

Abstract
TRPV1, a cloned capsaicin receptor, is a molecular sensor for detecting adverse stimuli and a key element for inflammatory nociception and represents biophysical properties of native channel. However, there seems to be a marked difference between TRPV1 and native capsaicin receptors in the pharmacological response profiles to vanilloids or acid. One plausible explanation for this overt discrepancy is the presence of regulatory proteins associated with TRPV1. Here, we identify Fas-associated factor 1 (FAF1) as a regulatory factor, which is coexpressed with and binds to TRPV1 in sensory neurons. When expressed heterologously, FAF1 reduces the responses of TRPV1 to capsaicin, acid, and heat, to the pharmacological level of native capsaicin receptor in sensory neurons. Furthermore, silencing FAF1 by RNA interference augments capsaicin-sensitive current in native sensory neurons. We therefore conclude that FAF1 forms an integral component of the vanilloid receptor complex and that it constitutively modulates the sensitivity of TRPV1 to various noxious stimuli in sensory neurons.
Keywords: TRPV1, FAF1, association, pharmacology, capsaicin, pain
Introduction
TRPV1 is expressed highly in small sensory neurons and is activated by capsaicin, heat, and acid (Caterina et al., 1997; Tominaga et al., 1998). Mice deficient in TRPV1 show reduced inflammatory hyperalgesia (Caterina et al., 2000; Davis et al., 2000), suggesting its role as a molecular sensor for detecting adverse stimuli during inflammation. In line with this notion, major inflammatory mediators such as bradykinin are now known to excite sensory neurons via the activation of TRPV1 (Premkumar and Ahern, 2000; Chuang et al., 2001; Shin et al., 2002).
TRPV1, a nonselective cation channel with a homotetrameric structure (Kedei et al., 2001; Kuzhikandathil et al., 2001), displays channel properties, such as channel conductance, current–voltage relationships, and ion selectivity identical to those of native capsaicin receptor in sensory neurons (Oh et al., 1996; Caterina et al., 1997). TRPV1 retains sensitization or desensitization properties similar to those observed with native capsaicin-sensitive currents in sensory neurons. TRPV1 elicits Ca2+-dependent desensitization or tachyphylaxis to repeated applications of capsaicin (Docherty et al., 1996; Bhave et al., 2002; Numazaki et al., 2003). Furthermore, inflammatory mediators augment the activity of TRPV1 via the action of protein kinase A or protein kinase C because they sensitize capsaicin-sensitive currents in sensory neurons (Premkumar and Ahern, 2000; Chuang et al., 2001).
Although TRPV1 represents channel property of native capsaicin-evoked currents in sensory neurons, there seems to be a marked difference between TRPV1 and native capsaicin receptors in terms of their pharmacological response profiles to vanilloids or acid (Oh et al., 1996; Baumann and Martenson, 2000; Shin et al., 2001). The cloned channel, TRPV1 is more sensitive to capsaicin than native channels in sensory neurons (Shin et al., 2001). TRPV1 is activated by acid, even at extreme levels (Tominaga et al., 1998). In contrast, native capsaicin receptors appear less sensitive to acid (Oh et al., 1996; Kwak et al., 1998; Baumann and Martenson, 2000). Thus, the presence of regulatory proteins associated with TRPV1 has been suggested to explain the pharmacological difference (Shin et al., 2001).
Numerous ion channels have auxiliary proteins that regulate trafficking or biophysical and pharmacological properties of pore-forming channel subunits. However, no associated proteins or subunit structures of TRPV1 that affect its channel properties have been reported to date. Recently, vesicular proteins, such as snapin and synaptotagmin IX, were reported to be associated with TRPV1 (Morenilla-Palao et al., 2004). But the vesicular proteins fail to affect channel property of TRPV1, although they augmented trafficking of TRPV1 to the plasma membrane. Therefore, we sought to identify the regulatory protein that controls the pharmacological profile of TRPV1.
Fas-associated factor 1 (FAF1) is an adapter protein that associates with Fas (Chu et al., 1995; Ryu et al., 2003; Kim et al., 2005). As an adaptor protein to Fas receptor, FAF1 is known to induce apoptosis or augment Fas-induced apoptosis. Because FAF1 does not contain the death domain, its function is not well characterized. In this study, we found that FAF1 interacts physically with TRPV1 and controls its activity constitutively.
Materials and Methods
Dorsal root ganglion cDNA λ expression library construction.
The SuperScript Lambda System (Invitrogen, Grand Island, NY) was used for cDNA synthesis and λ cloning. mRNAs were prepared from rat dorsal root ganglia (DRGs) by following the protocol in a FastTrack 2.0 kit (Invitrogen, Carlsbad, CA). The λ phage was packaged using Gigapack III Gold Extract (Stratagene, La Jolla, CA). A total of ∼3 × 106 independent clones were obtained. The λ-phage library was amplified and characterized as suggested by the manufacturer.
Screening for proteins associated with TRPV1.
Fragments of TRPV1 were cloned into pGEX-2TK (Amersham Biosciences, Uppsala, Sweden) in frame with glutathione S-transferase (GST) and protein kinase A-recognizing sequences. GST-ΔTRPV1 fusion proteins were expressed in Escherichia coli, purified with glutathione-Sepharose 4B, and 32P-labeled using the catalytic subunit of protein kinase A (bovine heart, 20 U; Sigma, St. Louis, MO) in a buffer containing 50 μCi of γ32P-ATP (10 μCi/μl). The DRG-cDNA λ-phage expression library incubated with Y1090 (ZL) was mixed with a top agarose, plated, and incubated at 42°C until tiny plaques became visible. The plates were overlaid and incubated with the isopropyl β-d-1-thiogalactopyranoside-soaked nitrocellulose membranes at 37°C overnight. The membranes were peeled off, washed, and probed with the 32P-labeled GST-ΔTRPV1 in PBS containing 5% nonfat milk and 0.02% Tween 20. The membranes were washed with PBS, dried, and exposed to film.
GST pull-down assay.
His-tagged fusion proteins of the N terminus (NTRPV1) and C terminus (CTRPV1) of TRPV1 were purified and incubated with GST-FAF1 fusion protein. Specifically bound proteins were purified and eluted from the glutathione-Sepharose beads with 10 mm of reduced glutathione in 50 mm Tris, pH 8.0. The eluted proteins were immunoblotted with mouse polyclonal antibody raised against rat NTRPV1 or CTRPV1 (Jung et al., 2002; Shin et al., 2002).
For localizing the TRPV1-interacting domain of FAF1 in vitro, human FAF1 and its mutants (FAF11–201, FAF1181–381, FAF1366–650, FAF11–81, FAF171–201, FAF1181–381, FAF1366–650, and FAF1Δ189–340) were cloned into pGEX4T-1 in frame with GST (Ryu et al., 1999, 2003). The GST fusion proteins were expressed and purified with glutathione-Sepharose beads. pcDNA-Myc·His-TRPV1 (2 μg) was translated in vitro using the TNT transcription/translation system kit (Promega, Madison, WI) in the presence of 10 μCi of [35S]methionine. Equal aliquots of 35S-labeled TRPV1 were incubated with either 1 μg of GST or GST-FAF1 fusion proteins in a buffer (50 mm HEPES, pH 7.6, 50 mm NaCl, 5 mm EDTA, 0.1% Nonidet P-40, and 10% glycerol) for 12 h at 4°C. The binding mixtures were then washed three times, separated by SDS-PAGE, and exposed to x-ray film.
Coimmunoprecipitation.
Lysates of DRG cells or human embryonic kidney (HEK) 293T cells transfected with rat TRPV1 alone or together with rat FAF1 were mixed with 10 μg of mouse polyclonal antiserum to rat CTRPV1 or goat antiserum to rat FAF1 (SC-1885; Santa Cruz Biotechnology, Santa Cruz, CA). Forty-eight hours after transfection, cells were harvested and solubilized with binding buffer (50 mm Tris-HCl, pH 7.4, 150 mm NaCl, 0.25% sodium deoxycholate, 1% Nonidet P-40, 2 mm phenylmethylsulfonylfluoride, 2 mm EDTA, and 5 mm iodoacetamide) for 20 min at room temperature. Cell debris was removed by centrifugation, and the supernatants were incubated 1 h at 4°C with mouse polyclonal antiserum to rat CTRPV1 (1:500) or goat antiserum to rat FAF1 (1:500, SC-1885; Santa Cruz Biotechnology). Then agarose-protein G beads (Pierce, Rockford, IL) were added and incubated for 2 h at 4°C. After centrifugation, the pellets were washed with binding buffer. Immunoprecipitated complexes were denatured with SDS-PAGE sample buffer (100°C for 5 min), separated by SDS-PAGE, and immunoblotted with specific antibodies.
Immunohistochemistry.
DRG neurons cultured on round coverslips were fixed with methanol at −20°C overnight. The coverslips were washed three times with ice-cold PBS and incubated with blocking buffer (5% BSA and 0.02% NaN3 in PBS) for 1 h at room temperature. The primary mouse polyclonal antibody raised against the rat TRPV1 peptide segment (TRPV12–199; 1:1000 dilution) and goat polyclonal antibody raised against rat FAF1 (1:400; Santa Cruz Biotechnology) were incubated overnight at 4°C. The two primary antibodies were bathed for 1 h at 37°C with Alexa Fluor 594-conjugated rabbit anti-mouse F(ab′)2 (diluted 1:200; Molecular Probes, Eugene, OR) and Alexa Fluor 488-conjugated rabbit anti-goat F(ab′)2 (diluted 1:200; Molecular Probes). After mounting, the cellular localization of TRPV1 and FAF1 was observed with an immunofluorescent confocal microscope (Zeiss, Thornwood, NY).
Biotinylation of cell-surface proteins.
Biotinylation of HEK 293T cell-surface proteins was performed with the Cell Surface Protein Biotinylation and Purification kit (Pierce). HEK 293T cells were transiently transfected using Lipofectamin (Invitrogen) with pCDNA3.1-TRPV1/myc-His alone or both pCDNA 3.1-TRPV1/myc-His and pCMV-FAF1. Transfected HEK293T cells were grown to confluence (90–95% confluent T75 flask, 48 h after transfection) and washed twice with ice-cold 1× PBS, pH 8.0. Cells were then incubated with 0.5 mg/ml EZ-Link NHS-SS-Biotin (Pierce) in ice-cold 1× PBS for 30 min at 4°C. The biotinylation reaction was terminated by adding quenching solution and rinsed with Tris-buffered saline. After quenching the reaction, cells were harvested and lysed in the lysis buffer containing protease inhibitors (Complete Mini; Roche, Mannheim, Germany). To improve solubilization, we sonicated the cells on ice. The cell lysates were centrifuged at 10,000 × g for 2 min at 4°C. The immobilized NeutrAvidin Gel slurry was used to adsorb the biotinylated membrane proteins in the supernatant. Gels were washed with wash buffer twice, and proteins were eluted from the NeutrAvidin Gel by heating at 95°C for 5 min in SDS-PAGE sample buffer.
Preparation of endoribonuclease-prepared small interfering RNA.
Endoribonuclease-prepared small interfering RNA (esiRNA) was prepared as described previously (Hammond et al., 2000). Briefly, using pcDNA-FAF1 as a template, a long double-stranded RNA that corresponded to nucleotides 339–689 of rat FAF1 was obtained from PCR-derived templates carrying the T7 promoter sequence (underlined) with the following primers: 5′-TAATACGACTCACTATAGGGTACTTGAAGACAGCTGTA-3′ and 5′-TAATACGACTCACTATAGGGTATGC-TTGTCAGATCATA-3′. The double-stranded RNA was digested with RNaseIII for 1 h at 37°C. The resultant short (12–30 bp) esiRNAs were precipitated with ethanol and labeled with Cy-3 using a Silencer siRNA labeling kit (Ambion, Austin, TX). esiRNAs were transfected into cultured DRG cells using Oligofectamine (Invitrogen, Groningen, Germany).
Reverse transcription-PCR.
First-strand cDNAs were reverse transcribed from total RNAs isolated from a FAF1 esiRNA-transfected cultured DRG neuron using the Power cDNA Synthesis kit (iNtRON Biotech, Ansan, Korea) with oligo(dT). PCR amplification (94°C for 3 min, then 94°C for 30 s, 60°C for 30 s, 72°C for 30 s at 25 cycles, and 72°C for 10 min) was performed using the GeneAmp PCR system 2700 (Applied Biosystems, Foster City, CA). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as a positive control.
Primary cultures of DRG cells.
Primary cultures of DRG cells were obtained for biochemical, immunohistochemical, and electrophysiolgical studies. Primary cultures of DRG cells were as described previously (Oh et al., 1996; Shin et al., 2002). Briefly, DRGs were dissected from all levels of the thoracic and lumbar spinal cords of neonatal rats and collected in cold culture medium (4°C). A mixture of DMEM and F-12 solution (Sigma) was used as the culture medium. In the culture medium, 10% fetal bovine serum (Invitrogen), 1 mm sodium pyruvate, 50–100 ng/ml nerve growth factor (Invitrogen), and 100 U/ml penicillin/streptomycin (Sigma) were added. Ganglia were washed with a mixture of DMEM and F-12 solution and incubated for 30 min in a warm (37°C) DMEM/F-12 mixture containing 1 mg/ml collagenase (Worthington, Freehold, NJ). The ganglia were then washed three times with Mg2+- and Ca2+-free Hank’s solution (Sigma), followed by incubation with gentle shaking for 30 min in warm Hank’s solution (37°C) that contained 2.5 mg/ml trypsin (Boehringer Mannheim, Indianapolis, IN). The solution was then centrifuged at 1000 rpm for 10 min, and the pellet was washed gently with the culture medium to inhibit trypsin activity. The pellet was then suspended in the culture medium, gently triturated with a fire-polished Pasteur pipette, and plated in small Petri dishes (35 × 12 mm). Cells were placed in a 37°C incubator in a 95% air/5% CO2 atmosphere. Cells were used 2–4 d after plating.
Current recording.
As a borosilicate glass pipette coated with Sylgard (Dow Corning, Midland, MI) touched the surface of a cell, a gigaseal was formed by gentle suction. After cell-attached patches were formed, the membrane in contact with the pipette was ruptured by applying suction to make an entire cell. After an entire cell was formed, the capacitive transients were canceled. For recording single-channel currents, inside-out or outside-out patches were formed in cultured DRG neurons, HEK 293T, or Chinese hamster ovarian (CHO) cells transfected with TRPV1 and/or FAF1 as described previously (Jung et al., 1999; Hwang et al., 2000). The control bath solution for single-channel current recording contained (in mm) 130 NaCl, 2 MgCl2, 5 EGTA, and 10 NaOH/HEPES, pH 7.2. The half-amplitude algorithm in FETCHAN (pClamp 9.0; Molecular Devices, Union City, CA) was used to detect open events. Channel activity was calculated as NPo, where N represents functional channels and Po represents channel open probability.
Results
Cloning of a TRPV1 association protein
To clone genes encoding proteins that are associated with TRPV1, a sensory neuron-specific cDNA expression library was constructed and screened by bait protein segments of TRPV1 (Fig. 1A). The TRPV1 segments were purified after expression in E. coli, tagged with 32P by protein kinase A, and used as radiolabeled probes. A λ-phage clone was identified that showed a specific binding signal during the library screening when a protein segment spanning the amino acids 243–440 in the N-terminal region of TRPV1 (TRPV1243–440) was used as a bait (Fig. 1A). Other protein segments such as TRPV11–135 (Fig. 1A) and a C-terminal cytosolic tail (TRPV1682–838) did not show any binding signal in the library screening (data not shown). The nucleotide sequence of the TRPV1-associated cDNA clone indicates that it corresponds to a gene segment of FAF1, originally cloned as a Fas-interacting protein (Chu et al., 1995). We then cloned the full-length cDNA of rat FAF1, which has 649 aa and a predicted molecular weight of 74 kDa. The deduced amino acid sequence of rat FAF1 showed 95 and 98% sequence identity with human and mouse FAF1, respectively (Chu et al., 1995; Ryu et al., 1999) (Fig. 1B).
Figure 1.

λ-phage display for cloning of a TRPV1 associated protein. A, A radiolabeled signal of TRPV1 (32P-TRPV1243–440) was detected in a FAF1-expressing clone during the initial screening of the sensory neuron-specific λ-phage expression library. 32P-TRPV11–135, as a negative control, did not show any binding signal. Right, A schematic drawing of TRPV1. Bait segments of TRPV1 are shown. Cross bars represent transmembrane domains. N, N terminus; C, C terminus. B, Deduced amino acid sequences of rat, human, and mouse FAF1. Identical amino acid residues are marked in black.
FAF1 interacts with TRPV1
To confirm the specific interaction of TRPV1 with FAF1, we performed GST pull-down analysis after expressing and purifying GST-FAF1 fusion proteins from E. coli. His-tagged N terminus (His-NTRPV1, TRPV11–440) or C terminus (His-CTRPV1, TRPV1682–838) was also expressed and purified from E. coli. Consistent with the results of λ-phage library screening, His-NTRPV1, but not His-CTRPV1, was copurified with GST-FAF1 (Fig. 2A).
Figure 2.

FAF1 associates with TRPV1 in vitro and in vivo. A, The N-terminal fragment of TRPV1 (NTRPV1, TRPV11–440), but not the C-terminal fragment (CTRPV1, TRPV1682–838), was pulled down by GST-FAF1. B, Coimmunoprecipitation of TRPV1 and FAF1 in HEK 293T cells. HEK 293T cells transfected with pcDNA-TRPV1 alone or pcDNA-TRPV1 and pCMV-FAF1 were immunoprecipitated either with antibody against FAF1 (left) or TRPV1 (right). Immunoprecipitates were immunoblotted by anti-TRPV1 (α-TRPV1) or anti-FAF1 (α-FAF1) antibody. Input indicates immunoblots of TRPV1 (left) or FAF1 (right) in cell lysates before immunoprecipitation. C, Coimmunoprecipitation of TRPV1 and FAF1 in DRG cells. DRG cultures were immunoprecipitated with preimmune serum (Pre-immune), α-FAF1, or α-TRPV1. Immunoprecipitates were then immunoblotted by α-TRPV1 or α-FAF1. i.p., Immunoprecipitated.
To determine further whether TRPV1 interacts with FAF1 in vivo, we coexpressed FAF1 and TRPV1 in HEK 293T cells (Fig. 2B). When cell lysates of HEK 293T cells transfected with TRPV1 and FAF1 were precipitated with anti-FAF1 antibody, TRPV1 was detected in the precipitates but not in the precipitates of cells transfected with TRPV1 alone. Conversely, when HEK 293T cells that were transfected with TRPV1 and FAF1 were precipitated with anti-TRPV1 antibody, FAF1 was detected in the precipitates but not in the immunoprecipitates of cells transfected with FAF1 alone. Thus, along with phage display, these results clearly suggest a strong physical interaction between TRPV1 and FAF1 in vitro as well as in vivo.
We then sought to determine whether the physical interaction occurred in sensory neurons. Indeed, coimmunoprecipitation of TRPV1 and FAF1 was also observed in DRG cultures. When lysates of primary cultures of DRG cells were immunoprecipitated with TRPV1 antibody, FAF1 was immunoblotted in the precipitates (Fig. 2C). Similarly, TRPV1 was found in the precipitates of DRG lysates that were immunoprecipitated with FAF1 antibody, confirming further the physical interaction between TRPV1 and FAF1 in sensory neurons.
To identify the region in FAF1 responsible for the interaction with TRPV1, we translated TRPV1 in a 35S-methionine-containing medium to make a radiolabeled probe. As shown in Figure 3A, the radiolabeled TRPV1 (35S-TRPV1) was copurified when it was incubated with GST-FAF1, GST-FAF11–201, GST-FAF171–201, or GST-FAF1Δ189–340. Because the peptide segment, FAF171–201, that contained the ubiquitin homologous domain (UB1) (Becker et al., 1997; Ryu et al., 1999; Kim et al., 2005), was copurified with 35S-TRPV1, this UB1 region appears to be responsible for the binding with TRPV1. Other regions, such as the death effecter domain interacting domain (Ryu et al., 2003) that is known to interact with Fas-associated death domain protein (FADD) or caspase-8, failed to be copurified with 35S-TRPV1 (Fig. 3A).
Figure 3.
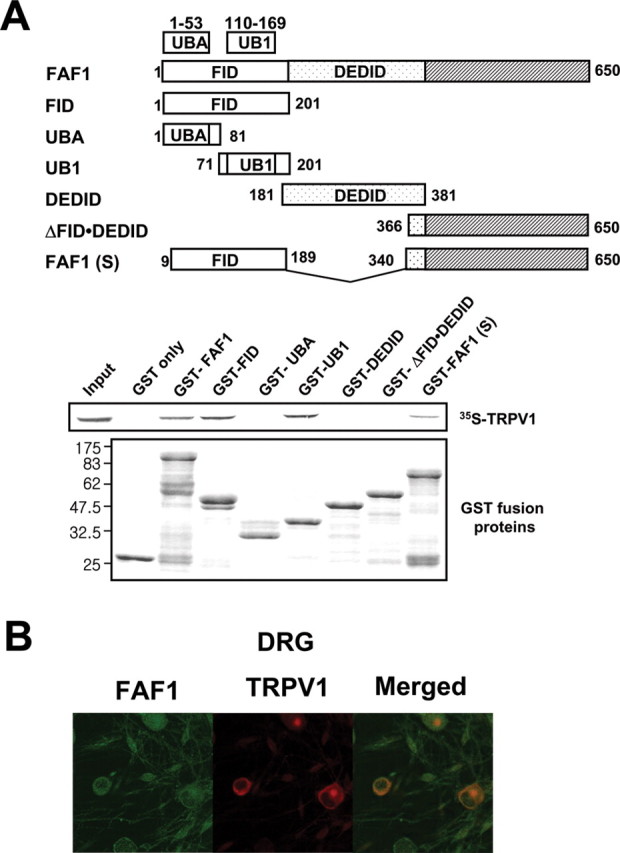
TRPV1 binds the ubiquitin homologous domain (UB1) region in FAF1. A, To identify the region in FAF1 responsible for the interaction with TRPV1, TRPV1 was translated in vitro in a 35S-methionine-containing medium to make a radiolabeled probe. Wild-type FAF1 and its mutants containing UB1 were copurified with 35S-TRPV1. The level of input proteins was identified by immunoblot with GST antibody. Schematic representation of the wild-type and deletion mutants of human FAF1. B, Coexpression of TRPV1 and FAF1 in sensory neurons. Cultured rat DRG neurons were incubated with mouse anti-TRPV1 and goat anti-FAF1 antisera, followed by Alexa Fluor 594-conjugated rabbit anti-mouse IgG and Alexa Fluor 488-conjugated rabbit anti-goat IgG. FID, Fas-interacting domain; DEDID, death effecter domain-interacting domain; UB1, ubiquitin homologous domain.
FAF1 and TRPV1 were coexpressed in sensory neurons (Fig. 3B). TRPV1 immunoreactivity was found in a subset of sensory neurons. However, dense FAF1 immunofluorescence was observed in sensory neurons as well as in satellite cells. FAF1 and TRPV1 immunoreactivity was present ubiquitously in the membrane and the cytosol but not observed in the nucleus (Frohlich et al., 1998; Olah et al., 2001). Among 358 DRG neurons counted, 339 (94.7%) neurons were FAF1 positive, whereas 144 (40.2%) neurons were TRPV1 positive. Among 339 FAF1-positive neurons, 125 (36.9%) cells were TRPV1 positive. In contrast, 125 (86.8%) cells were FAF1 positive among 144 TRPV1-positive neurons.
FAF1 controls pharmacological response of TRPV1 to capsaicin
Previously, we reported that the pharmacological profile of TRPV1 differs from those of native capsaicin receptors in sensory neurons (Shin et al., 2001). For example, TRPV1 is approximately three times as sensitive as native capsaicin receptors in sensory neurons in terms of capsaicin response (Shin et al., 2001). We therefore questioned whether the physical association between FAF1 and TRPV1 accounts for the pharmacological difference. To determine the pharmacological sensitivity of TRPV1 or TRPV1 and FAF1, 0.03–30 μm capsaicin was applied to the bath of inside-out membrane patches from DRG neurons, HEK-TRPV1, or HEK-FAF1/TRPV1 cells. As reported previously (Shin et al., 2001), the cloned capsaicin receptor, TRPV1, was approximately three times more sensitive to capsaicin than the native capsaicin receptor in sensory neurons, shifting the dose–response curve leftward from that of the native channel in sensory neurons (Fig. 4A). The half-maximal effective doses (EC50) of capsaicin in activating TRPV1 and native capsaicin receptor were 0.39 ± 0.05 μm (n = 11; mean ± SEM) and 1.11 ± 0.19 μm (n = 19), respectively. Surprisingly, cotransfection of TRPV1 with FAF1 reduced the sensitivity to capsaicin, shifting the dose–response curve to the right (i.e., back to that of the native capsaicin receptors in sensory neurons) (Fig. 4A). The EC50 value of capsaicin for HEK-FAF1/TRPV1 activation was 1.57 ± 0.37 μm (n = 9), significantly greater than that for HEK-TRPV1 (p < 0.01; ANOVA; Tukey’s post hoc test).
Figure 4.

FAF1 reduces capsaicin-induced responses of TRPV1. A, Dose–response relationships of capsaicin-induced single-channel currents recorded from HEK-TRPV1 (•; n = 11), HEK-FAF1/TRPV1 (○; n = 9), and DRG (▪; n = 19) neurons. B, FAF1 reduces capsaicin (CAP)-induced whole-cell currents. Right, A summary of capsaicin-induced whole-cell currents (Icap) from HEK-TRPV1 or HEK-FAF1/TRPV1 cells. *p < 0.001 (Student’s t test). C, FAF1 and TRPV1 coexpression reduces capsaicin-induced Ca2+ influx. Capsaicin (50 nm) was applied to HEK-TRPV1 (•; n = 20; arrow) or HEK-FAF1/TRPV1 (○; n = 20) cells loaded with Fluo3-AM. D, The stoichiometric relationship between capsaicin-induced Ca2+ influx and the ratios between FAF1 and TRPV1 cDNA amounts used for transfection of HEK 293T cells (n = 16–24; *p < 0.01; ANOVA; Tukey’s test). Error bars indicate SEM.
Reduction in the mean open time by FAF1 coexpression, but not the current amplitude, accounted for the reduction in channel activity. To acquire histograms of mean open times, we recorded single-channel currents having one or two levels of unitary currents from HEK-TRPV1 or HEK-FAF1/TRPV1 cells. The histogram of mean open times of single-channel currents activated by capsaicin was best fitted by two exponential functions (Kwak et al., 2000). Among the two mean open times (τo1 and τo2), FAF1 coexpression significantly reduced the mean open time of long opening (τo2) (6.33 ± 0.45 vs 4.00 ± 0.23 ms; n = 9). The mean open time of short opening (τo1) was not significantly different in both groups (0.84 ± 0.09 vs 0.67 ± 0.23 ms; n = 9). Coexpression of FAF1 did not affect the current amplitude of TRPV1 activated by 1 μm capsaicin. At −60 mV of the holding potential, mean current amplitudes of single-channel currents gated by capsaicin in HEK-TRPV1 and HEK-FAF1/TRPV1 cells were 2.62 ± 0.36 (n = 12) and 2.55 ± 0.13 pA (n = 8), respectively. FAF1 also increases the mean closed time for long closing (τc2) from 0.95 ± 0.42 to 3.31 ± 0.78 ms. Thus, FAF1 seems to reduce activity of TRPV1 via controlling the mean open and closed times of TRPV1.
The reduction in TRPV1 by FAF1 was also evident in entire cells. HEK 293T cells cotransfected with TRPV1 and FAF1 elicited significantly smaller current in response to 0.5 μm capsaicin than HEK-TRPV1 cells (Fig. 4B). A deletion mutant, FAF11–201 that copurified with TRPV1 failed to reduce the capsaicin-sensitive current of TRPV1 when coexpressed in HEK cells (Fig. 4B). FAF1- or mock-transfected HEK 293T cells showed no capsaicin-induced current even at 10 μm capsaicin (data not shown). The reduction in TRPV1 activity by FAF1 was also observed when Ca2+ influx induced by capsaicin was measured in HEK-TRPV1 and in HEK-FAF1/TRPV1 cells (Fig. 4C). Moreover, the reduction in TRPV1-induced Ca2+ mobilization was most prominent when the stoichiometric ratio between transfected amounts of FAF1 and TRPV1 cDNAs became 1:1 (Fig. 4D).
FAF1 controls pharmacological response of TRPV1 to acid
The functional association between FAF1 and TRPV1 was also tested for the response to acid. Because acid acts on the extracellular surface of TRPV1 (Jordt et al., 2000; Welch et al., 2000), acidic solutions at different degrees of acidity (pH 4.75–7.0) were applied to outside-out membrane patches. This time, CHO cells were chosen for the transfection of TRPV1 or TRPV1 and FAF1 because of the difficulty in forming outside-out patches from HEK 293T cells. After forming outside-out patches isolated from CHO-TRPV1, CHO-FAF1/TRPV1 cells, or cultured DRG neurons, 0.5–1 μm capsaicin was routinely applied to check for the presence of TRPV1 in these membrane patches. In DRG neurons, single-channel currents activated by capsaicin were also activated by applying acidic solution, pH 5.5, to the bath. Furthermore, the single-channel currents activated by acidic solution, pH 5.5, were significantly blocked (80.5 ± 0.05% reduction; n = 6) by 10 μm capsazepine, suggesting that the channel currents activated by acid were of TRPV1 origin (Tominaga et al., 1998). The current response of native capsaicin receptors in DRG neurons to acid was biphasic (Fig. 5A,B): single-channel currents increased steadily when the acidity of the bath solution was increased progressively to pH 5.5 but markedly reduced when the acidity of the bath solution exceeded pH 5.5. In contrast, the current response of TRPV1 to acid was monophasic: channel currents increased steadily even below a bath pH of 5.5. Strikingly, when TRPV1 was coexpressed with FAF1, a biphasic response profile to acid was observed, which exactly resembled the response to acid of the native capsaicin receptors in sensory neurons (Fig. 5A,B).
Figure 5.
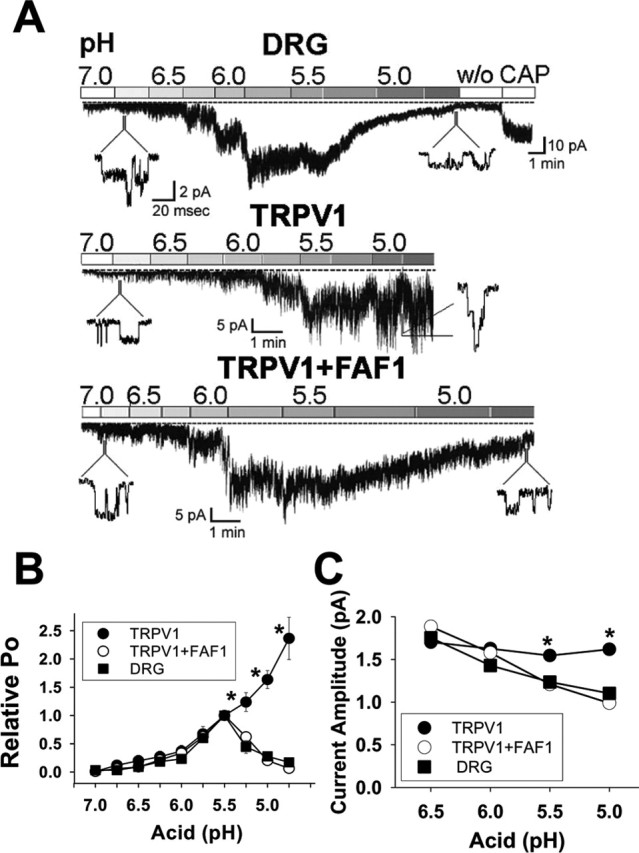
FAF1 reduces acid-induced responses of TRPV1. A, Current responses to acid of CHO-TRPV1, CHO-FAF1/TRPV1, and DRG cells. Acidic solutions of increasing acidity (pH 7.0–4.75) in increments of 0.25 of a pH unit were applied to outside-out patches isolated from DRG neurons or CHO-TRPV1 or CHO-FAF1/TRPV1. w/o, Washout; CAP, capsaicin (1 μm). B, The effect of FAF1 and TRPV1 coexpression on the open-channel probability (Po) of TRPV1. The Po at each pH was normalized to that of pH 5.5. Error bars indicate SEM. C, The effect of FAF1 and TRPV1 coexpression on the amplitude of single-channel currents activated by acid. Experimental numbers = 5–12. *p < 0.01 (ANOVA; Tukey’s test).
The biphasic change in the acid-induced macroscopic currents of native capsaicin receptors and of CHO-FAF1/TRPV1 primarily resulted from biphasic changes in open-channel probability (Po): a progressive increase in Po during the application of acidic solution to pH 5.5 was followed by a sharp reduction when the pH of the solution fell below 5.5. In contrast, Po of TRPV1 increased steadily when the pH fell to 4.75 (Fig. 5B). The reduction in the macroscopic currents of native capsaicin receptors in DRG neurons resulted also in part from the reduced single-channel current amplitude. As reported by Baumann and Martenson (2000), acidic solutions steadily reduced the current amplitudes of single-channel currents of native capsaicin receptor (Fig. 5C). Similarly, the amplitudes of single-channel currents recorded from CHO-FAF1/TRPV1 also declined significantly as the bath solution became more acidic. In contrast, the single-channel amplitudes of CHO-TRPV1 did not significantly decline on acidification (Fig. 5C).
FAF1 reduces the TRPV1 response to heat
We then tried to determine whether FAF1 controlled the sensitivity of TRPV1 to heat, another stimulus for the channel activation. Heat from 25 to 47.5°C was delivered to entire cells formed from HEK cells transfected with TRPV1 or TRPV1 and FAF1. In mock-transfected cells, application of heat from 25 to 47.5°C did not cause appreciable current (data not shown). In contrast, an increase in temperature of the bath solution caused channel currents in cells expressing HEK-TRPV1 or HEK-TRPV1/FAF1 (Fig. 6A). Coexpression of FAF1 did not affect the heat sensitivity of TRPV1. However, response currents of TRPV1 at 45 and 47.5°C from cells transfected with FAF1 and TRPV1 were significantly lower than those observed in TRPV1-transfected cells. The magnitudes of whole-cell currents at 45 and 47.5°C of HEK-TRPV1 cells were 981.1 ± 179.7 pA (n = 22) and 972.6 ± 174 pA (n = 20), respectively. Heat responses of HEK-TRPV1/FAF1 cells were 585.3 ± 85.7 pA (n = 28) and 491.8 ± 84.5 pA (n = 25). Thus, these results indicate that TRPV1 response to heat is reduced by FAF1.
Figure 6.

FAF1 reduces heat-induced current response of TRPV1. A, Whole-cell currents recorded from HEK-TRPV1 (triangle) and HEK-FAF1/TRPV1 (diamond) cells in a bath, the solution of which was heated from 25 to 47.5°C. *p < 0.05; **p < 0.01. B, Effects of FAF1 coexpression with ANKTM1 or ASIC3. Top, Whole-cell currents were recorded from HEK 293T cells transfected with either ANKTM1 alone or ANKTM1 and FAF1. Mustard oil (100 μm) was applied to both groups of cells. Note that the coexpression of FAF1 with ANKTM1 does not alter channel activity of ANKTM1. Calibration: 25 s, 200 pA. Bottom, Acidic (pH 5.5) solution was applied to HEK cells expressing ASIC3 alone or ASIC3 and FAF1. Also note that the coexpression of FAF1 with ASIC3 does not alter channel activity of ASIC3. Calibration: 5 s, 100 pA. The numbers in parentheses indicate the numbers of experiments. Error bars indicate SEM.
The reduction in channel activity by FAF1 was also tested in other channels. TRPA1 is a member of TRP channels activated by menthol, mustard oil, and tetrahydrocannabinol and is implicated in detection of noxious cold (Story et al., 2003; Jordt et al., 2004). We therefore tested whether FAF1 modulates channel activity of TRPA1. As reported by Jordt et al. (2004), application of mustard oil to TRPA1-transfected cells produced robust currents (Fig. 6B). However, unlike the case of TRPV1, FAF1 coexpression did not alter the current responses of TRPA1 to mustard oil. Similarly, FAF1 coexpression failed to modulate channel activity of ASIC3 evoked by acid (pH 5.0) application (Fig. 6B). Thus, FAF1 seems to regulate activity of TRPV1, not other channels.
FAF1 constitutively reduces TRPV1
To verify whether FAF1 is functional in vivo as a regulatory protein in native capsaicin receptor, expression of FAF1 in sensory neurons was silenced by RNA interference (RNAi) (Fire et al., 1998; Hammond et al., 2000). To have effective RNAi, we generated RNAi labeled with Cy-3 for easy detection of the transfected cells. Punctate Cy-3 fluorescence was visible in cultured DRG neurons 4–5 d after transfection of RNAi (Fig. 7B). After RNAi treatment, the mRNA level of FAF1 measured by reverse transcription-PCR (RT-PCR) was greatly depressed in sensory neurons, whereas FAF1 RNAi did not affect the message level of GAPDH. As shown in Figure 7A, application of 0.5 μm capsaicin evoked inward currents of 0.019 ± 0.005 nA/pF (n = 19). In contrast, silencing FAF1 by RNAi augmented the capsaicin-sensitive currents (0.101 ± 0.015 nA/pF, n = 11), eliciting an approximate fivefold increase in the whole-cell currents (Fig. 7B). Thus, these results suggest that FAF1 constitutively controls TRPV1 in vivo.
Figure 7.

Silencing of FAF1 by RNAi augments current response to capsaicin. A, Effect of FAF1 RNAi on 0.5 μm capsaicin (CAP) induced current response of sensory neurons. Whole-cell currents were recorded from control DRG neurons and neurons transfected with FAF1-targeted RNAi. B, A summary of capsaicin-induced whole-cell currents from control and RNAi-transfected neurons. *p < 0.001 (Student t test). The numbers in parentheses indicate the numbers of experiments. Right, A merged photomicrograph of fluorescent and phase-contrast images of DRG neurons transfected with the FAF1 RNAi that was labeled with Cy-3. Note the punctate Cy-3 fluorescence in the round soma of DRG neurons. Scale bar, 10 μm. Bottom, The level of mRNA of FAF1 was measured using RT-PCR in control and RNAi-treated groups of cultured DRG neurons. GAPDH was used as a positive control.
Effect of FAF1 on the cell-surface expression of TRPV1
Many proteins that are associated with ion channels affect the trafficking of pore-forming subunits (Anzai et al., 2002; Okuse et al., 2002). Furthermore, vesicular proteins, snapin and synaptotagmin IX, interact with TRPV1 to augment the TRPV1 expression in the membrane (Morenilla-Palao et al., 2004). Likewise, the downregulation of TRPV1 activity by FAF1 would result from the FAF1-dependent reduction in the expression of TRPV1 in the cell membrane. To determine whether FAF1 coexpression modulates TRPV1 expression in the membrane, we measured the expression level of TRPV1 in HEK cells by immunoblots after surface proteins were biotinylated and purified with streptavidin. As shown in Figure 8, the expression levels of TRPV1 in total cell lysates did not differ whether FAF1 was cotransfected or not. Similarly, FAF1 cotransfection failed to affect the expression levels of TRPV1 in the biotinylated fraction. These results suggest that FAF1 either does not affect degradation of TRPV1 or trafficking to the plasma membrane.
Figure 8.

FAF1 coexpression does not affect the overall levels of TRPV1 expression in the plasma membrane. The expression level of TRPV1 in HEK cells was measured by Western blots after surface proteins were biotinylated and purified with streptavidin (right).
Discussion
In the present study, we isolated a TRPV1-interacting protein, FAF1, which binds TRPV1 and reduces the sensitivity of TRPV1 to capsaicin, acid, and heat. Judging from the results from λ-phage display, GST pull down, or coimmunoprecipitation assay, FAF1 appears to maintain a strong and robust physical interaction with TRPV1. The interaction with FAF1 reduces its responses to capsaicin, acid, and heat and reinstalls the pharmacological properties of native capsaicin receptor in sensory neurons. The physico-functional interaction with FAF1 is physiologically relevant and constitutively active, because silencing TRPV1 gene increases its response to capsaicin. Thus, FAF1 seems to be an integral component of the capsaicin receptor complex in sensory neurons, controlling the activity of TRPV1. Because TRPV1 is a key molecule for the transduction of inflammatory pain (Caterina and Julius, 2001), elucidation of subunit structures of TRPV1 will provide a more detailed insight into nociception and pain treatment.
Because FAF1 was first identified as a Fas-associated protein, its role in Fas-induced cell death signaling has been suggested (Chu et al., 1995). FAF1 enhances Fas-induced cell death (Chu et al., 1995) as well as initiates apoptotic cell death by activating death signaling pathways (Ryu et al., 2003). Although FAF1 does not contain the typical death domain of many Fas-interacting proteins, it binds the death domain of Fas and induces apoptosis by interacting with FADD and caspase-8 that are known to be downstream death-signaling components of Fas (Ryu et al., 2003). In the present study, FAF1 binds the N terminus of TRPV1. Furthermore, TRPV1 binds the UB1 domain in FAF1 where Fas is known to bind (Becker et al., 1997; Ryu et al., 1999; Kim et al., 2005). The UB1 domain in FAF1 might be a region responsible for the interaction with other receptors. Thus, it is conceivable that FAF1 might mediate the downstream death signaling pathways initiated by TRPV1 as a receptor that lead to cell death. Because the expression of TRPV1 alone is sufficient to induce cell death when stimulated by TRPV1 agonists (Maccarrone et al., 2000; Shin et al., 2003), the involvement of FAF1 in TRPV1-dependent cell death is difficult to resolve. In addition, we expected that activation of Fas or TNF-α receptor (members of the TNF receptor family) (Suda et al., 1993; Enari et al., 1995) by its ligands would affect TRPV1 possibly via FAF1. However, we failed to see any change in current or membrane potentials when Fas antibody (Fas ligand) or TNF-α was applied to sensory neurons (data not shown). Thus, Fas does not appear to link to TRPV1 in sensory neurons.
Sensitivity to capsaicin of native capsaicin channels and heterologously expressed TRPV1 seems to be different: TRPV1 expressed in a heterologous system is much more sensitive to capsaicin than native capsaicin receptor. For example, the half-maximal concentrations (EC50) of capsaicin for activating native capsaicin-sensitive currents is ∼0.68–1.1 μm (Liu and Simon, 1996; Oh et al., 1996; Koplas et al., 1997; Liu et al., 1997; Jung et al., 1999). In contrast, EC50 values for activating TRPV1 expressed in mammalian cell lines are 0.1–0.25 μm, much lower than those for native capsaicin-sensitive currents in sensory neurons (Tominaga et al., 1998, 2001; Mohapatra and Nau, 2003). Similar results were also found in experiments measuring Ca2+ influx. EC50 values of capsaicin for causing Ca2+ influx in DRG neurons are 0.072- 0.2 μm (Wood et al., 1988; Cholewinski et al., 1993; Urban et al., 2000), whereas those in HEK-TRPV1 or CHO-TRPV1 cells are 0.016–0.098 μm (Gavva et al., 2004; Toth et al., 2004). The difference in potency stems from variation possibly raised by different experimental procedures performed in different laboratories. However, the potencies of capsaicin in activating both TRPV1 and native capsaicin receptor were compared by the same groups of researchers, minimizing the experimental variation among different laboratories. Vellani et al. (2001) found that TRPV1 expressed in HEK cells was much more sensitive to capsaicin than native channels in DRG neurons, because EC50 values of capsaicin for inducing inward currents in HEK-TRPV1 cells and in sensory neurons were 0.1 and 0.57 μm, respectively. Szallasi et al. (1999) observed a 10-fold difference in capsaicin sensitivity between native channel and TRPV1 in Ca2+ influx experiments (EC50 values: 0.3 vs 0.03 μm, respectively). Thus, TRPV1 seems more sensitive to capsaicin than native channels in sensory neurons. Variations in sensitizing or desensitizing cytosolic signals in sensory neurons and HEK or CHO cells that modulate TRPV1 activity would account for the difference in ligand sensitivity of TRPV1. However, in the present study, even in single-channel current recordings from an isolated membrane patch where cytosolic factors were mostly eliminated, we still observed an approximate threefold difference in capsaicin sensitivity (Fig. 4). Most notably, the difference in capsaicin sensitivity among native channels and TRPV1 was negated when it was coexpressed with FAF1. Furthermore, FAF1 silencing in sensory neurons by FAF1 RNAi augmented the capsaicin response (Fig. 7). Thus, it is likely that FAF1 actively controls TRPV1 in native sensory neurons as a negative regulator.
Acid, one of the major causes of the hyperalgesia induced by inflammation or ischemic injury, is known to excite sensory neurons (Steen et al., 1992; Reeh and Steen, 1996; Zeilhofer et al., 1997). Of the ion channels in sensory neurons that respond to acidic solution (Chen et al., 1998; Reeh and Kress, 2001), TRPV1 is a candidate transducer of acid-induced hyperalgesia (Caterina et al., 1997; Tominaga et al., 1998; Kress and Zeilhofer, 1999; Jung et al., 2002). As seen by others, TRPV1 activity increases as acidity of the solution increases up to pH 4.4 (Tominaga et al., 1998). However, unlike progressive activation of TRPV1, extreme acid reduces neural activity in sensory neurons (Steen et al., 1992; Zeilhofer et al., 1997). Thus, the apparent discrepancy between the response profile of TRPV1 and that of sensory neurons to acid suggests an obligatory regulation of TRPV1 by associated proteins. In the present study, the coexpression of TRPV1 and FAF1 reduces acid-induced response of TRPV1 and reasserts the response profiles of native capsaicin receptors to acid. It is not clear how FAF1 controls the sensitivity of TRPV1 to capsaicin, acid, and heat, although it is conceivable that FAF1 stabilizes TRPV1 from ligand activation.
The two vesicular proteins, snapin and synaptotagmin IX, are now known to interact with TRPV1. Using the yeast two-hybrid screening technique, Morenilla-Palao et al. (2004) cloned TRPV1-interacting proteins. Snapin and synaptotagmin IX associate and colocalize with TRPV1. The strong physical interaction would suggest a functional interaction with TRPV1. However, the two SNARE (soluble N-ethylmaleimide-sensitive factor attachment protein receptor) proteins failed to elicit modulatory action on TRPV1 properties because the overall channel properties were not changed after coexpression with TRPV1 (Morenilla-Palao et al., 2004). Instead, snapin and synaptotagmin IX augment TRPV1 expression in the plasma membrane in a protein kinase C-dependent manner. Thus, the interaction with two vesicular proteins is now thought to be implicated in protein kinase C-dependent exocytosis. In the present study, coexpression of TRPV1 with FAF1 elicited consistent change and reduction in its responses to capsaicin, acid, and heat. The reduction in the channel responses by FAF1 would be expected if FAF1 suppresses TRPV1 expression in the plasma membrane. However, coexpression of TRPV1 with FAF1 did not affect the expression level in the surface protein fraction (Fig. 8). Thus, FAF1 does not seem to affect the trafficking or expression of TRPV1 in the plasma membrane. Furthermore, TRPV1 binds FAF1 at the peptide segment (amino acids 71–201) that contains the ubiquitin homologous domain (UB1) (Fig. 3). Because the UB1 domain in FAF1 is known to bind proteasome after ubiquitination (Kleijnen et al., 2003), it is probable that FAF1 might cause degradation of TRPV1, resulting in the reduction in TRPV1 response. However, it is less likely that FAF1 induces TRPV1 degradation because it did not affect the overall expression level of TRPV1.
There is a wide variation in sensitivity to capsaicin among sensory neurons (Liu and Simon, 1996; Liu et al., 1997). The variation in capsaicin sensitivity is primarily attributable to the different levels of TRPV1 expression. Because FAF1 associates with and reduces the pharmacological responses of TRPV1 to ligands, FAF1 would in part account for the variation in capsaicin sensitivity. However, this by no means suggests that all the variation in capsaicin sensitivity is attributable to FAF1. Other factors such as sensitizing or desensitizing signals by phosphorylation or dephoshosphorylation would be more effective in determining the sensitivity.
In summary, we cloned a TRPV1-interacting protein, FAF1. Various in vivo and in vitro biochemical experiments, such as λ-phage display, GST pull down, or coimmunoprecipitation assay, exhibited robust and strong physical association of FAF1 with TRPV1. Expression of FAF1 with TRPV1 reduced its responses to all three types of stimuli: capsaicin, acid, and heat. In addition, the reduction by FAF1 was constitutively active. Because TRPV1 is a critical molecular transducer for mediating inflammatory pain, its regulation by FAF1 would contribute to understanding nociception as well as to designing and developing TRPV1 antagonists as novel analgesics.
Footnotes
*S.K. and C.K. contributed equally to this work.
This work was supported by the Creative Research Initiative Program of the Korean Ministry of Science and Technology and supported in part by the BK21 program of the Korean Ministry of Education and Human Resources Development.
References
- Anzai N, Deval E, Schaefer L, Friend V, Lazdunski M, Lingueglia E (2002). The multivalent PDZ domain-containing protein CIPP is a partner of acid-sensing ion channel 3 in sensory neurons. J Biol Chem 277:16655–16661. [DOI] [PubMed] [Google Scholar]
- Baumann TK, Martenson ME (2000). Extracellular protons both increase the activity and reduce the conductance of capsaicin- gated channels. In: J Neurosci 20: pp. RC80 (1–5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker K, Schneider P, Hofmann K, Mattmann C, Tschopp J (1997). Interaction of Fas(Apo-1/CD95) with proteins implicated in the ubiquitination pathway. FEBS Lett 412:102–106. [DOI] [PubMed] [Google Scholar]
- Bhave G, Zhu W, Wang H, Brasier DJ, Oxford GS, Gereau RWt (2002). cAMP-dependent protein kinase regulates desensitization of the capsaicin receptor (VR1) by direct phosphorylation. Neuron 35:721–731. [DOI] [PubMed] [Google Scholar]
- Caterina MJ, Julius D (2001). The vanilloid receptor: a molecular gateway to the pain pathway. Annu Rev Neurosci 24:487–517. [DOI] [PubMed] [Google Scholar]
- Caterina MJ, Schumacher MA, Tominaga M, Rosen TA, Levine JD, Julius D (1997). The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature 389:816–824. [DOI] [PubMed] [Google Scholar]
- Caterina MJ, Leffler A, Malmberg AB, Martin WJ, Trafton J, Petersen-Zeitz KR, Koltzenburg M, Basbaum AI, Julius D (2000). Impaired nociception and pain sensation in mice lacking the capsaicin receptor. Science 288:306–313. [DOI] [PubMed] [Google Scholar]
- Chen CC, England S, Akopian AN, Wood JN (1998). A sensory neuron-specific, proton-gated ion channel. Proc Natl Acad Sci USA 95:10240–10245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cholewinski A, Burgess GM, Bevan S (1993). The role of calcium in capsaicin-induced desensitization in rat cultured dorsal root ganglion neurons. Neuroscience 55:1015–1023. [DOI] [PubMed] [Google Scholar]
- Chu K, Niu X, Williams LT (1995). A Fas-associated protein factor, FAF1, potentiates Fas-mediated apoptosis. Proc Natl Acad Sci USA 92:11894–11898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang HH, Prescott ED, Kong H, Shields S, Jordt SE, Basbaum AI, Chao MV, Julius D (2001). Bradykinin and nerve growth factor release the capsaicin receptor from PtdIns(4,5)P2-mediated inhibition. Nature 411:957–962. [DOI] [PubMed] [Google Scholar]
- Davis JB, Gray J, Gunthorpe MJ, Hatcher JP, Davey PT, Overend P, Harries MH, Latcham J, Clapham C, Atkinson K, Hughes SA, Rance K, Grau E, Harper AJ, Pugh PL, Rogers DC, Bingham S, Randall A, Sheardown SA (2000). Vanilloid receptor-1 is essential for inflammatory thermal hyperalgesia. Nature 405:183–187. [DOI] [PubMed] [Google Scholar]
- Docherty RJ, Yeats JC, Bevan S, Boddeke HW (1996). Inhibition of calcineurin inhibits the desensitization of capsaicin-evoked currents in cultured dorsal root ganglion neurones from adult rats. Pflügers Arch 431:828–837. [DOI] [PubMed] [Google Scholar]
- Enari M, Hug H, Nagata S (1995). Involvement of an ICE-like protease in Fas-mediated apoptosis. Nature 375:78–81. [DOI] [PubMed] [Google Scholar]
- Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC (1998). Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 391:806–811. [DOI] [PubMed] [Google Scholar]
- Frohlich T, Risau W, Flamme I (1998). Characterization of novel nuclear targeting and apoptosis-inducing domains in FAS associated factor 1. J Cell Sci 111:2353–2363. [DOI] [PubMed] [Google Scholar]
- Gavva NR, Klionsky L, Qu Y, Shi L, Tamir R, Edenson S, Zhang TJ, Viswanadhan VN, Toth A, Pearce LV, Vanderah TW, Porreca F, Blumberg PM, Lile J, Sun Y, Wild K, Louis JC, Treanor JJ (2004). Molecular determinants of vanilloid sensitivity in TRPV1. J Biol Chem 279:20283–20295. [DOI] [PubMed] [Google Scholar]
- Hammond SM, Bernstein E, Beach D, Hannon GJ (2000). An RNA-directed nuclease mediates post-transcriptional gene silencing in Drosophila cells. Nature 404:293–296. [DOI] [PubMed] [Google Scholar]
- Hwang SW, Cho H, Kwak J, Lee SY, Kang CJ, Jung J, Cho S, Min KH, Suh YG, Kim D, Oh U (2000). Direct activation of capsaicin receptors by products of lipoxygenases: endogenous capsaicin-like substances. Proc Natl Acad Sci USA 97:6155–6160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordt SE, Tominaga M, Julius D (2000). Acid potentiation of the capsaicin receptor determined by a key extracellular site. Proc Natl Acad Sci USA 97:8134–8139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordt SE, Bautista DM, Chuang HH, McKemy DD, Zygmunt PM, Hogestatt ED, Meng ID, Julius D (2004). Mustard oils and cannabinoids excite sensory nerve fibres through the TRP channel ANKTM1. Nature 427:260–265. [DOI] [PubMed] [Google Scholar]
- Jung J, Hwang SW, Kwak J, Lee SY, Kang CJ, Kim WB, Kim D, Oh U (1999). Capsaicin binds to the intracellular domain of the capsaicin-activated ion channel. J Neurosci 19:529–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung J, Lee SY, Hwang SW, Cho H, Shin J, Kang YS, Kim S, Oh U (2002). Agonist recognition sites in the cytosolic tails of vanilloid receptor 1. J Biol Chem 277:44448–44454. [DOI] [PubMed] [Google Scholar]
- Kedei N, Szabo T, Lile JD, Treanor JJ, Olah Z, Iadarola MJ, Blumberg PM (2001). Analysis of the native quaternary structure of vanilloid receptor 1. J Biol Chem 276:28613–28619. [DOI] [PubMed] [Google Scholar]
- Kim HJ, Song EJ, Lee YS, Kim E, Lee KJ (2005). Human Fas-associated factor 1 interacts with heat shock protein 70 and negatively regulates chaperone activity. J Biol Chem 280:8125–8133. [DOI] [PubMed] [Google Scholar]
- Kleijnen MF, Alarcon RM, Howley PM (2003). The ubiquitin-associated domain of hPLIC-2 interacts with the proteasome. Mol Biol Cell 14:3868–3875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koplas PA, Rosenberg RL, Oxford GS (1997). The role of calcium in the desensitization of capsaicin responses in rat dorsal root ganglion neurons. J Neurosci 17:3525–3537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kress M, Zeilhofer HU (1999). Capsaicin, protons and heat: new excitment about nociceptors. Trends Pharmacol Sci 20:112–118. [DOI] [PubMed] [Google Scholar]
- Kuzhikandathil EV, Wang H, Szabo T, Morozova N, Blumberg PM, Oxford GS (2001). Functional analysis of capsaicin receptor (vanilloid receptor subtype 1) multimerization and agonist responsiveness using a dominant negative mutation. J Neurosci 21:8697–8706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwak J, Wang MH, Hwang SW, Kim TY, Lee SY, Oh U (2000). Intracellular ATP increases capsaicin-activated channel activity by interacting with nucleotide-binding domains. J Neurosci 20:8298–8304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwak JY, Jung JY, Hwang SW, Lee WT, Oh U (1998). A capsaicin-receptor antagonist, capsazepine, reduces inflammation-induced hyperalgesic responses in the rat: evidence for an endogenous capsaicin-like substance. Neuroscience 86:619–626. [DOI] [PubMed] [Google Scholar]
- Liu L, Simon SA (1996). Capsaicin-induced currents with distinct desensitization and Ca2+ dependence in rat trigeminal ganglion cells. J Neurophysiol 75:1503–1514. [DOI] [PubMed] [Google Scholar]
- Liu L, Lo Y, Chen I, Simon SA (1997). The responses of rat trigeminal ganglion neurons to capsaicin and two nonpungent vanilloid receptor agonists, olvanil and glyceryl nonamide. J Neurosci 17:4101–4111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maccarrone M, Lorenzon T, Bari M, Melino G, Finazzi-Agro A (2000). Anandamide induces apoptosis in human cells via vanilloid receptors. Evidence for a protective role of cannabinoid receptors. J Biol Chem 275:31938–31945. [DOI] [PubMed] [Google Scholar]
- Mohapatra DP, Nau C (2003). Desensitization of capsaicin-activated currents in the vanilloid receptor TRPV1 is decreased by the cyclic AMP-dependent protein kinase pathway. J Biol Chem 278:50080–50090. [DOI] [PubMed] [Google Scholar]
- Morenilla-Palao C, Planells-Cases R, Garcia-Sanz N, Ferrer-Montiel A (2004). Regulated exocytosis contributes to protein kinase C potentiation of vanilloid receptor activity. J Biol Chem 279:25665–25672. [DOI] [PubMed] [Google Scholar]
- Numazaki M, Tominaga T, Takeuchi K, Murayama N, Toyooka H, Tominaga M (2003). Structural determinant of TRPV1 desensitization interacts with calmodulin. Proc Natl Acad Sci USA 100:8002–8006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh U, Hwang SW, Kim D (1996). Capsaicin activates a nonselective cation channel in cultured neonatal rat dorsal root ganglion neurons. J Neurosci 16:1659–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuse K, Malik-Hall M, Baker MD, Poon WY, Kong H, Chao MV, Wood JN (2002). Annexin II light chain regulates sensory neuron-specific sodium channel expression. Nature 417:653–656. [DOI] [PubMed] [Google Scholar]
- Olah Z, Szabo T, Karai L, Hough C, Fields RD, Caudle RM, Blumberg PM, Iadarola MJ (2001). Ligand-induced dynamic membrane changes and cell deletion conferred by vanilloid receptor 1. J Biol Chem 276:11021–11030. [DOI] [PubMed] [Google Scholar]
- Premkumar LS, Ahern GP (2000). Induction of vanilloid receptor channel activity by protein kinase C. Nature 408:985–990. [DOI] [PubMed] [Google Scholar]
- Reeh PW, Kress M (2001). Molecular physiology of proton transduction in nociceptors. Curr Opin Pharmacol 1:45–51. [DOI] [PubMed] [Google Scholar]
- Reeh PW, Steen KH (1996). Tissue acidosis in nociception and pain. Prog Brain Res 113:143–151. [DOI] [PubMed] [Google Scholar]
- Ryu SW, Chae SK, Lee KJ, Kim E (1999). Identification and characterization of human Fas associated factor 1, hFAF1. Biochem Biophys Res Commun 262:388–394. [DOI] [PubMed] [Google Scholar]
- Ryu SW, Lee SJ, Park MY, Jun JI, Jung YK, Kim E (2003). Fas-associated factor 1, FAF1, is a member of Fas death-inducing signaling complex. J Biol Chem 278:24003–24010. [DOI] [PubMed] [Google Scholar]
- Shin CY, Shin J, Kim BM, Wang MH, Jang JH, Surh YJ, Oh U (2003). Essential role of mitochondrial permeability transition in vanilloid receptor 1-dependent cell death of sensory neurons. Mol Cell Neurosci 24:57–68. [DOI] [PubMed] [Google Scholar]
- Shin J, Cho H, Hwang SW, Jung J, Shin CY, Lee SY, Kim SH, Lee MG, Choi YH, Kim J, Haber NA, Reichling DB, Khasar S, Levine JD, Oh U (2002). Bradykinin-12-lipoxygenase-VR1 signaling pathway for inflammatory hyperalgesia. Proc Natl Acad Sci USA 99:10150–10155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin JS, Wang MH, Hwang SW, Cho H, Cho SY, Kwon MJ, Lee SY, Oh U (2001). Differences in sensitivity of vanilloid receptor 1 transfected to human embryonic kidney cells and capsaicin-activated channels in cultured rat dorsal root ganglion neurons to capsaicin receptor agonists. Neurosci Lett 299:135–139. [DOI] [PubMed] [Google Scholar]
- Steen KH, Reeh PW, Anton F, Handwerker HO (1992). Protons selectively induce lasting excitation and sensitization to mechanical stimulation of nociceptors in rat skin in vitro. J Neurosci 12:86–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Story GM, Peier AM, Reeve AJ, Eid SR, Mosbacher J, Hricik TR, Earley TJ, Hergarden AC, Andersson DA, Hwang SW, McIntyre P, Jegla T, Bevan S, Patapoutian A (2003). ANKTM1, a TRP-like channel expressed in nociceptive neurons, is activated by cold temperatures. Cell 112:819–829. [DOI] [PubMed] [Google Scholar]
- Suda T, Takahashi T, Golstein P, Nagata S (1993). Molecular cloning and expression of the Fas ligand, a novel member of the tumor necrosis factor family. Cell 75:1169–1178. [DOI] [PubMed] [Google Scholar]
- Szallasi A, Szabo T, Biro T, Modarres S, Blumberg PM, Krause JE, Cortright DN, Appendino G (1999). Resiniferatoxin-type phorboid vanilloids display capsaicin-like selectivity at native vanilloid receptors on rat DRG neurons and at the cloned vanilloid receptor VR1. Br J Pharmacol 128:428–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tominaga M, Caterina MJ, Malmberg AB, Rosen TA, Gilbert H, Skinner K, Raumann BE, Basbaum AI, Julius D (1998). The cloned capsaicin receptor integrates multiple pain-producing stimuli. Neuron 21:531–543. [DOI] [PubMed] [Google Scholar]
- Tominaga M, Wada M, Masu M (2001). Potentiation of capsaicin receptor activity by metabotropic ATP receptors as a possible mechanism for ATP-evoked pain and hyperalgesia. Proc Natl Acad Sci USA 98:6951–6956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toth A, Blumberg PM, Chen Z, Kozikowski AP (2004). Design of a high-affinity competitive antagonist of the vanilloid receptor selective for the calcium entry-linked receptor population. Mol Pharmacol 65:282–291. [DOI] [PubMed] [Google Scholar]
- Urban L, Campbell EA, Panesar M, Patel S, Chaudhry N, Kane S, Buchheit K, Sandells B, James IF (2000). In vivo pharmacology of SDZ 249–665, a novel, non-pungent capsaicin analogue. Pain 89:65–74. [DOI] [PubMed] [Google Scholar]
- Vellani V, Mapplebeck S, Moriondo A, Davis JB, McNaughton PA (2001). Protein kinase C activation potentiates gating of the vanilloid receptor VR1 by capsaicin, protons, heat and anandamide. J Physiol (Lond) 534:813–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch JM, Simon SA, Reinhart PH (2000). The activation mechanism of rat vanilloid receptor 1 by capsaicin involves the pore domain and differs from the activation by either acid or heat. Proc Natl Acad Sci USA 97:13889–13894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood JN, Winter J, James IF, Rang HP, Yeats J, Bevan S (1988). Capsaicin-induced ion fluxes in dorsal root ganglion cells in culture. J Neurosci 8:3208–3220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeilhofer HU, Kress M, Swandulla D (1997). Fractional Ca2+ currents through capsaicin- and proton-activated ion channels in rat dorsal root ganglion neurons. J Physiol (Lond) 503:67–78. [DOI] [PMC free article] [PubMed] [Google Scholar]