filopodia or spines (see Fig. 3). Hidden spines and filopodia that protruded either in back or in front of optical planes were not counted. The density of synaptic PSD-95 clusters and dendritic protrusions were expressed as the number per 10 μm dendritic length. Spine length and width were measured using Z-stacked DiI or GFP images as described previously (Pak et al., 2001).
Figure 3.

Abundant expression of TLCN in dendritic filopodia. A–F, Cultured hippocampal neurons were immunostained with anti-TLCN antibody at 10, 15, and 22 DIV. B, D, F, Merged images of GFP fluorescence (green) and TLCN immunolabeling (magenta). TLCN is located on numerous dendritic filopodia (B, D, arrowheads) and some spines (D, arrow) as well as dendritic shafts at early stages (10 and 15 DIV). TLCN is gradually excluded from mature mushroom-shaped spines at a later stage (22 DIV) (F, arrows). Scale bar, 2 μm. G, Densities of dendritic filopodia and spines at different developmental stages. The filled bars indicate the TLCN-positive dendritic protrusions. The column graphs show mean ± SEM of 10 neurons at each developmental time point. At all stages of development, the percentage of dendritic filopodia containing TLCN was significantly greater than that of spines (two-tailed t test; *p < 0.05; **p < 0.01).
Results
Morphological abnormalities of dendritic protrusions in TLCN-deficient mice
The dendrite-specific expression of TLCN prompted us to compare dendritic morphology of the telencephalic neurons between wild-type and TLCN-deficient mice. Consequently, we noticed subtle but significant morphological abnormalities of dendritic protrusions in TLCN-deficient mice. CA1 pyramidal neurons in the hippocampus of wild-type and TLCN-deficient mice were labeled with DiI, and the numbers of dendritic filopodia and spines in apical dendritic segments were quantified. At postnatal day 7 (P7) when massive spinogenesis is occurring in the hippocampal CA1 region (Fiala et al., 1998), the hippocampal neurons in wild-type mice abundantly possessed thin and long filamentous protrusions of dendrites, called dendritic filopodia (Fig. 1A, arrowheads). The density of dendritic filopodia was higher than that of spines in wild-type mice (Fig. 1C, open bars). In contrast, the majority of dendritic protrusions in TLCN-deficient mice were already spines at the same stage (Fig. 1B,C, filled bars).
Figure 1.

Altered morphology of dendritic protrusions in TLCN-deficient hippocampal slices. A, B, Two representative images of apical dendrites of DiI-labeled CA1 pyramidal neurons in wild-type (A) and TLCN-deficient (B) mice at P7. Scale bar, 4 μm. Arrowheads, dendritic filopodia; arrows, spines. C, Densities of filopodia and spines on apical dendrites of CA1 pyramidal neurons in wild-type mice (□; n = 3) and TLCN-deficient mice (▪; n = 3) at P7. Note that TLCN-deficient neurons show a significantly lower density of filopodia and a higher density of spines. *p < 0.05, **p < 0.01 (two-tailed t test). D, E, Two representative images of apical dendrites of DiI-labeled CA1 pyramidal neurons of wild-type (D) and TLCN-deficient (E) mice at the adult stage (10 weeks). Scale bar, 3 μm. F, Densities of filopodia and spines on apical dendrites of CA1 pyramidal neurons in wild-type (□) and TLCN-deficient (▪) mice at the adult stage. No difference in densities of dendritic protrusions is observed between wild-type mice (n = 3) and TLCN-deficient mice (n = 3). G, Cumulative frequency plot for spine length in adult CA1 neurons. No difference in distribution of spine length is observed between wild-type and TLCN-deficient mice. H, The mean spine length in adult CA1 neurons is comparable between TLCN-deficient mice and wild-type mice. I, Cumulative frequency plot for spine-head width in adult CA1 neurons. The proportion of wider spines is significantly greater in TLCN-deficient mice than in wild-type mice (Kolmogorov–Smirnov test; p < 0.01). J, The mean spine width in adult CA1 neurons is significantly wider in TLCN-deficient mice than in wild-type mice (two-tailed t test; **p < 0.01). G–J, Wild-type mice: n = 3567 spines; TLCN-deficient mice: n = 3552 spines. Error bars indicate SEM.
In adult mice, almost all dendritic protrusions were spines in both wild-type and TLCN-deficient neurons (Fig. 1D-1F). However, the width of spine heads was significantly larger in TLCN-deficient mice (Fig. 1I,J), whereas no difference was observed in the length of spines between wild-type and mutant mice (Fig. 1G,H). These results suggest that TLCN regulates morphology of dendritic protrusions in both the developing and mature brain.
TLCN is highly expressed in dendritic filopodia
To examine the detailed localization and functions of TLCN during synaptogenesis, we used cultured hippocampal neurons in the subsequent experiments. First, immunocytochemical staining was performed with an anti-TLCN antibody on cultured neurons at 14 d in vitro (DIV). TLCN was restricted to the MAP-2-positive somatodendritic compartment (Fig. 2A) but absent from the neurofilament-positive axonal compartment (Fig. 2F), confirming our previous report (Benson et al., 1998). In addition, a careful observation at higher magnification (Fig. 2B–E, 2G–J) revealed that TLCN was present in dendritic filopodia (Fig. 2E,J, arrowheads), as well as along the dendritic shafts. Cell-surface labeling of living hippocampal neurons with the antibody recognizing the extracellular region of TLCN showed a similar pattern to the labeling of the TLCN cytoplasmic region after cell fixation and permeabilization (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). This result clearly demonstrates that TLCN protein is present on the plasma membrane of dendritic filopodia and shafts.
Figure 2.
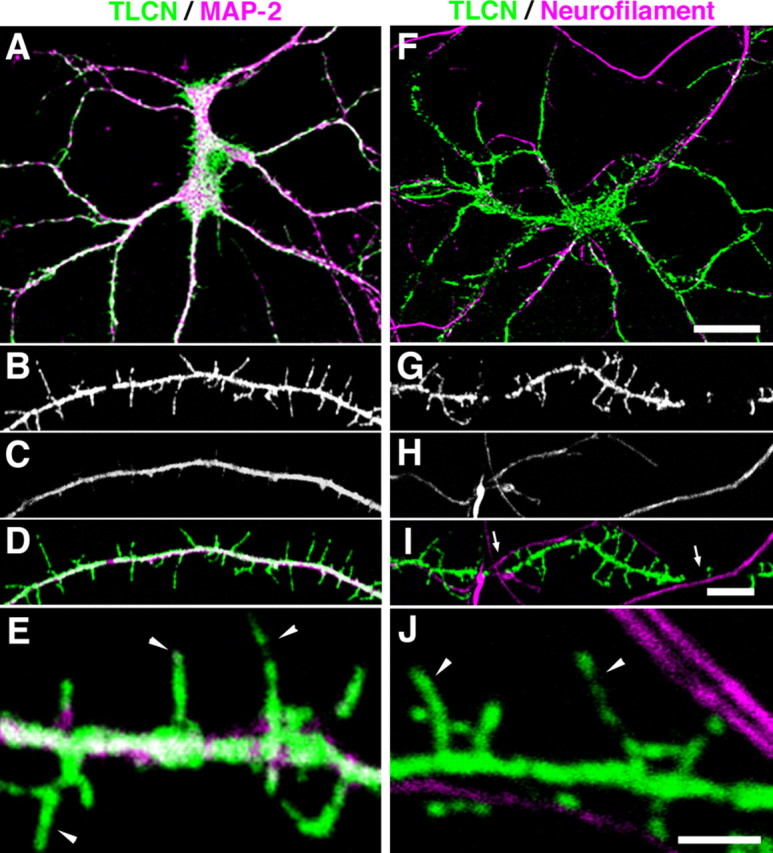
Dendrite-specific localization of TLCN in cultured hippocampal neurons. A–J, Cultured hippocampal neurons at 14 DIV were double-immunostained for TLCN and MAP-2 (dendritic marker) (A–E) or neurofilament (axonal marker) (F–J). In the merged images, TLCN (green) is colocalized with MAP-2 (A, D, E, magenta) but not with neurofilament (F, I, J, magenta). TLCN is frequently excluded from dendritic segments where axons are crossing over (I, arrows). High-power views (E, J) show localization of TLCN on dendritic filopodia (arrowheads). Scale bars: A, F, 30 μm; B–D, G–I, 5 μm; E, J, 2 μm.
Because dendritic filopodia are thought to be a precursor of the dendritic spine, we examined whether the filopodia-to-spine transition during synaptogenesis accompanies a change in TLCN localization. To clearly visualize the morphology of dendritic protrusions, cultured hippocampal neurons were transfected with membrane-targeted GFP and immunostained with anti-TLCN antibody after 24 h of transfection. At 10 DIV when the majority of dendritic protrusions were filopodia (Fig. 3B), TLCN was weakly expressed at punctate areas in the dendritic filopodia (66.7 ± 13.0%) as well as along the dendritic shafts (Fig. 3A,B). At 15 DIV when the density of filopodia reached maximum and spines started to appear (Fig. 3D), TLCN expression drastically increased and was found in most dendritic filopodia (89.2 ± 3.7%) and some spines (41.8 ± 4.4%) (Fig. 3C,D,G). At 22 DIV when the density of filopodia decreased and the density of spines markedly increased (Fig. 3F), TLCN was present in both types of protrusions (filopodia, 85.2 ± 6.8%; spines, 61.0 ± 8.0%) (Fig. 3E,G). Nevertheless, throughout the development of cultured hippocampal neurons, TLCN was detected in the majority of dendritic filopodia, whereas only in about one-half of the spines (Fig. 3G). These results suggest that the filopodia-to-spine transition is associated with a decrease in TLCN expression in dendritic protrusions.
Maturation of spiny synapses accompanies exclusion of TLCN
Nascent synapses are initially formed on highly motile dendritic filopodia (Saito et al., 1992; Ziv and Smith, 1996). Thereafter, the filopodia-to-spine transition takes place concomitant with maturation and stabilization of synapses. We therefore examined whether the change in TLCN expression on dendritic protrusions correlates with synapse maturation. To monitor synaptogenesis, we immunostained cultured hippocampal neurons with antibodies against synaptophysin (presynaptic marker) and PSD-95 (postsynaptic marker). Synaptic sites were defined as structures that contained PSD-95 clusters that were closely apposed to synaptophysin clusters (see Materials and Methods). In addition, we compared the localization of TLCN with that of αN-catenin, an intracellular linker molecule for classic cadherins (Hirano et al., 1992). It has been reported that the cadherin/αN-catenin complex is highly expressed in mature spines and plays a crucial role in stabilizing synapses (Uchida et al., 1996; Togashi et al., 2002; Abe et al., 2004). At 7 and 14 DIV, small PSD-95 clusters were observed along dendritic shafts and filopodia, some of which were nonsynaptic without apposed synaptophysin clusters (data not shown and Fig. 4B,D). More than one-half of the synaptic PSD-95 clusters (7 DIV, 57.6 ± 5.3%; 14 DIV, 54.9 ± 6.3%) overlapped with TLCN immunoreactivity (Fig. 4A,B,M,N). In contrast, at the same developmental stages, the expression of αN-catenin was very low and only slightly overlapped with synaptic PSD-95 clusters (7 DIV, 13.4 ± 1.3%; 14 DIV, 9.7 ± 1.3%) (Fig. 4C,D,M,N). At 21 DIV, synaptic PSD-95 clusters enlarged in size and increased in number (Fig. 4F,H,M). The number of TLCN-positive profiles still accounted for about one-half of the total synaptic clusters (46.4 ± 5.8%) (Fig. 4E,F,M,N). The size and number of αN-catenin clusters dramatically increased, and many of them overlapped with synaptic PSD-95 clusters (54.0 ± 5.2%) (Fig. 4G,H,M,N). At 28 DIV when synaptic maturation and stabilization further proceeded, the population of TLCN-positive synapses markedly decreased (31.1 ± 4.1%), whereas that of αN-catenin-positive synapses remained high (50.5 ± 3.8%) (Fig. 4I–N). These results suggest that TLCN is present at immature postsynaptic sites on dendritic filopodia at early stages of synaptogenesis and tends to be excluded from mature spiny synapses at later stages. Moreover, these data imply a switching of the cell adhesion system during synapse maturation: from TLCN in filopodia to cadherin/catenin in spines.
Figure 4.
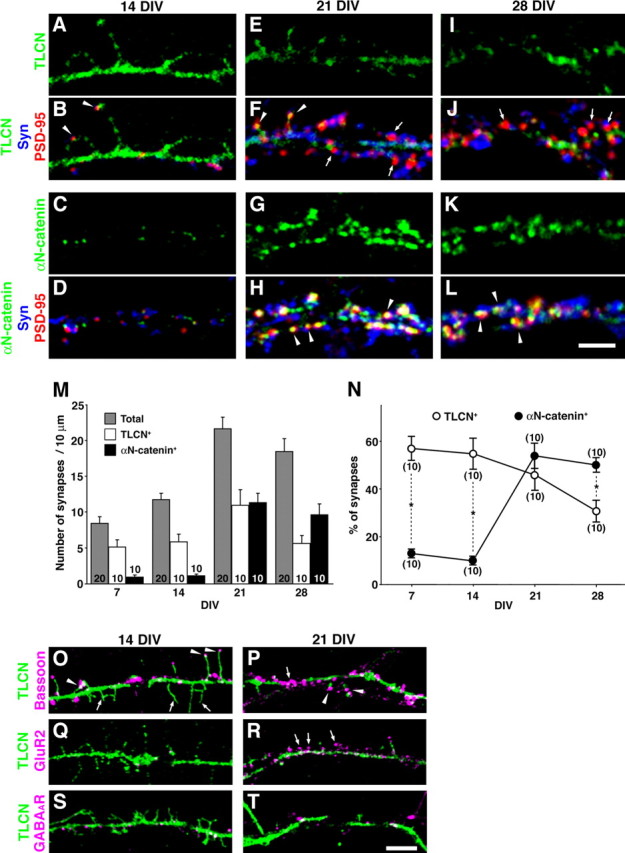
Gradual exclusion of TLCN from spines with maturation of synapses. A–L, Cultured hippocampal neurons at 14 DIV (A–D), 21 DIV (E–H), and 28 DIV (I–L) were triple-immunostained for TLCN (A, B, E, F, I, J, green) or αN-catenin (C, D, G, H, K, L, green) with PSD-95 (red) and synaptophysin (Syn) (blue). Synaptic sites having PSD-95 clusters apposing to synaptophysin clusters are frequently observed at 21 and 28 DIV. Note the different ontogenic appearance and localization between TLCN and αN-catenin. In F and J, arrowheads and arrows indicate TLCN-positive and -negative synaptic clusters, respectively. In H and L, arrowheads indicate αN-catenin-positive synapses. M, Densities of total (shaded bars), TLCN-positive (open bars), and αN-catenin-positive (filled bars) synaptic PSD-95 clusters at different developmental stages (7, 14, 21, and 28 DIV) in culture. The column graphs show mean ± SEM of 10 neurons at each developmental time point. The total number of synapses represents an average of 20 neurons. N, Percentage of TLCN-positive (○) and αN-catenin-positive (•) synaptic PSD-95 clusters at different developmental stages (7, 14, 21, and 28 DIV) in culture. The graphs show mean ± SEM of 10 neurons at each developmental time point. At early stages (7 and 14 DIV), the TLCN-positive synapses were more abundant than αN-catenin-positive synapses. At later stage (28 DIV), on the contrary, the percentage of αN-catenin-positive synapses surpassed that of TLCN-positive synapses (two-tailed t test; *p < 0.01). O, P, Double immunostaining of Bassoon (magenta) and TLCN (green). At 14 DIV, Bassoon-positive presynaptic clusters are frequently apposed to TLCN-positive filopodia (O, arrowheads). At 21 DIV, some Bassoon clusters were not associated with TLCN (P, arrow), whereas others still remained apposed to TLCN-positive dendritic protrusions (P, arrowheads). Q, R, Double immunostaining of GluR2 (magenta) and TLCN (green). GluR2 is hardly detectable at 14 DIV (Q) but accumulates in spines at 21 DIV at which TLCN is excluded (R, arrows). S, T, Double immunostaining of GABAA receptor (magenta) and TLCN (green). Clusters of GABAA receptor show no correlation with TLCN localization at both 14 DIV (S) and 21 DIV (T). Scale bars: A–L, 3 μm; O–T, 5 μm.
We frequently encountered TLCN-negative patches on dendritic shafts of cultured hippocampal neurons (Figs. 2, 4). A careful observation revealed that these TLCN-negative patches corresponded to the dendritic segments where the neurofilament-positive axons crossed over (Fig. 2G,H,I, arrows). Thus, it is likely that TLCN protein is excluded from the contact sites between dendrites and crossing axons as well as from mature dendritic spines contacting with presynaptic axons.
We further analyzed the dendritic localization of TLCN by comparison with Bassoon, GluR2, and GABAA receptors. Bassoon is a component of active zone vesicles, and its clusters are localized to both nascent and mature presynaptic sites. Bassoon clusters are detected in presynaptic sites at earlier stages than the appearance of synaptophysin clusters (tom Dieck et al., 1998; Friedman et al., 2000). At 14 DIV, some of the TLCN-positive filopodial tips were apposed to small Bassoon clusters (Fig. 4O, arrowheads) that could be considered as immature synapses, whereas there were also many TLCN-positive filopodia without any contact with axonal profiles (Fig. 4O, arrows). At 21 DIV, some Bassoon clusters were apposed to TLCN-positive dendritic protrusions and shafts (Fig. 4P, arrowheads), whereas the others were found on TLCN-negative segments of dendritic shafts (Fig. 4P, arrow). GluR2 is an AMPA receptor subunit present in mature postsynaptic sites, the appearance of which almost parallels that of PSD-95 and αN-catenin. At 14 DIV, GluR2 was barely detectable (Fig. 4Q). At 21 DIV, a marked increase in GluR2 expression was observed in mature spines, but the spiny GluR2 clusters were not colocalized with TLCN (Fig. 4R, arrows). These results support the view that TLCN is highly expressed in immature postsynaptic sites on dendritic filopodia apposed to Bassoon-positive nascent presynaptic sites but not present in mature postsynaptic sites that are positive for GluR2. In contrast, at inhibitory synapses, the GABAA receptor is localized to postsynaptic sites on the dendritic shaft and not on spines, at both 14 and 21 DIV. A subpopulation of GABAA receptor was found to colocalize with TLCN, but there appeared to be no relationship between TLCN distribution and inhibitory synapse maturation (Fig. 4S,T).
TLCN overexpression causes an increase in dendritic filopodia density
Enrichment of TLCN in dendritic filopodia and downregulation of TLCN from mature synapses imply that TLCN might play a role in the formation or maintenance of dendritic filopodia. To examine this possibility, we expressed rabbit TLCN in hippocampal neurons at 16–20 DIV when the filopodia-to-spine transition is actively taking place. Neuronal morphology was visualized by cotransfecting membrane-targeted GFP. After 48 h, mock-transfected neurons had a high density of mushroom-shaped spines (1.78 ± 0.08 per 10 μm; n = 5) and a low density of dendritic filopodia (0.29 ± 0.11 per 10 μm; n = 5) (Fig. 5A,B), as is the case with control neurons (Fig. 3G). In striking contrast, TLCN-overexpressing neurons showed a lower density of spines (0.90 ± 0.08 per 10 μm; n = 4; p = 0.0001) and a higher density of filopodia (1.73 ± 0.16 per 10 μm; n = 4; p = 0.0001) (Fig. 5D–F).
Figure 5.
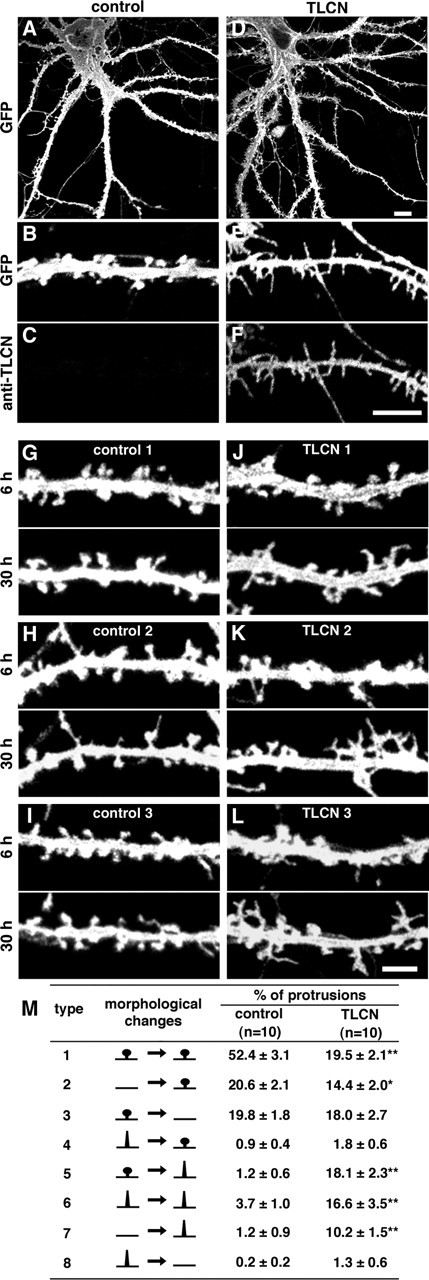
Increased formation of dendritic filopodia by TLCN overexpression. A, D, Low-power images of control (A) and TLCN-overexpressing (D) hippocampal neurons at 22 DIV. B, C, E, F, High-power views of control (B, C) and TLCN-overexpressing (E, F) dendrites at 22 DIV. B and E are images of GFP fluorescence, and C and F show exogenous expression of TLCN labeled with rabbit TLCN-specific antibody. A control dendrite bears mostly mushroom-shaped spines at this stage (B). In contrast, a TLCN-overexpressing dendrite exhibits numerous dendritic filopodia (E). G–I, Three examples of dendritic segments of control neurons at 6 and 30 h after mock transfection. J–L, Three examples of dendritic segments of TLCN-overexpressing neurons at 6 and 30 h after transfection. Scale bars: A, D, 10 μm; B, C, E, F, 5 μm; G–L, 3 μm. M, Percentage of different patterns of morphological changes that were detected by comparing the microscopic images at 6 and 30 h after transfection. *p < 0.05, **p < 0.01 (two-tailed t test). Type 1, Stable spines (spines at 6 h remain to be spines at 30 h); type 2, new spine formation from dendritic shafts; type 3, disappearance of spines; type 4, transformation from filopodia to spines; type 5, reverse transformation from spines to filopodia; type 6, stable filopodia; type 7, new filopodia formation from dendritic shafts; type 8, disappearance of filopodia. Data were collected from 10 neurons for each transfection (control, 547 protrusions; TLCN overexpression, 477 protrusions).
To address how TLCN overexpression induces an increase in the density of dendritic filopodia and a decrease in the density of spines, we imaged dendrites of cultured neurons at 6 and 30 h after transfection. Morphological changes of dendritic protrusions were classified into eight types and quantified (Fig. 5M). In mock-transfected neurons, stable spines (type 1, 52.4 ± 3.1%) accounted for about one-half of the dendritic protrusions, whereas filopodia formation either from spines (type 5, 1.2 ± 0.6%) or from dendritic shafts (type 7, 1.2 ± 0.9%) were rarely observed (Fig. 5G–I,M). In contrast, TLCN overexpression led to a significant decrease in stable spines (type 1, 19.5 ± 2.1%) and a dramatic increase in the reversion from spines to filopodia (type 5, 18.1 ± 2.3%), stable filopodia (type 6, 16.6 ± 3.5%), and newly formed filopodia (type 7, 10.2 ± 1.5%). Because the percentage of dendritic spines that disappeared after TLCN overexpression (type 3, 18.0 ± 2.7%) was almost similar to that in control (type 3, 19.8 ± 1.8%), TLCN overexpression does not change the half-life of dendritic spines. These results suggest that TLCN induces an increase in the number of filopodia through multiple mechanisms: the reverse transformation from spines to filopodia, the maintenance of pre-existing filopodia, and the elongation of new filopodia from dendritic shafts.
It has been reported that the filopodia-to-spine transition is accompanied by dynamic changes in the localization of various functional synaptic molecules (Hering and Sheng, 2001; Ehlers, 2002). For example, PSD-95 and F-actin exhibit increased expression and accumulation in mature spines, but not in filopodia, and play important roles in the stabilization of spiny synapses (Allison et al., 2000; El-Husseini et al., 2000; Marrs et al., 2001; Okabe et al., 2001). We therefore analyzed whether TLCN-induced filopodia formation correlates with the postsynaptic localization of PSD-95 and F-actin. In control neurons, dendritic spines were observed predominantly with punctate profiles of PSD-95 and F-actin that were apposed to synaptophysin clusters (Fig. 6A–D,I–L). In contrast, TLCN-overexpressing neurons exhibited diffuse PSD-95 and F-actin staining in dendritic protrusions with accumulation of synaptophysin clusters on dendritic shafts (Fig. 6E–H,M–P). Thus, TLCN overexpression reduced synaptic clustering of PSD-95 and F-actin, suggesting that TLCN might act as a negative regulator of postsynaptic targeting of the functional molecules.
Figure 6.
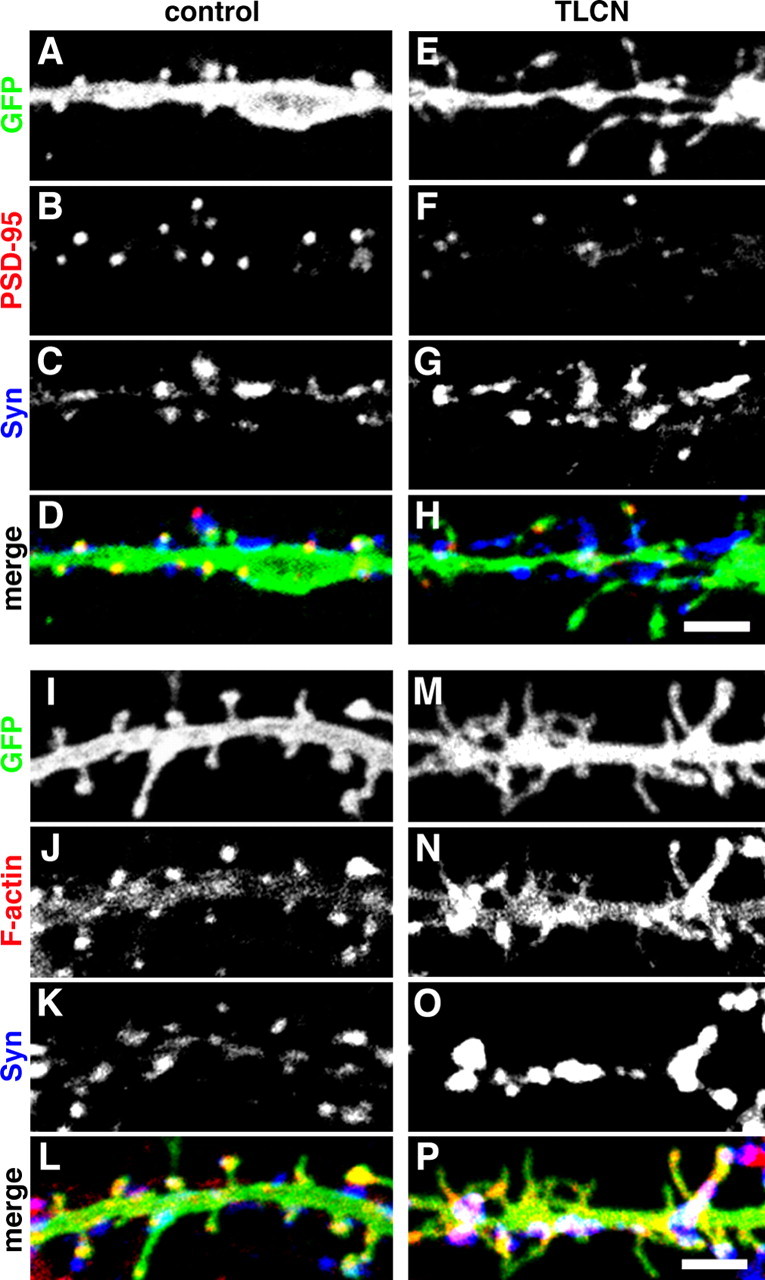
TLCN overexpression causes declustering of synaptic PSD-95 and F-actin. A–H, Representative images of control (A–D) and TLCN-overexpressing (E–H) dendrites triple-labeled for GFP (A, E; green in D, H), PSD-95 (B, F; red in D, H), and synaptophysin (C, G; blue in D, H). I–P, Representative images of control (I–L) and TLCN-overexpressing (M–P) dendrites triple-labeled for GFP (I, M; green in L, P), phalloidin (J, N; red in L, P), and synaptophysin (K, O; blue in L, P). Scale bars, 3 μm. Syn, Synaptophysin.
TLCN is a type I integral membrane protein that consists of a long extracellular region (825 aa) with nine tandemly arranged Ig-like domains, a transmembrane region (26 aa) and a short cytoplasmic tail (61 aa) (Fig. 7A). Thus, TLCN expressed on the surface membrane of dendritic filopodia might interact with some unknown ligand(s) present in the extracellular matrix or on incoming axons and send signals via its cytoplasmic domain to the interior of dendritic filopodia to maintain the filopodia or to reduce the filopodia-to-spine transformation. To examine this possibility, we transfected cultured hippocampal neurons with truncated or chimeric mutants of TLCN. Immunostaining of the transfected cells with anti-rabbit TLCN or anti-CD8 antibodies confirmed that the expression levels of wild-type and mutant proteins were comparable. Dendritic morphology was assessed by cotransfection of membrane-targeted GFP. A deletion mutant, TLCNΔ52, which lacks most of the cytoplasmic region (the C-terminal 52 aa were deleted) (Fig. 7A), did not induce any change in the density of spines or filopodia (Fig. 7B,C). Moreover, another deletion mutant, TLCNΔ4, which lacks only four amino acids at the C terminus (Fig. 7A), also showed no effect (Fig. 7B,C), suggesting that the cytoplasmic region of TLCN, especially the C terminus, is essential for the formation of dendritic filopodia. Because the C-terminal amino acid sequence of TLCN (-L-T-S-A in rabbit and human TLCN; -L-T-S-S in mouse TLCN) shows a similarity to the consensus sequence of the PDZ (PSD-95/discs large/zona occludens-1)-binding motif (-X-T/S-X-V), TLCN may interact with some intracellular signaling and/or scaffolding molecules with PDZ domains. A chimeric molecule (CD8/TLCN) was further constructed composed of the extracellular and transmembrane regions of an unrelated immune molecule, CD8, and the cytoplasmic region of TLCN (Fig. 7A). This CD8/TLCN chimera expressed in hippocampal neurons also failed to induce any apparent changes in morphology of dendritic protrusions (Fig. 7B,C), suggesting the importance of the extracellular region of TLCN. Together, these results indicate that both the extracellular and cytoplasmic regions of TLCN are required for the morphological changes in dendritic protrusions induced by TLCN overexpression.
Figure 7.
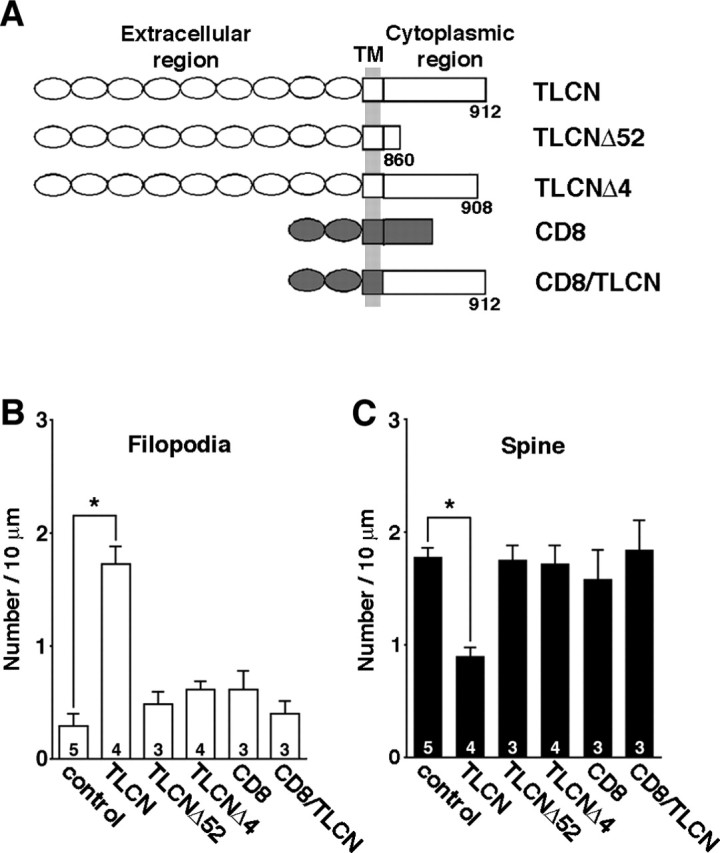
Full-length TLCN is required for filopodia formation. A, A schematic diagram of molecules exogenously expressed in hippocampal neurons. B, C, Densities of dendritic filopodia (B) and spines (C) in cultured hippocampal neurons transfected with individual constructs shown in A. Note that only full-length TLCN causes morphological transformation of dendritic protrusions. Data were collected from >10 neurons. The numbers on the bars indicate the total number of individual experiments for each construct. Error bars indicate SEM. *p < 0.01 (two-tailed t test). TLCN, Full-length rabbit TLCN; TLCNΔ52, TLCN lacking C-terminal 52 aa in the cytoplasmic region; TLCNΔ4, TLCN lacking C-terminal 4 aa in the cytoplasmic region; CD8, full-length human CD8; CD8/TLCN, a chimeric molecule consisting of the extracellular and transmembrane (TM) regions of CD8 and the cytoplasmic region of TLCN.
Rapid maturation of spines in TLCN-deficient neurons
The gain-of-function experiment (overexpression of TLCN) revealed direct involvement of TLCN in dendritic morphogenesis: an increase in the densitiy of filopodia and a concomitant decrease in the density of spines. Next, we explored in detail the loss-of-function of TLCN in vitro by culturing hippocampal neurons from TLCN-deficient mice (Nakamura et al., 2001). Because TLCN is expressed in 99% of neurons containing CaMKII in the hippocampus (Benson et al., 1998), we compared the dendritic morphology of CaMKII-positive neurons of wild-type and TLCN-deficient mice (Fig. 8B,D). At 10 DIV, the spine density of the TLCN-deficient neurons was significantly higher than that of wild-type neurons (Fig. 8F), whereas no difference was observed in the density of filopodia (Fig. 8E). In addition, the TLCN-deficient neurons contained larger PSD-95 clusters throughout the development (data not shown).
Figure 8.

Acceleration of spine maturation in TLCN-deficient neurons in vitro. A–D, Representative images of dendrites of cultured hippocampal neurons at 15 DIV from wild-type (+/+; A, B) and TLCN-deficient (−/−; C, D) mice. A, C, Merged images of GFP fluorescence (green) and synaptophysin (magenta) immunolabeling. B, D, Same fields as A and C, respectively, stained with anti-CaMKII antibody. Scale bar, 1 μm. E, F, Densities of filopodia (E) and spines (F) of wild-type (□) and TLCN-deficient (▪) neurons at different developmental stages (10, 15, and 22 DIV). *p < 0.05, **p < 0.01 (two-tailed t test). G, Cumulative frequency plot for spine length in 22 DIV neurons. No difference in distribution of spine length is observed between wild-type and TLCN-deficient mice. H, The mean spine length in 22 DIV neurons is comparable between TLCN-deficient mice and wild-type mice. I, Cumulative frequency plot for spine-head width in 22 DIV neurons. The proportion of wider spines is significantly greater in TLCN-deficient mice than in wild-type mice (Kolmogorov–Smirnov test; p < 0.01). J, The mean spine width in 22 DIV neurons is significantly wider in TLCN-deficient mice than in wild-type mice (two-tailed t test; **p < 0.01). G–J, Data were collected from >10 neurons in three to four independent experiments (wild-type spines, n = 231; TLCN-deficient spines, n = 278). Error bars indicate SEM.
At 15 DIV when wild-type neurons still possess numerous dendritic filopodia (Fig. 8A, arrowheads), the TLCN-deficient neurons showed a lower density of filopodia and a higher density of spines (Fig. 8C) than wild-type neurons (Fig. 8E,F). Furthermore, in TLCN-deficient neurons, most synaptophysin clusters made contact with spine heads (Fig. 8C, arrows), whereas they were mostly apposed to the dendritic shaft in wild-type neurons (Fig. 8A). Thus, TLCN-deficient neurons exhibited fewer filopodia and a faster increase in functional spines during dendritic development.
At 22 DIV, there was no longer any significant difference in the density of dendritic protrusions between wild-type and TLCN-deficient neurons (Fig. 8E,F). However, the width of spine heads was significantly larger in TLCN-deficient neurons than in wild-type neurons (Fig. 8I,J), whereas there was no difference in spine length (Fig. 8G,H). These results are reminiscent of the in vivo situation (Fig. 1) and suggest that TLCN may play a role as a negative regulator of spine maturation.
Discussion
Developing neurons: TLCN preserves dendritic filopodia
Synaptogenesis proceeds through dynamic and sequential steps: filopodial extension from dendritic shafts, initial contact between filopodia and axons, transformation from filopodia to spines, and stabilization of mature synapses. Dendritic filopodia are abundantly found in immature neurons during development including the “critical period.” Filopodia are highly motile, as if they are searching for axon terminals to make appropriate synaptic connections (Ziv and Smith, 1996; Fiala et al., 1998; Lendvai et al., 2000). Additionally, recent time-lapse imaging studies have revealed a reversion from spines to filopodia (Dunaevsky et al., 1999; Marrs et al., 2001). Therefore, filopodia may play important roles not only in the initiation of synaptic contacts but also in the refinement of functional synapses in the developing brain.
Here, we showed that TLCN is located in dendritic filopodia and gradually excluded from spines in the course of synaptogenesis. In addition, the presence of TLCN on dendritic filopodia before the initial contact with axons suggests that TLCN might play a role at the very early steps of synaptogenesis. Furthermore, overexpression of TLCN induced a dramatic increase in filopodia and a concomitant decrease in spines. Conversely, TLCN-deficient neurons showed a lower density of filopodia and a rapid increase in spines during development both in vitro and in vivo. From these results, we speculate that TLCN controls dendritic morphogenesis through the promotion and preservation of filopodia as well as slowing their maturation into spines (supplemental Fig. 2, available at www.jneurosci.org as supplemental material).
What is the physiological meaning of filopodia maintenance by TLCN? Although the function of dendritic filopodia are still enigmatic, they are dynamic and transient structures that may hold a “trial period” for synapse formation. A highly motile property of filopodia will be beneficial for dendrites to directly interact with a number of incoming axons and increase the opportunities to select proper presynaptic partners. TLCN may function as a “softener” that endows plastic and flexible properties to dendritic protrusions, for example by enabling activity-dependent rearrangement of synapses. To elucidate the function of TLCN in vivo, the electrophysiological analysis has been performed on synapse maturation during the visual cortex development of wild-type and TLCN-deficient mice. Interestingly, TLCN-deficient mice exhibit an accelerated maturation of receptive field properties (ocular dominance plasticity and orientation preference) in the primary visual cortex (T. K. Hensch, personal communication). Thus, TLCN is certainly involved in the synapse development in vivo. The preservation of filopodia by TLCN may be an important step for the formation of functional neural circuits. Because of the absence of this step in TLCN-deficient mice, spine formation is accelerated and the resulting synapses may rapidly become hard-wired.
Involvement of TLCN in plasticity of developing synapses may be limited to excitatory synapses, because TLCN dynamics was associated with the clustering of GluR2 and PSD-95 into spines, but not with the localization of GABAA receptors on dendritic shafts. This view is supported by our previous immunoelectron microscopic study of the olfactory bulb, demonstrating the specific localization of TLCN at postsynaptic membranes of excitatory synapses, but not of inhibitory synapses, in dendrodendritic reciprocal contacts between mitral cells and granule cells (Murakami et al., 1991).
Mature neurons: TLCN modulates spine morphology and synaptic plasticity
In the adult brain, neuronal dendrites are equipped with numerous spines and only few filopodia (Grutzendler et al., 2002). However, it has been reported that mature dendrites undergo morphological changes on various stimuli, such as widening or narrowing of spine heads and elimination or formation of synapses (Grutzendler et al., 2002; Trachtenberg et al., 2002; Petrak et al., 2005). The expression level of TLCN dramatically increases postnatally and reaches a peak at ∼1–2 weeks when dendritic filopodia are abundant in the telencephalon. However, a high level of TLCN expression is still observed in the adult brain despite the rare occurrence of dendritic filopodia (Mori et al., 1987; Yoshihara et al., 1994).
Although we showed a gradual exclusion of TLCN from spines during development, it is not removed completely from all spines but remains in ∼60% of them (Fig. 3G). This is consistent with our previous electron microscopic observation that TLCN is localized to peripostsynaptic regions in heads and necks of spines in adult hippocampal neurons (Sakurai et al., 1998). Additionally, we found that the heads of spines are wider in mature neurons of TLCN-deficient mice compared with wild-type both in vitro and in vivo. Nakamura et al. (2001) reported that TLCN-deficient mice show an enhancement of hippocampal LTP and spatial memory performance, which may appear contradictory to the present results on a functional role of filopodial TLCN in maintenance of plasticity during development. There are, however, at least two possible explanations based on a correlation between the spine size and the synaptic strength. One explanation is that the spine enlargement in TLCN-deficient mice may afford stronger synaptic transmission during the maintenance phase of LTP. This view is supported by recent reports that the width of spine heads correlates with the synapse efficacy (Matsuzaki et al., 2001; Kasai et al., 2003). Large-head spines are stable and contribute to strong synaptic connections, whereas small-head spines are motile and unstable. Another possibility is that synaptic transmission is enhanced through facilitated spine stabilization after LTP in TLCN-deficient mice. It has been described that LTP accompanies structural changes of synapses such as new spine formation and spine-head enlargement (Engert and Bonhoeffer, 1999; Maletic-Savatic et al., 1999; Toni et al., 1999; Ostroff et al., 2002; Matsuzaki et al., 2004). We demonstrated that TLCN-deficient mice showed accelerated spine maturation during development and that, once spines are formed, the spine width became significantly larger than in wild type. A similar event may occur in the process of morphological alteration of spines after the tetanic stimulation in TLCN-deficient mice, leading to enhanced LTP.
Thus, TLCN may determine the functional state of spines also in the adult brain, allowing flexibility for subtle refinement of spine morphology, motility, and synaptic efficacy. The telencephalon-specific expression of TLCN may ensure plasticity in the telencephalic neurons that take charge of higher brain functions such as learning, memory, and emotion.
Switching of cell adhesion systems during synaptogenesis
TLCN is a unique neuronal member of the ICAM family. Dynamic localization of TLCN in dendritic protrusions during neuronal synaptogenesis is reminiscent of that of ICAM-1 in the formation of immunological synapses. The immunological synapse is a specialized and central site where T lymphocytes and antigen-presenting cells make an organized interface in the nanometer scale gap (Bromley et al., 2001). The immunological synapse formation proceeds with several steps including cell polarization, initial adhesion, and engagement of T-cell receptors (TCRs) and peptide-bearing major histocompatibility complex (MHC) molecules (Dustin and Chan, 2000). These steps require a series of dynamic activities of various cell recognition/adhesion molecules. Particularly, ICAM-1 on T-lymphocytes plays a major role at the initial adhesion process by interacting with its counter-receptor lymphocyte function-associated antigen-1 (LFA-1) integrin on the antigen-presenting cells. Thereafter, ICAM-1 and LFA-1 are excluded from the central synaptic region of TCR/MHC–peptide interaction and stay at the peripheral margin of the mature immunological synapses (Bromley et al., 2001). Thus, ICAM-1 and TLCN (ICAM-5) play similar roles at early steps in the formation of immunological and neuronal synapses, respectively, providing a new and strong molecular link between the immune and nervous systems.
Various cell–cell interactions in developing and mature brains are mediated in part by adhesion molecules that are classified into several families. Particularly, the Ig superfamily and the cadherin family are most diverse with respect to their structures and functions. This study demonstrates a “switching” of these two cell adhesion systems in dendritic protrusions. TLCN is abundant on dendritic filopodia and downregulated in mature spines, whereas αN-catenin is reciprocally upregulated as synapses mature. The spine density is altered in opposite directions by manipulation of TLCN and cadherin/catenin expression levels. The overexpression of TLCN causes a decrease in spines, whereas TLCN-deficient neurons show an increase. Conversely, the overexpression of αN-catenin induces an increase in spines, whereas both a forced expression of dominant-negative N-cadherin and a deficiency of the αN-catenin gene cause a spine decrease (Togashi et al., 2002; Abe et al., 2004).
Although TLCN may function as a softener of synapses by maintaining dynamic and plastic properties of dendritic filopodia, cadherins and associated proteins (αN-catenin and β-catenin) in spines may act as “stabilizers” of synaptic contacts (supplemental Fig. 2, available at www.jneurosci.org as supplemental material) (Bozdagi et al., 2000; Togashi et al., 2002; Abe et al., 2004). Thus, two distinct families of cell adhesion molecules, the Ig superfamily and the cadherin superfamily, play different roles at different states of synapse maturation. We assume that TLCN mediates the initial loose adhesion between dendritic filopodia and nascent axons and the cadherin/catenin system takes charge of firm adhesion between spines and presynaptic terminals.
In addition to N-cadherin, several cell-surface molecules promote spine maturation and synapse formation/stabilization, including EphB2/ephrin-B, EphA4/ephrin-A3, β3-integrins, syndecan-2, neurexin/neuroligin, nectins, and SynCAM (Ethell and Yamaguchi, 1999; Scheiffele et al., 2000; Chavis and Westbrook, 2001; Biederer et al., 2002; Mizoguchi et al., 2002; Murase et al., 2002; Murai et al., 2003). In contrast, TLCN is the first example of a cell-surface molecule that preserves dendritic filopodia and slows spine maturation (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). The balance between accelerators and brakes on spine maturation would decide the capacity for synaptic plasticity of functional neural circuits.
Footnotes
This work was supported in part by the Special Coordination Funds for Promoting Science and Technology from the Japan Science and Technology Corporation and by a Grant-in-Aid for Scientific Research on Priority Areas (Advanced Brain Science Project and Molecular Brain Science) from the Ministry of Education, Culture, Sports, Science, and Technology of Japan to (Y.Y.). H.M. was supported by a grant from the Junior Research Associate Program in RIKEN. We thank Dr. T. K. Hensch for critical reading of this manuscript, Dr. M. Takeichi for the gift of anti-αN-catenin antibody, Dr. A. Miyawaki for the gift of fluorescent protein cDNAs, and Dr. S. Usui, I. Kawabata, and S. Mitsui for technical support.
References
- Abe K, Chisaka O, Van Roy F, Takeichi M (2004). Stability of dendritic spines and synaptic contacts is controlled by alphaN-catenin. Nat Neurosci 7:357–363. [DOI] [PubMed] [Google Scholar]
- Allison DW, Chervin AS, Gelfand VI, Craig AM (2000). Postsynaptic scaffolds of excitatory and inhibitory synapses in hippocampal neurons: maintenance of core components independent of actin filaments and microtubules. J Neurosci 20:4545–4554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson DL, Yoshihara Y, Mori K (1998). Polarized distribution and cell type-specific localization of telencephalin, an intercellular adhesion molecule. J Neurosci Res 52:43–53. [DOI] [PubMed] [Google Scholar]
- Biederer T, Sara Y, Mozhayeva M, Atasoy D, Liu X, Kavalali ET, Sudhof TC (2002). SynCAM, a synaptic adhesion molecule that drives synapse assembly. Science 297:1525–1531. [DOI] [PubMed] [Google Scholar]
- Bozdagi O, Shan W, Tanaka H, Benson DL, Huntley GW (2000). Increasing numbers of synaptic puncta during late-phase LTP: N-cadherin is synthesized, recruited to synaptic sites, and required for potentiation. Neuron 28:245–259. [DOI] [PubMed] [Google Scholar]
- Bromley SK, Burack WR, Johnson KG, Somersalo K, Sims TN, Sumen C, Davis MM, Shaw AS, Allen PM, Dustin ML (2001). The immunological synapse. Annu Rev Immunol 19:375–396. [DOI] [PubMed] [Google Scholar]
- Chavis P, Westbrook G (2001). Integrins mediate functional pre- and postsynaptic maturation at a hippocampal synapse. Nature 411:317–321. [DOI] [PubMed] [Google Scholar]
- Dunaevsky A, Tashiro A, Majewska A, Mason C, Yuste R (1999). Developmental regulation of spine motility in the mammalian central nervous system. Proc Natl Acad Sci USA 96:13438–13443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dustin ML, Chan AC (2000). Signaling takes shape in the immune system. Signaling takes shape in the immune system. Cell 103:283–294. [DOI] [PubMed] [Google Scholar]
- Ehlers MD (2002). Molecular morphogens for dendritic spines. Trends Neurosci 25:64–67. [DOI] [PubMed] [Google Scholar]
- El-Husseini AE, Schnell E, Chetkovich DM, Nicoll RA, Bredt DS (2000). PSD-95 involvement in maturation of excitatory synapses. Science 290:1364–1368. [PubMed] [Google Scholar]
- Engert F, Bonhoeffer T (1999). Dendritic spine changes associated with hippocampal long-term synaptic plasticity. Nature 399:66–70. [DOI] [PubMed] [Google Scholar]
- Ethell IM, Yamaguchi Y (1999). Cell surface heparan sulfate proteoglycan syndecan-2 induces the maturation of dendritic spines in rat hippocampal neurons. J Cell Biol 144:575–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ethell IM, Irie F, Kalo MS, Couchman JR, Pasquale EB, Yamaguchi Y (2001). EphB/syndecan-2 signaling in dendritic spine morphogenesis. Neuron 31:1001–1013. [DOI] [PubMed] [Google Scholar]
- Fiala JC, Feinberg M, Popov V, Harris KM (1998). Synaptogenesis via dendritic filopodia in developing hippocampal area CA1. J Neurosci 18:8900–8911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman HV, Bresler T, Garner CC, Ziv NE (2000). Assembly of new individual excitatory synapses: time course and temporal order of synaptic molecule recruitment. Neuron 27:57–69. [DOI] [PubMed] [Google Scholar]
- Grutzendler J, Kasthuri N, Gan WB (2002). Long-term dendritic spine stability in the adult cortex. Nature 420:812–816. [DOI] [PubMed] [Google Scholar]
- Harris KM, Kater SB (1994). Dendritic spines: cellular specializations imparting both stability and flexibility to synaptic function. Annu Rev Neurosci 17:341–371. [DOI] [PubMed] [Google Scholar]
- Hayflick JS, Kilgannon P, Gallatin WM (1998). The intercellular adhesion molecule (ICAM) family of proteins. New members and novel functions. Immunol Res 17:313–327. [DOI] [PubMed] [Google Scholar]
- Hering H, Sheng M (2001). Dendritic spines: structure, dynamics and regulation. Nat Rev Neurosci 2:880–888. [DOI] [PubMed] [Google Scholar]
- Hirano S, Kimoto N, Shimoyama Y, Hirohashi S, Takeichi M (1992). Identification of a neural alpha-catenin as a key regulator of cadherin function and multicellular organization. Cell 70:293–301. [DOI] [PubMed] [Google Scholar]
- Imamura K, Mori K, Oka S, Watanabe Y (1990). Variations by layers and developmental changes in expression of telencephalin in the visual cortex of cat. Neurosci Lett 119:118–121. [DOI] [PubMed] [Google Scholar]
- Irwin SA, Galvez R, Greenough WT (2000). Dendritic spine structural anomalies in fragile-X mental retardation syndrome. Cereb Cortex 10:1038–1044. [DOI] [PubMed] [Google Scholar]
- Kasai H, Matsuzaki M, Noguchi J, Yasumatsu N, Nakahara H (2003). Structure-stability-function relationships of dendritic spines. Trends Neurosci 26:360–368. [DOI] [PubMed] [Google Scholar]
- Kaufmann WE, Moser HW (2000). Dendritic anomalies in disorders associated with mental retardation. Cereb Cortex 10:981–991. [DOI] [PubMed] [Google Scholar]
- Lendvai B, Stern EA, Chen B, Svoboda K (2000). Experience-dependent plasticity of dendritic spines in the developing rat barrel cortex in vivo. Nature 404:876–881. [DOI] [PubMed] [Google Scholar]
- Maletic-Savatic M, Malinow R, Svoboda K (1999). Rapid dendritic morphogenesis in CA1 hippocampal dendrites induced by synaptic activity. Science 283:1923–1927. [DOI] [PubMed] [Google Scholar]
- Marrs GS, Green SH, Dailey ME (2001). Rapid formation and remodeling of postsynaptic densities in developing dendrites. Nat Neurosci 4:1006–1013. [DOI] [PubMed] [Google Scholar]
- Matsuzaki M, Ellis-Davies GC, Nemoto T, Miyashita Y, Iino M, Kasai H (2001). Dendritic spine geometry is critical for AMPA receptor expression in hippocampal CA1 pyramidal neurons. Nat Neurosci 4:1086–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuzaki M, Honkura N, Ellis-Davies GC, Kasai H (2004). Structural basis of long-term potentiation in single dendritic spines. Nature 429:761–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitsui S, Saito M, Hayashi K, Mori K, Yoshihara Y (2005). A novel phenylalanine-based targeting signal directs telencephalin to neuronal dendrites. J Neurosci 25:1122–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizoguchi A, Nakanishi H, Kimura K, Matsubara K, Ozaki-Kuroda K, Katata T, Honda T, Kiyohara Y, Heo K, Higashi M, Tsutsumi T, Sonoda S, Ide C, Takai Y (2002). Nectin: an adhesion molecule involved in formation of synapses. J Cell Biol 156:555–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori K, Fujita SC, Watanabe Y, Obata K, Hayaishi O (1987). Telencephalon-specific antigen identified by monoclonal antibody. Proc Natl Acad Sci USA 84:3921–3925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murai KK, Nguyen LN, Irie F, Yamaguchi Y, Pasquale EB (2003). Control of hippocampal dendritic spine morphology through ephrin-A3/EphA4 signaling. Nat Neurosci 6:153–160. [DOI] [PubMed] [Google Scholar]
- Murakami F, Tada Y, Mori K, Oka S, Katsumaru H (1991). Ultrastructural localization of telencephalin, a telencephalon-specific membrane glycoprotein, in rabbit olfactory bulb. Neurosci Res 11:141–145. [DOI] [PubMed] [Google Scholar]
- Murase S, Mosser E, Schuman EM (2002). Depolarization drives beta-catenin into neuronal spines promoting changes in synaptic structure and function. Neuron 35:91–105. [DOI] [PubMed] [Google Scholar]
- Nakamura K, Manabe T, Watanabe M, Mamiya T, Ichikawa R, Kiyama Y, Sanbo M, Yagi T, Inoue Y, Nabeshima T, Mori H, Mishina M (2001). Enhancement of hippocampal LTP, reference memory and sensorimotor gating in mutant mice lacking a telencephalon-specific cell adhesion molecule. Eur J Neurosci 13:179–189. [DOI] [PubMed] [Google Scholar]
- Niwa H, Yamamura K, Miyazaki J (1991). Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene 108:193–200. [DOI] [PubMed] [Google Scholar]
- Oka S, Mori K, Watanabe Y (1990). Mammalian telencephalic neurons express a segment-specific membrane glycoprotein, telencephalin. Neuroscience 35:93–103. [DOI] [PubMed] [Google Scholar]
- Okabe S, Miwa A, Okado H (1999). Alternative splicing of the C-terminal domain regulates cell surface expression of the NMDA receptor NR1 subunit. J Neurosci 19:7781–7792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okabe S, Miwa A, Okado H (2001). Spine formation and correlated assembly of presynaptic and postsynaptic molecules. J Neurosci 21:6105–6114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostroff LE, Fiala JC, Allwardt B, Harris KM (2002). Polyribosomes redistribute from dendritic shafts into spines with enlarged synapses during LTP in developing rat hippocampal slices. Neuron 35:535–545. [DOI] [PubMed] [Google Scholar]
- Pak DT, Yang S, Rudolph-Correia S, Kim E, Sheng M (2001). Regulation of dendritic spine morphology by SPAR, a PSD-95-associated RapGAP. Neuron 31:289–303. [DOI] [PubMed] [Google Scholar]
- Petrak LJ, Harris KM, Kirov SA (2005). Synaptogenesis on mature hippocampal dendrites occurs via filopodia and immature spines during blocked synaptic transmission. J Comp Neurol 484:183–190. [DOI] [PubMed] [Google Scholar]
- Rao A, Kim E, Sheng M, Craig AM (1998). Heterogeneity in the molecular composition of excitatory postsynaptic sites during development of hippocampal neurons in culture. J Neurosci 18:1217–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito Y, Murakami F, Song WJ, Okawa K, Shimono K, Katsumaru H (1992). Developing corticorubral axons of the cat form synapses on filopodial dendritic protrusions. Neurosci Lett 147:81–84. [DOI] [PubMed] [Google Scholar]
- Sakurai E, Hashikawa T, Yoshihara Y, Kaneko S, Satoh M, Mori K (1998). Involvement of dendritic adhesion molecule telencephalin in hippocampal long-term potentiation. NeuroReport 9:881–886. [DOI] [PubMed] [Google Scholar]
- Sala C, Piech V, Wilson NR, Passafaro M, Liu G, Sheng M (2001). Regulation of dendritic spine morphology and synaptic function by Shank and Homer. Neuron 31:115–130. [DOI] [PubMed] [Google Scholar]
- Scheiffele P, Fan J, Choih J, Fetter R, Serafini T (2000). Neuroligin expressed in nonneuronal cells triggers presynaptic development in contacting axons. Cell 101:657–669. [DOI] [PubMed] [Google Scholar]
- Togashi H, Abe K, Mizoguchi A, Takaoka K, Chisaka O, Takeichi M (2002). Cadherin regulates dendritic spine morphogenesis. Neuron 35:77–89. [DOI] [PubMed] [Google Scholar]
- tom Dieck S, Sanmarti-Vila L, Langnaese K, Richter K, Kindler S, Soyke A, Wex H, Smalla KH, Kampf U, Franzer JT, Stumm M, Garner CC, Gundelfinger ED (1998). Bassoon, a novel zinc-finger CAG/glutamine-repeat protein selectively localized at the active zone of presynaptic nerve terminals. J Cell Biol 142:499–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toni N, Buchs PA, Nikonenko I, Bron CR, Muller D (1999). LTP promotes formation of multiple spine synapses between a single axon terminal and a dendrite. Nature 402:421–425. [DOI] [PubMed] [Google Scholar]
- Trachtenberg JT, Chen BE, Knott GW, Feng G, Sanes JR, Welker E, Svoboda K (2002). Long-term in vivo imaging of experience-dependent synaptic plasticity in adult cortex. Nature 420:788–794. [DOI] [PubMed] [Google Scholar]
- Uchida N, Honjo Y, Johnson KR, Wheelock MJ, Takeichi M (1996). The catenin/cadherin adhesion system is localized in synaptic junctions bordering transmitter release zones. J Cell Biol 135:767–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshihara Y, Mori K (1994). Telencephalin: a neuronal area code molecule? Neurosci Res 21:119–124. [DOI] [PubMed] [Google Scholar]
- Yoshihara Y, Oka S, Nemoto Y, Watanabe Y, Nagata S, Kagamiyama H, Mori K (1994). An ICAM-related neuronal glycoprotein, telencephalin, with brain segment-specific expression. Neuron 12:541–553. [DOI] [PubMed] [Google Scholar]
- Yuste R, Bonhoeffer T (2004). Genesis of dendritic spines: insights from ultrastructural and imaging studies. Nat Rev Neurosci 5:24–34. [DOI] [PubMed] [Google Scholar]
- Ziv NE, Smith SJ (1996). Evidence for a role of dendritic filopodia in synaptogenesis and spine formation. Neuron 17:91–102. [DOI] [PubMed] [Google Scholar]
- Zuo Y, Lin A, Chang P, Gan WB (2005). Development of long-term dendritic spine stability in diverse regions of cerebral cortex. Neuron 46:181–189. [DOI] [PubMed] [Google Scholar]