

Abstract
Antidepressant- and cocaine-sensitive serotonin (5-hydroxytryptamine, 5-HT) transporters (SERTs) dictate clearance of extracellular 5-HT after release. To explore protein kinase C-mediated SERT regulation, we generated a stable human SERT (hSERT)-expressing cell line (293-hSERT) and evaluated modulation of 5-HT activity via studies of 5-HT flux, hSERT-mediated currents under voltage clamp, and surface distribution of SERT protein. 293-hSERT cells exhibit saturable, high-affinity, and antidepressant-sensitive 5-HT uptake as well as hSERT-dependent whole-cell currents. In these cells, the protein kinase C activator β-PMA caused a time-dependent reduction in 5-HT uptake capacity (Vmax) after acute application and a reduction in SERT-mediated currents. Effects of β-PMA were mimicked by the phorbol ester β-PDBu, were not observed with the inactive α-isomers, and could be blocked by treatment of cells with the protein kinase C inhibitor staurosporine. Biotinylation/immunoblot analyses showed that activity reductions are paralleled by a staurosporine-sensitive loss of surface SERT protein. These data indicate that altered surface abundance, rather than reduced catalytic transport efficiency, mediates acute PKC-dependent modulation of 5-HT uptake.
Keywords: serotonin, serotonin transporter, antidepressant, protein kinase C, regulation, phosphorylation, protein trafficking
Presynaptic, sodium-, and chloride-dependent serotonin (5-hydroxytryptamine, 5-HT) transporters (SERTs) clear extracellular 5-HT after release from serotonergic terminals (Kuhar et al., 1972; Gershon and Jonakait, 1979; Rudnick and Clark, 1993; Fuller, 1994). SERTs also are expressed on a number of specialized non-neuronal cells, including platelets (Rudnick, 1977), placental syncytiotrophoblasts (Balkovetz et al., 1989), intestinal crypt epithelial cells (Wade et al., 1996), and adrenal chromaffin cells (Blakely et al., 1995). SERTs are high-affinity targets in vivo for tricyclic antidepressants such as imipramine, the serotonin-selective-reuptake inhibitors (SSRIs), and nonselective stimulants such as cocaine and amphetamine (Reith, 1988; Fuller, 1994;Barker and Blakely, 1995). These substances are believed to exert at least a portion of their physiological actions by elevating extracellular 5-HT concentrations and potentiating synaptic 5-HT actions. Prolonged treatment of rats with antidepressants can downregulate 5-HT clearance capacity (Piñeyro et al., 1994) and modulate raphe neuron firing rates and 5-HT receptor sensitivity (de Montigny et al., 1990; Wade et al., 1996), suggesting an interdependence among the magnitude of 5-HT release, rates of SERT-mediated 5-HT clearance, and physiological actions of 5-HT.
The degree to which endogenous regulation of SERT expression or activity contributes to in vivo modulation of amine signaling presently is unknown. In vitro, SERT gene expression is regulated by both cAMP-dependent and -independent pathways (Ramamoorthy et al., 1993a, 1995), and in vivo can be influenced by antidepressants and steroid hormones (Lesch et al., 1993; Blakely et al., 1996). Multiple reports also have demonstrated a capacity of SERTs to be regulated rapidly after acute elevation/depletion of intracellular Ca2+ (Nishio et al., 1995), treatment with calmodulin inhibitors (Jayanthi et al., 1994), or via activation of protein kinase C (PKC) (Myers et al., 1989; Anderson and Horne, 1992; Miller and Hoffman, 1994; Ramamoorthy et al., 1995) and NOS/cGMP pathways (Launay et al., 1994; Miller and Hoffman, 1994). The major kinetic alterations observed to date are reductions or elevations of transport capacity (Vmax) rather than a change in apparent 5-HT affinity (KM orKI). In neuronal, platelet, and mast cell preparations, however, mechanistic interpretations of altered SERT activity are complicated by possibilities of parallel effects of modulators on vesicular amine storage pools (Rudnick and Clark, 1993;Nakanishi et al., 1995). In addition, lack of SERT-directed antibodies has precluded attempts to monitor SERT protein in regulatory paradigms.
Recently, we and others have cloned cDNAs encoding SERTs from multiple species, including human, and demonstrated functional expression of antidepressant and cocaine-sensitive 5-HT uptake in mammalian, insect, and amphibian cells (Blakely et al., 1991a; Hoffman et al., 1991;Ramamoorthy et al., 1993c; Corey et al., 1994b; Demchyshyn et al., 1994; Chang et al., 1996). SERTs have been found not only to mediate 5-HT accumulation after heterologous expression but also to exhibit channel-like behavior, with nonstoichiometric ion flow gated by 5-HT evident (Mager et al., 1994). Structurally, SERTs are members of the γ-aminobutyric acid/norepinephrine transporter (GAT/NET) gene family, composed of amino acid, biogenic amine, osmolyte, and nutrient transporters (Amara and Kuhar, 1993; Uhl and Johnson, 1995). Sequence-based topology predictions indicate that SERTs and homologs are integral membrane glycoproteins with cytoplasmic NH2 and COOH termini and 12-membrane-spanning domains (TMDs). To date, little is understood regarding the nature of SERT protein assembly, trafficking, or the importance of post-translational processing. Evidence suggests that native SERTs may not be monomeric (Cesura et al., 1983; Habert et al., 1986; Ramamoorthy et al., 1993b) and that SERTs exhibit cell-specific patterns of N-glycosylation (Qian et al., 1995b). N-Glycosylation has been found to be important for biogenic amine transporter expression of activity, although not ligand recognition (Tate and Blakely, 1994; Melikian et al., 1996). Canonical sites for protein kinases on presumed cytoplasmic domains (Blakely et al., 1991a;Miller and Hoffman, 1994) raise the possibility that SERTs, like other membrane transporters and ion pumps (Bertorello et al., 1991; Casado et al., 1993; Hoffman et al., 1994), may be regulated by phosphorylation. Recent studies (Qian et al., 1995a) reveal that SERT cytoplasmic domains are substrates for PKC and PKA in vitro, although the relationship of these observations to activity modulation in vivo is presently unclear. Other transporters, such as the insulin-regulated glucose transporter GLUT4, exhibit hormone-dependent transporter recruitment to surface membranes (Kasanicki and Pilch, 1990) with consequent changes in sugar transport capacity.
The availability of biogenic amine transporter cDNAs has allowed for the construction of stable cell lines for direct evaluation of neurotransmitter influx and efflux in the absence of a vesicular release pathway (Gu et al., 1993). In the present report, we use HEK-293 cells stably transfected with hSERT cDNA (293-hSERT) to establish a paradigm for acute PKC-mediated regulation of 5-HT transport and 5-HT-gated currents. Our studies reveal a significant and specific downregulation of SERT activity by PKC activators, revealed kinetically as a reduction in 5-HT uptake capacity. hSERT-mediated currents, recorded under voltage clamp also are reduced, suggesting functional silencing of hSERT-associated channels, loss of transporter from the cell surface, or both. Immunoblots performed with SERT-specific antibodies (Qian et al., 1995b) on total and surface-biotinylated fractions of 293-hSERT membranes indicate no loss of SERT protein as a consequence of PKC activation, but rather reveal a redistribution of transporter protein from the cell surface.
MATERIALS AND METHODS
Production of 293-hSERT cells. AHindIII/XbaI fragment containing the hSERT cDNA (Ramamoorthy et al., 1993c) was released from pBluescript SKII− and subcloned intoHindIII/XbaI-digested pRc/CMV (Invitrogen, San Diego, CA), placing hSERT expression under the dual control of the CMV promoter and the T7 RNA polymerase promoter. We validated functional expression of the construct in transiently transfected HeLa cells with vaccinia-T7 expression, as previously described (Blakely et al., 1991b;Ramamoorthy et al., 1993c). To generate stably transfected cells, we transfected hSERT/pcDNA3 as a liposome suspension with Lipofectin (Life Technologies, Gaithersburg, MD) into HEK-293 cells (ATCC) cultured and selected in 250 μg/ml Geneticin (G418), as previously described (Galli et al., 1995). Individual cells were used to generate clonal lines. Multiple lines tested positive for paroxetine-sensitive [3H]5-HT uptake, and the clone that displayed the highest 5-HT uptake (m3) was expanded for the experiments reported here.
Assay of 5-HT uptake and antagonist binding. 293-hSERT and nontransfected HEK-293 cells initially were passaged in DMEM culture media containing 10% fetal bovine serum, 2 mm glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin, with 250 mg/l G418 added for 293-hSERT cells. Levels of 5-HT uptake tended to become reduced with subsequent passages in standard media, but we found that this could be stabilized primarily via the use of either dialyzed fetal calf serum (Sigma, St. Louis, MO) or serum allowed to stand at 4°C for several days, suggesting the presence of an unstable, inhibitory activity of mass <10 kDa. We suspected 5-HT from the serum might accumulate inside cells and reduce viability, thereby selecting for low transporter-expressing cells, but we failed to reduce the trend toward low uptake levels by growth in the presence of a SERT antagonist (10 μm citalopram). Examination of SERT immunostaining also revealed that changes in expression reflected reduced immunoreactivity throughout the entire population rather than a loss of SERT-expressing cells from a nonclonal population. SERT downregulation by serum, like PMA effects, occurs after acute application and thus may reflect endogenous HEK-293 receptor activation. For uptake studies, 293-hSERT cells were plated at 100,000 cells/well on poly-d-lysine-coated (0.1 mg/ml), 24 well tissue culture plates 2 d before experiments. At assay, the medium was removed by aspiration, and cells were washed with 2 ml of Krebs–Ringer’s-HEPES (KRH) medium containing (in mm): 130 NaCl, 1.3 KCl, 2.2 CaCl2, 1.2 MgSO4, 1.2 KH2PO4, 10 HEPES, pH 7.4. Then cells were preincubated in KRH containing 1.8 gm/l glucose, 100 μmpargyline (Sigma), and 100 μm ascorbic acid (Sigma) with or without antagonists and/or various PKC modulators. PMA and phorbol 12,13-dibutyrate (PDBu) isomers and staurosporine were obtained from Sigma. 5-HT transport assays (10 min at 37°C) were initiated by the addition of [3H]5-HT (5-hydroxy-[3H] tryptamine trifluoroacetate, ∼100 Ci/mmol; Amersham, Arlington Heights, IL) and terminated by three rapid washes with 37°C KRH containing 1 mm imipramine (Sigma). Similar assays were conducted with [3H]leucine (20 nm; DuPont NEN, Boston, MA), [3H]glutamate (100 nm; Amersham), [3H]alanine (100 nm; Amersham), or [3H]glycine (1 μm; DuPont NEN). Cells were lysed in Optiphase Supermix scintillation cocktail (Wallac, Gaithersburg, MD) and accumulated radioactivity directly quantified in a microplate liquid scintillation counter (Microbeta, Wallac). Some extracts were prepared as SDS lysates and used for protein determinations (Bradford assay, Bio-Rad, Richmond, CA), the results of which demonstrated no alteration in well protein content by PKC modulators. Nonspecific [3H]5-HT uptake, defined as the accumulation in the presence of excess unlabeled RTI-55 or paroxetine (see legends), was subtracted from total uptake to define hSERT-specific accumulation. Nonspecific uptake for amino acid substrates was defined with parallel assays on ice (leucine) or with choline-Cl-substituted KRH (glutamate, glycine, alanine). In transport assays conducted to mimic the configuration used for biotinylation studies, cells were plated on poly-d-lysine-coated six well plates at a density of 500,000 cells/well 48 hr before assay. Assays were performed in a total assay volume of 5 ml for 10 min essentially as described above, with radioactivity from SDS-lysed cells quantitated by liquid scintillation spectrometry (LS6000IC, Beckman, Fullerton, CA). Nonspecific uptake was defined as the uptake in the presence of 100 nm–1 μm paroxetine or 1 μmof RTI55, and these data were subtracted from total counts to yield specific uptake. For radioligand binding assays, total cell membranes were prepared from cells grown to confluence in 150 mm tissue culture dishes, as previously described (Galli et al., 1995). Protein concentrations were determined by Bradford assay (Bio-Rad), and hSERT density was assessed with [125I] RTI-55 [3β-(4-iodophenyl)-tropane-2β-carboxylic acid methylester tartrate, 2200 Ci/mmol; Dupont NEN]. Initial studies with 293-hSERT cell membranes demonstrated linearity of specific binding up to 10 μg of membrane protein/tube, and subsequent assays used 7.5 μg/tube. Assays, performed in triplicate, with various concentrations of [125I]RTI55 were initiated with the addition of membranes in binding buffer (100 mm NaCl and 50 mm Tris, pH 7.4) and terminated after 1 hr incubation in room temperature by rapid filtration (Brandel, Gaithersburg, MD) over GF/B glass fiber filters (Whatman, Maidstone, UK), presoaked in 0.5% polyethylenimine (Sigma). Filters were washed in ice-cold binding buffer, and bound radioactivity was measured by gamma emission spectrometry (Gamma 5500B, Beckman). Nonspecific binding, defined as the binding in the presence of 1 μm paroxetine, was subtracted from total binding to define specific binding. Nonlinear curve fits of data (Kaleidagraph) for uptake, binding, and currents (see below) used the generalized Michaelis–Menten model V = Vmax[S]n/[S]n+ [K]n, substituting Band Bmax for binding isotherms and Iand Imax for currents.
hSERT immunoblots and biotinylation. hSERT protein was detected in transfected 293-hSERT membranes with CT-2 antibody directed at the rat SERT COOH terminus as previously described (Qian et al., 1995b). Briefly, membrane extracts were separated by 10% SDS-PAGE and transferred for 16 hr at 150 mA onto PVDF membrane (Millipore, Bedford, MA), blocked with 5% nonfat dry milk in PBS, and probed with CT-2 at 1 μg/ml. Bound antibody was visualized with HRP-conjugated goat anti-rabbit antibody (1:10,000; Bio-Rad), and immunoreactive bands were visualized by ECL (Amersham). In biotinylation studies, cells were seeded on poly-d-lysine-coated six well plates at 500,000 cells/well 48 hr before treatments, washed with 37°C KRH, and incubated with vehicle, phorbol 12-myristate 13-acetate (β-PMA), and/or staurosporine for 40 min in KRH at 37°C for the times indicated. Cells were washed quickly with warm KRH and then treated with sulfosuccinimido-NHS-biotin (1.5 mg/ml; Pierce, Rockford, IL) at 4°C for 1 hr in PBS/Ca-Mg containing (in mm): 138 NaCl, 2.7 KCl, 1.5 KH2PO4, 9.6 Na2HPO4, 1 MgCl2, and 0.1 CaCl2, pH 7.3. Biotinylating reagents were removed by washing with 100 mm glycine in PBS/Ca-Mg twice, the reaction was quenched further by incubation with 100 mmglycine for 30 min, and then cells were washed with PBS/Ca-Mg rapidly three times before lysis with 250 μl/well radioimmunoprecipitation assay (RIPA) buffer containing (in mm): 10 Tris, pH 7.4, 150 NaCl, and 1 EDTA with 0.1% SDS, 1% Triton X-100, 1% sodium deoxycholate, supplemented with protease inhibitors (1 μg/ml aprotinin, 1 μg/ml leupeptin, 1 μm pepstatin, 1 mg/ml soybean trypsin inhibitors, 1 mm iodoacetamide, and 250 μm PMSF) for 1 hr at 4°C with constant shaking. Lysates were centrifuged at 20,000 × g for 30 min at 4°C, and supernatants were incubated with monomeric avidin beads (175 μl of beads/1250 μl of supernatant; Pierce) for 1 hr at room temperature. Beads were washed three times with RIPA, and adsorbed proteins were eluted with 50 μl of Laemmli loading buffer (62.5 mm Tris-HCl, pH 6.8, 20% glycerol, 2% SDS, 5% βME, and 5% bromophenol blue) for 30 min at room temperature. Then 40 μl of total cell lysate, lysates after incubation with avidin beads, last wash, and the bead eluate (50 μl) were separated by SDS-PAGE (10%) and immunoblotted with CT-2 antibody (1 μg/ml) with 1:3000 goat anti-rabbit HRP-conjugated secondary antibody. Subsequently, blots were stripped (62.5 mm Tris-HCl, pH 6.8, 2% SDS, and 100 mm βME) for 30 min at 50°C, washed with PBS twice for 10 min, reblocked in 5% dry milk for 1 hr, and probed with anti-calnexin (1:1000; Stressgen, Victoria, BC, Canada), followed by goat anti-rabbit HRP-conjugated secondary antibody (1:3000). Immunoreactive bands were visualized by ECL on hypersensitive ECL film (Amersham), and scanned bands were quantitated with ImageQuant (Molecular Dynamics, Sunnyvale, CA). Exposures were precalibrated to insure quantitation within the linear range of the film. Values of hSERT surface protein were normalized by levels of calnexin immunoreactivity in total cell extracts to preclude errors accompanying with sample loading and transfer. We also confirmed findings of hSERT surface reductions by using [125I]protein-A (DuPont NEN) detection of bound CT-2 with direct PhosphorImager quantitation (Molecular Dynamics) (data not shown).
Whole-cell patch-clamp recording of hSERT currents.293-hSERT cells and HEK-293 cells were plated at a density of 10,000 cells/35 mm culture dish in normal culture media. Transporter currents were recorded with the patch-clamp technique in whole-cell configuration (Galli et al., 1995) at room temperature in external media of containing (in mm): 120 NaCl, 4.7 KCl, 1.2 KH2PO4, 2.2 CaCl2, 10 HEPES, and 10 glucose, pH 7.35 (300 mOsm). Bath solutions were changed by a gravity pump at a rate of 1 ml/min. Seals were obtained, and cells were washed three times with bath solution. 5-HT and SERT antagonists were added to the bath to achieve the concentrations detailed in the figures in the presence of 100 μm pargyline and 100 μmascorbic acid. The pipette solution for whole-cell recording contained (in mm): 120 KCl, 0.1 CaCl2, 2 MgCl2, 1.1 EGTA, 10 HEPES, and 30 glucose, pH 7.35 (270 mOsm). Free Ca2+ in the pipette was 0.1 μm. Patch electrodes (1–3 μm in diameter and 2–4 MΩ) were pulled from borosilicate glass (Garner Glass Company) with a programmable puller (Sachs-Flaming, P87; Sutter Instruments, Novato, CA). After a seal was formed, the conductance was examined in series with the capacitance, which is ∼0.1 μS. Voltage ramps and steps were applied, and currents were recorded with an Axon 200A voltage clamp (Axon Instruments, Foster City, CA) at a band width of 1000 Hz and stored for analysis on videotape (Panasonic, Secaucus, NJ). To remove low frequency drift from difference currents (Figs. 5A,B,6B,C), we low-pass filtered at 10 Hz and subtracted this filtered trace from the originals. The filtered difference currents were then reset to the mean value.
Fig. 5.
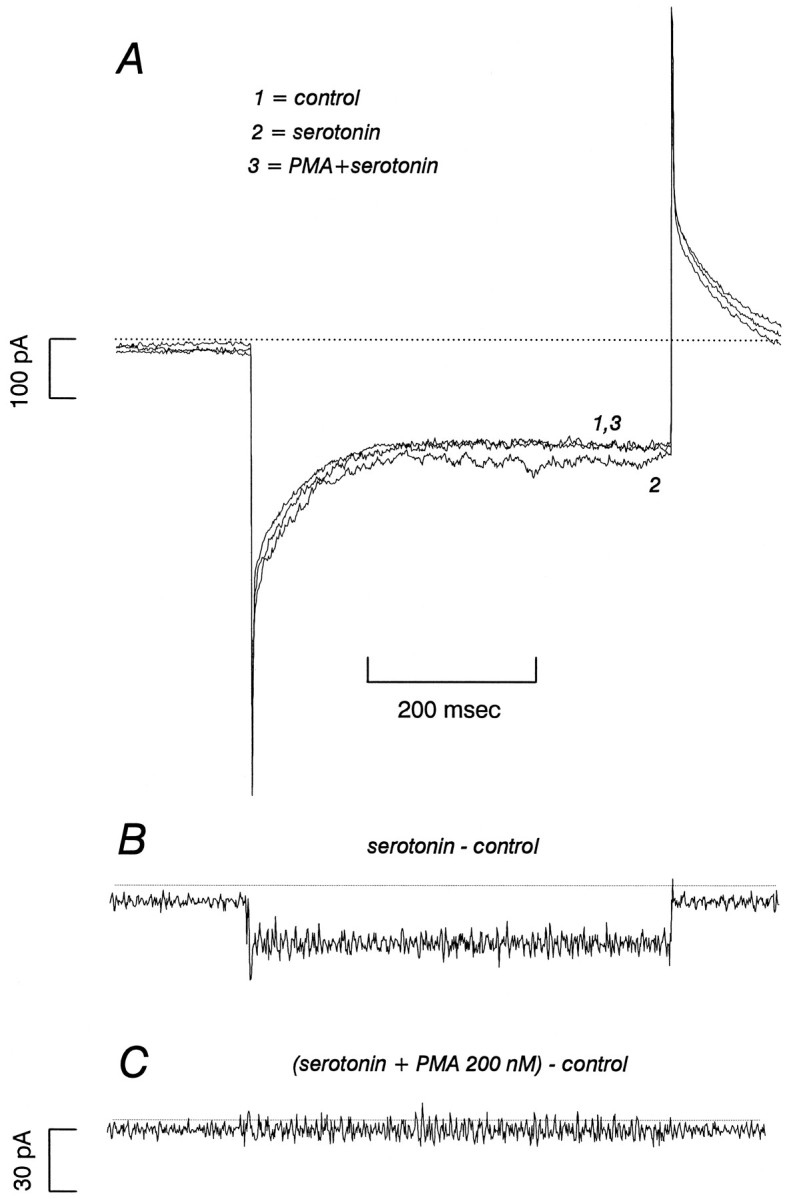
β-PMA reduces SERT-mediated 5-HT currents in 293-hSERT cells. A, 293-hSERT cells were held at −40 mV and stepped to −120 mV for 500 msec with a 5 sec rest interval between pulses. After addition of 1 μm 5-HT, the current increased and reached a steady state in three or four steps. β-PMA (0.2 μm) added in the presence of 1 μm 5-HT always reduced the 5-HT-induced current by >75% within ∼5 min.B, hSERT-mediated currents from the cell recorded inA are revealed by subtraction of 5-HT-induced current from currents in the absence of 5-HT. C, The same subtraction procedure as in B after the addition of 0.2 μm β-PMA. In this experiment, β-PMA essentially abolished the 5-HT-induced current.
Fig. 6.
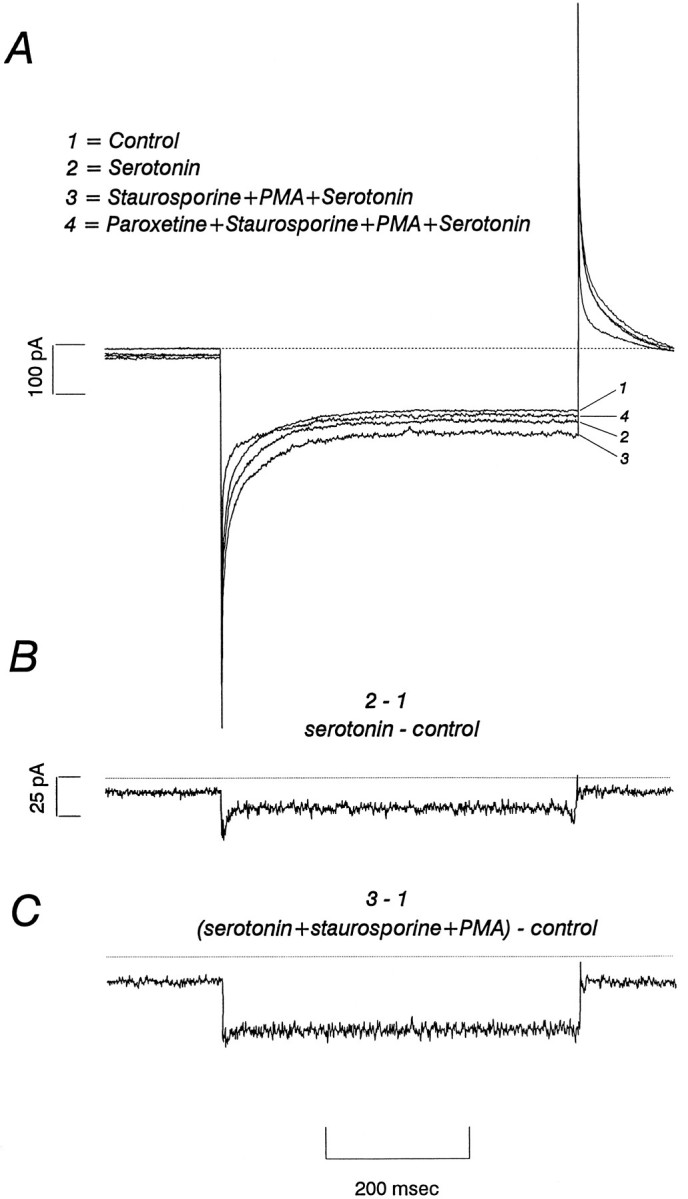
Staurosporine blocks the effect of β-PMA on 5-HT-induced currents. A, The protocol applied is that described in Figure 5. hSERT-mediated currents were induced by addition of (B) 1 μm 5-HT, and then (C) β-PMA (0.2 μm) was added in the presence of staurosporine (1 μm). Unlike the addition of β-PMA alone (Fig. 5), β-PMA plus staurosporine failed to reduce 5-HT-induced currents in all cells tested. In this experiment, staurosporine increased the 5-HT-induced current. The current seems to be hSERT-mediated, because 1 μm paroxetine reduced the 5-HT current by 80%.
RESULTS
Stable expression of human SERT in HEK-293 cells
Antibody CT-2, directed at the rat SERT COOH terminus (Qian et al., 1995b), detects expression of a ∼96 kDa protein on immunoblots of membranes prepared from stably transfected cells that is absent from parental HEK-293 cells (Fig.1A). The size of the hSERT band is consistent with the mobility of N-glycosylated hSERT protein (Ramamoorthy et al., 1993c) and is similar to that observed for N-glycosylated SERT in rat (Qian et al., 1995b) and human platelets (Qian and Blakely, unpublished data). Membranes prepared from 293-hSERT cells, but not parental cells, displayed saturable binding of the hSERT antagonist [125I] RTI-55 (Boja et al., 1992) with aKD of 0.26 nm and aBmax of 1.2 pmol/mg of membrane protein (Fig.1B). A conversion of thisBmax, based on protein yield per cell, gives an average value of ∼7 × 104 [125I]RTI55 binding sites per cell, although expression levels were found to vary considerably with passage in standard media (see Materials and Methods). 293-hSERT cells also exhibit saturable [3H]5-HT transport (Fig. 1C) with a KM of 0.26 ± 0.03 μm, a Vmax of 11.9 ± 0.3 pmol/min/106 cells, and a Hill coefficient (n) of 1.6 ± 0.1. Assuming all SERTs are available for transport, we can estimate a 5-HT translocation rate from the ratioVmax/Bmax = 1.7 molecules of 5-HT per transporter per second. [3H]5-HT transport in 293-hSERT cells was found to be sensitive to compounds known to inhibit [3H] 5-HT uptake in native tissues (Wielosz et al., 1976;Segonzac et al., 1987) and transiently transfected HeLa cells (Ramamoorthy et al., 1993c; Barker et al., 1994), including the tricyclic imipramine and the SSRIs paroxetine and citalopram. In keeping with previous studies (Boja et al., 1992; Barker et al., 1994), RTI-55 exhibited nearly 1000 times greater potency for inhibition of [3H]5-HT uptake over the parent compound cocaine.
Fig. 1.

Characterization of 293-hSERT cells.A, Immunoblot of 293-hSERT membranes with SERT antibody CT-2 (1 μg/ml). A single 96 kDa band is evident that is not present in parental HEK-293 cells. Each lane contains 50 μg of total protein extract. B, Equilibrium binding of [125I]RTI-55 to 293-hSERT cell membranes. Membranes (7.5 μg) were incubated with various concentrations of RTI-55 for 1 hr at room temperature. Nonspecific binding was defined by incubations in the presence of 1 μm paroxetine, and data were subtracted from total bound to define specific binding. 293-hSERT cell membranes bind [125I]RTI-55 with a KD of 0.26 nm and a Bmax of 1.2 pmol/mg protein, which, on the basis of membrane recovery, converts to ∼7 × 104 sites/cell. The figure shown is a representative single binding isotherm, performed in triplicate, plotted ± SD. Two other replicates gave essentially equivalentKD and Bmaxvalues. C, 5-HT uptake by 293-hSERT cells. 293-hSERT cells exhibit saturable 5-HT uptake, with aVmax of 11.9 pmol/min/106 cells, a KM of 0.26 μm, and a Hill coefficient (n) of 1.6. Uptake was conducted in triplicate, plotted ± SD, and repeated once at this passage, although equivalent KM values were obtained in five other experiments. Nonspecific uptake was defined as the uptake in the presence of 1 μm paroxetine and subtracted from total uptake. D, Antagonist sensitivity of 5-HT uptake in 293-hSERT cells. [3H]5-HT uptake (20 nm), assayed in triplicate and repeated with equivalent results, was inhibited by paroxetine (IC50 = 0.15 nm), RTI55 (IC50 = 1.7 nm), imipramine (IC50 = 3.9 nm), citalopram (IC50 = 7.4 nm), and cocaine (IC50 = 0.3 μm). Nonspecific uptake was defined as the uptake in the presence of 1 μm of paroxetine (for RTI55, citalopram, imipramine, and cocaine inhibition) or 1 μm of RTI55 (for paroxetine inhibition) and subtracted from total uptake. Values are plotted as a percentage of specific 5-HT uptake obtained in the absence of antagonists and curve fit to a three-parameter logistic equation, % inhib = 100/(1 + (IC50/[I]n).
Antidepressant-sensitive, 5-HT-gated currents mediated by hSERT
rSERT expressed in Xenopus laevis oocytes (Mager et al., 1994) and hNET expressed in HEK-293 cells (Galli et al., 1995,1996) reveal channel-like behavior in addition to an amine flux pathway. Similarly, 293-hSERT cells showed an inward current at hyperpolarized membrane potentials in the presence of 5-HT (Fig.2A) that can be blocked by 1 μm paroxetine (Fig. 2B). No paroxetine-sensitive 5-HT currents are observed in parental HEK-293 cells (data not shown). At −120 mV, the whole-cell current induced by 1 μm 5-HT was typically ∼30 pA. When 5-HT concentration in the bath was increased incrementally from 0.03 to 1 μmand results of voltage sweeps were normalized to the inward current obtained at 1 μm and −100 mV, aKM of 0.21 ± 0.05 μm and a Hill coefficient (n) of 1.4 ± 0.2 (n = 5) were obtained. 5-HT uptake mediated by SERTs is absolutely dependent on extracellular Na+ with a presumed coupling stoichiometry of 1 Na+ per 1 5-HT (Rudnick and Clark, 1993). As inXenopus oocytes (Mager et al., 1994), the 5-HT-induced current in 293-hSERT cells also was found to be dependent on external Na+ concentration. Complete Li+ substitution for Na+ eliminated the 5-HT-induced current in 293-hSERT cells (data not shown). When external Na+ was varied from 0 to 130 mm by equimolar substitution with Li+, 5-HT-induced currents rose monotonically, with fits of the data yielding a Na+ KM of 15.7 ± 5.1 mm and a Hill coefficient (n) of 0.9 ± 0.3 (n = 4). Finally, we tested the dose dependency and specificity of an antagonist block of 5-HT-gated currents in 293-hSERT cells. The SSRIs paroxetine and citalopram blocked the 5-HT-induced current with KI (1.4 ± 0.2 and 2.9 ± 0.6 nm, respectively), as expected from whole-cell 5-HT transport studies. Desipramine, a high-affinity hNET antagonist and a weak hSERT antagonist (Pacholczyk et al., 1991; Barker and Blakely, 1995) failed to block the 5-HT-gated currents at 1 μm(data not shown). Together these data reveal that hSERT expressed in stably transfected cells mediates antidepressant-sensitive, 5-HT-gated currents as well as saturable high-affinity 5-HT transport.
Fig. 2.
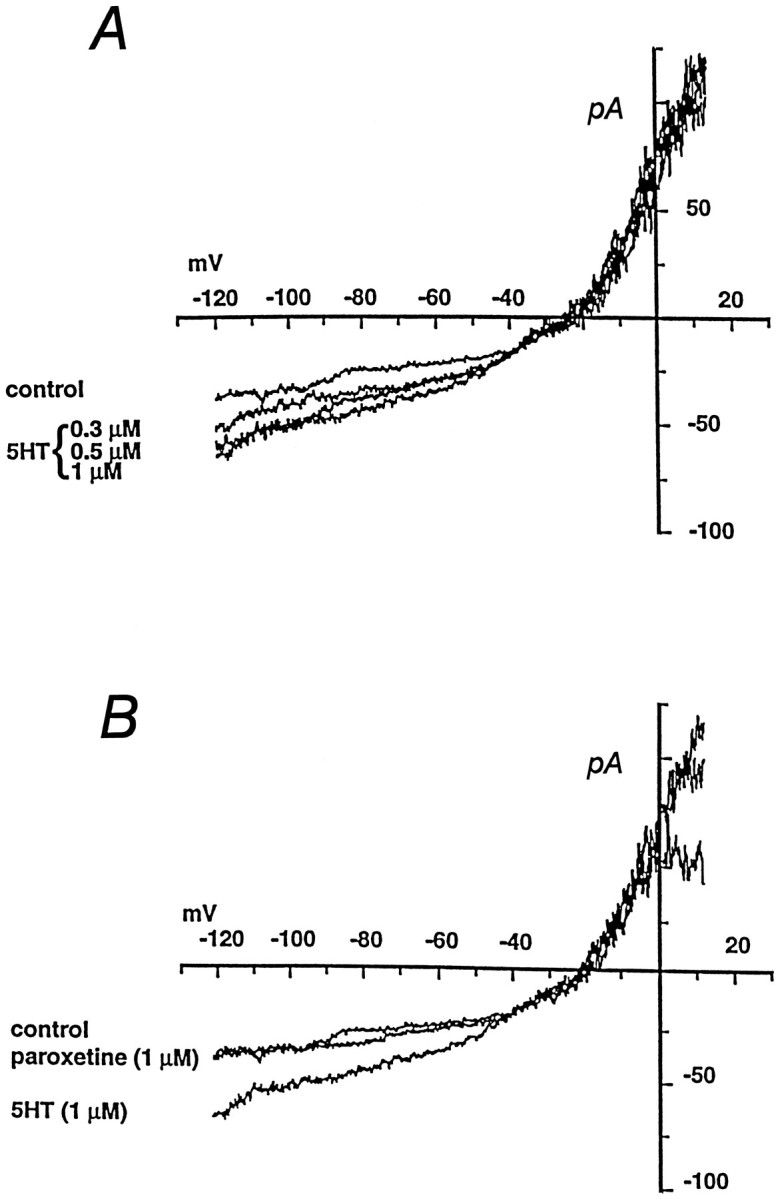
5-HT-induced currents in 293-hSERT cells.A, Cells were clamped in whole-cell mode and stimulated with a 7 sec ramp from −120 mV to 20 mV before (control) or after addition of increasing concentrations of 5-HT to the bath. 5-HT-induced currents are not observed in nontransfected HEK-293 cells. Averaged data from experiments on five cells reveal a saturable 5-HT-induced current with a KM = 0.21 ± 0.05 μmand n = 1.4 ± 0.2. B, Paroxetine (1 μm) blocks the 5-HT-induced current in 293-hSERT cells. Currents were evoked by 1 μm 5-HT as inA, before (control) or after application of paroxetine to the bath. In dose–response studies on five cells, paroxetine inhibited the 5-HT-induced current with aKI of 1.4 ± 0.2 nm.
Regulation of SERT transport activity by PKC activators
Treatment of 293-hSERT cells with the PKC activator phorbol 12-myristate 13-acetate (β-PMA) decreased 5-HT uptake in a time- and concentration-dependent manner. (Fig. 3A,B). Reductions were apparent with 5 min of 1 μm β-PMA pretreatment with maximal reductions (43 ± 5%) observed with 30 min of pretreatment. When β-PMA pretreatment time was fixed at 30 min, reductions were observed first at 10 nm β-PMA and essentially had plateaued at 40–50% reduction by 1 μm. If we define the maximal effect as the decrease caused by 10 μm PMA, an EC50 of 20 nm is obtained. Accumulation of [3H]leucine via endogenous transport systems was unaffected by β-PMA treatments (106 ± 4%), although several other endogenous Na+-dependent transport systems were found to be downregulated in these cells to variable extents after β-PMA treatment (alanine 34 ± 1%, glutamate 74 ± 5%, and glycine 35 ± 7% decrease relative to control). Quantitatively equivalent reductions in 5-HT uptake also were induced by β-PMA in the presence of 1 mm ouabain, a Na/K ATPase inhibitor. Kinetically, the reduction in SERT activity at 1 μm β-PMA is revealed in saturation kinetic experiments to arise from a 46% reduction in Vmax (Fig.3C) with little or no contribution from a change in 5-HTKM (0.39 μm control vs 0.27 μm β-PMA treated; n = 3). The effects of β-PMA were stereospecific (Fig.4A), because the PKC-inactive isomer α-PMA was inactive at similar concentrations and a similar stereospecificity was evident for the phorbol analog phorbol 12,13-dibutyrate (PDBu). Importantly, the effects of β-PMA (1 μm) on SERT activity were found to be blocked by coapplication of staurosporine (1 μm; Fig.4B), a membrane-permeant inhibitor of PKC (Tamaoki et al., 1986). Staurosporine itself did not cause appreciable effects on 5-HT uptake in these cells, nor was the vehicle for β-PMA (ethanol) or the vehicle for staurosporine (DMSO) active on 5-HT uptake at equivalent dilutions (data not shown).
Fig. 3.
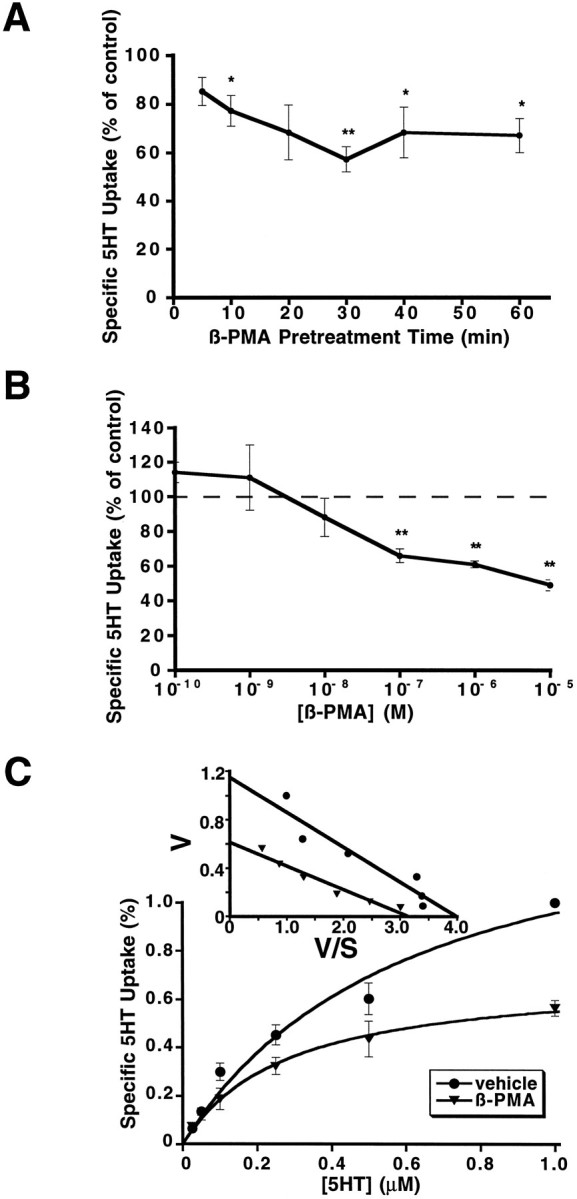
Regulation of 5-HT uptake in 293-hSERT cells by β-PMA. A, Time course of the inhibition of 5-HT uptake by β-PMA. Cells were preincubated with 1 μm β-PMA for the times indicated and then assayed for 5-HT (1 μm, 10 min) uptake, as described in Materials and Methods. Maximal inhibition of 5-HT uptake by β-PMA was observed after 30 min of pretreatment.B, Dose–response curve of β-PMA effects on 5-HT uptake in 293-hSERT cells. Cells were preincubated with various concentrations of β-PMA for 30 min, followed by 10 min 5-HT uptake assays. C, Kinetic analysis of the effect of β-PMA on 5-HT uptake in 293-hSERT cells. Cells were preincubated with 1 μm PMA or vehicle for 20 min before 10 min uptake assay at various concentrations of 5-HT, as indicated. Parallel assays were performed in the presence of 100 nm paroxetine to define specific 5-HT uptake. Velocities measured were normalized to transport rates at 1 μm to compensate for varying expression levels across passages (see Materials and Methods). β-PMA caused a 47% decrease of 5-HT transport Vmax with little change in KM (0.39 μm in control vs 0.28 μm in β-PMA-treated cells).Inset shows an Eadie–Hofstee transformation of the data. For A–C, data presented are the averaged data from three separate experiments performed with six replicates per concentration. *p < 0.05; **p< 0.01; two-sided Student’s t test.
Fig. 4.
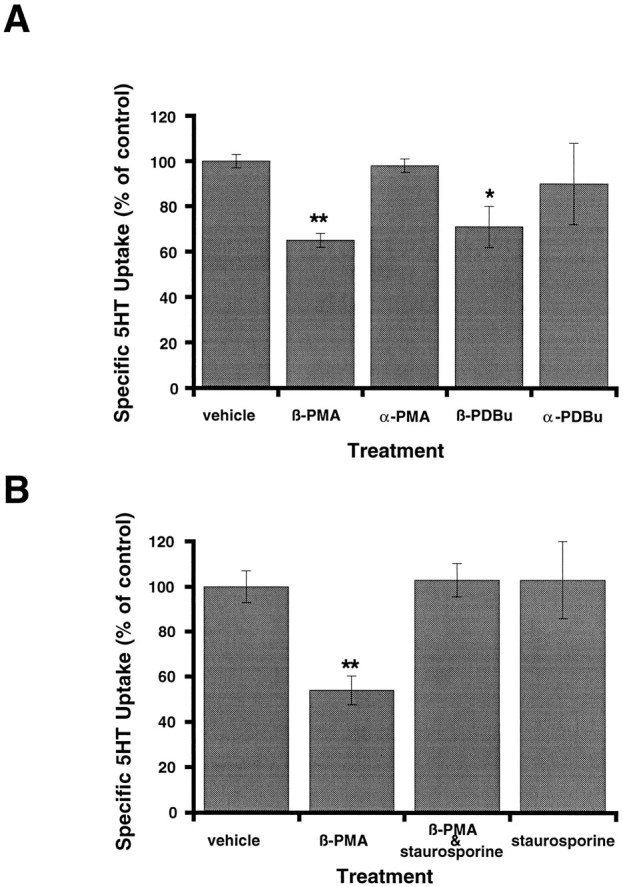
Stereospecificity and staurosporine sensitivity of phorbol ester-induced reductions in 5-HT uptake. A, The effect of phorbol esters on 5-HT uptake in 293 cells is stereospecific. Cells were preincubated separately with α or β stereoisomers of PMA and PDBu for 30 min before uptake assays with [3H]5-HT (1 μm, 10 min). PKC-inactive α isomers were ineffective in reducing 5-HT uptake, unlike the β forms. Data presented are mean ± SEM of three experiments performed in triplicate.B, The effect of β-PMA on 5-HT uptake in 293-hSERT cells can be blocked by staurosporine. Cells were preincubated with 1 μm β-PMA, 1 μm staurosporine, or 1 μm β-PMA plus 1 μm staurosporine for 30 min. β-PMA (1 μm) significantly inhibited 5-HT uptake, but it became ineffective in the presence of 1 μmstaurosporine. Staurosporine alone (1 μm) had no effect on 5-HT uptake. Data presented are the mean ± SEM of three experiments performed in quadruplicate. Nonspecific uptake forA and B was defined as the uptake in the presence of 100 nm paroxetine and subtracted from total accumulation to yield specific uptake. *p < 0.05; **p < 0.01; two-sided Student’s ttest.
PKC-mediated regulation of hSERT currents
Although hSERT is predicted to be an electroneutral transporter with ion-coupled 5-HT transport insensitive to membrane potential (Cool et al., 1990; Ramamoorthy et al., 1992), the presence of 5-HT-gated currents in 293-hSERT cells presented an opportunity to repeat our experiments in single voltage-clamped cells with control over membrane potential and a clamp over the transmembrane Na+-gradient. 293-hSERT cells were clamped at −40 mV and stepped in the presence or absence of 1 μm 5-HT to −120 mV, and the difference currents attributable to hSERT were obtained by subtraction (Fig. 5). Subsequently, 200 nmβ-PMA was added to the bath, and the step was repeated. In all five cells tested, β-PMA caused a marked reduction in 5-HT-induced current (up to 100%) within ∼5 min. In experiments in which only a partial current block was achieved, residual current was eliminated by subsequent addition of 5 μm paroxetine. Bath application of 1 μm staurosporine before β-PMA treatment eliminated the reduction in 5-HT-gated current (n = 5 cells) and, in some cases, potentiated the hSERT currents (Fig. 6). Staurosporine application alone had negligible effects on whole-cell currents recorded in the same paradigm in HEK-293 parental cells (data not shown).
PKC activation reduces hSERT surface abundance
Reductions in 293-hSERT 5-HT transport capacity and whole-cell currents are consistent with either a functional silencing of cell surface hSERTs and/or a loss of transporter protein from surface membranes. To evaluate this issue, we treated cells with 1 μm β-PMA for 40 min (30 min pretreatment, 10 min uptake) and then blotted membrane extracts of some wells with CT-2 antibody to assess loss of total hSERT protein. In addition, we performed cell surface biotinylation (Sargiacomo et al., 1989; Gottardi et al., 1995) experiments with the membrane-impermeant biotinylating reagent sulfosuccinimido-NHS-biotin to determine whether hSERT surface protein was diminished by β-PMA treatments. Biotinylation per se did not affect SERT immunoreactivity (data not shown). On the basis of the amount of SERT immunoreactivity present in the nonbiotinylated fraction, we estimate ∼75% of SERT protein reaches the cell surface in these cells. Under the larger culture format used to prepare total and biotinylated membrane extracts, 30 min of 1 μmβ-PMA pretreatment induced a 27 ± 2% (n = 3) decrease of 5-HT uptake. Results from immunoblot experiments are shown in Figure 7. β-PMA induced no change (101 ± 6% of control) in the amount of hSERT-immunoreactive protein in total extracts of 293-hSERT cells. However, the PKC activator induced a statistically significant decrease (31 ± 10%) in hSERT protein recovered from biotinylated fractions and a consistent increase (143 ± 16%) in transporter recovered from nonbiotinylated fractions (Fig. 7A,C). Calnexin, an endoplasmic reticulum membrane protein (Wada et al., 1991), was not biotinylated in this paradigm (Fig. 7B), providing validation of membrane impermeability and surface labeling of the biotinylating reagent. The effect of β-PMA on cell surface SERT density could be blocked by cotreatment with staurosporine (Fig. 8). In these experiments, 1 μm β-PMA reduced surface hSERT to 55% of control levels and increased the amount of hSERT protein in the nonbiotinylated fractions (166% of control) but had little effect (106% of control) in the presence of 1 μm staurosporine. As with 5-HT uptake, staurosporine alone had no effect on surface hSERT protein (103% of control) or the level of nonbiotinylated hSERT (92%). Together, these findings suggest that activation of PKC in 293-hSERT cells induces a reduction in hSERT-mediated 5-HT uptake and currents via a reduction in cell surface abundance of transporter protein.
Fig. 7.
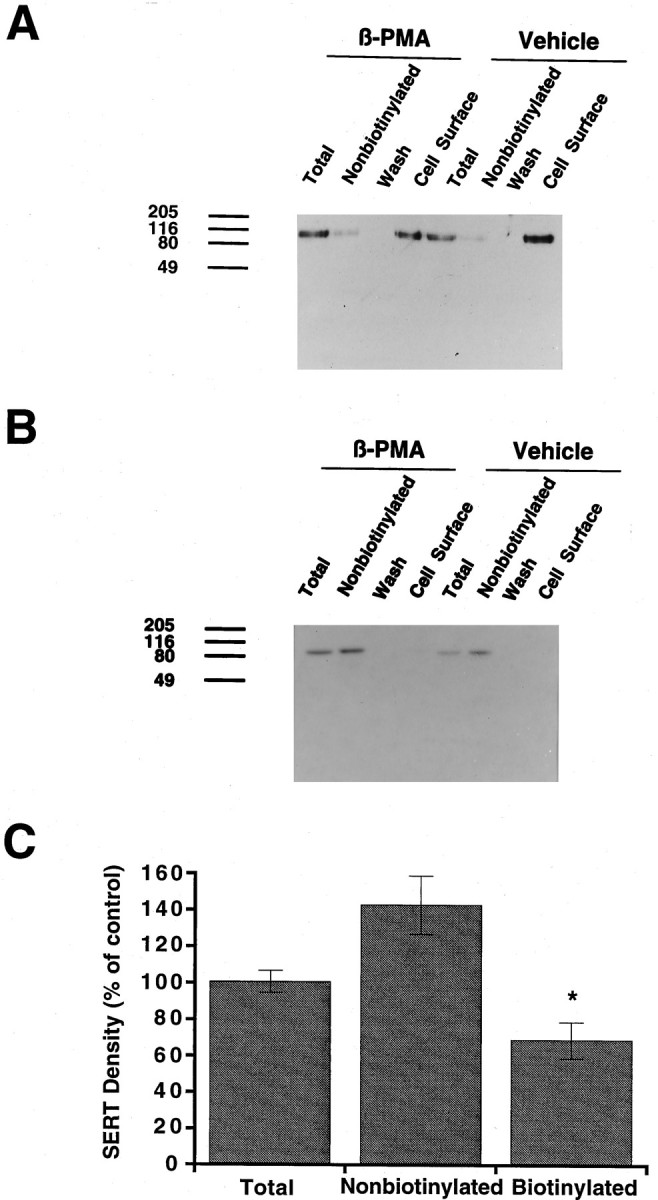
Effect of β-PMA on cell surface hSERT density. A, SERT immunoblot of total, nonbiotinylated, and biotinylated (cell surface) protein in 293-hSERT cells. Cells were treated with 1 μm β-PMA or vehicle for 40 min before biotinylation with sulfo-NHS-biotin. Aliquots (40 μl) of total, nonbiotinylated, and wash fractions were loaded, whereas the entire eluate (50 μl) from the streptavidin beads was loaded as the biotinylated sample and the blots were probed with CT-2 antibody (1 μg/ml). B, The same blot that was stripped and probed with anti-calnexin antibody to identify the endoplasmic reticular membrane protein calnexin. Calnexin immunoreactivity was observed only in the total and nonbiotinylated lanes and was used to normalize hSERT immunoreactivity between β-PMA-treated and untreated cells.C, Averaged quantitation of β-PMA effects on hSERT total, nonbiotinylated, and surface density. Immunoblots from three separate biotinylation experiments were scanned densitometrically, SERT values were normalized with calnexin immunoreactivity, and mean values were plotted ± SEM. Data are expressed as a percentage of vehicle-treated cells (control). Asterisk indicates a statistically significant reduction in biotinylated hSERT protein (p < 0.05; Student’s ttest). Two additional experiments to test staurosporine effects (Fig.8) gave consistent results, as did direct visualization of CT-2 immunoreactivity with [125I]protein-A.
Fig. 8.

Staurosporine blocks β-PMA-induced reduction in hSERT surface abundance. A, Biotinylation experiments were performed with or without 1 μm β-PMA, as in Figure7, and blotted with CT-2 antibody. Separate wells were coincubated with 1 μm staurosporine. B, Calnexin immunoreactivity of the same blot as in A after stripping. C, Quantitation of the effect of staurosporine on total and biotinylated (cell surface) hSERT. CT-2 immunoreactive bands were scanned, and density values were normalized for calnexin immunoreactivity in the same cell extracts. Data are expressed as a percentage of values obtained with vehicle-treated cells (control). Staurosporine alone had little or no effect on total or surface hSERT protein, but its presence blocked the β-PMA-induced reduction in hSERT surface protein. The experiment was performed twice, yielding identical results.
DISCUSSION
HEK-293 cells stably transfected with hSERT cDNA exhibit saturable 5-HT transport with a 5-HT KM comparable to that observed in human platelets and placental syncytiotrophoblasts (Rudnick, 1977; Balkovetz et al., 1989) and rank order antagonist sensitivity, as described in native and transiently transfected cells (Barker and Blakely, 1995). Membranes from these cells exhibit a single SERT-immunoreactive species and bind the cocaine analog [125I]RTI-55 with high affinity. Significant levels of transporter expression in transfected HEK-293 cells, a host cell well suited to whole-cell patch-clamp analysis, permitted us not only to investigate the effects of PKC activation on 5-HT uptake and hSERT protein distribution but also to corroborate our findings by using 5-HT-gated currents as an index of membrane transporter activity. Our studies demonstrate that phorbol ester application reduces both 5-HT accumulation and 5-HT-gated currents, an effect we show likely to be mediated by protein kinase C and attributable to altered surface abundance of SERT protein. We observed that several endogenous Na+-dependent transport systems in HEK-293 cells are sensitive to PKC activation, to varying extents. However, effects on 5-HT uptake still could be observed in the presence of ouabain, a Na/K ATPase inhibitor. Thus, PKC-mediated downregulation of the Na/K ATPAse and consequent shifts in the Na+ gradient are unlikely to be responsible for the reduction in 5-HT uptake. The lack of effect of β-PMA on Na+-independent leucine transport suggests that hSERT can take advantage of specific pathways for PKC-mediated membrane protein internalization present in HEK-293 cells not available to all membrane transport systems. Homologous GABA (Corey et al., 1994a), dopamine (DA) (Kitayama et al., 1994), and glycine transporters (Sato et al., 1995) expressed in transfected cells also exhibit specific PKC-mediated alterations in transport capacity, suggesting that a common mechanism may apply to members of the GAT/NET gene family.
The currents we record are significantly larger than anticipated if charge movement follows predicted stoichiometry for ion-coupled 5-HT flux. For example, a turnover rate of 1 5-HT+/sec applied to the transporter density we estimate of 7 × 104carriers/cell would yield only ∼0.01 pA of whole-cell current, whereas we record up to 30 pA of whole-cell current at −120 mV. This is even more striking when one considers that the presumed stoichiometry for coupled 5-HT movement (1 Na+:1Cl−:1 5-HT+in/1K+ out per cycle) should result in no net charge movement (Ramamoorthy et al., 1992; Rudnick and Clark, 1993). Similar observations of nonstoichiometric ion flow in rat SERT cRNA-injectedXenopus oocytes (Mager et al., 1994) suggest the presence of a 5-HT-gated ion pore at some stage in the SERT transport cycle or the occurrence of channel activity in a subset of transporter proteins (Cammack and Schwartz, 1996). Our determination of a rate of 5-HT translocation by hSERT of ∼1/sec agrees with previous estimates (Talvenheimo et al., 1979; Ross and Hall, 1983) and lends further support to the view that currents measured reflect nonstoichiometric channel-like ion flow gated by 5-HT. As with oocytes, we obtain no reversal of the 5-HT-gated current at negative potentials consistent with a requirement for cytoplasmic substrate to activate an intracellular gate (Cammack et al., 1994; Risso et al., 1996). Nonetheless, channel activation with external 5-HT has essentially the same properties as 5-HT transport, including saturable dependence on 5-HT and Na+ and sensitivity to SERT-specific antagonists. Importantly, the presence of hSERT-mediated currents allows PKC effects on hSERT to be corroborated in single voltage-clamped cells to reduce concerns of altered membrane potential. The greater influence of β-PMA on SERT-mediated currents over 5-HT uptake requires further study, although it may signal additional effects of the PKC activator on plasma membrane-resident SERTs than accounted for by surface redistribution. Alternatively, this simply may reflect sampling bias inherent in the single-cell assay conditions of voltage-clamp studies relative to the population measurements inherent in determinations of 5-HT uptake. We did find evidence for reduction in activity of other endogenous transport systems (although not for leucine transport); however, the large pipette volume adds additional constraints to attempts to explain the reduction in 5-HT currents (and presumably transport) completely via gross alterations in the Na+gradient. To our knowledge, this is the first report of SERT-associated currents in mammalian cells and the first application of whole-cell recording techniques to study amine transporter regulation.
Our findings for loss of surface SERT protein after PKC activation seem to reflect transporter redistribution rather than irreversible loss of transporter protein via degradation. Thus, we find a consistent increase in SERT material eluting with nonbiotinylated protein and no change in the total SERT pool under conditions in which SERT protein in surface membranes is depleted. These findings are in agreement with previous studies examining binding of lipophilic antagonists to amine transporters, in which no reduction in total transporter density has been detected after PKC activation (Anderson and Horne, 1992; Kitayama et al., 1994; Miller and Hoffman, 1994). We used cell surface biotinylation rather than whole-cell binding assays, because the available SERT radioligands are highly lipophilic and we wished to follow SERT distribution rather than label all accessible SERT proteins. Recently, Corey and coworkers have reached similar conclusions of transporter redistribution concerning PKC-mediated regulation of homologous GAT1 GABA transporters expressed inXenopus laevis oocytes (Corey et al., 1994a), Treatment of oocytes with PKC activators induces a movement of GAT1 protein from intracellular to surface membranes, in this case effecting anincrease in transporter expression, suggesting that the direction of redistribution is host cell-dependent, rather than transporter-dependent. The latter studies also used intracellular injections of PKC activators, and thus the population of PKCs activated may be distinct from those activated in our studies. Further studies are required to determine whether reduced hSERT insertion in or increased removal from the plasma membrane follows PKC activation to account for a net loss of surface carriers. However, similar effects of PKC activators on endogenous mammalian SERTs and 293-hSERT cells suggest that the mechanisms we have described may correspond to pathways for transporter regulation in less accessible preparations, including neuronal terminals and platelets, in vivo.
The presence of conserved phosphorylation sites for PKC and other kinases (Ramamoorthy et al., 1993c; Miller and Hoffman, 1994) on SERTs raises the possibility for transporter phosphorylation in parallel with or as a trigger for transporter redistribution. Preliminary in vivo phosphorylation experiments in 293-hSERT cells reveal the transporter to be a target for endogenous protein kinases activated by β-PMA (Ramamoorthy and Blakely, unpublished data). Although a member of a distinct transporter gene family, glutamate transporters seem to be phosphorylated in parallel with PKC-mediated upregulation of transport activity (Casado et al., 1993). It is not understood whether these phosphorylation events trigger, or follow, surface redistribution or whether or not PKC directly phosphorylates these carriers. We have observed that the NH2 and COOH termini expressed as fusion proteins are substrates for purified PKC and PKA, but not PKG, although canonical sites do not seem to be responsible for phosphate incorporation (Qian et al., 1995a). Canonical PKC sites also do not seem to be responsible for PKC-mediated alterations in GABA (Corey et al., 1994a) or glycine (Sato et al., 1995) transport, suggesting that if direct transporter phosphorylation is a requirement for changes in activity, it may occur through kinases downstream of PKC or at noncanonical PKC sites. Future studies of SERT currents in cell-detached patches (Galli et al., 1996) perfused with activated protein kinases may allow us to determine whether phosphorylation modifies SERT function in parallel with changes in surface density. Regardless, the kinetic similarities of PKC-mediated reductions in transport capacity evident for multiple transporters in the GAT/NET gene family suggest a common mechanism involving surface redistribution.
Our studies have been conducted in a heterologous expression system for technical advantages; however, SERTs in platelets (Anderson and Horne, 1992; Launay et al., 1994), mast cells (Miller and Hoffman, 1994), pulmonary endothelial cells (Myers et al., 1989), placental cells (Ramamoorthy et al., 1995), and CNS neurons (Anderson et al., 1995;Miller and Hoffman, 1995) are subject to acute regulation by second-messenger-dependent pathways. In particular, downregulation of SERT activity by PKC activators is a consistent finding, arising in most cases from a reduction in 5-HT transportVmax. Controls to explore indirect contributions of the vesicular 5-HT storage efflux pathway or altered Na+gradients have suggested that a significant fraction of the PKC effect reflects alterations in SERT protein or its distribution (Anderson and Horne, 1992; Miller and Hoffman, 1994; Ramamoorthy et al., 1995). Elevation of intraneuronal calcium after sustained depolarization and/or occupancy of phospholipase C-coupled receptors activates and translocates PKC (2211) and could contribute to activity-dependent modulation of 5-HT uptake capacity via SERT protein redistribution. Post-translational events such as we have described may serve as one mechanism for short-term functional plasticity at serotonergic terminals. Multiple receptors on raphe neurons exist to activate PKC via phospholipase C activation (Maeno et al., 1993; Mansour et al., 1994; Pieribone et al., 1994) that may influence SERT distribution as well as affect 5-HT release and firing rates. Clearly, additional studies are required with native SERT-expressing cells to assess role of kinase activation in vivo. Because cyclic AMP-dependent and -independent pathways also influence SERT gene expression (Ramamoorthy et al., 1993a, 1995), regulated SERT expression is likely to play a significant role in establishing quantitatively appropriate levels of extracellular 5-HT in vivo (Blakely et al., 1996). Recent studies with DA transporter knockout animals (Giros et al., 1996) that demonstrate compensatory changes in catecholamine biosynthesis and receptor expression, as well as behavioral deficits, underscore the significance of matching levels of neurotransmitter transport activity to neuronal excitation and release.
Footnotes
These studies were supported through National Institute on Drug Abuse Grant DA-07390 to R.D.B. and National Association for Research into Schizophrenia and Affective Disorders Established Investigator Awards to R.D.B. and L.J.D. We thank Drs. J. Justice and M. Owens, Emory University, for providing cocaine and paroxetine, respectively; Dr. J. Hytell, Lundbeck Company (Copenhagen, Denmark), for providing citalopram; Dr. F. I. Carroll, Research Triangle Institute (Research Triangle Park, NC), for providing unlabeled RTI-55; Dr. L. Limbird for initial donation of calnexin antibody; and Drs. E. Barker, S. Apparsundaram, and H. C. Hartzell for review of this manuscript.
Correspondence should be addressed to Dr. Randy D. Blakely, Center for Molecular Neuroscience, MRBII, Room 419, Vanderbilt School of Medicine, Nashville, TN 37232-6600.
REFERENCES
- 1.Amara SG, Kuhar MJ. Neurotransmitter transporters: recent progress. Annu Rev Neurosci. 1993;16:73–93. doi: 10.1146/annurev.ne.16.030193.000445. [DOI] [PubMed] [Google Scholar]
- 2.Anderson GM, Horne WC. Activators of protein kinase C decrease serotonin transport in human platelets. Biochim Biophys Acta. 1992;1137:331–337. doi: 10.1016/0167-4889(92)90154-4. [DOI] [PubMed] [Google Scholar]
- 3.Anderson GM, Vaccadino FM, Hall LM. Short-term regulation of glial and neuronal serotonin uptake. Soc Neurosci Abstr. 1995;21:344.16. [Google Scholar]
- 4.Balkovetz DF, Tiruppathi C, Leibach FH, Mahesh VB, Ganapathy V. Evidence for an imipramine-sensitive serotonin transporter in human placental brush-border membranes. J Biol Chem. 1989;264:2195–2198. [PubMed] [Google Scholar]
- 5.Barker EL, Blakely RD. Norepinephrine and serotonin transporters: molecular targets of antidepressant drugs. In: Bloom FE, Kupfer DJ, editors. Psychopharmacology: the fourth generation of progress. Raven; New York: 1995. pp. 321–333. [Google Scholar]
- 6.Barker EL, Kimmel HL, Blakely RD. Chimeric human and rat serotonin transporters reveal domains involved in recognition of transporter ligands. Mol Pharmacol. 1994;46:799–807. [PubMed] [Google Scholar]
- 7.Bertorello AM, Aperia A, Walaas SI, Nairn AC, Greengard P. Phosphorylation of the catalytic subunit of Na+K+-ATPase inhibits the activity of the enzyme. Proc Natl Acad Sci USA. 1991;88:11359–11362. doi: 10.1073/pnas.88.24.11359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Blakely RD, Berson HE, Fremeau RT, Jr, Caron MG, Peek MM, Prince HK, Bradley CC. Molecular cloning and functional expression of a rat brain serotonin transporter. Nature. 1991a;354:66–70. doi: 10.1038/354066a0. [DOI] [PubMed] [Google Scholar]
- 9.Blakely RD, Clark JA, Rudnick G, Amara SG. Vaccinia-T7 RNA polymerase expression system: evaluation for the expression cloning of plasma membrane transporters. Anal Biochem. 1991b;194:302–308. doi: 10.1016/0003-2697(91)90233-j. [DOI] [PubMed] [Google Scholar]
- 10.Blakely RD, Wise KR, Schroeter S. Localization of the serotonin transporter to rodent adrenal chromaffin cells: support for a role of 5-HT in adrenal physiology. Soc Neurosci Abstr. 1995;21:316.1. [Google Scholar]
- 11.Blakely RD, Ramamoorthy S, Qian Y, Schroeter S, Bradley CC. Regulation of antidepressant-sensitive serotonin transporters. In: Reith MEA, editor. Neurotransmitter transporters: structure, function, and regulation. Humana; Totowa, NJ: 1996. pp. 29–71. [Google Scholar]
- 12.Boja JW, Mitchell WM, Patel A, Kopajtic TA, Carroll FI, Lewin AH, Abraham P, Kuhar MJ. High-affinity binding of [125I]RTI-55 to dopamine and serotonin transporters in rat brain. Synapse. 1992;12:27–36. doi: 10.1002/syn.890120104. [DOI] [PubMed] [Google Scholar]
- 13.Cammack JN, Schwartz EA. Channel behavior in a gamma-aminobutyrate transporter. Proc Natl Acad Sci USA. 1996;93:723–727. doi: 10.1073/pnas.93.2.723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cammack JN, Rakhilin SV, Schwartz EA. A GABA transporter operates asymmetrically with variable stoichiometry. Neuron. 1994;13:949–960. doi: 10.1016/0896-6273(94)90260-7. [DOI] [PubMed] [Google Scholar]
- 15.Casado M, Bendahan A, Zafra F, Danbolt NC, Aragon C, Gimenez C, Kanner BI. Phosphorylation and modulation of brain glutamate transporters by protein kinase C. J Biol Chem. 1993;268:27313–27317. [PubMed] [Google Scholar]
- 16.Cesura AM, Muller K, Peyer M, Pletscher A. Solubilization of imipramine-binding protein from human blood platelets. Eur J Pharmacol. 1983;96:235–242. doi: 10.1016/0014-2999(83)90312-6. [DOI] [PubMed] [Google Scholar]
- 17.Chang AS, Chang SM, Starnes DM, Schroeter S, Blakely RD (1996) Molecular cloning and expression of the mouse serotonin transporter. Mol Brain Res, in press. [DOI] [PubMed]
- 18.Cool DR, Leibach FH, Ganapathy V. Modulation of serotonin uptake kinetics by ions and ion gradients in human placental brush-border membrane vesicles. Biochemistry. 1990;29:1818–1822. doi: 10.1021/bi00459a022. [DOI] [PubMed] [Google Scholar]
- 19.Corey JL, Davidson N, Lester HA, Brecha N, Quick MW. Protein kinase C modulates the activity of a cloned γ-aminobutyric acid transporter expressed in Xenopus oocytes via regulated subcellular redistribution of the transporter. J Biol Chem. 1994a;269:14759–14767. [PubMed] [Google Scholar]
- 20.Corey JL, Quick MW, Davidson N, Lester HA, Guastella J. A cocaine-sensitive Drosophila serotonin transporter: cloning, expression, and electrophysiological characterization. Proc Natl Acad Sci USA. 1994b;91:1188–1192. doi: 10.1073/pnas.91.3.1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Demchyshyn LL, Pristupa ZB, Sugamori KS, Barker EL, Blakely RD, Wolfgang WJ, Forte MA, Niznik HB. Cloning, expression, and localization of a chloride-facilitated, cocaine-sensitive serotonin transporter from Drosophila melanogaster. Proc Natl Acad Sci USA. 1994;91:5158–5162. doi: 10.1073/pnas.91.11.5158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.de Montigny C, Chaput Y, Blier P. Modification of serotonergic neuron properties by long-term treatment with serotonin reuptake blockers. J Clin Psychiatry. 1990;51:4–8. [PubMed] [Google Scholar]
- 23.Fuller RW. Uptake inhibitors increase extracellular serotonin concentration measured by brain microdialysis. Life Sci. 1994;55:163–167. doi: 10.1016/0024-3205(94)00876-0. [DOI] [PubMed] [Google Scholar]
- 24.Galli A, DeFelice LJ, Duke BJ, Moore KR, Blakely RD. Sodium-dependent norepinephrine-induced currents in norepinephrine transporter transfected HEK-293 cells blocked by cocaine and antidepressants. J Exp Biol. 1995;198:2197–2212. doi: 10.1242/jeb.198.10.2197. [DOI] [PubMed] [Google Scholar]
- 25.Galli A, Blakely RD, DeFelice LJ. Norepinephrine transporters have channel modes of conduction. Proc Natl Acad Sci USA. 1996;93:8671–8676. doi: 10.1073/pnas.93.16.8671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gershon MD, Jonakait GM. Uptake and release of 5-hydroxytryptamine by enteric serotonergic neurons: effects of fluoxetine (Lilly 110140) and chlorimipramine. Br J Pharmacol. 1979;66:7–9. doi: 10.1111/j.1476-5381.1979.tb16089.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Giros B, Jaber M, Jones SR, Wigthman RM, Caron MG. Hyperlocomotion and indifference to cocaine and amphetamine in mice lacking the dopamine transporter. Nature. 1996;379:606–612. doi: 10.1038/379606a0. [DOI] [PubMed] [Google Scholar]
- 28.Gottardi CJ, Dunbar LA, Caplan MJ. Biotinylation and assessment of membrane polarity: caveats and methodological concerns. Am J Physiol. 1995;268:F285–F295. doi: 10.1152/ajprenal.1995.268.2.F285. [DOI] [PubMed] [Google Scholar]
- 29.Gu H, Wall SC, Rudnick G. Stable expression of biogenic amine transporters reveals differences in inhibitor sensitivity, kinetics, and ion dependence. J Biol Chem. 1993;269:7124–7130. [PubMed] [Google Scholar]
- 30.Habert E, Graham D, Langer SZ. Solubilization and characterization of the 5-hydroxytryptamine transporter complex from rat cerebral cortical membranes. Eur J Pharmacol. 1986;122:197–204. doi: 10.1016/0014-2999(86)90103-2. [DOI] [PubMed] [Google Scholar]
- 31.Hoffman BJ, Mezey E, Brownstein MJ. Cloning of a serotonin transporter affected by antidepressants. Science. 1991;254:579–580. doi: 10.1126/science.1948036. [DOI] [PubMed] [Google Scholar]
- 32.Hoffman PW, Ravindran A, Huganir RL. Role of phosphorylation in desensitization of acetylcholine receptors expressed in Xenopus oocytes. J Neurosci. 1994;14:4185–4195. doi: 10.1523/JNEUROSCI.14-07-04185.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jayanthi LD, Ramamoorthy S, Mahesh VB, Leibach FH, Ganapathy V. Calmodulin-dependent regulation of the catalytic function of the human serotonin transporter in placental choriocarcinoma cells. J Biol Chem. 1994;269:14424–14429. [PubMed] [Google Scholar]
- 34.Kasanicki MA, Pilch PF. Regulation of glucose-transporter function. Diabetes Care. 1990;13:219–227. doi: 10.2337/diacare.13.3.219. [DOI] [PubMed] [Google Scholar]
- 35.Kitayama S, Dohi T, Uhl G. Phorbol esters alter functions of the expressed dopamine transporter. Eur J Pharmacol Mol Pharmacol Sect. 1994;268:115–119. doi: 10.1016/0922-4106(94)90180-5. [DOI] [PubMed] [Google Scholar]
- 36.Kuhar MJ, Roth RH, Aghajanian GK. Synaptosomes from forebrains of rats with midbrain raphe lesions: selective reduction of serotonin uptake. J Pharmacol Exp Ther. 1972;181:36–45. [PubMed] [Google Scholar]
- 37.Launay J, Bondoux D, Oset-Gasque M, Emami S, Mutel V, Haimart M, Gespach C. Increase of human platelet serotonin uptake by atypical histamine receptors. Am J Physiol. 1994;266:526–536. doi: 10.1152/ajpregu.1994.266.2.R526. [DOI] [PubMed] [Google Scholar]
- 38.Lesch KP, Aulakh CS, Wolozin BL, Tolliver TJ, Hill JL, Murphy DL. Regional brain expression of serotonin transporter mRNA and its regulation by reuptake inhibiting antidepressants. Mol Brain Res. 1993;17:31–35. doi: 10.1016/0169-328x(93)90069-2. [DOI] [PubMed] [Google Scholar]
- 39.Maeno H, Kiyama H, Tohyama M. Distribution of the substance P receptor (NK-1 receptor) in the central nervous system. Mol Brain Res. 1993;18:43–58. doi: 10.1016/0169-328x(93)90172-l. [DOI] [PubMed] [Google Scholar]
- 40.Mager S, Min C, Henry DH, Chavkin C, Hoffman BJ, Davidson N, Lester HA. Conducting states of a mammalian serotonin transporter. Neuron. 1994;12:845–859. doi: 10.1016/0896-6273(94)90337-9. [DOI] [PubMed] [Google Scholar]
- 41.Mansour A, Fox CA, Burke S, Meng F, Thompson RC, Akil H, Watson SJ. Mu, delta, and kappa opioid receptor mRNA expression in the rat CNS: an in situ hybridization study. J Comp Neurol. 1994;350:412–438. doi: 10.1002/cne.903500307. [DOI] [PubMed] [Google Scholar]
- 42.Melikian HE, Ramamoorthy S, Tate CG, Blakely RD. N-glycosylation-deficient human norepinephrine transporter: transport deficits arise from a combination of decreased protein stability, reduced surface abundance, and compromised amine translocation independent of ligand recognition. Mol Pharmacol. 1996;50:266–276. [PubMed] [Google Scholar]
- 43.Miller KJ, Hoffman BJ. Adenosine A3 receptors regulate serotonin transport via nitric oxide and cGMP. J Biol Chem. 1994;269:27351–27356. [PubMed] [Google Scholar]
- 44.Miller KJ, Hoffman BJ. 5-HT2 receptors increase 5-HT transport via nitric oxide and cGMP. Soc Neurosci Abstr. 1995;21:344.4. [Google Scholar]
- 45.Myers CL, Lazo JS, Pitt BR. Translocation of protein kinase C is associated with inhibition of 5-HT uptake by cultured endothelial cells. Am J Physiol. 1989;257:L253–L258. doi: 10.1152/ajplung.1989.257.4.L253. [DOI] [PubMed] [Google Scholar]
- 46.Nakanishi N, Onozawa S, Matsumoto R, Hasegawa H, Yamada S. Cyclic AMP-dependent modulation of vesicular monoamine transport in pheochromocytoma cells. J Neurochem. 1995;64:600–607. doi: 10.1046/j.1471-4159.1995.64020600.x. [DOI] [PubMed] [Google Scholar]
- 47.Nishio H, Nezasa K, Nakata Y. Role of calcium ion in platelet serotonin uptake regulation. Eur J Pharmacol Mol Pharmacol Sect. 1995;288:149–155. doi: 10.1016/0922-4106(95)90189-2. [DOI] [PubMed] [Google Scholar]
- 48.Pacholczyk T, Blakely RD, Amara SG. Expression cloning of a cocaine- and antidepressant-sensitive human noradrenaline transporter. Nature. 1991;350:350–354. doi: 10.1038/350350a0. [DOI] [PubMed] [Google Scholar]
- 49.Pieribone VA, Nicholas AP, Dagerlind A, Hokfelt T. Distribution of alpha 1 adrenoreceptors in rat brain revealed by in situ hybridization experiments utilizing subtype-specific probes. J Neurosci. 1994;14:4252–4268. doi: 10.1523/JNEUROSCI.14-07-04252.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Piñeyro G, Blier P, Dennis T, de Montigny C. Desensitization of the neuronal 5-HT carrier following long-term blockade. J Neurosci. 1994;14:3036–3047. doi: 10.1523/JNEUROSCI.14-05-03036.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Qian Y, Melikian HE, Moore KR, Duke BJ, Blakely RD. Phosphorylation of serotonin transporter domains and their role of phosphorylation in acute transporter regulation. Soc Neurosci Abstr. 1995a;21:344.7. [Google Scholar]
- 52.Qian Y, Melikian HE, Rye DB, Levey AI, Blakely RD. Identification and characterization of antidepressant-sensitive serotonin transporter proteins using site-specific antibodies. J Neurosci. 1995b;15:1261–1274. doi: 10.1523/JNEUROSCI.15-02-01261.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ramamoorthy S, Cool DR, Leibach FH, Mahesh VB, Ganapathy V. Reconstitution of the human placental 5-hydroxytryptamine transporter in a catalytically active form after detergent solubilization. Biochem J. 1992;286:89–95. doi: 10.1042/bj2860089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ramamoorthy S, Cool DR, Mahesh VB, Leibach FH, Melikian H, Blakely RD, Ganapathy V. Regulation of the human serotonin transporter: cholera toxin-induced stimulation of serotonin uptake in human placental choriocarcinoma cells is accompanied by increased serotonin transporter mRNA levels and serotonin transporter-specific ligand binding. J Biol Chem. 1993a;268:21626–21631. [PubMed] [Google Scholar]
- 55.Ramamoorthy S, Leibach FH, Mahesh VB, Ganapathy V. Partial purification and characterization of the human placental serotonin transporter. Placenta. 1993b;14:449–461. doi: 10.1016/s0143-4004(05)80465-5. [DOI] [PubMed] [Google Scholar]
- 56.Ramamoorthy S, Bauman AL, Moore KR, Han H, Yang-Feng T, Chang AS, Ganapathy V, Blakely RD. Antidepressant- and cocaine-sensitive human serotonin transporter: molecular cloning, expression, and chromosomal localization. Proc Natl Acad Sci USA. 1993c;90:2542–2546. doi: 10.1073/pnas.90.6.2542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ramamoorthy JD, Ramamoorthy S, Papapetropoulos A, Catravas JD, Leibach FH, Ganapathy V. Cyclic AMP-independent up-regulation of the human serotonin transporter by staurosporine in choriocarcinoma cells. J Biol Chem. 1995;270:17189–17195. doi: 10.1074/jbc.270.29.17189. [DOI] [PubMed] [Google Scholar]
- 58.Reith MEA. Cocaine receptors on monoamine transporters and sodium channels. NIDA Res Monogr. 1988;88:23–43. [PubMed] [Google Scholar]
- 59.Risso S, DeFelice LJ, Blakely RD. Whole-cell patch-clamp detection of γ-aminobutyric acid transporter currents in GAT1-transfected HeLa cells. J Physiol (Lond) 1996;490:691–702. doi: 10.1113/jphysiol.1996.sp021178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ross SB, Hall H. Maximal turnover number of the membranal serotonin carrier in rat synaptosomes in vitro. Acta Physiol Scand. 1983;118:185–187. doi: 10.1111/j.1748-1716.1983.tb07260.x. [DOI] [PubMed] [Google Scholar]
- 61.Rudnick G. Active transport of 5-hydroxytryptamine by plasma membrane vesicles isolated from human blood platelets. J Biol Chem. 1977;252:2170–2174. [PubMed] [Google Scholar]
- 62.Rudnick G, Clark J. From synapse to vesicle: the reuptake and storage of biogenic amine neurotransmitters. Biochim Biophys Acta. 1993;1144:249–263. doi: 10.1016/0005-2728(93)90109-s. [DOI] [PubMed] [Google Scholar]
- 63.Sargiacomo M, Lisante M, Graeve L, Le Bivic A, Rodriguez-Boulan E. Integral and peripheral protein composition of the apical and basolateral membrane domains in MDCK cells. J Membr Biol. 1989;107:277–286. doi: 10.1007/BF01871942. [DOI] [PubMed] [Google Scholar]
- 64.Sato K, Ralf A, Betz H, Schloss P. Modulation of a recombinant glycine transporter (Glyt1b) by activation of protein kinase C. J Neurochem. 1995;65:1967–1973. doi: 10.1046/j.1471-4159.1995.65051967.x. [DOI] [PubMed] [Google Scholar]
- 65.Segonzac A, Shoemaker H, Langer SL. Temperature dependence of drug interaction with the platelet 5-hydroxytryptamine transporter: a clue to the imipramine selectivity paradox. J Neurochem. 1987;48:331–337. doi: 10.1111/j.1471-4159.1987.tb04098.x. [DOI] [PubMed] [Google Scholar]
- 66.Talvenheimo J, Nelson PJ, Rudnick G. Mechanism of imipramine inhibition of platelet 5-hydroxytrytamine transport. J Biol Chem. 1979;254:4631–4635. [PubMed] [Google Scholar]
- 67.Tamaoki T, Nomoto H, Takahashi I, Kato Y, Morimoto M, Tomita F. Staurosporine, a potent inhibitor of phospholipid/Ca2+-dependent kinase. Biochem Biophys Res Commun. 1986;135:397–402. doi: 10.1016/0006-291x(86)90008-2. [DOI] [PubMed] [Google Scholar]
- 68.Tate CG, Blakely RD. The effect of N-linked glycosylation on activity of the Na+- and Cl−-dependent serotonin transporter expressed using recombinant baculovirus in insect cells. J Biol Chem. 1994;269:26303–26310. [PubMed] [Google Scholar]
- 69.Uhl GR, Johnson PS. Neurotransmitter transporters: three important gene families for neuronal function. J Exp Biol. 1995;196:229–236. doi: 10.1242/jeb.196.1.229. [DOI] [PubMed] [Google Scholar]
- 70.Wada I, Rindress D, Cameron PH, Ou W, Doherty JJ, Louvard D, Bell AW, Dignard D, Thomas DY, Bergeron JM. SSRα and associated calnexin are major calcium binding proteins of the endoplasmic reticulum membrane. J Biol Chem. 1991;266:19599–19610. [PubMed] [Google Scholar]
- 71.Wade PR, Chen J, Jaffe B, Kassem IS, Blakely RD, Gershon MD. Localization and function of a 5-HT transporter in crypt epithelia of the gastrointestinal tract. J Neurosci. 1996;16:2352–2364. doi: 10.1523/JNEUROSCI.16-07-02352.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wielosz M, Salmona M, De Gaetano G, Garattini S. Uptake of 14C-5-hydroxytryptamine by human and rat platelets and its pharmacological inhibition. Naunyn Schmiedebergs Arch Pharmacol. 1976;296:59–65. doi: 10.1007/BF00498840. [DOI] [PubMed] [Google Scholar]