

Abstract
Ca2+ influx through skeletal muscle Ca2+channels and the force of contraction are increased in response to β-adrenergic stimulation and high-frequency electrical stimulation. These effects are thought to be mediated by cAMP-dependent phosphorylation of the skeletal muscle Ca2+ channel. Modulation of the cloned skeletal muscle Ca2+ channel by cAMP-dependent phosphorylation and by depolarizing prepulses was reconstituted by transient expression in tsA-201 cells and compared to modulation of the native skeletal muscle Ca2+ channel as expressed in mouse 129CB3 skeletal muscle cells. The heterologously expressed Ca2+ channel consisting of α1, α2δ, and β subunits gave currents that were similar in time course, current density, and dihydropyridine sensitivity to the native Ca2+ channel. cAMP-dependent protein kinase (PKA) stimulation by Sp-5,6-DCl-cBIMPS (cBIMPS) increased currents through both native and expressed channels two- to fourfold. Tail currents after depolarizations to potentials between −20 and +80 mV increased in amplitude and decayed more slowly as either the duration or potential of the depolarization was increased. The time- and voltage-dependent slowing of channel deactivation required the activity of PKA, because it was enhanced by cBIMPS and reduced or eliminated by the peptide PKA inhibitor PKI (5–24) amide. This voltage-dependent modulation of the cloned skeletal muscle Ca2+ channel by PKA also required anchoring of PKA by A-Kinase Anchoring Proteins because it was blocked by peptide Ht 31, which disrupts such anchoring. The results show that the skeletal muscle Ca2+ channel expressed in heterologous cells is modulated by PKA at rest and during depolarization and that this modulation requires anchored protein kinase, as it does in native skeletal muscle cells.
Keywords: L-type Ca2+ channel, cAMP-dependent protein kinase, skeletal muscle, heterologous expression
In vertebrate skeletal muscle, synaptic transmission from motor nerve endings initiates a sodium-dependent action potential. L-type Ca2+ channels in the transverse tubule membranes are activated by depolarization and serve two critical functions. Within milliseconds these Ca2+ channels undergo a conformational change that activates the sarcoplasmic reticulum Ca2+ release channels via a physical coupling mechanism, resulting in Ca2+ release and muscle contraction (Chandler et al., 1976; Tanabe et al., 1990; Ríos et al., 1991; Lu et al., 1994). Calcium entry through the channels activates much more slowly and is thought to replenish intracellular Ca2+ (Oz and Frank, 1991). Activation of cAMP-dependent protein kinase (PKA) increases this current (Schmid et al., 1985; Arreola et al., 1987), resulting in increased contractile force (Arreola et al., 1987; Huerta et al., 1991). Basal levels of PKA activity are also required for voltage-dependent potentiation, which enhances Ca2+ entry through L-type channels (Sculptoreanu et al., 1993a,b; Bourinet et al., 1994). Strong depolarizations or high-frequency stimulation typical of a tetanus potentiate Ca2+ currents during subsequent depolarizations (Sculptoreanu et al., 1993b). Potentiated channels deactivate slowly, as evidenced by slowed tail currents after potentiation (Sculptoreanu et al., 1993b; Fleig and Penner, 1995). Much of the Ca2+ entry occurs during these tail currents because of the larger driving force at negative potentials. Potentiation requires anchoring of PKA (Johnson et al., 1994) by A-Kinase Anchoring Proteins (AKAPs) (for review, see Scott and McCartney, 1994). The complex regulation of current through skeletal muscle Ca2+channels by protein phosphorylation and membrane depolarization contributes in a critical way to the overall regulation of muscle contraction in response to motor nerve stimulation.
Calcium channels purified from skeletal muscle are a complex of five subunits: a pore-forming α1 subunit of 190 kDa in association with an intracellular β subunit, a transmembrane disulfide-linked glycoprotein dimer of α2 and δ subunits, and a transmembrane glycoprotein γ subunit (for review, seeHofmann et al., 1994; Catterall, 1995). The α1 subunit (Tanabe et al., 1987) forms a functional voltage-gated Ca2+channel on expression in muscle or heterologous cells (Tanabe et al., 1988, 1990; Perez-Reyes et al., 1989). The β and γ subunits are products of separate genes, whereas the α2δ subunits are encoded by a single gene and formed by post-translational processing (Hofmann et al., 1994; Isom et al., 1994). The α2δ and β subunits modulate the function of L-type Ca2+ channels, whereas the γ subunit, which is unique to skeletal muscle, does not have substantial effects (for review, seeHofmann et al., 1994; Isom et al., 1994).
Skeletal muscle Ca2+ channels are poorly expressed in heterologous cells, and regulation of channels composed of cloned subunits has not been successfully reconstituted. Molecular analysis of the complex regulation of skeletal muscle Ca2+ channels by phosphorylation, membrane depolarization, and association with AKAPs requires expression of the cloned cDNAs for the Ca2+channel subunits, kinases, and AKAPs in a heterologous cell system to allow study of the functional and regulatory properties of wild-type and mutant forms of the proteins. In this report, we describe successful transient expression of the skeletal muscle Ca2+channel in a heterologous cell line and show that the regulatory properties of the channel composed of cloned α1, α2δ, and β subunits are comparable to those of native skeletal muscle Ca2+ channels. Surprisingly, as in native skeletal muscle cells, anchoring of PKA by AKAPs is required for voltage-dependent potentiation of the expressed skeletal muscle Ca2+ channel, implying that the nonmuscle cell has the capability of anchoring PKA near the heterologously expressed Ca2+ channel.
MATERIALS AND METHODS
Molecular biology. cDNAs encoding the α1 and α2δ1 subunits cloned from rabbit skeletal muscle (Ellis et al., 1988) were provided by Drs. Steven B. Ellis, Michael M. Harpold (Salk Institute Biotechnology/Industrial Associates, Inc.) and Arnold Schwartz (University of Cincinnati College of Medicine). Two overlapping fragments of α1 cDNA (pSKMCaCHα1.7 and 1.8) were assembled, and the entire 5′-noncoding region was excised to yield the construct inserted into the expression plasmid ZemRVSP6 (West et al., 1992) derived from Zem 228 (Dr. Eileen Mulvihill, Zymogenetics Corp.).
Cell culture and expression of channel subunits. Mouse skeletal muscle myotubes were differentiated from an immortalized mouse skeletal muscle myoblast line, 129CB3 (Pinçon-Raymond et al., 1991). Myoblasts were grown in DMEM supplemented with 10% fetal bovine serum (Hyclone, Logan, UT) in a 5% CO2 incubator at 37°C. On reaching confluence, myoblasts fused to form myotubes in ∼7 d. Nearly spherical myotubes smaller than 50 μm were chosen for recording.
The α1 subunit of the rabbit skeletal muscle Ca2+ channel (Ellis et al., 1988) in the ZemRVSP6 vector was expressed with the β1b (Pragnell et al., 1991) in the pMT-2 vector (Genetics Institute, Cambridge, MA) and α2δ1 subunits in the ZemRVSP6 vector. cDNAs encoding these channel subunits and the CD8 antigen (EBO-pCD-Leu2, American Type Culture Collection, Rockville, MD) were transfected into tsA-201 cells by CaPO4 precipitation as described (Margolskee et al., 1994). tsA-201 cells, a subclone of the human embryonic kidney cell line HEK293, which expresses SV40 T antigen (a gift of Dr. Robert Dubridge, Cell Genesis, Foster City, CA), were maintained in a monolayer culture in DMEM/F-12 medium (Life Technologies, Gaithersburg, MD) supplemented with 10% fetal bovine serum (Hyclone), and incubated at 37°C in 10% CO2. Seventy-five percent confluent cultures of tsA-201 cells in 35 mm dishes were transfected with a total of 4 μg of DNA containing an equimolar ratio of the three-channel subunit cDNAs and 0.8 μg of CD8 cDNA to mark the successfully transfected cells. After addition of CaPO4-DNA, cells were incubated overnight at 37°C in 5% CO2. Twenty hours after transfection, the cells were removed from culture dishes using 2 mm EDTA in PBS and replated at low density for electrophysiological analysis. Transfectants were selected by fluorescent antibody labeling (phycoerythrin-labeled anti-CD8, Sigma, St. Louis, MO) followed by viewing, using an epifluorescence microscope (Nikon Diaphot, rhodamine filters), or by labeling with anti-CD8-coated beads (Dynal, Great Neck, NY).
Electrophysiology. Barium currents through skeletal muscle Ca2+ channels were recorded using the whole-cell configuration of the patch-clamp technique. Patch electrodes were pulled from VanWaters & Rogers micropipettes and fire-polished to produce an inner tip diameter of 4–6 μm. Currents were recorded using a List EPC-7 patch-clamp amplifier and filtered at 2 kHz (8-pole Bessel filter, −3 dB). Data were acquired using Fastlab software (Indec Systems). Voltage-dependent currents have been corrected for leak using an on-line P/4 subtraction paradigm. The extracellular (bath) saline contained (in mm): 150 Tris, 2 MgCl2, 10 BaCl2; pH was adjusted to 7.3 with methanesulfonic acid. The intracellular (patch electrode) saline contained (in mm): 130N-methyl-d-glucamine, 10 EGTA, 60 HEPES, 2 MgATP, 1 MgCl2; pH was adjusted to 7.3 with methanesulfonic acid. All experiments were performed at room temperature (20–23°C). No nonlinear outward currents were detected under these conditions.
Ca2+-activated Cl− currents are a potential concern in measurements of tail currents in skeletal muscle cells and possibly in tsA-201 cells. Contamination of Ca2+ channel tail current measurements by Ca2+-activated Cl− currents was prevented by using low chloride intracellular and extracellular salines yielding a calculated Cl− reversal potential (zero-current potential) of −63 mV, near the potential (−80 mV) at which Ca2+ channel tail currents were measured. The reversal potential of Ba2+ tail currents was more than +30 mV, consistent with relatively pure Ba2+ permeation, and identical tail currents were observed when Na+ carried current through the channel or when the Ca2+ buffer EGTA (10 mm) in the intracellular saline was replaced with 10 mm BAPTA.
Sp-5,6-DCl-cBIMPS (cBIMPS) (BioLog Life Sciences Institute) was stored frozen in a 100 mm DMSO stock. The appropriate amount of this stock was either added directly to the recording chamber or first diluted to 6× its final concentration in extracellular saline before addition. Ht 31 peptide (Carr et al., 1992) was synthesized and purified by HPLC in the University of Washington Molecular Pharmacology Facility with the following sequence: Asp-Leu-Ile-Glu-Glu-Ala-Ala-Ser-Arg-Ile-Val-Asp-Ala-Ala-Val-Ile-Glu-Gln-Val-Lys-Ala-Ala-Gly-Ala-Tyr. Ht-31P peptide containing proline residues substituted for the isoleucine residues at positions 10 and 16 was synthesized and purified using the same methods. Ht 31 peptides and PKI (5–24) amide (LC Laboratories) were stored frozen in 1 mm stocks made from intracellular saline without MgATP and diluted to final concentrations (100 or 500 μm and 10 μm, respectively) in intracellular saline containing MgATP.
RESULTS
Expression and characterization of skeletal muscle Ca2+ channels
Ba2+ currents through native Ca2+ channels in mouse skeletal muscle-derived 129CB3 cells were first detectable during test depolarizations to −20 mV (Fig.1A). These currents rose slowly to maximum over several hundred milliseconds, and large tail currents were recorded on repolarization from positive test potentials. Peak currents were recorded at +10 mV (Fig. 1D). Application of the dihydropyridine channel activator Bay K 8644 substantially accelerated activation of these Ca2+ channels, slowed the tail currents approximately sixfold after pulses to the peak current potential, and shifted the voltage dependence of channel activation ∼10 mV in the negative direction so that peak currents were recorded at 0 mV (Fig.1B,D).
Fig. 1.

Comparison of heterologously expressed skeletal muscle Ca2+ channels and native skeletal muscle channels. Ca2+ channel current–voltage relationships (using Ba2+ as the charge carrier) were determined during 500 msec depolarizations from a holding potential of −80 mV. Test pulses between −40 and +60 mV were applied in 10 mV increments every 6 sec. A, Native Ca2+ channel current traces from a mouse 129CB3 skeletal muscle cell recorded in the absence of Bay K 8644. B, Currents from a different mouse skeletal muscle cell recorded in the presence of 10 μm Bay K 8644.C, Average current traces from the cloned rabbit skeletal muscle Ca2+ channel expressed in tsA-201 cells. Mean currents from 13 cells giving measurable currents during the pulse in the presence of 10 μm Bay K 8644 are shown.D, Mean current–voltage relations (±SE) for each of the three conditions shown in A–C: control native channel (circles, six cells), native channel with Bay K 8644 (squares, four cells), and channel expressed in tsA-201 cells in the presence of 10 μm Bay K 8644 (triangles, 29 cells). Peak currents were measured during 500 msec depolarizations and normalized before they were averaged.
Most recordings from tsA-201 cells expressing cDNAs encoding skeletal muscle Ca2+ channel α1, β1b, and α2δ subunits yielded no detectable current during depolarizing test pulses, but tail currents were observed on repolarization to −80 mV in 20% (3/15) of cells. Inclusion of 10 μm Bay K 8644 in the extracellular saline increased the percentage of cells with detectable currents during test depolarizations to 35% (29/82) of cells, averaging 24 ± 5 pA peak current (±SE), and tail currents were detectable in >95% of all cells. Averaged current traces from 13 of these cells are shown in Figure 1C. The expressed Ca2+ channel activated at more positive test pulse potentials than the native Ca2+channel current recorded with or without Bay K 8644 and peak current was recorded at +20 mV. Because application of Bay K 8644 to the native Ca2+ channel shifted the current–voltage relation by −10 to −15 mV (Fig. 1D), it is possible that in the absence of Bay K 8644, activation of expressed Ca2+channels requires depolarizations approaching the reversal potential and that current through the channel during test pulses was rarely observed because of the reduced driving force.
Ba2+ currents were 10-fold larger in Bay K 8644-treated 129CB3 cells expressing native Ca2+ channels than in cells expressing cloned skeletal muscle Ca2+ channels (Fig. 1). Part of the difference in current size between expressed and native channels is attributable to the difference in the sizes of the two cell types. The membrane capacitance of cells expressing the cloned skeletal muscle channel was 16 ± 2 pF compared with 42 ± 12 pF for skeletal muscle cells, consistent with a 2.6-fold larger surface area. In the presence of Bay K 8644, mean current densities were 7.6 pA/pF for native and 1.8 pA/pF for the expressed channel. Because of the difference in driving force at the peak current potential discussed above, current densities measured for tail currents at −80 mV were substantially closer in magnitude (see below).
Tail currents of native and expressed skeletal muscle Ca2+ channels
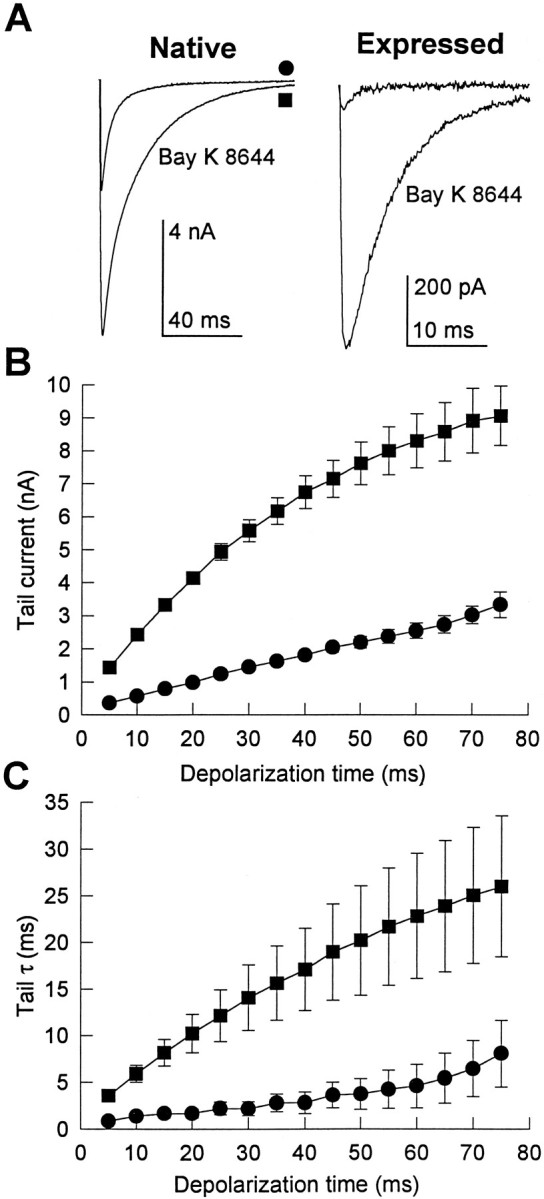
Tail currents that flow during deactivation of skeletal muscle Ca2+ channels on repolarization carry more Ca2+into the cell than do currents during the brief depolarization (<3 msec) caused by an action potential (Sculptoreanu et al., 1993b). Therefore, regulation of tail current amplitude and time course is more important for intracellular Ca2+ homeostasis in skeletal muscle than regulation of currents during depolarizations. Figure2A shows tail currents from one skeletal muscle cell (without Bay K 8644) and an average of tail currents through heterologously expressed channels from 28 cells in 10 μm Bay K 8644, illustrating the increase in tail current amplitude with increasing strength of depolarization. The tail currents from native and expressed channels behaved similarly in most respects. Repolarization to −80 mV after 500 msec depolarizations to potentials that increased from −40 and +80 mV elicited tail currents that increased in amplitude with increasing depolarization (Fig.2A,C). In addition, the time course of tail current decay became slower with increasing depolarization (Fig.2B,C). In Figure 2B, currents have been scaled to the same peak amplitude to demonstrate this slowing. Between 0 mV and +60 mV, the mean deactivation rate slowed 2.3 ± 0.8-fold for the native channel and 2.8 ± 0.7-fold for the expressed channel (Fig. 2C).
Fig. 2.

Voltage-dependent properties of native and expressed Ca2+ channel tail currents. Native and expressed channels were activated during 500 msec depolarizations ranging from −40 to +80 mV in 10 mV increments. Tail currents reflecting channel deactivation were recorded on repolarization to the holding potential of −80 mV. Native channels were recorded in the absence of Bay K 8644, whereas the expressed channel was recorded in the presence of 10 μm Bay K 8644. A, The amplitude of both native (left) and expressed tail currents (right) increased progressively as the magnitude of depolarization increased. The native currents were obtained from a single cell after depolarizations ranging from −40 to +80 mV and are representative of results from nine similar experiments. The expressed currents are the mean data from 28 cells after depolarizations ranging from −10 to +80 mV. B, Currents from the same cells shown in A were scaled to compare the time course of channel deactivation. Tail current time course progressively slowed as the magnitude of depolarization increased. C, Voltage dependence of peak tail current amplitude (open triangles) and single-exponential decay time constant (filled circles) are compared in native (left) and expressed (right) channels. Data represent means (±SE) from 9 and 28 cells, respectively.
The dependence of tail current activation on the duration of the preceding test depolarization was characterized by applying depolarizations to +80 mV of variable duration and recording tail currents after returning to the holding potential of −80 mV. As the duration of the depolarization was increased, tail currents became larger and slower for both native and expressed channels (Fig.3A,B). After 50 msec depolarizations to +80 mV, native Ca2+ channel tail currents were 5.3 ± 0.5-fold larger than after 5 msec depolarizations, whereas expressed Ca2+ channel tail currents were 6.2 ± 1.7-fold larger (Fig. 3C). Although the activation kinetics of native and expressed channels are similar for modest depolarizations, the time-dependent increase in tail current amplitude as the duration of pulses to +80 mV was increased followed different time courses (Fig.3C). For native cells, tail currents continued to increase with pulse durations up to 75 msec. The amplitudes of the tail currents for the expressed channel increased more rapidly with depolarization duration but approached a maximum beyond 30 msec (31 ± 16% increase from 75 to 100 msec; six cells).
Fig. 3.

Time-dependent properties of native and expressed Ca2+ channel tail currents. Tail currents were recorded in the presence of 10 μm Bay K 8644 after depolarizations to +80 mV that increased in duration from 5 to 75 msec in 5 msec increments. Examples of tail currents flowing on repolarization to −80 mV from native (A) and expressed (B) channels are shown. The smallest and fastest currents in each example correspond to tails after 5 msec depolarizations, whereas the largest and slowest currents were recorded after 75 msec depolarizations. Dashed lines show single-exponential fits to the 5 and 75 msec tail currents in these examples, giving time constants, respectively, of 3.2 and 42.4 msec for native and 3.7 and 19.4 msec for expressed. C, Mean time course of the increase in tail current amplitude recorded as shown inA and B are compared. Symbols represent mean ± SE for native (circles, nine cells) and expressed (triangles, 23 cells) Ca2+channels. D, Mean time course of the increase in tail current decay time constant from the same cells shown inC.
Normalized for capacitance, the tail currents after 75 msec depolarizations to +80 mV from tsA-201 cells expressing the cloned Ca2+ channel were approximately half as large as those of native Ca2+ channels in skeletal muscle cells (99 ± 26 pA/pF for expressed, 214 ± 66 pA/pF for native). If native and expressed channels have a similar probability of being open at this time, these numbers indicate that there are about half as many channels per unit membrane area expressed in tsA-201 cells as there are native channels in 129CB3 cells.
The rate of tail current decay was fit approximately by a single exponential as shown by the dashed lines in the examples (Fig.3A,B). The fit to a single exponential was exact for smaller tail currents but was only approximate for the largest ones, possibly because the voltage clamp was not precise for the largest tail currents. The mean time constants of tail current decay at −80 mV after 5 msec depolarizations were similar for the native (τ = 2.9 ± 0.4 msec) and expressed channels (τ = 3.6 ± 1.2 msec) (Fig. 3C,D). Tail current decay time constants for both native and expressed channels increased for depolarizations between 5 and 75 msec (Fig. 3D). Native tail current slowed 7.3 ± 2.1-fold between 5 and 75 msec depolarizations, whereas the tail current of the expressed channel slowed 4.4 ± 1.1-fold over the same range. Longer depolarization (500 msec compared with 75 msec) slowed tail current decay 52% more in native channels and 14% more in expressed channels. Although the absolute values of the time constants were different for the native and expressed channels, the time course of slowing with longer depolarizations was similar.
Effect of the dihydropyridine agonist Bay K 8644 on tail currents
Because Bay K 8644 was required to study the expressed Ca2+ channel, its effects on the depolarization-induced slowing of tail current were investigated. Examples of Bay K 8644 application (10 μm) to both native and expressed channels are shown in Figure 4A. Bay K 8644 both increased and slowed the decay of tail currents through native Ca2+ channels in skeletal muscle cells and through expressed Ca2+ channels. Tail current through native channels measured after a 100 msec depolarization to +60 mV increased 2.8 ± 0.3-fold in five cells and slowed 6.5 ± 3.4-fold (from τ = 4.3 msec to τ = 28 msec) in the presence of Bay K 8644. Tail currents through expressed channels in three cells increased 7.7 ± 3.5-fold and slowed 6.1 ± 1.9-fold (from τ = 1.9 msec to τ = 11.5 msec).
Fig. 4.
Effect of Bay K 8644 on tail currents.A, Tail currents from native and expressed Ca2+ channel are shown before and after application of 10 μm Bay K 8644. Tail currents result from 100 msec depolarizations to +60 mV followed by repolarization to −80 mV and are traces from single cells representative of three (Expressed) or five (Native) similar experiments. B, C, The mean time-dependent increases in tail current amplitude (B) and decay time constant (C) were determined for native channels in the absence (circles, six cells) and presence (squares, nine cells) of Bay K 8644. Tails currents were measured at −80 mV after depolarizations of the duration indicated on the abscissa to +80 mV.
To determine the effect of Bay K 8644 on the depolarization-induced slowing of tail current, tail currents through native Ca2+channels were compared in the absence and presence of Bay K 8644. In the absence of Bay K 8644, tail current amplitude (Fig.4B) and decay time constant (Fig. 4C) increased nearly linearly with increasing pulse duration. Substantially larger and slower tail currents were observed in the presence of Bay K 8644 (Fig. 4B,C), but the fold increase was similar in the presence or absence of the drug. Tail current amplitude increased 9.3 ± 1.1-fold as the pulse duration increased from 5 to 75 msec compared with 9.5 ± 4.2-fold in Bay K 8644 (Fig.4B). The decay time constant of tail currents increased 6.4 ± 0.7-fold over the same range of pulse durations compared with 7.3 ± 2.1-fold in Bay K 8644 (Fig. 4C). Thus, in spite of its large effects on the absolute amplitude and decay time constant of tail currents, Bay K 8644 did not significantly alter the relative increase in tail current amplitude or time constant caused by increasing the duration of the test depolarizations.
Modulation by cAMP-dependent phosphorylation
To examine the effect of stimulation of PKA on cloned skeletal muscle Ca2+ channels, the membrane-permeant nonhydrolyzable cAMP analog cBIMPS was used (Sandberg et al., 1991). Application of 25 μm cBIMPS greatly stimulated both native and expressed currents. Figure 5 shows examples of Ba2+currents through native (A) and expressed (B) skeletal muscle Ca2+ channels before and 3 min after application of cBIMPS. Peak Ca2+ channel current–voltage relationships were shifted toward negative potentials in the presence of cBIMPS for both the native (Fig. 5C) and expressed (Fig.5D) skeletal muscle Ca2+ channels. In many of the 129CB3 cells expressing native Ca2+ channels, the effect of cBIMPS was transient and dissipated after several minutes (Fig. 5C). This reversal of the effect may have been attributable to phosphatase activity, because it was prevented by phosphatase inhibitors (data not shown). No such reversal was observed for skeletal muscle channels expressed in tsA-201 cells. PKA stimulation increased expressed current by 127 ± 55% in tsA-201 cells (n = 10 cells). Application of cBIMPS had no effect on membrane capacitance in either cell type.
Fig. 5.

Effects of PKA stimulation on native and expressed Ca2+ channel currents. The membrane-permeant, nonhydrolyzable cAMP analog cBIMPS (25 μm) was used to stimulate PKA. A, B, Examples of native (A) and expressed (B) currents before and 3 min after cBIMPS application are shown. Currents were evoked by 500 msec depolarizations to −10 (Native) or +10 mV (Expressed) from a holding potential of −80 mV.C, D, Examples of current–voltage relations for native (C) and expressed (D) currents are representative of results from 4 and 10 cells, respectively. Control (circles); cBIMPS, 3 min (triangles). For the native channels, current–voltage relationships measured 3 min (cBIMPS early) and 5 min (cBIMPS late) after cBIMPS application are shown.
Effect of PKA stimulation on tail currents
As expected from the increased Ca2+ current during depolarizing test pulses in the presence of activators of PKA (Fig. 5), cBIMPS increased native Ca2+ channel tail currents 2.2 ± 0.5-fold in six cells and increased tail currents through expressed channels 3.5 ± 0.6-fold in 16 cells (Fig.6A). The time course of the increase in tail current amplitude after depolarizing pulses of increasing duration was accelerated substantially by cBIMPS (Fig.6B). Tail current amplitudes for native Ca2+ channels increased 5.3 ± 0.6-fold in cBIMPS, comparable to the 6.4 ± 0.7-fold increase observed under control conditions. For expressed channels, tail current amplitudes were already 6.5-fold larger after 5 msec pulses in the presence of cBIMPS and increased only 2.1-fold further with longer depolarizing pulses in the presence of cBIMPS. This is consistent with rapid activation of tail currents through the expressed channel in the presence of cBIMPS by the 5 msec time point.
Fig. 6.
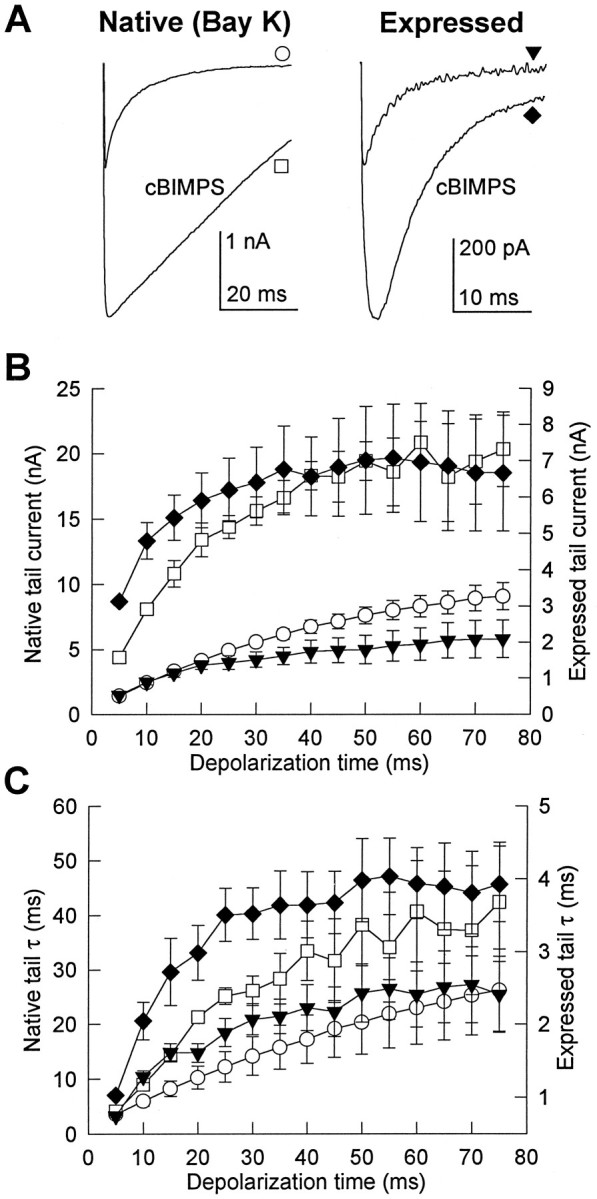
Effect of PKA stimulation on Ca2+ channel tail currents. cBIMPS (25 μm) was used to stimulate PKA. A, Tail currents from native and expressed Ca2+ channels are shown before and after application of cBIMPS. Tail currents were recorded at −80 mV after 100 msec depolarizations to +60 mV and are traces from single cells representative of 5 (Native) or 16 (Expressed) similar experiments. B, C, The mean time-dependent increases in tail current amplitude (B) and decay time constant (C) in the presence of 25 μm cBIMPS were determined after depolarizations to +80 mV as the duration of the depolarizations was increased from 5 to 75 msec in 5 msec increments. Native Ca2+ channel (three cells) and expressed Ca2+channel (six cells) are shown using the same symbols as inA.
As illustrated in Figure 3, tail current decay was slowed progressively as the duration of the preceding depolarizing pulse was increased in control conditions. Activation of PKA with cBIMPS slowed the decay of Ca2+ channel tail currents for all depolarization durations tested (Fig. 6C). As depolarization duration was increased from 5 to 75 msec, the decay time constant increased 9.1 ± 3.6-fold in the presence of cBIMPS compared with 7.3 ± 2.1-fold in control for the native channel and increased 3.9 ± 0.5-fold in the presence of cBIMPS and 3.4 ± 0.5-fold in control for the expressed channel. Thus, although cBIMPS resulted in slower tail currents for both native and expressed channels, the relative slowing induced by prepulses remained similar after PKA stimulation by cBIMPS.
Effect of PKA inhibition on tail currents
The quantitative differences between the effects of prepulses and treatment with cBIMPS on native and expressed Ca2+ channels may be attributable to different levels of endogenous phosphorylation in tsA-201 cells in comparison to native myocytes. To assess the importance of basal phosphorylation, the specific inhibitor of PKA, PKI (5–24) amide (10 μm), was included in the patch electrode saline when recording from skeletal muscle cells in the absence of Bay K 8644 or from cloned Ca2+ channels expressed in tsA-201 cells in the presence of 10 μm Bay K 8644. Tail currents were measured after depolarizations of increasing duration to +80 mV. Examples of these recordings are shown in Figure7A,B. The tail current amplitude increased normally with pulse duration in the presence of PKI (Fig.7D); however, this was not accompanied by the strong slowing in the decay time constant of the tail currents that was observed in control (Fig. 7E). The increase in decay time constant with prepulses of increasing duration was substantially reduced by PKI for expressed channels and eliminated for native channels (Fig.7E).
Fig. 7.

Effect of PKA inhibition on native and expressed Ca2+ channel tail currents. The peptide PKA inhibitor PKI (5–24) amide (10 μm) was included in the patch electrode saline and allowed to diffuse into the cell for 5 min before tail currents were measured. A, B, Tail currents measured at −80 mV in the presence of PKI from a 129CB3 skeletal muscle cell (A) and a tsA-201 cell expressing skeletal muscle Ca2+ channels (B) after 5–75 msec depolarizations to +80 mV in 5 msec increments. The smallest current in each series corresponds to the tail after a 5 msec depolarization, whereas the largest current was measured after a 75 msec depolarization. C, The PKA activator cBIMPS (25 μm) was applied to a skeletal muscle cell containing PKI in this example representative of three experiments. Tail currents were recorded at −80 mV after 100 msec depolarizations to +60 mV. D, E, The mean relative time-dependent increases in tail current amplitude (D) and decay time constant (E) in the presence of PKI were recorded at −80 mV after 5–75 msec depolarizations to +80 mV in 5 msec increments. Native Ca2+channel was recorded in the absence of Bay K 8644 (control:circles, six cells; PKI: squares, four cells) and expressed Ca2+ channel was recorded in the presence of 10 μm Bay K 8644 (control:triangles, 23 cells; PKI: diamonds, four cells).
The absolute amplitudes of tail currents measured after 5 msec depolarizations were reduced in PKI by 67% in skeletal muscle cells (359 ± 150 pA to 117 ± 43 pA) and by 84% in the tsA-201 cells expressing cloned channels (335 ± 141 pA to 54 ± 16 pA). Surprisingly, the decay time constants of these smaller tail currents recorded after short depolarizations in the presence of PKI were not significantly different from those measured in control cells (native control: 0.85 ± 0.28 msec, six cells; native PKI: 0.57 ± 0.15 msec, four cells; expressed control: 2.9 ± 0.4 msec, 23 cells; expressed PKI: 3.4 ± 1.0 msec, four cells). Only the slowing observed with increasing prepulse duration was affected. This suggests that basal phosphorylation of the channel does not alter tail current kinetics and that the phosphorylation event that slows tail currents after strong prepulses is distinct from the one that maintains channel activity.
Experiments with PKI also provided an opportunity to examine whether the effects of cBIMPS on Ca2+ channel function were entirely dependent on PKA activation. One example of three experiments in which skeletal muscle cells perfused with PKI were exposed to cBIMPS is shown in Figure 7C. cBIMPS application in the presence of PKI decreased tail currents by 13 ± 14% and had no effect on the current time course. Thus, the effects of cBIMPS on Ca2+channel function are attributable entirely to activation of PKA.
Role of PKA anchoring in Ca2+channel modulation
Previous work on time- and voltage-dependent potentiation of skeletal muscle Ca2+ channel activity revealed that anchoring of PKA near Ca2+ channels was required for rapid modulation by voltage-dependent phosphorylation (Johnson et al., 1994). PKA is localized through association of the cAMP-binding regulatory subunit RII with AKAPs (Scott and McCartney, 1994). A peptide containing the critical RII-binding region of a human thyroid AKAP, Ht 31, dissociated RII from all known AKAPs (Carr et al., 1992). AKAPs are present in all tissues studied, including kidney, from which tsA-201 cells are derived. Moreover, blot overlay assays with labeled RII revealed several AKAP bands in protein extracts of tsA-201 cells (P. C. Gray, W. A. Catterall, B. J. Murphy, unpublished results). Because modulation of the cloned skeletal muscle Ca2+ channel expressed in tsA-201 cells was time- and voltage-dependent and required phosphorylation by PKA as in skeletal muscle cells, these results suggested that PKA might also be anchored to AKAPs in this heterologous expression system. We used the Ht 31 peptide to detect the involvement of AKAPs in the time-dependent slowing of tail currents of expressed Ca2+ channels. Ht 31 reduced the slowing of tail current deactivation to a level not significantly different from that measured in PKI (Fig. 8A, compare with Fig.7B). As the preceding depolarization was increased from 5 to 75 msec, tail currents slowed 2.0 ± 0.2-fold in the presence of 100 μm Ht 31 in comparison with 2.2 ± 0.3-fold in the presence of PKI and 4.5 ± 1.1-fold in control (Fig.8D). A higher concentration of Ht 31 (500 μm) had no greater effect, suggesting that 100 μm Ht 31 is a saturating concentration. In preliminary experiments, we have also studied the effects of N-terminal myristoylated Ht 31, which is expected to be more effective in inhibition of phosphorylation of membrane-bound substrates. A concentration of 1 μm myristoylated Ht 31 is sufficient to prevent the slowing of tail currents in response to depolarizations of increasing duration (B. Johnson, J. Scott, T. Scheuer, and W. A. Catterall, unpublished results). Together with the experiments with unmodified Ht 31 (Fig. 8), these results support the conclusion that Ht 31 peptides act specifically to prevent Ca2+ channel modulation by blocking PKA binding to AKAPs.
Fig. 8.

Role of PKA anchoring in modulation of the expressed skeletal muscle Ca2+ channel. A peptide derived from a human thyroid PKA anchoring protein, Ht 31, was applied at 100 μm through the patch electrode to disrupt PKA anchoring.A, Tail currents measured in the presence of Ht 31 peptide in a tsA-201 cell expressing the skeletal muscle Ca2+ channel. Tail currents were recorded at −80 mV after 5–75 msec depolarizations to +80 mV in 5 msec increments. The smallest current in this series corresponds to the tail current from a 5 msec depolarization, whereas the largest current was measured after a 75 msec depolarization. This example is representative of five experiments. B, Tail currents from the expressed Ca2+ channel exposed to Ht 31 are shown before and after application of 25 μm cBIMPS. Tail currents followed 100 msec depolarizations to +60 mV and are traces from a single cell representative of eight similar experiments. C, Example of voltage-dependent potentiation of the expressed Ca2+channel in the presence of Ht 31, before and after application of cBIMPS. Currents shown were recorded during 300 msec depolarizations to 0 mV. The smallest current represents a control trace in which no conditioning pulse was applied, whereas the two larger currents were preceded by a 200 msec depolarization to +80 mV. A 20 msec recovery period at −60 mV separated the conditioning and test pulses. The largest current was recorded in the presence of 25 μmcBIMPS. D, Mean relative time-dependent increases in tail current decay time constant in the absence (circles, 23 cells) and presence of Ht 31 peptide either without (filled squares, 10 cells) or with 25 μm cBIMPS (diamonds, 5 cells) were recorded at −80 mV after 5–75 msec depolarizations to +80 mV in 5 msec increments. The control peptide Ht 31P was tested under identical conditions at a concentration of 500 μm (open squares, 7 cells). E, Model of PKA interaction with the skeletal muscle Ca2+ channel and modulation by Ht 31 and cBIMPS. Top, Rapid Ca2+ channel phosphorylation responsible for voltage- and time-dependent channel modulation requires PKA anchoring through association of the RII regulatory subunit and an AKAP. Bottom, Ht 31 peptide dissociates RII from the endogenous AKAP, reducing the local concentration of PKA. The cAMP analog cBIMPS dissociates the PKA catalytic subunit from RII and partially restores a high concentration of active catalytic subunits.
AKAPs require an amphipathic α helix to bind to Ca2+channels, and insertion of proline residues disrupts the α helix and prevents binding. To test the specificity of Ht 31, we analyzed the effects of Ht 31P, an identical peptide with proline residues substituted for isoleucines in positions 10 and 16 (Carr et al., 1992). Tail currents were slowed normally by prepulses of increasing length in the presence of 500 μm Ht 31P (Fig.8D). These results support the conclusion that the effect of the Ht 31 peptide in reducing potentiation of Ca2+ currents results from inhibition of AKAP interaction with PKA.
In contrast to experiments with PKI (e.g., Fig. 7C), cells containing Ht 31 showed normal stimulation by 25 μmcBIMPS (Fig. 8B). Application of cBIMPS to Ht 31-containing cells increased tail currents 3.4 ± 0.6-fold in eight cells, compared with 3.6 ± 0.5-fold in 25 control cells. cBIMPS also partially restored prepulse-dependent slowing of tail currents with a time course similar to that observed in control cells to which cBIMPS was applied (Fig. 8C,D, diamonds). Unlike direct inhibition of PKA catalytic subunits with PKI, Ht 31 dissociates RII from its membrane- or channel-associated AKAP, effectively reducing the local concentration of PKA near the channel. cBIMPS application evidently dissociates catalytic subunits from RII subunits throughout the cell and restores the local concentration of activated PKA near the Ca2+ channel. These experiments support the specificity of the effects of Ht 31 because they can be overcome by general activation of PKA.
Our experiments indicate that the physical proximity between PKA and Ca2+ channels found in skeletal muscle cells also occurs when the skeletal muscle Ca2+ channel is expressed in a heterologous cell type and that this localization requires an endogenous AKAP (Fig. 8E, top). When PKA is generally activated, the requirement for AKAP binding is overcome by the high level of free activated PKA, and Ca2+ channel regulation is restored (Fig. 8E, bottom). It is surprising that this result is observed in a heterologous cell type that does not usually express Ca2+ channels. Evidently, the interactions necessary for AKAP localization of PKA near Ca2+ channels are present in nonexcitable cells, which do not express Ca2+ channels.
DISCUSSION
Functional expression of the skeletal muscle L-type Ca2+ channel
Unlike the other cloned Ca2+ channel α1subunits, the α1 subunit of the skeletal muscle channel expresses poorly in Xenopus oocytes, and only small Ca2+ or Ba2+ currents are observed infrequently in stably transfected mammalian cell lines (Perez-Reyes et al., 1989;Lacerda et al., 1991; Varadi et al., 1991). We demonstrate here that transient expression of cDNA encoding the α1 subunit of the skeletal muscle Ca2+ channel with α2δ and β subunits in tsA-201 cells consistently yields voltage-gated Ba2+ currents with a tail current density approaching that recorded from a skeletal muscle cell line. This is consistent with the high level of dihydropyridine binding to tsA-201 cells expressing skeletal muscle Ca2+ channels (Peterson and Catterall, 1995). This expression system gives Ca2+ channel currents that are similar in time course (Fig. 1A–C) and voltage dependence (Fig. 1D) to those reported previously for acutely isolated preparations of skeletal muscle cells (Beam and Knudson, 1988; Gonoi and Hasegawa, 1988; Dirksen and Beam, 1995), for expression of cloned Ca2+ channels in dysgenic myotubes (Tanabe et al., 1988), and for stable expression in mammalian cell lines (Perez-Reyes et al., 1989; Lacerda et al., 1991; Varadi et al., 1991). The voltage dependence of activation of the expressed Ca2+ channel was 10–15 mV more positive than native mouse skeletal muscle Ca2+ channels in 129CB3 cells or native rabbit skeletal muscle Ca2+ channel (Johnson et al., 1994), and the time course of activation was approximately threefold slower when measured in the presence of Bay K 8644 (Fig. 1). The source of these differences is unknown but likely possibilities include cell type-specific differences in post-translational modification, differences in modulation by phosphorylation or G-proteins, and the absence of interacting muscle-specific proteins.
Ca2+ channels expressed in tsA-201 cells also respond normally to dihydropyridine Ca2+ channel modulators. Dihydropyridine binding to skeletal muscle Ca2+ channels expressed in these cells has normal affinity, is enhanced by Ca2+ similarly to native skeletal muscle Ca2+channels, and is inhibited by competition with the dihydropyridine agonist Bay K 8644 (Peterson and Catterall, 1995; Peterson et al., 1996). Our results show that Bay K 8644 increased channel activity three- to sixfold, slowed deactivation six- to sevenfold, and shifted channel voltage dependence ∼15 mV toward more negative potentials for both native and expressed channels. Bay K 8644 also induced voltage-dependent slowing of tail current that was resistant to inhibition of PKA activity by PKI or Ht 31 peptides (Figs.7E, 8D). Such voltage-dependent modulation of channels in the presence of Bay K 8644 has been described previously and is thought to arise from dihydropyridine interaction with multiple channel open states (Nakayama and Brading, 1995; Fass and Levitan, 1996). Although Bay K 8644 has strong effects, Ca2+ channel activity is not stimulated and tail current deactivation is not slowed to an extent that occludes further increases caused by phosphorylation by PKA or potentiation by prepulses (Fig. 4).
Effect of PKA phosphorylation on the cloned skeletal muscle L-type Ca2+ channel
Compared with the cardiac Ca2+ channel (McDonald et al., 1994), relatively little is known about the effects of phosphorylation on skeletal muscle Ca2+ channel function. When the purified skeletal muscle channel is reconstituted into lipid vesicles, phosphorylation by PKA increases channel activity two- to eightfold as measured by single-channel recording, Ca2+indicators, or 45Ca2+ influx (Curtis and Catterall, 1986; Flockerzi et al., 1986; Glossmann et al., 1987; Nunoki et al., 1989; Pelzer et al., 1989; Chang et al., 1991;Mundiña-Weilenmann et al., 1991a,b).45Ca2+ influx into cultured rat skeletal muscle cells is increased by PKA-stimulating agents, including the β-adrenergic agonist isoproterenol and dibutyryl cAMP (Schmid et al., 1985), and Ca2+ channel current in frog skeletal muscle is increased 50–70% on application of adrenaline, cAMP analogs, or activated PKA (Arreola et al., 1987; Kokate et al., 1993). Our results show that PKA stimulation by cBIMPS increases current through both native and expressed skeletal muscle Ca2+ channels two- to fourfold (Fig. 5) and increases the amplitude of tail currents by a similar factor (Fig. 6A,B). PKA stimulation slowed channel deactivation of both native and expressed channels (Fig.6A,C). Inhibition of basal PKA activity by PKI decreased absolute tail current amplitudes and inhibited depolarization-induced slowing of tail current decay for both native and expressed channels (Fig. 7). Thus, basal activity of PKA in skeletal muscle cells and in tsA-201 cells modulates Ca2+channels as it does in other cells (Armstrong and Eckert, 1987; Lory and Nargeot, 1992; Wang et al., 1993; Allen and Chapman, 1995), and this basal activity is sufficient to slow deactivation of channels expressed in tsA-201 cells; however, this basal PKA activity is far from maximal, because PKA stimulation with cBIMPS causes large increases in current. Thus, in both skeletal muscle cells and in tsA-201 cells, the unstimulated level of PKA activity affects both prepulse-dependent potentiation and the overall level of Ca2+ channel current such that bidirectional modulation of Ca2+ channels by increases or decreases in PKA activity is possible.
Voltage-dependent potentiation of the cloned skeletal muscle Ca2+ channel
Activation of Ca2+ channels in skeletal muscle cells is greatly enhanced after trains of short depolarizations or single longer depolarizations (Sculptoreanu et al., 1993b). Similar strong depolarizations increased the amplitude of tail currents and slowed their decay severalfold for both native (Sculptoreanu et al., 1993b;Fleig and Penner, 1995) and expressed channels (Fig. 3). Both prepulse-dependent potentiation (Sculptoreanu et al., 1993b) and slowing of the tail currents were increased and accelerated by activation of PKA (Fig. 6) and reduced by its inhibition (Fig. 7). These results are consistent with a model (Sculptoreanu et al., 1993b) in which phosphorylation by PKA during the depolarizing prepulses causes slowed decay of tail currents. The prepulse-dependent slowing of channel deactivation is likely to be the basis for prepulse and frequency-dependent potentiation of skeletal muscle Ca2+channel current attributable to PKA phosphorylation (Sculptoreanu et al., 1993b; Johnson et al., 1994). Similarly, incomplete deactivation is thought to underlie prepulse-dependent speeding of activation in frog muscle (Feldmeyer et al., 1990, 1992; Garcia et al., 1990). Potentiation of L-type Ca2+ channels and slowing of their deactivation would increase Ca2+ influx during tetanic stimulation of muscle, augment intracellular Ca2+ (Arreola et al., 1987; Oz and Frank, 1991; Kokate et al., 1993; Sculptoreanu et al., 1993b), and contribute to the increase in Ca2+ release that is observed after β-adrenergic stimulation.
Slowing of tail currents after depolarization in L-type Ca2+ channels has been attributed to reopening of inactivated channels on repolarization (Slesinger and Lansman, 1991), opening of new channels on repolarization (Artalejo et al., 1991; Fleig and Penner, 1995), or mode-2 gating characterized by enhanced opening and slower closing (Pietrobon and Hess, 1990; Nakayama and Brading, 1993). The latter two schemes predict a single, well defined second time constant in exponential fits to the decay of the tail currents. Attempts to fit our tail currents after pulses of increasing duration (Fig. 3A,B) with sums of two exponentials of fixed time constant did not reveal evidence for a second decay time constant. This is especially clear from tail currents of intermediate size where the fit to a single exponential is exact. Thus, adequate definition of the kinetic transitions underlying tail current slowing will require further study. Whatever its kinetic basis, our data demonstrate that heterologous expression of the skeletal muscle Ca2+channel α1s subunit yields channels whose deactivation tail currents are substantially slowed after strong depolarizations.
Molecular determinants of Ca2+ channel modulation by phosphorylation
Prepulse-dependent modulation of tail current decay kinetics occurs at basal levels of PKA activity (Figs. 6C,7E), whereas large increases in Ca2+ current amplitude (Figs. 5, 6B, 8B) require activation of PKA with cBIMPS. Thus, these two aspects of Ca2+ channel function may be regulated by phosphorylation of distinct sites. At least four serines in the α1 subunit of purified skeletal muscle Ca2+ channels are phosphorylated by PKA under various conditions (O’Callahan and Hosey, 1988; Röhrkasten et al., 1988; Lai et al., 1990; Rotman et al., 1992, 1995). In addition, modulation of skeletal muscle Ca2+ channels may involve protein kinases in addition to PKA (Ma et al., 1992; Gutiérrez et al., 1994). The reconstitution of the major features of PKA modulation in a heterologous expression system described in this paper provides a tool for studying the molecular basis of skeletal muscle Ca2+ channel modulation by PKA and other kinases.
Role of PKA anchoring in Ca2+ channel modulation by phosphorylation
A surprising result of this study was that kinase anchoring near the Ca2+ channel, found previously to be critical for voltage-dependent channel potentiation in skeletal muscle cells, was also reconstituted in this expression system (Fig. 8). PKA is specifically localized to different cellular compartments through RII regulatory subunit binding to AKAPs (Scott and McCartney, 1994). Application of a peptide, Ht 31, which disrupts AKAP-RII binding (Carr et al., 1992), was found in earlier experiments to reduce the activity of glutamate receptors in hippocampal neurons (Rosenmund et al., 1994) and to disrupt voltage-dependent potentiation of the skeletal muscle Ca2+ channel in primary skeletal muscle cells and in the 129CB3 cell line used here (Johnson et al., 1994). When the Ht 31 peptide was applied to the cloned skeletal muscle Ca2+channel transiently expressed in tsA-201 cells, time- and voltage-dependent slowing of tail current deactivation was reduced to a level not significantly different from that measured in PKI. PKA stimulation by cBIMPS still increased channel activity and partially restored voltage-dependent potentiation (Fig. 8B–D), indicating that Ht 31 reduces the local PKA concentration near the channel but does not inhibit PKA activity directly. Future experiments will determine whether PKA is specifically targeted to the Ca2+ channel itself, implying that the Ca2+channel is an AKAP or an AKAP binding protein, or is simply concentrated at the plasma membrane by AKAPs.
Footnotes
This work was supported in part by a research grant from the Muscular Dystrophy Association to W.A.C. and by a postdoctoral research fellowship from the Muscular Dystrophy Association to B.D.J. We thank Drs. K. Campbell, S. Ellis, M. Harpold, A. Schwartz, and T. Snutch for cDNA clones and Drs. M. Pinçon-Raymond and F. Rieger for the 129CB3 cell line.
Correspondence should be addressed to Dr. William A. Catterall, Department of Pharmacology, Box 357280, University of Washington, Seattle, WA 98195-7280.
REFERENCES
- 1.Allen TJA, Chapman RA. The effect of a chemical phosphatase on single Ca2+ channels and the inactivation of whole-cell Ca2+ current from isolated guinea-pig ventricular myocytes. Pflügers Arch. 1995;430:68–80. doi: 10.1007/BF00373841. [DOI] [PubMed] [Google Scholar]
- 2.Armstrong D, Eckert R. Voltage-activated Ca2+ channels that must be phosphorylated to respond to membrane depolarization. Proc Natl Acad Sci USA. 1987;84:2518–2522. doi: 10.1073/pnas.84.8.2518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arreola J, Calvo J, García MC, Sánchez JA. Modulation of Ca2+ channels of twitch skeletal muscle fibres of the frog by adrenaline and cyclic adenosine monophosphate. J Physiol (Lond) 1987;393:307–330. doi: 10.1113/jphysiol.1987.sp016825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Artalejo CR, Dahmer MK, Perlman RL, Fox AP. Facilitation of Ca2+ current in bovine chromaffin cells is due to recruitment of a second type of whole-cell current with novel properties. J Physiol (Lond) 1991;432:681–707. doi: 10.1113/jphysiol.1991.sp018406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beam KG, Knudson M. Calcium currents in embryonic and neonatal mammalian skeletal muscle. J Gen Physiol. 1988;91:781–789. doi: 10.1085/jgp.91.6.781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bourinet E, Charnet P, Tomlinson WJ, Stea A, Snutch TP, Nargeot J. Voltage-dependent facilitation of a neuronal α1C L-type Ca2+ channel. EMBO J. 1994;13:5032–5039. doi: 10.1002/j.1460-2075.1994.tb06832.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carr DW, Hausken ZE, Fraser IDC, Stofko-Hahn RE, Scott JD. Association of the type-II cAMP-dependent protein kinase with a human thyroid RII-anchoring protein: cloning and characterization of the RII-binding domain. J Biol Chem. 1992;267:13376–13382. [PubMed] [Google Scholar]
- 8.Catterall WA. Structure and function of voltage-gated ion channels. Annu Rev Biochem. 1995;64:493–531. doi: 10.1146/annurev.bi.64.070195.002425. [DOI] [PubMed] [Google Scholar]
- 9.Chandler WK, Schneider MF, Rakowski RF. Effects of glycerol treatment and maintained depolarization on charge movement in muscle. J Physiol (Lond) 1976;254:285–316. doi: 10.1113/jphysiol.1976.sp011233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chang CF, Gutiérrez LM, Mundiña-Weilenmann C, Hosey M. Dihydropyridine-sensitive Ca2+ channels from skeletal muscle. II. Functional effects of differential phosphorylation of channel subunits. J Biol Chem. 1991;266:16395–16400. [PubMed] [Google Scholar]
- 11.Curtis BM, Catterall WA. Phosphorylation of the Ca2+ antagonist receptor of the voltage-sensitive Ca2+ channel by cAMP-dependent protein kinase. Proc Natl Acad Sci USA. 1985;82:2528–2532. doi: 10.1073/pnas.82.8.2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Curtis BM, Catterall WA. Reconstitution of the voltage-gated Ca2+ channel purified from skeletal muscle transverse tubules. Biochemistry. 1986;23:2113–2118. doi: 10.1021/bi00359a002. [DOI] [PubMed] [Google Scholar]
- 13.Dirksen RT, Beam KG. Single Ca2+ channel behavior in native skeletal muscle. J Gen Physiol. 1995;105:227–247. doi: 10.1085/jgp.105.2.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ellis SB, Williams ME, Ways NR, Brenner R, Sharp AH, Leung AT, Campbell KP, Mckenna E, Koch WJ, Hui A, Schwartz A, Harpold MM. Sequence and expression of mRNAs encoding the α1 and α2 subunits of a DHP-sensitive Ca2+ channel. Science. 1988;241:1661–1664. doi: 10.1126/science.2458626. [DOI] [PubMed] [Google Scholar]
- 15.Fass DM, Levitan ES. L-type Ca2+ channels access multiple open states to produce two components of Bay K 8644-dependent current in GH3 cells. J Gen Physiol. 1996;108:13–26. doi: 10.1085/jgp.108.1.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Feldmeyer D, Melzer W, Pohl B, Zöllner P. Fast gating kinetics of the slow Ca2+ current in cut skeletal muscle fibres of the frog. J Physiol (Lond) 1990;425:347–367. doi: 10.1113/jphysiol.1990.sp018107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Feldmeyer D, Melzer W, Pohl B, Zöllner P. Modulation of Ca2+ current gating in frog skeletal muscle by conditioning depolarization. J Physiol (Lond) 1992;457:639–653. doi: 10.1113/jphysiol.1992.sp019399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fleig A, Penner R. Excessive repolarization-dependent Ca2+ currents induced by strong depolarizations in rat skeletal myoballs. J Physiol (Lond) 1995;489:44–53. doi: 10.1113/jphysiol.1995.sp021028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Flockerzi V, Oeken HJ, Hofmann F, Pelzer D, Cavalíe A, Trautwein W. Purified dihydropyridine-binding site from skeletal muscle T-tubules is a functional Ca2+ channel. Nature. 1986;323:66–68. doi: 10.1038/323066a0. [DOI] [PubMed] [Google Scholar]
- 20.Garcia J, Avila-Sakar AJ, Stefani E. Repetitive stimulation increases the activation rate of skeletal muscle Ca2+ currents. Pflügers Arch. 1990;416:210–212. doi: 10.1007/BF00370245. [DOI] [PubMed] [Google Scholar]
- 21.Glossmann H, Striessnig J, Hymel L, Schindler H. Purified L-type Ca2+ channels: only one single polypeptide (alpha 1-subunit) carries the drug receptor domains and is regulated by protein kinases. Biomed Biochim Acta. 1987;46:S351–356. [PubMed] [Google Scholar]
- 22.Gonoi T, Hasegawa S. Post-natal disappearance of transient Ca2+ channels in mouse skeletal muscle: effects of denervation and culture. J Physiol (Lond) 1988;401:617–637. doi: 10.1113/jphysiol.1988.sp017183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gutiérrez LM, Zhao X, Hosey M. Protein kinase C-mediated regulation of L-type Ca2+ channels from skeletal muscle requires phosphorylation of the α1 subunit. Biochem Biophys Res Commun. 1994;202:857–865. doi: 10.1006/bbrc.1994.2009. [DOI] [PubMed] [Google Scholar]
- 24.Hofmann F, Biel M, Flockerzi V. Molecular basis for Ca2+ channel diversity. Annu Rev Neurosci. 1994;17:399–418. doi: 10.1146/annurev.ne.17.030194.002151. [DOI] [PubMed] [Google Scholar]
- 25.Huerta M, Muniz J, Trujillo X, Lomeli J. Adrenergic modulation of the K+ contractures in tonic skeletal muscle fibers of the frog. Jpn J Physiol. 1991;41:851–860. doi: 10.2170/jjphysiol.41.851. [DOI] [PubMed] [Google Scholar]
- 26.Isom LL, De Jongh KS, Catterall WA. Auxiliary subunits of voltage-gated ion channels. Neuron. 1994;12:1183–1194. doi: 10.1016/0896-6273(94)90436-7. [DOI] [PubMed] [Google Scholar]
- 27.Johnson BD, Scheuer T, Catterall WA. Voltage-dependent potentiation of L-type Ca2+ channels in skeletal muscle cells requires anchored cAMP-dependent protein kinase. Proc Natl Acad Sci USA. 1994;91:11492–11496. doi: 10.1073/pnas.91.24.11492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kokate TG, Heiny JA, Sperelakis N. Stimulation of the slow Ca2+ current in bullfrog skeletal muscle fibers by cAMP and cGMP. Am J Physiol. 1993;265:C47–53. doi: 10.1152/ajpcell.1993.265.1.C47. [DOI] [PubMed] [Google Scholar]
- 29.Lacerda AE, Kim HS, Ruth P, Perez-Reyes E, Flockerzi V, Hofmann F, Birnbaumer L, Brown AM. Normalization of current kinetics by interaction between the alpha 1 and beta subunits of the skeletal muscle dihydropyridine-sensitive Ca2+ channel. Nature. 1991;352:527–530. doi: 10.1038/352527a0. [DOI] [PubMed] [Google Scholar]
- 30.Lai Y, Seagar MJ, Takahashi M, Catterall WA. Cyclic AMP-dependent phosphorylation of two size forms of alpha 1 subunits of L-type Ca2+ channels in rat skeletal muscle cells. J Biol Chem. 1990;265:20839–20848. [PubMed] [Google Scholar]
- 31.Lory P, Nargeot J. Cyclic AMP-dependent modulation of cardiac Ca2+ channels expressed in Xenopus laevis oocytes. Biochem Biophys Res Commun. 1992;182:1059–1065. doi: 10.1016/0006-291x(92)91839-i. [DOI] [PubMed] [Google Scholar]
- 32.Lu X, Xu L, Meissner G. Activation of the skeletal muscle Ca2+ release channel by a cytoplasmic loop of the dihydropyridine receptor. J Biol Chem. 1994;269:6511–6516. [PubMed] [Google Scholar]
- 33.Ma J, Gutiérrez LM, Hosey M, Rios E. Dihydropyridine-sensitive skeletal muscle Ca2+ channels in polarized planar bilayers. 3. Effects of phosphorylation by protein kinase C. Biophys J. 1992;63:639–647. doi: 10.1016/S0006-3495(92)81634-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Margolskee RF, McHendry-Rinde B, Horn R. Panning transfected cells for electrophysiological studies. Biotechniques. 1994;15:906–911. [PubMed] [Google Scholar]
- 35.McDonald TF, Pelzer S, Trautwein W, Pelzer DJ. Regulation and modulation of Ca2+ channels in cardiac, skeletal, and smooth muscle cells. Physiol Rev. 1994;74:365–507. doi: 10.1152/physrev.1994.74.2.365. [DOI] [PubMed] [Google Scholar]
- 36.Mundiña-Weilenmann C, Chang CF, Gutiérrez LM, Hosey MM. Demonstration of the phosphorylation of dihydropyridine-sensitive Ca2+ channels in chick skeletal muscle and the resultant activation of the channels after reconstitution. J Biol Chem. 1991a;266:4067–4073. [PubMed] [Google Scholar]
- 37.Mundiña-Weilenmann C, Ma J, Rios E, Hosey M. Dihydropyridine-sensitive skeletal muscle Ca2+ channels in polarized planar bilayers. 2. Effects of phosphorylation by cAMP-dependent protein kinase. Biophys J. 1991b;60:902–909. doi: 10.1016/S0006-3495(91)82124-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nakayama S, Brading AF. Evidence for multiple open states of the Ca2+ channel in smooth muscle cells isolated from the guinea-pig detrusor. J Physiol (Lond) 1993;471:87–105. doi: 10.1113/jphysiol.1993.sp019892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nakayama S, Brading AF. Interaction of Ca2+ agonist and depolarization on Ca2+ channel current in guinea-pig detrusor cells. J Gen Physiol. 1995;106:1211–1224. doi: 10.1085/jgp.106.6.1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nunoki K, Florio V, Catterall WA. Activation of purified Ca2+ channels by stoichiometric protein phosphorylation. Proc Natl Acad Sci USA. 1989;86:6816–6820. doi: 10.1073/pnas.86.17.6816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.O’Callahan CM, Hosey M. Multiple phosphorylation sites in the 165-kilodalton peptide associated with dihydropyridine-sensitive Ca2+ channels. Biochemistry. 1988;27:6071–6077. doi: 10.1021/bi00416a036. [DOI] [PubMed] [Google Scholar]
- 42.Oz M, Frank GB. Decrease in the size of tetanic responses produced by nitrendipine or by extracellular Ca2+ ion removal without blocking twitches or action potentials in skeletal muscle. J Pharmacol Exp Ther. 1991;257:575–581. [PubMed] [Google Scholar]
- 43.Pelzer D, Grant AO, Cavalie A, Pelzer S, Sieber M, Hofmann F, Trautwein W. Calcium channels reconstituted from the skeletal muscle dihydropyridine receptor protein complex and its alpha 1 peptide subunit in lipid bilayers. Ann NY Acad Sci. 1989;560:138–154. doi: 10.1111/j.1749-6632.1989.tb24091.x. [DOI] [PubMed] [Google Scholar]
- 44.Perez-Reyes E, Kim HS, Lacerda AE, Horne W, Wei XY, Rampe D, Campbell KP, Brown AM, Birnbaumer L. Induction of Ca2+ currents by the expression of the alpha 1 subunit of the dihydropyridine receptor from skeletal muscle. Nature. 1989;340:233–236. doi: 10.1038/340233a0. [DOI] [PubMed] [Google Scholar]
- 45.Peterson BZ, Catterall WA. Calcium binding in the pore of L-type Ca2+ channels modulates high affinity dihydropyridine binding. J Biol Chem. 1995;270:18201–18204. doi: 10.1074/jbc.270.31.18201. [DOI] [PubMed] [Google Scholar]
- 46.Peterson BZ, Tanada T, Catterall WA. Molecular determinants of high affinity dihydropyridine binding in L-type Ca2+ channels. J Biol Chem. 1996;271:5293–5296. doi: 10.1074/jbc.271.10.5293. [DOI] [PubMed] [Google Scholar]
- 47.Pietrobon D, Hess P. Novel mechanism of voltage dependent gating in L-type Ca2+ channels. Nature. 1990;346:651–655. doi: 10.1038/346651a0. [DOI] [PubMed] [Google Scholar]
- 48.Pinçon-Raymond M, Vicar P, Bois P, Chassande O, Romey G, Varadi G, Li ZL, Lazdunski M, Rieger F, Paulin D. Conditional immortalization of normal and dysgenic mouse muscle cells by the SV40 large T antigen under the vimentin promoter control. Dev Biol. 1991;148:517–528. doi: 10.1016/0012-1606(91)90270-d. [DOI] [PubMed] [Google Scholar]
- 49.Pragnell M, Sakamoto J, Jay SD, Campbell KP. Cloning and tissue-specific expression of the brain Ca2+ channel β-subunit. FEBS Lett. 1991;291:253–258. doi: 10.1016/0014-5793(91)81296-k. [DOI] [PubMed] [Google Scholar]
- 50.Ríos E, Ma J, González A. The mechanical hypothesis of excitation-contraction (EC) coupling in skeletal muscle. J Muscle Res Cell Motil. 1991;12:127–135. doi: 10.1007/BF01774031. [DOI] [PubMed] [Google Scholar]
- 51.Röhrkasten A, Meyer HE, Nastainczyk W, Sieber M, Hofmann F. cAMP-dependent protein kinase rapidly phosphorylates serine-687 of the skeletal muscle receptor for Ca2+ channel blockers. J Biol Chem. 1988;263:15325–15329. [PubMed] [Google Scholar]
- 52.Rosenmund C, Carr DW, Bergeson SE, Nilaver G, Scott JD, Westbrook GL. Anchoring of protein kinase A is required for modulation of AMPA/kainate receptors on hippocampal neurons. Nature. 1994;368:853–856. doi: 10.1038/368853a0. [DOI] [PubMed] [Google Scholar]
- 53.Rotman EI, De Jongh KS, Florio V, Lai Y, Catterall WA. Specific phosphorylation of a COOH-terminal site of the full length form of the alpha-1 subunit of the skeletal muscle Ca2+ channel by cAMP-dependent protein kinase. J Biol Chem. 1992;267:16100–16105. [PubMed] [Google Scholar]
- 54.Rotman EI, Murphy BJ, Catterall WA. Sites of selective cAMP-dependent phosphorylation of the L-type Ca2+ channel α1 subunit from intact rabbit skeletal muscle myotubes. J Biol Chem. 1995;270:16371–16377. doi: 10.1074/jbc.270.27.16371. [DOI] [PubMed] [Google Scholar]
- 55.Sandberg M, Butt E, Nolte C, Fischer L, Halbrugge M, Beltman J, Jahnsen T, Genieser HG, Jastorff B, Walter U. Characterization of Sp-5,6-dichloro-1-beta-d-ribofuranosylbenzimidazole-3′,5′-monophosphorothioate (Sp-5,6-DCl-cBiMPS) as a potent and specific activator of cyclic-AMP-dependent protein kinase in cell extracts and intact cells. Biochem J. 1991;279:521–527. doi: 10.1042/bj2790521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schmid A, Renaud JF, Lazdunski M. Short term and long term effects of beta-adrenergic effectors and cyclic AMP on nitrendipine-sensitive voltage-dependent Ca2+ channels of skeletal muscle. J Biol Chem. 1985;260:13041–13046. [PubMed] [Google Scholar]
- 57.Scott JD, McCartney S. Localization of A-kinase through anchoring proteins. Mol Endocrinol. 1994;8:5–11. doi: 10.1210/mend.8.1.8152430. [DOI] [PubMed] [Google Scholar]
- 58.Sculptoreanu A, Rotman E, Takahashi M, Scheuer T, Catterall WA. Voltage-dependent potentiation of the activity of cardiac L-type Ca2+ channel alpha-1 subunits due to phosphorylation by cAMP-dependent protein kinase. Proc Natl Acad Sci USA. 1993a;90:10135–10139. doi: 10.1073/pnas.90.21.10135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sculptoreanu A, Scheuer T, Catterall WA. Voltage-dependent potentiation of L-type Ca2+ channels due to phosphorylation by cAMP-dependent protein kinase. Nature. 1993b;364:240–243. doi: 10.1038/364240a0. [DOI] [PubMed] [Google Scholar]
- 60.Slesinger PA, Lansman JB. Reopening of Ca2+ channels in mouse cerebellar neurons at resting membrane potentials during recovery from inactivation. Neuron. 1991;7:755–762. doi: 10.1016/0896-6273(91)90278-8. [DOI] [PubMed] [Google Scholar]
- 61.Tanabe T, Takeshima H, Mikami A, Flockerzi V, Takahashi H, Kangawa K, Kojima M, Matsuo H, Hirose T, Numa S. Primary structure of the receptor for Ca2+ channel blockers from skeletal muscle. Nature. 1987;328:313–318. doi: 10.1038/328313a0. [DOI] [PubMed] [Google Scholar]
- 62.Tanabe T, Beam KG, Powell JA, Numa S. Restoration of excitation-contraction coupling and slow Ca2+ current in dysgenic muscle by dihydropyridine receptor complementary DNA. Nature. 1988;336:134–139. doi: 10.1038/336134a0. [DOI] [PubMed] [Google Scholar]
- 63.Tanabe T, Mikami A, Numa S, Beam KG. Cardiac-type excitation-contraction coupling in dysgenic skeletal muscle injected with cardiac dihydropyridine receptor cDNA. Nature. 1990;244:451–453. doi: 10.1038/344451a0. [DOI] [PubMed] [Google Scholar]
- 64.Varadi G, Lory P, Schultz D, Varadi M, Schwartz A. Acceleration of activation and inactivation by the beta subunit of the skeletal muscle Ca2+ channel. Nature. 1991;352:159–162. doi: 10.1038/352159a0. [DOI] [PubMed] [Google Scholar]
- 65.Wang Y, Townsend C, Rosenberg RL. Regulation of cardiac L-type Ca2+ channels in planar lipid bilayers by G proteins and protein phosphorylation. Am J Physiol. 1993;264:C1473–1479. doi: 10.1152/ajpcell.1993.264.6.C1473. [DOI] [PubMed] [Google Scholar]
- 66.West JW, Scheuer T, Maechler L, Catterall WA. Efficient expression of rat brain type IIA Na+ channel alpha subunits in a somatic cell line. Neuron. 1992;8:59–70. doi: 10.1016/0896-6273(92)90108-p. [DOI] [PubMed] [Google Scholar]