

Abstract
Regulator of G-protein signaling 9-1 (RGS9-1) and RGS9-2 are highly related RGS proteins with distinctive C termini arising from alternative splicing of RGS9 gene transcripts. RGS9-1 is expressed in photoreceptors where it functions as a regulator of transducin. In contrast, RGS9-2 is abundantly expressed in the brain, especially in basal ganglia, where its specific function remains poorly understood. To gain insight into the function of RGS9-2, we screened a human cDNA library for potential interacting proteins. This screen identified a strong interaction between RGS9-2 and α-actinin-2, suggesting a possible functional relationship between these proteins. Consistent with this idea, RGS9-2 and α-actinin-2 coimmunoprecipitated after coexpression in human embryonic kidney 293 (HEK-293) cells. Furthermore, endogenous RGS9-2 and α-actinin-2 could also be coimmunoprecipitated from extracts of rat striatum, an area highly enriched in both these proteins. These results supported the idea that RGS9-2 and α-actinin-2 could act in concert in central neurons. Like α-actinin-2, RGS9-2 coimmunoprecipitated NMDA receptors from striatal extracts, suggesting an interaction between RGS9-2, α-actinin-2, and NMDA receptors. Previous studies have shown that α-actinin mediates calcium-dependent inactivation of NMDA receptors. In HEK-293 cells expressing NMDA receptors, expression of RGS9-2 significantly modulated this form of NMDA receptor inactivation. Furthermore, this modulation showed remarkable preference for NMDA receptor inactivation mediated by α-actinin-2. Using a series of deletion constructs, we localized this effect to the RGS domain of the protein. These results identify an unexpected functional interaction between RGS9-2 and α-actinin-2 and suggest a potential novel role for RGS9-2 in the regulation of NMDA receptor function.
Keywords: RGS9, α-actinin-2, NMDA receptors, striatum, calcium-dependent inactivation, protein–protein interactions, RGS9-2
Introduction
Regulator of G-protein signaling (RGS) proteins are characterized by a conserved RGS domain that can serve as a GTPase-activating protein (GAP) for the α subunit of heterotrimeric G-proteins. RGS proteins constitute a diverse family unified by the presence of an RGS domain but diverging through the inclusion of additional domains that may specify interactions with varying signaling partners (Hollinger and Hepler, 2002). One of the larger proteins of the RGS family is RGS9, which exists in two alternatively spliced forms known as RGS9-1 and RGS9-2 (Granneman et al., 1998; Rahman et al., 1999; Zhang et al., 1999). RGS9 is a modular protein comprised of four identifiable domains: a conserved Disheveled, egl10, pleckstrin (DEP) domain, a G-γ-like (GGL) domain, an RGS domain, and a C-terminal tail that differs between RGS9-1 and RGS9-2 (see Fig. 1A). Most of our understanding of the function of RGS9 come from studies on RGS9-1, which is abundantly expressed in the retina, in which it functions as a GAP for transducin (Zhang et al., 1999; Hollinger and Hepler, 2002; Wensel, 2002). In contrast, much less is known about the function of RGS9-2, which is expressed predominantly in the brain, and more specifically in the basal ganglia (Granneman et al., 1998; Rahman et al., 1999, 2003). Previous studies have shown that RGS9-2 associates in central neurons with two binding partners, the atypical G-protein β subunit Gβ5 (Chen et al., 2003; Martemyanov et al., 2005) and the syntaxin family protein R7BP (Drenan et al., 2005; Martemyanov et al., 2005), and that these proteins contribute to the folding and stability of RGS9-2, its membrane targeting, and its ability to translocate from the membrane to the nucleus (Chen et al., 2003; Bouhamdan et al., 2004; Drenan et al., 2005). In an effort to gain additional insight into the function of RGS9-2 at a cellular/molecular level, our laboratory has begun a screen to identify additional proteins capable of interacting with RGS9-2 in the brain. Here, we report that RGS9-2 binds the cytoskeleton protein α-actinin-2 and that, through this interaction, it can regulate α-actinin-2-mediated NMDA receptor (NMDAR) inactivation.
Figure 1.
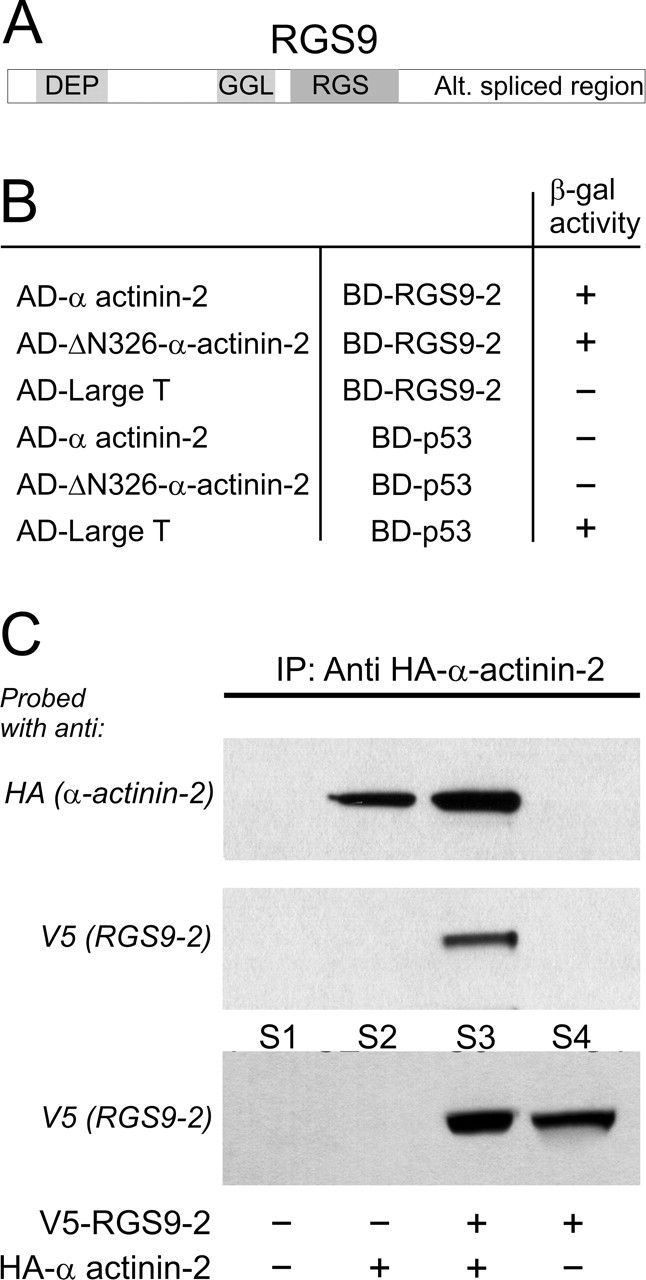
Brain-specific RGS9-2 binds the cytoskeletal protein α-actinin-2. A, Domain structure of RGS9: DEP domain, GGL domain, RGS domain, and a C terminus that is alternatively spliced. B, A human brain cDNA library was screened in a yeast two-hybrid assay using full-length RGS9-2 as bait. The initial screening identified a strong interaction of RGS9-2 with a truncated α-actinin-2 construct (designated as ΔN-326-α-actinin-2). This interaction was confirmed using full-length α-actinin-2. No interaction was seen between RGS9-2 and the large T antigen or between α-actinin-2 and p53 (negative controls), although the large T antigen interacted with p53 (positive control). C, RGS9-2 and α-actinin-2 physically interact when coexpressed in HEK-293 cells. Cells were transfected with plasmids encoding HA-tagged α-actinin-2 and V5-tagged RGS9-2, either individually or in combination (1:1 ratio). Robust RGS9-2 expression was evident in the supernatant (S) from cells transfected with V5-tagged RGS9 or V5-tagged RGS9 plus HA-tagged α-actinin-2 (bottom panel, S4 and S3, respectively). HA-tagged α-actinin-2 was immunoprecipitated only from cells transfected with HA-tagged α-actinin-2 or HA-tagged α-actinin-2 plus V5-tagged RGS9-2 (top panel, lanes 2 and 3, respectively) but not from cells transfected solely with V5-tagged RGS9-2 (lane 4) or mock transfected (lane 1). Immunoprecipitation of HA-tagged α-actinin-2 coimmunoprecipitated V5-tagged RGS9-2 from cells cotransfected with V5-tagged RGS9-2 and HA-tagged α-actinin-2 (middle panel, lane 3) but not from singly- or mock-transfected cells (middle panel, lanes 1, 2, and 4). This experiment was replicated four times with comparable results.
Materials and Methods
Yeast two-hybrid screen.
Yeast two-hybrid screenings were performed using the BD Biosciences Matchmaker 2 system (Clontech, Palo Alto, CA). Full-length RGS9-2 cDNA was amplified by PCR with RGS9-2-specific primers and cloned into GAL4 DNA-binding domain plasmid pGBKT7. The RGS9-2-pGBKT7 construct was transformed into yeast strain AH109 and shown not to elicit autoactivation of the GAL4 promoter using a β-galactosidase colorimetric filter assay. Screening of the human brain cDNA library was performed as recommended by the manufacturer. Briefly, AH109 was transfected with RGS9-2-pGBKT7 and mated with yeast strain Y187 containing a library of human brain cDNAs cloned into a GAL4 activating-domain plasmid pACT2 (Clontech). The mated yeast were plated onto plates lacking tryptophan, leucine, histidine, and adenine. After 10 d incubation at 30°C, interaction was determined by β-galactosidase colorimetric filter assay. Plasmid DNA from positive clones was isolated and transformed into bacteria. The inserts of positive clones were identified by PCR and automated sequencing. Full-length α-actinin-2 cDNA was PCR-amplified and cloned into the prey vector pGADT7 for subsequent analyses.
DNA constructs.
cDNAs encoding rat RGS9-2, different RGS9-2 domains (DEP, RGSd, and RGS-PRO), and α-actinin-1, -2, and -4 were subcloned into the pcDNA6 vector containing a C terminus V5 tag (Invitrogen, Carlsbad, CA) by PCR using specific primers. The RGS-PRO truncated protein includes amino acids 295–677 and thus lacks both the DEP and GGL domains. The RGSd truncated protein includes amino acids 297–421. The DEP domain includes amino acids 1–112. The cDNAs encoding Kir3.1 and Kir3.2 (Kovoor et al., 2000) (gifts from Dr. H. A. Lester, California Institute of Technology, Pasadena, CA), 5-HT1A receptor (Evans et al., 2001) (a gift from Dr. W. Clark, University of Texas, San Antonio, TX), NR1 and NR2A (Boeckman and Aizenman, 1994) (a gift from Dr. E. Aizenman, University of Pittsburgh, Pittsburgh, PA), hemagglutinin (HA)-α-actinin-2 (Zhou et al., 1999) (a gift from Dr. J. Chen, University of California, San Diego, La Jolla, CA), and RGS4 (Granneman et al., 1998) (a gift from Dr. J. Granneman, Wayne State University, Detroit, MI) have been described previously. The cDNA for enhanced green fluorescent protein (EGFP) was obtained from Clontech, that for Gβ5 in pcDNA3.1 was from Guthrie cDNA Resources (Sayre, PA), and those for α-actinin-4 (accession number BC013616) and α-actinin-1 cDNA (accession number BC003576) were obtained from the National Institutes of Health Mammalian Gene Collection.
Cell cultures and transfections.
Human embryonic kidney 293 (HEK-293) and HEK-293T cells (American Type Culture Collection, Manassas, VA) were maintained in DMEM supplemented with 10% fetal calf serum. Transfection of HEK-293 cells was conducted using Lipofectamine 2000 (Invitrogen) and a fixed amount of DNA (24 μg of DNA/100 mm plate for biochemical experiments or 4.25 μg/35 mm plate for electrophysiological experiments). Transfections involving multiple plasmids used equal total amounts of plasmid DNA, replacing each construct with empty vector (pcDNA3) as needed to generate the appropriate groups. For electrophysiological experiments, EGFP (0.25 μg) was included in the transfection to identify transfected cells. After 24–48 h, cells were harvested for immunoprecipitation (IP) and immunoblotting or used in electrophysiological experiments.
Immunoprecipitation and immunoblotting.
Transfected HEK-293 cells or striatal tissue were lysed with the IP buffer (1% Triton X-100, 50 mm Tris·Cl, pH 7.4, 300 mm NaCl, 5 mm EDTA, 0.02 sodium azide), and the lysate was centrifuged to pellet the cell debris. The resulting supernatant was then used for Western blotting or immunoprecipitation. For Western blotting, after adding the loading buffer, 50 μg of protein lysate was electrophoresed using a 10% Bis-Tris gel (Invitrogen) and then blotted onto nitrocellulose membranes. Membranes were blocked overnight at 4°C with 5% nonfat dry milk and then incubated overnight at 4°C, with goat anti-α-actinin (1:300; N19; Santa Cruz Biotechnology, Santa Cruz, CA) or rabbit anti-α-actinin-2 (1:1000) (a gift from Dr. Alan Beggs, Harvard Medical School, Boston, MA) (Dunah et al., 2000). Membranes were incubated with HRP-conjugated secondary antibody (1:15,000 goat anti-rabbit or rabbit anti-goat; Sigma, St. Louis, MO) for 1 h at room temperature, and the signal was detected using a chemiluminescence reagent. For immunoprecipitation, the lysate was incubated overnight with anti-HA (Roche Applied Science, Indianapolis, IN), anti-RGS9-2 (directed against the RGS9-2-specific C terminus region) (Bouhamdan et al., 2004), anti-NR1 (Chemicon, Temecula, CA) or anti-dopamine- and cAMP-regulated phosphoprotein of 32 kDa (DARPP-32) antibody (Santa Cruz Biotechnology) with 0.1% BSA overnight at 4°C with gentle rotation. Protein A-Sepharose beads (30 μl; Sigma) were added and incubated for 2 h at 4°C with mixing. After centrifugation, immunoprecipitation pellets were washed five times using ice-cold wash buffer (50 mm Tris·Cl, pH 7.4, 5 mm EDTA, 300 mm NaCl, 0.1% Triton X-100, 0.02% sodium azide) with a brief centrifugation each time in a refrigerated microcentrifuge. Samples from the pellets were resuspended in the loading buffer. Protein lysates (immunoprecipitates or 50 μg of protein from the supernatant) were electrophoresed using Nupage 10% Bis-Tris gel (Invitrogen) and then were blotted onto nitrocellulose membranes (Bio-Rad, Hercules, CA). Membranes were blocked and then incubated for 1 h at room temperature with mouse monoclonal anti-V5 antibody (1:5000; Invitrogen), goat anti-RGS9 antibody (directed against the RGS9 N terminus region; 1:300; T19; Santa Cruz Biotechnology), mouse anti-NR1 antibody (1:300; Chemicon), rabbit anti-α-actinin-2 (1:500), rabbit anti-DARPP-32 antibody (1:300; Santa Cruz Biotechnology), or mouse monoclonal anti-HA antibody (1:3000). Membranes were then incubated with HRP-conjugated secondary antibody (1:15,000 goat anti-mouse, goat anti-rabbit, or rabbit anti-goat; Sigma) for 1 h at room temperature. Signal was detected using a chemiluminescence reagent (Perkin-Elmer, Boston, MA).
Immunohistochemistry.
Immunohistochemical studies were performed as described previously (Bouhamdan et al., 2004). Briefly, striatal tissue sections were postfixed with 4% paraformaldehyde (for α-actinin-2) or ethanol (for RGS9-2) before preincubation in blocking solution (5% normal goat serum, 5% donkey serum, 2% BSA) and overnight incubation with primary antibodies directed against α-actinin-2 (1:2000) (Dunah et al., 2000) or RGS9-2 (1:1000) (Bouhamdan et al., 2004). Sections were subsequently incubated with goat anti-rabbit secondary antibody (1:2000 and 1:1000, respectively; Vector Laboratories, Burlingame, CA) followed by ABC reagent (Vector Laboratories) and DAB (Sigma).
Electrophysiological studies.
Whole-cell voltage-clamp recordings were performed 24–48 h after transfection. The recording chamber was continuously perfused (2–3 ml/min) at room temperature with an extracellular solution containing the following (in mm): 150 NaCl, 5 KCl, 2 CaCl2, 5 HEPES, 10 glucose, pH 7.3, 300–310 mOsm. Patch pipettes were pulled from 1.2 mm outer diameter borosilicate glass (Sutter Instruments, Novato, CA) and had resistances of 3–5 MΩ when filled with an intracellular solution containing the following (in mm): 140 CsCl, 10 EGTA, 15 HEPES, 4 Mg-ATP, 0.5 GTP, pH 7.3 (Zhang et al., 1998). 5-HT1A receptor-activated potassium current were recorded using an extracellular solution containing the following (in mm): 111.5 NaCl, 30.4 KCl, 1.8 CaCl2, 0.53 MgCl2, 5 HEPES, pH 7.3. For these experiments, the recording pipette solution contained the following (in mm): 130 KMeSO4, 5 KCl, 1 MgCl2, 0.02 EGTA, 10 HEPES, 4 MgATP, 0.5 GTP, pH 7.3 with KOH, 290–300 mOsm. Recordings were obtained using an EPC-7 patch-clamp amplifier (HEKA Elektronik, Lambrecht, Germany), whereas data acquisition used a Digidata 1200 (Molecular Devices, Foster City, CA) under the control of pClamp 8 (Molecular Devices). Cells were held at −50 mV for NMDA-induced current or at −90 mV for Kir3 currents. They were locally perfused using a short length of PE50 tubing attached to a miniature manifold that allowed for solution exchange under the control of an array of electronically controlled valves (Warner Instruments, Hamden, CT). Cells were continuously perfused with the appropriate bath media, transiently switching to the experimental solution as needed under computer control. Glycine (100 μm) was added to the solutions to prevent glycine-dependent desensitization. Statistical comparisons were made against the appropriate controls (e.g., pcDNA3, α-actinin-2, or wild-type RGS9-2) transfected in parallel on sister cultures. The extent of NMDA current inactivation was measured as the percent inactivation, as defined by the reduction in current amplitude from the initial peak detected at the beginning of the NMDA application to the steady-state level at the end of the application, expressed as a percentage of the peak current (Krupp et al., 1999). All data are presented as mean ± SEM, and statistical comparisons used one-way ANOVAs followed by Dunnett’s post hoc tests. Statistical comparison between the effects of different α-actinin isoforms used a two-way ANOVA followed by protected pairwise multiple comparisons (Fisher’s least significant difference).
Results
As a first step toward identifying interacting proteins for brain-specific RGS9-2, we screened a human brain cDNA library using full-length RGS9-2 as bait in a yeast two-hybrid assay. This screen identified seven independent clones, the most interesting of which, based on its brain colocalization with RGS9-2 (see below), coded for α-actinin-2. The initial yeast two-hybrid screen detected an interaction between RGS9-2 and a truncated α-actinin-2 extending from amino acid R327 to the C terminus (designated as ΔN-326-α-actinin-2). Subsequent work confirmed a robust interaction between RGS9-2 and a full-length α-actinin-2 prey (Fig. 1B). No interaction was seen between RGS9-2 and large T antigen or between α-actinin-2 and p53 (negative controls), whereas the large T antigen strongly interacted with p53 (positive control) (Fig. 1B).
To assess the significance of the interaction between RGS9-2 and α-actinin-2, we examined the ability of these proteins to coimmunoprecipitate after expression in HEK-293 cells. Cells were transfected with plasmids encoding HA-tagged α-actinin-2 and V5-tagged RGS9-2 either individually or in combination (1:1 ratio). As illustrated in Figure 1C, robust RGS9-2 expression was detected in the supernatant of cells transfected with V5-tagged RGS9-2 (bottom panel, S3 and S4) but not in cells transfected only with HA-tagged α-actinin-2 or mock-transfected cells (S2 and S1). Similarly, HA-tagged α-actinin-2 was immunoprecipitated from cells transfected with HA-tagged α-actinin-2 (Fig. 1C, top panel, lanes 2 and 3, respectively) but not from cells transfected only with V5-tagged RGS9-2 or mock-transfected cells (lanes 4 and 1). Most importantly, immunoprecipitation of HA-tagged α-actinin-2 coimmunoprecipitated V5-tagged RGS9-2 from extracts derived from cells cotransfected with V5-tagged RGS9-2 and HA-tagged α-actinin-2 (Fig. 1C, middle panel, lane 3) but not from those derived from singly or mock-transfected cells (lanes 1, 2 and 4). Conversely, immunoprecipitation of V5-tagged RGS9-2 coimmunoprecipitated HA-tagged α-actinin-2 only from cells cotransfected with V5-tagged RGS9-2 and HA-tagged α-actinin-2 (data not shown). Together, these results demonstrate a physical interaction between RGS9-2 and α-actinin-2 in mammalian HEK-293 cells.
The experiment above indicated that RGS9-2 and α-actinin-2 can physically interact when expressed in the same cell. If these proteins are to act together in the brain, however, they must be coexpressed in at least a subpopulation of central neurons. Previous studies have mapped the distribution of RGS9-2 (Granneman et al., 1998; Rahman et al., 1999) and α-actinin-2 (Wyszynski et al., 1997; Dunah et al., 2000) in the brain, and a comparison of these distributions reveals a striking anatomical overlap, with very high levels of expression in the striatum (illustrated in Fig. 2A), somewhat lower levels of expression in hippocampus and cortex, and generally very low levels of expression elsewhere in the brain. Most importantly, at least in striatum, both RGS9-2 and α-actinin-2 are expressed medium spiny neurons, the predominant cellular elements in this region (Dunah et al., 2000; Rahman et al., 2003). As such, these observations suggest coexpression of these proteins at the cellular level. To further examine the possibility of an interaction of these proteins in central neurons, we used coimmunoprecipitation under non-denaturing conditions to assess whether endogenous RGS9-2 and α-actinin-2 could physically interact. To that effect, we used an antibody directed against the carboxyl tail of RGS9-2 (Bouhamdan et al., 2004) to “pull down” RGS9-2 from striatal tissue. As illustrated in Figure 2B, column 1, probing the immunoprecipitated material with a different anti-RGS9 antibody, directed against the DEP domain of the protein (T19 Ab), revealed a band of Mr ∼82 kDa, indicating effective pull-down of endogenous RGS9-2. Reprobing the immunoprecipitated material with a highly specific antibody against α-actinin-2 (Wyszynski et al., 1997, 1998) identified a band of ∼100 kDa (Fig. 2B), indicating the coimmunoprecipitation of α-actinin-2 with RGS9-2. In contrast, probing with an antibody against DARPP-32, another cytosolic protein abundantly expressed in medium spiny neurons (Ouimet et al., 1984), did not reveal detectable amounts of DARPP-32 in the RGS9-2 immunoprecipitate (Fig. 2B, column 1). Similarly, immunoprecipitation of DARPP-32 failed to coimmunoprecipitate either RGS9-2 or α-actinin-2 (Fig. 2B, column 2). These results supported the idea that RGS9-2 and α-actinin-2 physically interact in vivo and thus could act in concert as functional partners in central neurons.
Figure 2.
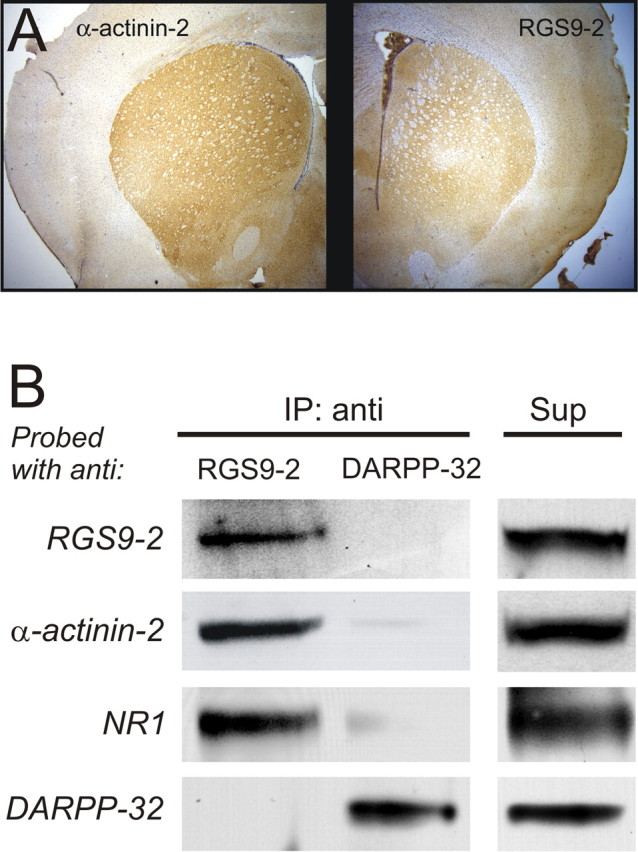
RGS9-2 and α-actinin-2 are coexpressed and interact in striatum. A, RGS9-2 and α-actinin-2 exhibit a similar distribution in the rat forebrain, with robust expression in striatum. B, Coimmunoprecipitation of RGS9-2, α-actinin-2, and NMDA receptor subunit NR1 from striatum. Antibodies detect robust expression of these proteins plus DARPP-32 in striatal supernatant (Sup). Immunoprecipitation of endogenous RGS9-2 from striatal extract results in coimmunoprecipitation of α-actinin-2 and NR1, but not DARPP-32 (column 1). Conversely, immunoprecipitation of DARPP-32 failed to coimmunoprecipitate either RGS9-2, α-actinin-2, or NR1 (column 2). This experiment was replicated three times with comparable results.
Previous work has shown that α-actinin-2 associates with NMDARs in central neurons (Wyszynski et al., 1997; Dunah et al., 2000). Therefore, we next examined whether RGS9-2 could also interact with NMDARs. As illustrated in Figure 2B, column 1, reprobing with an anti-NR1 antibody revealed that the RGS9-2 antibody coimmunoprecipitated not only α-actinin-2 but also the NR1 subunit of the NMDAR. The specificity of this protein–protein interaction was supported again by the fact that an anti-DARPP-32 antibody did not appreciably coimmunoprecipitate NR1 (Fig. 2B, column 2). Previous studies have shown that α-actinin-2 is a key component of the macromolecular ensemble that mediates the calcium-dependent inactivation (CDI) of NMDARs (Zhang et al., 1998; Krupp et al., 1999). Therefore, the physical interaction of RGS9-2, NMDARs, and α-actinin-2 detected above raised the possibility that RGS9-2 could regulate this physiological phenomenon.
CDI is robustly expressed in HEK-293 cells expressing NMDARs, and this model system has been used extensively to characterize this phenomenon (Zhang et al., 1998; Krupp et al., 1999). Therefore, we next examined whether RGS9-2 could regulate CDI of NMDA currents in HEK-293 cells. As previously reported by others (Zhang et al., 1998; Krupp et al., 1999), a brief application of NMDA (50 μm) to HEK-293 cells transfected with the NR1 and NR2A subunits of the NMDA receptor elicits a (+)-5-methyl-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5,10-imine maleate (MK-801)- and carboxypiperazin-4-yl-propyl-1-phosphonic acid (CPP)-sensitive inward current (MK-801, 10 μm, n = 5; CPP, 50 μm, n = 6) (data not shown) that shows pronounced inactivation (Fig. 3A1). This inactivation reflects predominantly CDI, because little if any inactivation is seen when calcium is removed from the extracellular media (n = 4) (data not shown). The robust expression of CDI in HEK-293 cells allowed us to test for a possible role for RGS9-2 in this phenomenon.
Figure 3.

Effect of RGS9-2 on NMDAR CDI in HEK-293 cells. A1, CDI in a HEK-293 cell transfected with the NR1 and NR2A subunits and pcDNA3. A2, In a different cell, transfection with α-actinin-2 does not affect CDI. A3, Transfection of RGS9-2, however, results in an inhibition of CDI. A4, An even stronger suppression of CDI is seen after cotransfection of RGS9-2 and α-actinin-2. B, Summary plot comparing the magnitude of CDI, expressed as percent inhibition of the peak NMDA current under control conditions. n = 36–44 cells per group; *p < 0.01 against pcDNA3; **p < 0.01 against α-actinin-2 alone. C, Expression of Gβ5 does not modify the ability of RGS9-2 to regulate CDI (n = 16–18 cells per group). Error bars indicate SE.
To test the hypothesis that RGS9-2 could regulate CDI through an interaction with α-actinin-2, we next coexpressed these proteins in HEK-293 cells in conjunction with NMDARs. As illustrated in Figure 3, A4 and B, cotransfection of RGS9-2 and α-actinin-2 resulted in a robust suppression of CDI, whereas expression of α-actinin-2 alone had no detectable effect (Fig. 3A2,B). These results supported the idea of a functional interaction between RGS9-2, α-actinin-2, and NMDARs. Surprisingly, however, transfection with RGS9-2 alone, although still eliciting a significant suppression of CDI (Fig. 3A3,B), was much less effective than the cotransfection of RGS9-2 and α-actinin-2. Because this observation suggested a specific effect of the transfected α-actinin-2 vis-à-vis endogenous α-actinins, we next examined the basis for this observation.
Previous studies have identified Gβ5 as a key factor stabilizing RGS9-2 (Chen et al., 2003). Although many cells, including HEK-293, express Gβ5 endogenously, it is possible that, under our experimental conditions, Gβ5 could be limiting for the availability of RGS9-2. If α-actinin-2 stabilized RGS9-2, thus essentially substituting for Gβ5 and leading to higher effective concentrations of RGS9-2 inside the cell, this could explain the paradoxical facilitation by α-actinin-2 on the effect of RGS9-2. We tested this idea by cotransfecting Gβ5 and RGS9-2, a manipulation that results in an increase in the availability of functional RGS9-2 (Kovoor et al., 2000). As illustrated in Figure 3C, expression of Gβ5, however, had no detectable effect on the ability of RGS9-2 either alone or coexpressed with α-actinin-2 to modulate CDI. As such, these results did not support the idea that Gβ5 is limiting under our experimental conditions, and suggest that a stabilizing effect is unlikely to account for the facilitatory effect of α-actinin-2 on RGS9-2 function.
In previous work, it has been assumed that CDI in HEK-293 cells is mediated solely by α-actinin-2 (Zhang et al., 1998). This idea is based on the ability of the spectrin domain of α-actinin-2 to interact with the C-terminal tail of NR1 (Wyszynski et al., 1997) and on the expression of endogenous α-actinin-2 in HEK-293 cells (Zhang et al., 1998). However, this idea has not been tested experimentally, and the high degree of conservation between the different α-actinin isoforms suggests that other isoforms may also be able to mediate CDI. Because it turns out that HEK-293 cells express at least two α-actinin isoforms (Li et al., 2005), a selective interaction between RGS9-2 and α-actinin-2 would be able to account for the synergistic effect obtained by cotransfection, especially if CDI was mediated predominantly by other α-actinin isoforms under basal conditions (i.e., in the absence of RGS9-2).
As a first step in testing this idea, we determined the expression of different α-actinin isoform mRNAs in HEK-293 cells using semiquantitative RT-PCR. Under our experimental conditions, HEK cells expressed α-actinin-4 mRNA most robustly, with lower expression levels of α-actinin-1 and α-actinin-2 (relative abundance ratio, 3.2/1.9/1.0) (data not shown). In contrast, α-actinin-3 was not detectable in these same samples, consistent with its restricted expression in skeletal muscle cells (MacArthur and North, 2004). Because the key issue is whether these α-actinin isoforms interact with NMDA receptors under our experimental conditions, we next tested the ability of α-actinin-1, -2, and -4 to interact with NMDARs using coimmunoprecipitation from cells transfected with NR1, NR2A, RGS9-2, and V5-tagged α-actinin-1, -2, or -4. As illustrated in Figure 4A1, immunoprecipitation with an anti-NR1 antibody coimmunoprecipitated V5-tagged α-actinin-4 and also, to a somewhat lesser degree, V5-tagged α-actinin-2. In contrast, NR1 immunoprecipitation resulted in the coimmunoprecipitation of only trace amounts of V5-tagged α-actinin-1. These observations, namely that α-actinin-4 is expressed much more robustly than α-actinin-2 at the mRNA level and that NR1 associates with α-actinin-4 more avidly than with α-actinin-2, suggest that α-actinin-4 is the predominant mediator of CDI in this experimental system.
Figure 4.
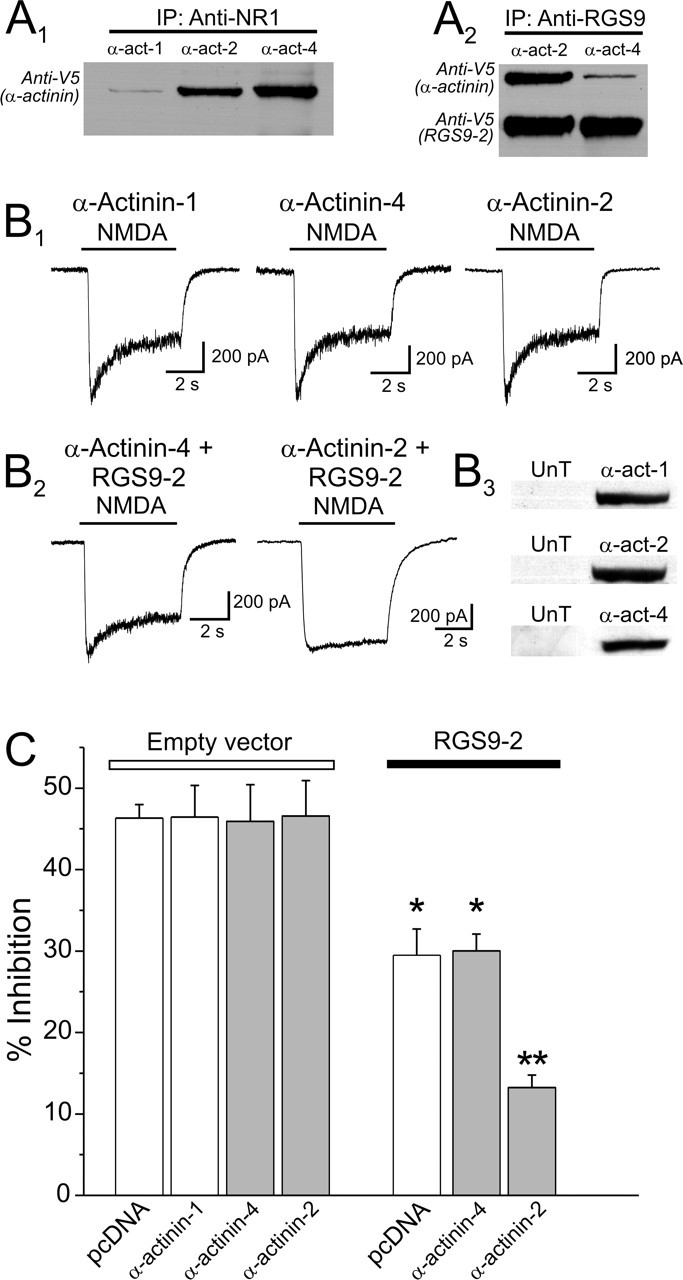
Effect of different α-actinin isoforms on the ability of RGS9-2 to modulate CDI. A1, α-Actinin-4 or α-actinin-2 interact with NR1. Cells were transfected with plasmids encoding NR1, NR2A, RGS9-2, and V5-tagged α-actinin-1, -2, or -4. NMDARs were immunoprecipitated using an anti-NR1 antibody, and an anti-V5 antibody was used to probe for the coimmunoprecipitation of each of the α-actinin isoforms. Immunoprecipitation of NR1 effectively coimmunoprecipitated α-actinin-4 and α-actinin-2 but only a trace amount of α-actinin-1. A2, In these same cells, immunoprecipitation of RGS9-2 preferentially coimmunoprecipitated α-actinin-2 vis-à-vis α-actinin-4. B1, Overexpression of α-actinin-1, α-actinin-4, or α-actinin-2 does not affect the expression of CDI in HEK-293 cells. B2, Expression of α-actinin-4 had no detectable effect on the ability of RGS9-2 to modulate CDI. In contrast, expression of α-actinin-2 markedly facilitated the ability of RGS9-2 to modulate NMDAR CDI. B3, Western blot confirming the overexpression of the different α-actinin isoforms in the HEK-293 cells used for these experiments. An anti-α-actinin-2 antibody and an anti-pan α-actinin antibody were used for this experiment. Endogenous α-actinins were below the level of detection for the probing conditions. C, Plot summarizing the results of this experiment. pcDNA3, 23 cells; α-actinin-1, 17 cells; α-actinin-4, 13 cells; α-actinin-2, 8 cells; RGS9-2, 15 cells; RGS9-2 plus α-actinin-1, 14 cells; RGS9-2 plus α-actinin-4, 16 cells; RGS9-2 plus α-actinin-2, 20 cells. *p < 0.01 against the respective control group; **p < 0.01 against α-actinin-2 alone and also against RGS9, RGS9-2 plus α-actinin-1, or RGS9-2 plus α-actinin-4 (ANOVA, followed by protected t test). UnT indicates mock-transfected cells. Error bars indicate SE.
If, as outlined above, RGS9-2 modulated CDI by acting preferentially on α-actinin-2 vis-à-vis α-actinin-4, this could explain the paradoxical facilitation of RGS9-2 by α-actinin-2. We tested this hypothesis by comparing the ability of α-actinin-2 and α-actinin-4 to facilitate the effect of RGS9-2. As illustrated in Figure 4, B1 and C, overexpression of α-actinin-1 (Fig. 4B3), which interacts with NR1 only very weakly (Fig. 4A1), had no effect on CDI. Furthermore, overexpression of either α-actinin-2 or α-actinin-4 (Fig. 4B3), which do interact with NR1 (Fig. 4A1), also failed to modulate CDI. We interpret these results to indicate that both α-actinin-2 and α-actinin-4 can support CDI and that these are endogenously expressed at or near saturating concentrations with respect to CDI under our experimental conditions. Consistent with the view that α-actinin-4 mediates CDI under basal conditions, overexpression of α-actinin-4 had no detectable impact on the effect of RGS9-2 (Fig. 4B2,C). In contrast, expression of α-actinin-2 resulted in a profound facilitation in the ability of RGS9-2 to modulate CDI (Fig. 4B2,C). Consistent with these electrophysiological findings, RGS9-2 was able to coimmunoprecipitate α-actinin-2 much more effectively than α-actinin-4 (Fig. 4A2). Combined, these results support the idea that RGS9-2 preferentially interacts with α-actinin-2 and that this interaction can regulate NMDAR CDI.
To better understand the mechanism whereby RGS9-2 regulates CDI, we sought to identify the protein domain(s) involved in this effect. To that end, we compared the effects of a series of RGS9-2 deletion constructs. As illustrated in Figure 5, A1, A2, and B, deletion of the N-terminal DEP and GGL domains of RGS9-2 resulted in a truncated protein (RGS-PRO) that significantly inhibited CDI and was nearly as effective as full-length RGS9-2. Conversely, transfection of a construct encoding the isolated DEP domain of RGS9-2 failed to suppress CDI (Fig. 5A4,B), although the DEP domain was robustly expressed in these cells as assessed by Western blotting (data not shown). These converging experiments suggested that the DEP domain of RGS9-2 was unlikely to play an essential role in the regulation of NMDARs. Deletion of the N-terminal DEP domain and the proline-rich C-terminal domain resulted in a construct containing predominantly, but not exclusively, the catalytic domain of RGS9-2 (RGS9-d). This isolated domain was still capable of significantly inhibiting CDI (Fig. 5A3,B), although in a somewhat attenuated manner. Consistent with these results, immunoprecipitation of α-actinin-2 was found to coimmunoprecipitate both the RGS-Pro and RGS9-d truncated forms of RGS9-2 (data not shown). These results identified the RGS9-2 catalytic core and adjacent sequences as central to the interaction between RGS9-2 and α-actinin-2 that leads to the regulation of CDI of NMDARs.
Figure 5.
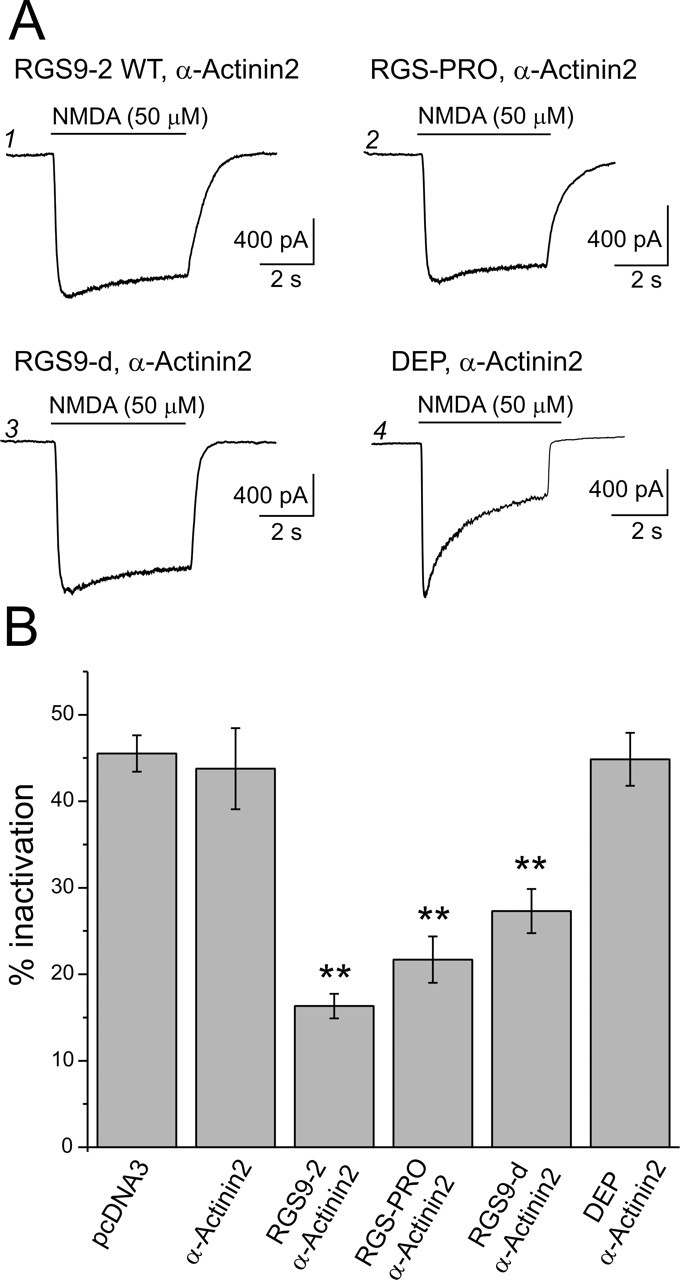
The catalytic domain of RGS9-2 mediates the inhibition of CDI. A1, NMDAR current in a cell transfected with RGS9-2 and α-actinin-2 shows little inactivation, indicating a suppression of CDI. A2, A3, A similar suppression of CDI is seen in cells transfected with RGS-PRO and α-actinin-2 or RGS9-d (the catalytic domain of RGS9-2) and α-actinin-2. A4, In contrast, a cell transfected with the isolated DEP domain of RGS9-2 and α-actinin-2 exhibits CDI comparable with that seen in control cells. B, Summary plot comparing the magnitude of CDI under control conditions (pcDNA3 transfection, n = 10; α-actinin-2 and pcDNA3, n = 8) and that observed after transfection with RGS9-2 plus α-actinin-2 (n = 33), RGS-PRO plus α-actinin-2 (n = 12), RGS9-d plus α-actinin-2 (n = 21), and isolated DEP domain of RGS9-2 plus α-actinin-2 (n = 16). **p < 0.01 compared with α-actinin-2. Error bars indicate SE.
The identification of the RGS domain of RGS9-2 as sufficient to regulate CDI raised the possibility that this effect could reflect a nonspecific increase in GAP activity within the cells. To test this possibility, we took advantage of the homology between the RGS domain of RGS9 and RGS4 (De Vries et al., 2000). As illustrated in Figure 6A, transfection of RGS9-2 or RGS4 resulted in comparable effects on the deactivation rate of Kir3.1/Kir3.2 currents elicited by activating a Gαi-coupled serotonin receptors (5-HT1A) (Granneman et al., 1998). In contrast, only RGS9-2 was capable of suppressing CDI (Fig. 6B). These results indicate that the effect of RGS9-2 on CDI does not simply reflect a generalized nonspecific increase in RGS-mediated GAP activity within the cell.
Figure 6.
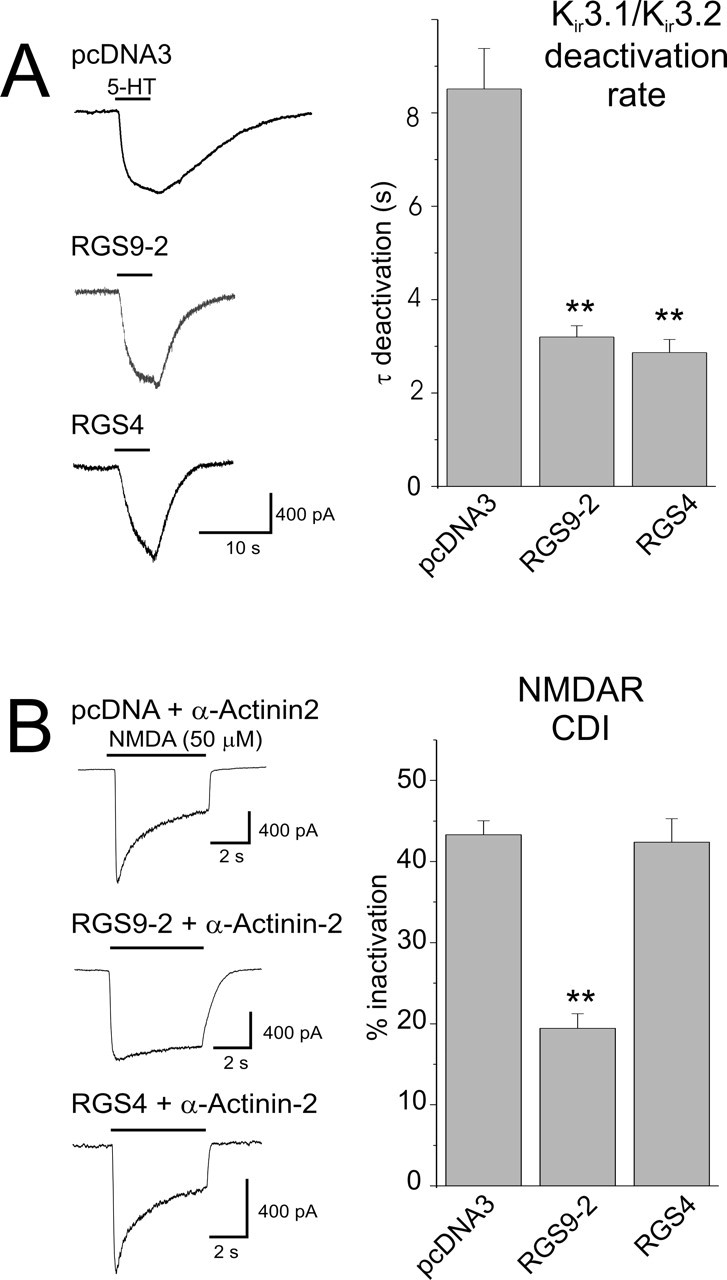
Comparison of the effect of RGS9-2 and RGS4 on Kir3 potassium current and NMDAR CDI. A, In cells transfected with the Gαi-coupled 5-HT1A receptor plus Kir3.1 and Kir3.2 potassium channels, administration of 5-HT (10 μm) elicits a current that deactivates slowly on removal of the agonist. As reported previously, transfection of either RGS9-2 or RGS4 significantly accelerates the deactivation rate of this current by increasing the GTPase activity of the G-protein (Granneman et al., 1998). pcDNA, n = 10; RGS9-2, n = 19; RGS4, n = 9. **p < 0.01. B, Despite the comparable effect of RGS9-2 and RGS4 on Kir3 currents, only RGS9-2 suppressed CDI. pcDNA3, n = 29; RGS9-2, n = 18; RGS4, n = 18. **p < 0.01 compared with pcDNA3. Error bars indicate SE.
Discussion
In the present study, we have identified an interaction between RGS9-2 and α-actinin-2 and shown that this interaction can regulate NMDAR function. An interaction between RGS9-2 and α-actinin-2 was initially detected in a yeast two-hybrid screen using full-length RGS9-2 as bait. This interaction was subsequently confirmed using coimmunoprecipitation from HEK-293 cells cotransfected with RGS9-2 and α-actinin-2.
Multiple independent lines of evidence support the idea that RGS9-2 interacts with α-actinin-2 in central neurons and that this interaction may be functionally significant. First, RGS9-2 and α-actinin-2 exhibit striking macroscopic colocalization in the CNS. In particular, both of these proteins exhibit robust expression in the striatum (Wyszynski et al., 1997; Granneman et al., 1998; Rahman et al., 1999; Dunah et al., 2000; the present study), with lower levels of expression in hippocampus and cortex, and little expression elsewhere in the brain. In striatum, both RGS9-2 (Rahman et al., 2003) and α-actinin-2 (Dunah et al., 2000) are expressed in medium spiny neurons, the principal cell type within this structure, consistent with a possible interaction. As a biochemical correlate of these anatomical data, we now show that endogenous RGS9-2 and α-actinin-2 can be coimmunoprecipitated from striatal tissue, supporting the idea that these proteins can physically interact in central neurons.
Second, previous studies have shown that α-actinin-2 is a key component of the macromolecular complex mediating CDI of NMDAR (Zhang et al., 1998; Krupp et al., 1999; Dunah et al., 2000). Furthermore, at least in striatum, immunoprecipitation of RGS9-2 results in the coimmunoprecipitation of NMDARs as well as α-actinin-2. These results raise the possibility that RGS9-2 could regulate NMDAR function through its interaction with α-actinin-2. Therefore, we tested this idea in HEK-293 cells. As previously reported by others (Zhang et al., 1998; Krupp et al., 1999), NMDARs in this model system express robust CDI, which can be readily quantified. Expression of RGS9-2 in these cells resulted in a significant suppression of CDI, thus supporting the functional significance of the interaction between RGS9-2 and α-actinin and suggesting a possible role for RGS9-2 in the regulation of NMDARs.
Surprisingly, the present results revealed an unanticipated selectivity in the ability of RGS9-2 to regulate CDI based on the specific α-actinin isoform involved in this process. HEK-293 cells express α-actinin-1, -2, and -4, with α-actinin-4 representing the predominant isoform, whereas both α-actinin-2 and α-actinin-4 appear capable of interacting significantly with NMDARs. Overexpression of α-actinin-4 elicits no detectable effect on CDI or on the ability of RGS9-2 to suppress CDI, whereas overexpression of α-actinin-2 has no basal effect on CDI, but markedly facilitates the ability of RGS9-2 to suppress CDI. Because α-actinins are generally thought to function as homodimers, the simplest explanation for these findings is that, under basal conditions, CDI is mediated predominantly by the more abundantly expressed α-actinin-4 and that overexpression of α-actinin-2 leads to the replacement of this endogenous α-actinin-4 by α-actinin-2, leading to a sensitization of CDI to the effects of RGS9-2. As such, these results point to a surprising selectivity in the interaction between RGS9-2 and α-actinin-2 in the context of NMDAR CDI. Consistent with these conclusions, which are based on physiological experiments, coimmunoprecipitation experiments confirmed a marked selectivity in the ability of RGS9-2 to interact with α-actinin-2 vis-à-vis α-actinin-4. A selective interaction between RGS9-2 and α-actinin-2 is of considerable physiological interest given the robust colocalization of these proteins within the striatum (Dunah et al., 2000).
Previous studies have shown that RGS9-2 is a multifunctional protein composed of at least four functional domains. Therefore, in a last series of experiments, we used a series of deletion constructs to identify the domain within RGS9-2 responsible for the interaction with α-actinin-2. Deletion of the DEP and GGL domains of RGS9-2 failed to impair the ability of RGS9-2 to suppress CDI, whereas expression of the DEP domain alone had essentially no effect. This indicated that the DEP domain of RGS9-2 was not an essential participant in the interaction with α-actinin-2. Similarly, deletion of the DEP and GGL domains as well as the alternatively spliced, RGS9-2-specific C-terminal domain resulted in only a modest attenuation of the effect of RGS9-2. These results indicated a key role for the RGS domain of RGS9-2 in mediating the interaction of this protein with α-actinin-2.
The requirement for the RGS domain raised the possibility that the effect of CDI could simply reflect a nonspecific increase in RGS-mediated GAP activity inside the cell. However, expression of RGS4, at levels comparable with those of RGS9-2, as assayed by their regulation of Kir3 potassium currents, was without effect on CDI. These results indicate that the effect of RGS9-2 on CDI did not reflect a nonspecific increase in GAP activity within the cell, but rather a selective effect of RGS9-2, most likely mediated by its interaction with α-actinin-2. These findings, however, do not rule out that the GAP activity of RGS9-2 may be necessary for the ability of this protein to regulate α-actinin-2 function.
Previous work identified Gβ5 as an essential partner for the functional expression of RGS9-1 (Makino et al., 1999; Chen et al., 2003). In the current study, RGS9-2 was capable of regulating CDI without the additional coexpression of Gβ5, and overexpression of Gβ5 had no detectable effect on this response. This is consistent with previous electrophysiological studies showing robust R7 family RGS protein-mediated responses in the absence of coexpression of Gβ5 (Granneman et al., 1998; Saitoh et al., 1999; Rahman et al., 2003). This may reflect the expression of endogenous Gβ5 in HEK-293 cells (Zhang et al., 2000). Recent work has identified a syntaxin family protein termed R7BP as an additional functional partner for RGS9-2 (Drenan et al., 2005; Martemyanov et al., 2005). Additional studies will be required to test for a possible role of R7BP in the effects of RGS9-2 on NMDAR CDI.
In a previous study, we reported on the ability of RGS9-2 to localize to the nucleus, a function that is mediated by the alternatively spliced, brain-specific C-terminal domain of RGS9-2 (Bouhamdan et al., 2004). This is consonant with the growing realization that RGS proteins of the R7 family, including RGS9-2, can translocate to the nucleus (Hepler, 2005), where they may regulate gene transcription (Bouhamdan et al., 2004; Hepler, 2005). Interestingly, recent studies have begun to delineate a similar role for α-actinins (Gettemans et al., 2005). Combined with the present findings, these results begin to sketch a bifunctional role for RGS9-2 as both a participant in membrane signaling and as a regulator of nuclear function. Such bifunctionality is reminiscent of the dual roles of β-catenin, which functions both in cell adhesion processes and also as a transcriptional regulator (Harris and Peifer, 2005). As such, these data suggest a role for RGS9-2, perhaps in conjunction with α-actinin-2, linking events at the plasma membrane with functional changes in the nucleus.
RGS proteins were initially isolated as regulators of heterotrimeric G-protein signaling (Hollinger and Hepler, 2002). In this context, the identification of α-actinin-2 as a binding and functional partner for RGS9-2 might appear to be surprising. However, recent work has hinted at a broader role for RGS proteins beyond the unidimensional regulator of signaling by Gα at the cell membrane (De Vries et al., 2000; Hollinger and Hepler, 2002). In this broader context, and given the known functions of α-actinins (Otey and Carpen, 2004), the present results raise the possibility that RGS9-2 may function in association with the actin cytoskeleton to regulate NMDARs. Interestingly, there is now growing evidence that RGS9-2 plays an important role in dopaminergic signaling within the basal ganglia (Rahman et al., 2003; Kovoor et al., 2005). The present results raise the intriguing possibility that RGS9-2 may do so, at least in part, by virtue of its interaction with α-actinin-2 and NMDARs.
Footnotes
*M.B. and H.-D.Y. contributed equally to this work.
This work was supported by National Institutes of Health Grants MH60854 and MH43985. We thank Drs. G. Kapatos and L. Swick for their helpful advice.
References
- Boeckman FA, Aizenman E (1994). Stable transfection of the NR1 subunit in Chinese hamster ovary cells fails to produce a functional N-methyl-d-aspartate receptor. Neurosci Lett 173:189–192. [DOI] [PubMed] [Google Scholar]
- Bouhamdan M, Michelhaugh SK, Calin-Jageman I, Ahern-Djamali S, Bannon MJ (2004). Brain-specific RGS9-2 is localized to the nucleus via its unique proline-rich domain. Biochim Biophys Acta 1691:141–150. [DOI] [PubMed] [Google Scholar]
- Chen CK, Eversole-Cire P, Zhang H, Mancino V, Chen YJ, He W, Wensel TG, Simon MI (2003). Instability of GGL domain-containing RGS proteins in mice lacking the G protein beta-subunit Gbeta5. Proc Natl Acad Sci USA 100:6604–6609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Vries L, Zheng B, Fischer T, Elenko E, Farquhar MG (2000). The regulator of G protein signaling family. Annu Rev Pharmacol Toxicol 40:235–271. [DOI] [PubMed] [Google Scholar]
- Drenan RM, Doupnik CA, Boyle MP, Muglia LJ, Huettner JE, Linder ME, Blumer KJ (2005). Palmitoylation regulates plasma membrane-nuclear shuttling of R7BP, a novel membrane anchor for the RGS7 family. J Cell Biol 169:623–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunah AW, Wyszynski M, Martin DM, Sheng M, Standaert DG (2000). α-Actinin-2 in rat striatum: localization and interaction with NMDA glutamate receptor subunits. Brain Res Mol Brain Res 79:77–87. [DOI] [PubMed] [Google Scholar]
- Evans KL, Cropper JD, Berg KA, Clarke WP (2001). Mechanisms of regulation of agonist efficacy at the 5-HT(1A) receptor by phospholipid-derived signaling components. J Pharmacol Exp Ther 297:1025–1035. [PubMed] [Google Scholar]
- Gettemans J, Van Impe K, Delanote V, Hubert T, Vandekerckhove J, De Corte V (2005). Nuclear actin-binding proteins as modulators of gene transcription. Traffic 6:847–857. [DOI] [PubMed] [Google Scholar]
- Granneman JG, Zhai Y, Zhu Z, Bannon MJ, Burchett SA, Schmidt CJ, Andrade R, Cooper J (1998). Molecular characterization of human and rat RGS-9L, a novel splice variant enriched in dopamine target regions, and chromosomal localization of the RGS 9 gene. Mol Pharmacol 54:687–694. [PubMed] [Google Scholar]
- Harris TJ, Peifer M (2005). Decisions, decisions: beta-catenin chooses between adhesion and transcription. Trends Cell Biol 15:234–237. [DOI] [PubMed] [Google Scholar]
- Hepler JR (2005). R7BP: a surprising new link between G proteins, RGS proteins, and nuclear signaling in the brain. Sci STKE 2005:e38. [DOI] [PubMed] [Google Scholar]
- Hollinger S, Hepler JR (2002). Cellular regulation of RGS proteins: modulators and integrators of G protein signaling. Pharmacol Rev 54:527–559. [DOI] [PubMed] [Google Scholar]
- Kovoor A, Chen CK, He W, Wensel TG, Simon MI, Lester HA (2000). Co-expression of Gbeta5 enhances the function of two Ggamma subunit-like domain-containing regulators of G protein signaling proteins. J Biol Chem 275:3397–3402. [DOI] [PubMed] [Google Scholar]
- Kovoor A, Seyffarth P, Ebert J, Barghshoon S, Chen CK, Schwarz S, Axelrod JD, Cheyette BN, Simon MI, Lester HA, Schwarz J (2005). D2 dopamine receptors colocalize regulator of G-protein signaling 9-2 (RGS9-2) via the RGS9 DEP domain, and RGS9 knock-out mice develop dyskinesias associated with dopamine pathways. J Neurosci 25:2157–2165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krupp JJ, Vissel B, Thomas CG, Heinemann SF, Westbrook GL (1999). Interactions of calmodulin and alpha-actinin with the NR1 subunit modulate Ca2+-dependent inactivation of NMDA receptors. J Neurosci 19:1165–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Montalbetti N, Shen PY, Dai XQ, Cheeseman CI, Karpinski E, Wu G, Cantiello HF, Chen XZ (2005). Alpha-actinin associates with polycystin-2 and regulates its channel activity. Hum Mol Genet 14:1587–1603. [DOI] [PubMed] [Google Scholar]
- MacArthur DG, North KN (2004). A gene for speed? The evolution and function of alpha-actinin-3. BioEssays 26:786–795. [DOI] [PubMed] [Google Scholar]
- Makino ER, Handy JW, Li T, Arshavsky VY (1999). The GTPase activating factor for transducin in rod photoreceptors is the complex between RGS9 and type 5 G protein beta subunit. Proc Natl Acad Sci USA 96:1947–1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martemyanov KA, Yoo PJ, Skiba NP, Arshavsky VY (2005). R7BP, a novel neuronal protein interacting with RGS proteins of the R7 family. J Biol Chem 280:5133–5136. [DOI] [PubMed] [Google Scholar]
- Otey CA, Carpen O (2004). Alpha-actinin revisited: a fresh look at an old player. Cell Motil Cytoskeleton 58:104–111. [DOI] [PubMed] [Google Scholar]
- Ouimet CC, Miller PE, Hemmings HC Jr, Walaas SI, Greengard P (1984). DARPP-32, a dopamine- and adenosine 3′:5′-monophosphate-regulated phosphoprotein enriched in dopamine-innervated brain regions. III. Immunocytochemical localization. J Neurosci 4:111–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman Z, Gold SJ, Potenza MN, Cowan CW, Ni YG, He W, Wensel TG, Nestler EJ (1999). Cloning and characterization of RGS9-2: a striatal-enriched alternatively spliced product of the RGS9 gene. J Neurosci 19:2016–2026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman Z, Schwarz J, Gold SJ, Zachariou V, Wein MN, Choi KH, Kovoor A, Chen CK, DiLeone RJ, Schwarz SC, Selley DE, Sim-Selley LJ, Barrot M, Luedtke RR, Self D, Neve RL, Lester HA, Simon MI, Nestler EJ (2003). RGS9 modulates dopamine signaling in the basal ganglia. Neuron 38:941–952. [DOI] [PubMed] [Google Scholar]
- Saitoh O, Kubo Y, Odagiri M, Ichikawa M, Yamagata K, Sekine T (1999). RGS7 and RGS8 differentially accelerate G protein-mediated modulation of K+ currents. J Biol Chem 274:9899–9904. [DOI] [PubMed] [Google Scholar]
- Wensel TG (2002). RGS9-1 phosphorylation and Ca2+ Adv Exp Med Biol 514:125–129. [DOI] [PubMed] [Google Scholar]
- Wyszynski M, Lin J, Rao A, Nigh E, Beggs AH, Craig AM, Sheng M (1997). Competitive binding of alpha-actinin and calmodulin to the NMDA receptor. Nature 385:439–442. [DOI] [PubMed] [Google Scholar]
- Wyszynski M, Kharazia V, Shanghvi R, Rao A, Beggs AH, Craig AM, Weinberg R, Sheng M (1998). Differential regional expression and ultrastructural localization of α-actinin-2, a putative NMDA receptor-anchoring protein, in rat brain. J Neurosci 18:1383–1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JH, Lai Z, Simonds WF (2000). Differential expression of the G protein beta(5) gene: analysis of mouse brain, peripheral tissues, and cultured cell lines. J Neurochem 75:393–403. [DOI] [PubMed] [Google Scholar]
- Zhang K, Howes KA, He W, Bronson JD, Pettenati MJ, Chen C, Palczewski K, Wensel TG, Baehr W (1999). Structure, alternative splicing, and expression of the human RGS9 gene. Gene 240:23–34. [DOI] [PubMed] [Google Scholar]
- Zhang S, Ehlers MD, Bernhardt JP, Su CT, Huganir RL (1998). Calmodulin mediates calcium-dependent inactivation of N-methyl-d-aspartate receptors. Neuron 21:443–453. [DOI] [PubMed] [Google Scholar]
- Zhou Q, Ruiz-Lozano P, Martone ME, Chen J (1999). Cypher, a striated muscle-restricted PDZ and LIM domain-containing protein, binds to alpha-actinin-2 and protein kinase C. J Biol Chem 274:19807–19813. [DOI] [PubMed] [Google Scholar]