

Abstract
We present evidence that the electrical excitability of the terminals of a group of spinal premotor interneurons can be increased after stimulation of sensory afferents. The interneurons were located in the midlumbar segments of the spinal cord and had projections to the lower lumbar motor nuclei. Thresholds for antidromic activation of a substantial number of interneurons were reduced after electrical stimulation of group II muscle afferents. Several observations suggest that the excitability changes are unlikely to have arisen from electrotonic spread of depolarization from the interneuron soma to its terminals or by environmental changes in the vicinity of the terminals related to neuronal activity. A particularly interesting possibility is that the excitability of the central terminals of the interneurons is increased because they are depolarized by a mechanism similar to that acting at the terminals of primary sensory afferents (primary afferent depolarization, PAD), which accompanies one type of presynaptic inhibition. This type of presynaptic action has been shown in premotor interneurons in the lamprey but not in the mammalian spinal cord. From our observations the organization of the systems generating excitability changes at the interneuron terminals seem in general to parallel the organization of the systems generating PAD at afferent terminals, raising the possibility that common principles might underlie the operation of this form of presynaptic control.
Keywords: spinal cord, interneuron, presynaptic inhibition, primary afferent depolarization, muscle afferent, terminal excitability
One of the earliest electrical phenomena described in the CNS was the depolarization in dorsal roots on stimulation of sensory afferents (Barron and Matthews, 1938). Primary afferent depolarization (PAD) has subsequently been shown to be associated with one form of presynaptic inhibition of the terminals of the afferents, mediated by axo-axonic synapses (Eccles et al., 1961; Rudomin, 1990). This mechanism is highly conserved across different phyla. For example, depolarization of proprioceptive afferent terminals has recently been described in crustaceans (Cattaert et al., 1992) and insects (Burrows and Laurent, 1993) as well as in mammals. There is indirect evidence that a similar phenomenon may also occur in man (Hultborn et al., 1987; Rudomin et al., 1991). Precisely how this leads to presynaptic inhibition is a matter of debate. It has been established that PAD in mammals is generated by the activation of GABAA receptors (Rudomin et al., 1981; Curtis and Lodge, 1982) and involves the opening of chloride channels in the terminal. This increases membrane conductance, and as a result action potentials invading the terminal may be “shunted,” reducing the depolarization they produce at the terminal and thus transmitter release. Alternatively, the conductance change may be insufficient to affect spike invasion, but the depolarization might be important, for example, by inactivating voltage-gated calcium channels in the terminal (Graham and Redman, 1994; Zhang and Jackson, 1995). Activation of GABAAreceptors might also lead to branch point failure in the preterminal axon (Wall, 1994).
Recent evidence indicated that this same form of presynaptic depolarization occurs at the terminals of central neurons as well as primary afferents, at the terminals of premotor interneurons in the lamprey spinal cord (Alford et al., 1991), and at neurosecretory terminals of the rat posterior pituitary (Zhang and Jackson, 1995). Detailed searches for such a phenomenon have failed to reveal an action at the terminals of descending fibers in the mammalian spinal cord (Rudomin et al., 1981; Curtis and Malik, 1984; Curtis et al., 1984).
One of the most widely used techniques to detect PAD is the excitability testing method (Wall, 1958; Curtis and Lodge, 1982,Rudomin, 1990). This relies on an increased susceptibility of terminal axon branches to activation by externally applied electrical stimuli when the terminal is depolarized. In this report we provide evidence that the excitability of the terminals of a group of premotor interneurons in the mammalian spinal cord can be increased by conditioning stimuli. The interneurons are located in the midlumbar segments of the cat spinal cord. Neurons in these segments include excitatory and inhibitory premotor interneurons with projections to hindlimb motoneurons in the L7–S1 segments (Cavallari et al., 1987). Many have a characteristic monosynaptic input from group II muscle afferents (Edgley and Jankowska, 1987b). One possible explanation of these excitability changes would be that a terminal depolarization, like that occurring in PAD, occurs in the interneuron terminals within the motor nuclei.
A preliminary report has been published previously (Aggelopoulos et al., 1995).
MATERIALS AND METHODS
The experiments were performed on 11 adult cats. Anesthesia was induced with an intramuscular injection of ketamine and xylazine (25 and 1 mg/kg, respectively). Subsequently anesthesia was maintained either with halothane in an O2/N2O mixture during surgery and chloralose after surgery (two experiments), with intravenous doses of sodium pentobarbitone (2–3 mg/kg) during surgery followed by intravenous α-chloralose after surgery (four experiments), or with intravenous doses of sodium pentobarbitone throughout (five experiments). In cats in which sodium pentobarbitone was used to maintain anesthesia, dose rates ranged from 2.5 to 5.2 mg · /kg−1 · hr−1. In cats in which chloralose was given, dose rates ranged from 3.5 to 6.4 mg · /kg−1 · hr−1. The rates used were sufficient to maintain full general anesthesia. Arterial and venous cannulae were inserted for administration of fluids and blood pressure monitoring. A number of hindlimb nerves were exposed and dissected free for stimulation. Branches of the femoral nerve [quadriceps (including rectus femoris, Q) and sartorius (including medial and lateral branches, SART)] were mounted in tunnel electrodes. Branches of the sciatic nerve [posterior biceps and semitendinosus (PBST)], anterior biceps and semimembranosus (SMAB), gastrocnemius-soleus (GS), the nerves to tibialis anterior and extensor digitorum longus (TA-EDL), and in some experiments the superficial peroneal (SP), and lateral and caudal cutaneous sural nerves (SUR) were also transected and ligated for mounting on silver hook electrodes. The spinal cord was exposed from L3 to the cauda equina by laminectomy. The spinal cord and dissected nerves were immersed in a mineral oil pool maintained at 37.5°C.
During recording the animals were paralyzed with pancuronium (doses of 1–2 mg/kg, i.v.) and artificially ventilated, and a bilateral pneumothorax was made to minimize thoracic movements. We ensured that the anesthesia was adequate during paralysis by checking at regular intervals that neither heart rate nor blood pressure were altered in response to noxious stimuli and that the pupils were not dilated. At the end of the experiment, a lethal dose of barbiturate was given intravenously.
Recording. Extracellular and occasionally intra-axonal or intracellular records were made from single interneurons in the midlumbar segments (L4 and rostral L5) using glass electrodes. We searched for neurons lying close to the large extracellular fields generated by stimulation of group II afferents in the quadriceps nerve in the intermediate zone of the gray matter (Edgley and Jankowska, 1987b). These were evoked by stimulation of the quadriceps nerve at 5× threshold for the most excitable fibers (5T). Actions were defined as originating from group I afferents if they appeared with stimuli of <1.5T where the group I volley was maximal, and they were attributed to group II afferents if they appeared with stimuli >2T (Edgley and Jankowska, 1987a; Lundberg et al., 1987). Neurons with descending projections to the region of the GS and/or PBST motor nuclei located ∼25 mm further caudally in caudal L7/rostral S1 were identified by antidromic activation after stimuli (0.2 msec square pulses) delivered through a fine epoxylite-insulated tungsten microelectrode (125 μm shank diameter, tips exposed to ∼10 μm, impedances 100–300 kΩ). This electrode was positioned in the motor nuclei at the beginning of the recording session, guided by antidromic field potentials evoked by stimulation of muscle nerves (GS and PBST). Currents of up to 50 μA were used as search stimuli. Wherever possible, antidromic activation was verified with collision, i.e., spikes evoked from the motor nucleus were abolished if spontaneous or orthodromically evoked spikes occurred with a delay of less than the response latency from the motor nucleus stimulus. However, very few of the interneurons discharged spontaneously, and many discharged rarely or not at all orthodromically. The short antidromic latencies made also collision difficult. We therefore took cells to be antidromically activated if they were activated with fixed latency and followed a train of stimuli (three to four stimuli at 333 Hz or above) (Lipski, 1982).
When an antidromically driven interneuron was found, the point of lowest threshold in the motor nuclei for antidromic activation was determined by moving this electrode and the stimulus current set just subthreshold. These currents ranged from 4 to 60 μA, the large majority being <20 μA. We took care to ensure that “anodic block” was not responsible for failure to antidromically activate the neurons (Curtis and Lodge, 1982). In practice we rarely encountered this problem, probably because we used tungsten-stimulating electrodes with larger tips and lower tip current densities than the glass electrodes used by Curtis and Lodge (1982). Conditioning stimuli to various hindlimb nerves were then delivered at various times before the test stimulus. Excitability of the cells was quantified in terms of the firing index, the probability of a response occurring. With no response, the firing index is 0; with a response on every stimulus delivery, the firing index is 1. Changes in firing index with conditioning stimulation were measured (Rudomin and Jankowska, 1981). Repetitive testing with interspersed blocks of conditioned and unconditioned stimuli was carried out in all cases to ensure that the excitability changes were reproducible. We considered excitability to have increased only if the firing index was reliably increased by >0.1 in multiple tests. To estimate central latencies and to set the strength of conditioning stimuli, we placed a silver ball electrode on the cord dorsum at the L6–L7 junction.
RESULTS
Reliable excitability changes have been observed in 70 midlumbar interneurons. These were drawn from a large sample of interneurons tested (>120). Our approach, however, cannot reveal whether this reflects the true frequency of occurrence of this phenomenon. All of the neurons were antidromically activated from the lower lumbar motor nuclei and were located in the intermediate zone of segments L4 or rostral L5. Antidromic latencies ranged from 0.5 to 2.2 msec, with a mean of 0.96 msec. This mean is skewed by a small number of longer values (only two neurons had antidromic latencies >1.5 msec). The median value was 0.86 msec. Allowing 0.1 msec for utilization time, we may estimate that the conduction velocities of the interneuron axons range from 12 to 62 m/sec, with a mean of 29 m/sec.
Our basic observation is that stimuli that were subthreshold for antidromically activating midlumbar neurons when delivered alone were able to elicit antidromic spikes when delivered after a conditioning stimulus (Rudomin and Jankowska, 1981). An illustration of this phenomenon from an intra-axonal recording is shown in Figure1. In this case a stimulus of 11 μA delivered in the motor nuclei was subthreshold (Fig. 1, top), although increasing the intensity of this stimulus to 12 μA or greater did produce antidromic activation (not shown). The same stimulus was suprathreshold when preceded 35 msec earlier by a single stimulus to the Q nerve (5T) delivered (Fig. 1, bottom). Antidromic responses to stimulation in the motor nuclei after the conditioning stimulus are shown on a faster timebase in the inset. In this case the conditioning stimulus orthodromically activated the interneuron, driving one or two spikes at 4–10 msec after the stimulus (central latency 3–9 msec). In most cases the changes in excitability were substantial, as in the case of Figure 1. With single conditioning, stimuli to the firing index of Q group II afferents was increased from near zero to >0.8 in 38 of 60 (63%) tested interneurons. In all interneurons the terminal excitability could be increased by a single conditioning stimulus. Failure of the unconditioned stimulus to antidromically activate the cell was not a problem of invasion of the soma: in every case, higher stimulus intensities reliably produced antidromic activation.
Fig. 1.

Changes in threshold for antidromic activation after conditioning afferent stimulation. The recordings shown are intra-axonal records (three traces superimposed) from a midlumbar interneuron. In the top traces, stimuli of 11 μA delivered to the lower lumbar motor nuclei (MN) were subthreshold for antidromic activation. The same stimuli evoke antidromic spikes when preceded, 35 msec earlier, by a single stimulus to the quadriceps nerve at 5× threshold (Q 5T). This conditioning stimulus generates one to two orthodromic spikes at short latency. The inset shows the antidromic spikes on a faster timebase, where they can be distinguished from the stimulus artifact.
These excitability changes were not a simple expression of the supernormal period that follows impulse conduction along an axon (Swadlow et al., 1980), because many of the interneurons in which terminal excitability was increased were not orthodromically discharged by the conditioning stimulus (e.g., Figs. 2, 3). The most frequently tested nerve was Q: 20 of the 60 interneurons in which the terminal excitability was increased were never orthodromically activated by the conditioning stimulus. This was also the case for other nerves: 14 of 29 neurons in which terminal excitability was increased by conditioning stimulation of SART and 15 of 19 neurons conditioned by stimulation of TA-EDL were never orthodromically driven by conditioning stimuli, which did increase terminal excitability. Furthermore, similar conditioning stimuli sometimes evoked substantial orthodromic discharges, which were not accompanied by excitability changes at the terminals.
Fig. 2.
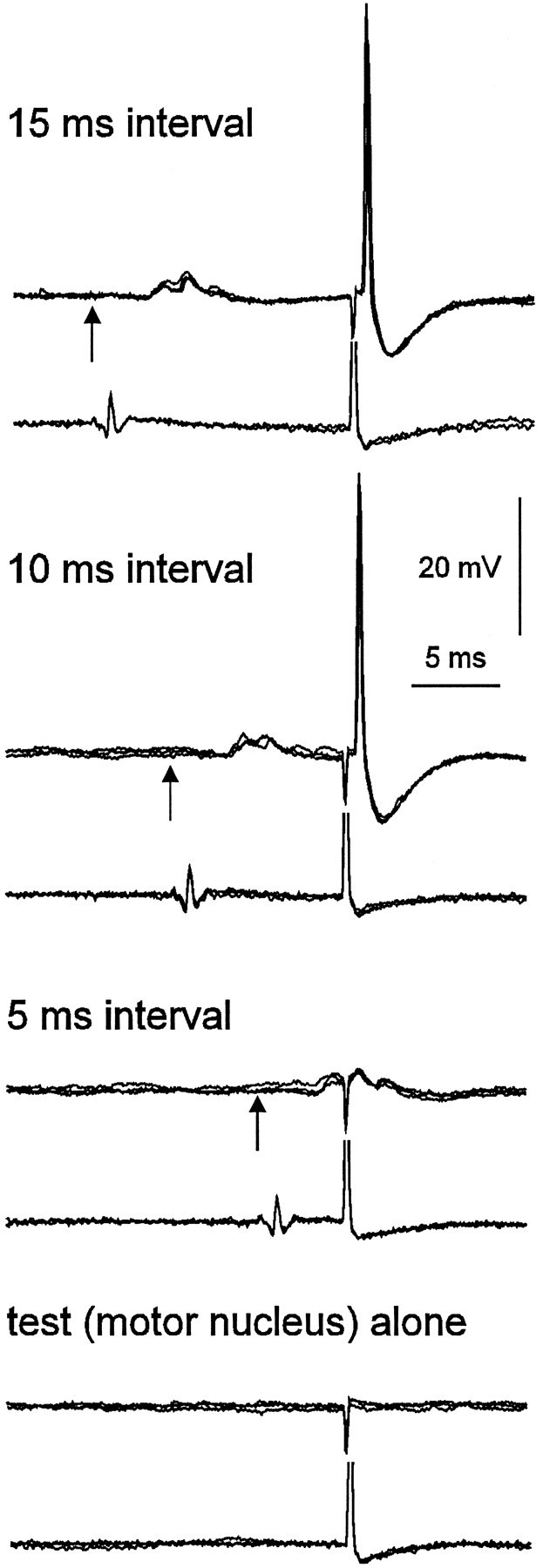
Intracellular recordings from a midlumbar interneuron. Four sets of records each show three superimposed sweeps. In each record, the top trace is an intracellular recording, and the bottom trace a recording from the cord dorsum at the L6–L7 junction. A 9 μA test stimulus was delivered to the motor nucleus in all traces. Antidromic spikes were evoked when the interval between the conditioning stimulus to the quadriceps nerve (5T) preceded the test stimulus by 10 and 15 msec but not 5 msec. Arrowsindicate the onset of the conditioning stimulus.
Fig. 3.

Time course of the excitability changes. In the top panel are extracellular records from a midlumbar interneuron with corresponding cord dorsum records. The top traces show that conditioning stimuli to the TA-EDL nerve at 5T delivered 30 msec before a 6 μA stimulus to the motor nuclei increased terminal excitability to give consistent antidromic activation (firing index = 1). Conditioning stimuli to the same nerve at 2.5T did not increase the firing index above 0. The graph below summarizes the time course and stimulus intensity dependence of this effect. The firing indices for stimuli delivered at different conditioning–test intervals are plotted for three different strengths of conditioning stimulation (5T, 3T, and 2T) as well as in the absence of conditioning stimulation (broken line). Firing indices were calculated from the frequency of antidromic activation in batches of at least 20 stimulus presentations at each strength and at each time point.
Orthodromic activation of some of the neurons raises the possibility that the excitability changes might occur by simple passive spread of somatic depolarization through the axon to the site of activation at the terminals. Even in those neurons that were not discharged by the conditioning stimulus, subthreshold EPSPs may have occurred. Theoretically, spread of depolarization over a distance of 25 mm or more is unlikely (see Discussion). A number of other observations also suggest that this was not the case. Figure 2 shows an intracellular recording from a midlumbar interneuron. This interneuron was excited but not discharged by quadriceps group II afferents. The EPSPs can be seen and as described previously for midlumbar interneurons (Edgley and Jankowska, 1987b) are short latency and fast rising, and they decay within ∼10 msec. In this case a stimulus of 9 μA to the motor nucleus was ineffective when delivered alone or during the EPSP (Fig.2, bottom two traces), but was effective when delivered later, after the EPSP had declined (Fig. 2, top two traces). Additionally, a number (11) of our recordings were classed as intra-axonal, in that a negative membrane potential with fast, positive-going spikes were recorded (often including orthodromic spikes, as in the case of Fig. 1), but the EPSPs or IPSPs were either very small or could not be seen. Our inability to detect EPSPs or IPSPs spreading from soma to the axon at a site likely to be close to its origin suggests that depolarization is unlikely to spread 25 mm further caudally to the motor nuclei.
Whenever the recording conditions were sufficiently stable we examined the time course of the excitability changes by varying the conditioning–test stimulus interval. In most cases the excitability changes were largest between 10 and 30 msec after a single conditioning stimulus. The graph in Figure 3 illustrates the time course for one interneuron with conditioning stimulation to the TA-EDL nerve (5T). Stimuli delivered 10 msec after the conditioning volley were ineffective, whereas stimuli at 15–30 msec always antidromically activated the interneuron. Stimuli at intervals >30 msec produced antidromic activation with a firing index <1. With this method, it is time-consuming and difficult to determine the time courses of the excitability changes to a resolution of less than a few milliseconds. Time courses or at least partial time courses were determined for the excitability changes that followed conditioning stimulation of Q in 35 of the interneurons. In five of the neurons, the onset of the excitability change occurred within a central delay of 5 msec, and in an additional 15 cells it was between 5 and 10 msec. Among these same cells, the longest interval at which excitability changes were seen after a single conditioning stimulus was 55 msec. These time courses were all examined after single conditioning stimuli.
Origin of the excitability changes
By varying the stimulus intensity and by stimulating different nerves, we have examined the origin of the excitability changes. In Figure 3, the probabilities of antidromic activation after conditioning stimuli to TA-EDL of 2T, 3T, and 5T and at different intervals are plotted. Stimuli of 5T (which would excite almost all group II afferents) produced large changes in the probabilities of antidromic activation (Fig. 3, squares), whereas stimuli of 2T (which would activate only the most excitable group II afferents) produced much smaller changes in excitablity (Fig. 3, circles). Thus it was possible to attribute the major part of the excitability change to effects evoked by stimulation of group II afferents. In 34 cells conditioned by quadriceps in which stimulus intensity was varied, excitability changes were evoked only by stimuli above 1.5T and in all cases were largest after stimuli exceeding 2T. Similarly, excitability changes induced in five cells after stimuli to SART and in four cells after stimuli to TA-EDL required stimuli >1.6T. In one neuron, excitability changes were apparently evoked by stimulation of Q at strengths <1.5T, but in this case much larger changes were evoked by stimuli >2T. It is possible that group I afferents evoke smaller excitability changes than group II afferents, but the method we have used in these experiments may be too insensitive to detect them. In addition, trains of stimuli to group I afferents may evoke excitability changes.
We have not surveyed the effectiveness of different nerves exhaustively, but a number of points can be made. The proportions of cells in which excitability changes were evoked by stimulation of different nerves varied widely: Q, SART, and TA-EDL all evoked excitability changes in >70% of the neurons. In contrast, stimulation of PBST, SMAB, and GS, all of which have afferents terminating in the region of the motor nuclei from which the interneurons were antidromically activated, evoked excitability changes in <7% of the neurons. Furthermore, stimulation of these latter nerves never increased firing indices beyond 0.7, whereas Q, SART, and TA-EDL increased firing indices to > 0.8 in 68%, 36%, and 46% of cells, respectively.
Stimulation of cutaneous afferents could also increase terminal excitability in some cases, but was rarely as effective as stimulation of group II afferents. Stimulation of the SUR nerve (single stimulus at 5T) increased the terminal excitability in 15 of 28 cells, but only in four of these (14%) did the firing index increase to >0.8. Similar results were obtained for the SP nerve: terminal excitability was increased in 15 of 25 tested interneurons, but the firing index was increased to >0.8 in only five (20%) of these cells.
Excitability increases induced by intraspinal stimuli
If terminal excitability changes are evoked via the activation of intraspinal neuronal circuitry, then one might expect that direct activation of the circuitry by intraspinal stimulation would also evoke excitability changes. This has been shown to be the case (Rudomin et al., 1981). Intraspinal stimuli that are subthreshold for activation of a terminal can increase the excitability of terminals of group I afferents, which is where PAD occurs, but they do not increase the excitability of rubrospinal terminals in the spinal intermediate zone, at which PAD does not occur. Subthreshold intraspinal stimuli can also evoke increases in terminal excitability at midlumbar interneuron terminals. Figure 4 illustrates this phenomenon for one such cell. Trains of stimuli (10 stimuli at 5 or 10 msec intervals) were tested for 14 interneurons in which terminal excitability could be increased by conditioning stimulation of Q or TA-EDL at 5T. In nine (64%) of these, the firing index was increased from <0.1 for the first stimulus to >0.8 for the later stimuli. Illustrative graphs of these changes for three interneurons are shown in Figure 4. In four other neurons the firing index increased to >0.5. These changes occurred despite the fact that the initial stimuli were subthreshold for spike generation. Similar behavior was seen in three of six interneurons in which we did not detect terminal excitability changes after nerve stimulation.
Fig. 4.

Excitability increases brought about by intraspinal stimuli at intensities subthreshold for spike generation. The left panel shows the initial stimuli of a train delivered to the motor nucleus (23 μA). The plots show for three different cells the firing index for each successive stimulus in a train (5 msec intervals). In each case, the firing index increases with successive stimuli from close to 0 to close to 1.
DISCUSSION
Our results show that increases in the excitability of the terminals of midlumbar interneurons were commonly evoked by conditioning stimuli to group II afferents. A number of possible mechanisms might underlie these excitability changes. One is that the conditioning stimulus may have evoked changes in the ionic environment around the terminals in the motor nuclei, perhaps by increasing extracellular potassium concentrations (Rudomin et al., 1981; Schmied and Fetz, 1987). For example, excitability increases can be generated by trains of stimuli to sensory nerves at rubrospinal terminals in the intermediate zone of the spinal cord that are unlikely to have arisen from a PAD-like mechanism (Rudomin and Jankowska, 1981; Rudomin et al., 1981). This is unlikely to be the explanation in our case, because excitability changes were either not evoked or rarely evoked by conditioning stimuli that evoke large responses within the region of the terminals. The interneurons were antidromically activated from their terminals within the GS and PBST motor nuclei, yet terminal excitability was rarely increased by stimuli to the GS or PBST nerves. Such stimuli result in near-synchronous antidromic activation of the motoneurons and activity in Ia and group II afferents, which terminate close to our stimulating electrode in the motor nuclei. In agreement with this, large (>1 mV) extracellular field potentials were evoked by these stimuli in the region of the tip of the stimulating electrode (these fields were used to position the stimulating electrode within the motor nuclei). Excitability changes were evoked, however, by stimuli to Q, SART, and TA-EDL nerves, which do not evoke visible field potentials in these motor nuclei. Furthermore, we used single stimuli in these experiments, and changes in potassium accumulation usually require repetitive stimulation. For example, Kriz et al. (1974) did not find substantial changes in potassium concentration in the ventral horn after single stimuli to sensory nerves (for discussion, see Rudomin, 1990). Accumulation of potassium ions should be largest around the terminals of the afferents, but we know of no evidence that group II afferents from Q or SART project to the lower lumbar motor nuclei. On the other hand, group I and II afferents from GS and the hamstring nerves have dense terminations in this area but do not evoke excitability changes.
A second possibility is that depolarization evoked at the interneuron cell body may have spread to the terminals electrotonically. This would be consistent with the observation that the excitability increases were most effectively evoked from afferents that most commonly excite midlumbar interneurons (i.e., Q, SART, and TA-EDL) (Edgley and Jankowska, 1987b); however, this is unlikely for a number of reasons. First, the distance between cell body and the terminals was large: from direct measurements in these experiments this distance was at least 20 mm and more realistically 25–30 mm. Although we have no information on the length constants of the axons of midlumbar interneurons, they are unlikely to amount to more than a few millimeters. The mean conduction velocity of the axons of our sample of interneurons was 29 m/sec. This would be consistent with quite small axons, which is in good agreement with direct observations from intracellularly stained axons of midlumbar interneurons, which rarely exceed 4 μm in diameter (Bras et al., 1989). There is little published information on the length constants of central neurons. Central portions of abducens motoneuron axons (which are substantially larger than the axons of midlumbar interneurons) have been estimated to have length constants in the region of 1.7 mm (Gogan et al., 1983). If we assume that the axons of midlumbar interneurons have similar length constants and that these apply over the full length of the axon, then the somatic potential changes 20 mm from the soma should decline toe−20/1.7 or 7.77 × 10−6 of their initial value. If the distance exceeds 20 mm or the interneuron axon space constants are smaller than those of motoneuron axons, then the decrement would be much greater. Postsynaptic potentials can clearly extend for at least several millimeters along motoneuron axons, because they can be detected in ventral root recordings made close to the root entry with a sucrose-gap technique (Luscher et al., 1980;Brink et al., 1981). Passively conducted potentials should decrement much more in interneurons axons, because the axons should not only be smaller but should taper toward their terminals. We have attempted to alter antidromic thresholds of intracellularly recorded motoneurons with conditioning stimuli that evoked large somatic EPSPs or IPSPs. However, we were unable to alter the excitability of the axons (unpublished observations), suggesting that these postsynaptic potentials did not extend as far as 20 mm along the axon. Excitability changes seen in interneuron terminals were also of later onset and longer duration than intracellularly recorded EPSPs in midlumbar interneurons (e.g., Fig. 2). Passive conduction of somatic potentials into axons should include some delays. For midlumbar interneurons it is difficult to assess what these might be. In sucrose-gap recordings of electrotonically conducted potentials from the soma to the ventral root origin along motoneuron axons, the delay was estimated to be <1.5 msec (Luscher et al., 1980; Brink et al., 1981). The smaller size and greater length of interneuron axons (together with likely terminal branching and tapering) might delay the potentials more, but these features would also act to limit the spread of potentials along axons. The absence of EPSPs and IPSPs in intra-axonal or intradendritic recordings from midlumbar interneurons also implies that somatic synaptic potentials spread only a limited distance along the axon.
A third possibility to explain the excitability changes is that a process akin to PAD (i.e., terminal depolarization induced by activation of GABAA receptors at axo-axonic terminals) may occur at the terminals of midlumbar interneurons. Presynaptic depolarization has already been demonstrated at the terminals of central neurons, on premotor interneurons in the lamprey spinal cord (Alford et al., 1991), and at the terminals of neurosecretory neurons in the rat posterior pituitary (Zhang and Jackson, 1995). To our knowledge, our data provide the first indications that such an action may occur at spinal interneuron terminals in the mammalian spinal cord. The observation that trains of intraspinal stimuli at intensities subthreshold for antidromic activation of the terminal were often very effective in increasing terminal excitability might also support this possibility. Intraspinal stimuli have been shown to increase the excitability of group I afferent fibers in the motor nuclei and in the intermediate zone, but they were never seen at rubrospinal terminals in the intermediate zone (Rudomin et al., 1981). This and other evidence was used to differentiate between a nonspecific action on the rubrospinal terminals and a selective presynaptic action on the Ia fibers. The presence of facilitation by intraspinal stimuli at the terminals of interneurons would be consistent with an action specific to the interneuron terminals. One characteristic feature of PAD is its prolonged duration. Our observations do not demonstrate excitability changes, which are as long lasting as changes in afferents that have been described. Our method may have limited our ability to detect the full time course of the excitability changes, because we used single stimuli and would have seen only the peak of the excitability change. However, the peak of the excitability changes in response to single group I stimuli in Ia afferents is at ∼30 msec (Curtis and Lodge, 1982), which fits well with our observations on interneuron terminals.
In relation to this possibility, it is interesting to note that the pattern of terminal excitability changes may generally resemble the patterns of PAD on sensory terminals. Afferents of a particular class frequently give rise to mutual PAD; for example, Ia muscle afferents depolarize Ia afferents, whereas Ib and cutaneous afferents depolarize Ib afferents (Rudomin, 1990), group II afferents depolarize group II afferents (Riddell et al., 1995), and cutaneous afferents of a specific modality depolarize other afferents of the same modality (Schmidt, 1971). In locusts, it has been shown that particular groups of chordotonal organ afferents exert their strongest depolarization on afferents of similar properties (Burrows and Matheson, 1994). In these experiments, effective sources of excitability changes at the terminals of midlumbar interneurons (group II afferents of Q, SART, and TA-EDL) are also common sources of excitatory input to the interneurons (Edgley and Jankowska, 1987b, their Fig. 6). This also parallels the effectiveness of these nerves in evoking PAD at the terminals of group II primary afferents in the midlumbar segments (Riddell et al., 1995, their Figs. 7, 8). Thus, if the excitability changes are produced by a presynaptic depolarizing mechanism, similar principles may underlie the depolarization of midlumbar interneurons and primary afferents.
Footnotes
We thank the Wellcome Trust for their support (project number 045832/z/95).
Correspondence should be addressed to S. A. Edgley, Department of Anatomy, University of Cambridge, Downing Street, Cambridge CB2 3DY, UK.
REFERENCES
- 1.Aggelopoulos NC, Chakrabarty S, Edgley SA. Evoked changes in the excitability of presynaptic terminals of cat midlumbar premotor interneurons. J Physiol (Lond) 1995;487:P71. doi: 10.1523/JNEUROSCI.17-04-01512.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alford S, Christenson J, Grillner S. Presynaptic GABAA and GABAB receptor mediated phasic modulation in axons of spinal motor interneurons. Eur J Neurosci. 1991;3:107–117. doi: 10.1111/j.1460-9568.1991.tb00071.x. [DOI] [PubMed] [Google Scholar]
- 3.Barron DH, Matthews BHC. The interpretation of potential changes in the spinal cord. J Physiol (Lond) 1938;92:276–321. doi: 10.1113/jphysiol.1938.sp003603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bras H, Cavallari P, Jankowska E, Kubin L. Morphology of midlumbar interneurons relaying information from group II muscle afferents in the cat spinal cord. J Comp Neurol. 1989;290:1–15. doi: 10.1002/cne.902900102. [DOI] [PubMed] [Google Scholar]
- 5.Brink E, Jankowska E, McCrea D, Skoog B. Use of sucrose gap for recording postsynaptic population potentials evoked by single interneurones in spinal motoneurones. Brain Res. 1981;223:165–169. doi: 10.1016/0006-8993(81)90817-9. [DOI] [PubMed] [Google Scholar]
- 6.Burrows M, Laurent G. Synaptic potentials in the central terminals of locust proprioceptive afferents generated by other afferents from the same sense organ. J Neurosci. 1993;13:808–819. doi: 10.1523/JNEUROSCI.13-02-00808.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Burrows M, Matheson T. A presynaptic gain-control mechanism among sensory neurons of a locust leg proprioceptor. J Neurosci. 1994;14:272–282. doi: 10.1523/JNEUROSCI.14-01-00272.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cattaert D, El-Manira A, Clarac F. Direct evidence for presynaptic inhibitory mechanisms in crayfish sensory afferents. J Neurophysiol. 1992;67:610–624. doi: 10.1152/jn.1992.67.3.610. [DOI] [PubMed] [Google Scholar]
- 9.Cavallari P, Edgley SA, Jankowska E. Post-synaptic actions of midlumbar interneurons on motoneurons of hind-limb muscles in the cat. J Physiol (Lond) 1987;389:675–689. doi: 10.1113/jphysiol.1987.sp016677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Curtis DR, Lodge DR. The depolarization of feline ventral horn group Ia spinal afferent terminations by GABA. Brain Res. 1982;46:215–233. doi: 10.1007/BF00237180. [DOI] [PubMed] [Google Scholar]
- 11.Curtis DR, Malik R. The effect of GABA on lumbar terminations of rubrospinal neurons in the cat spinal cord. Proc R Soc Lond [Biol] 1984;223:25–33. doi: 10.1098/rspb.1984.0080. [DOI] [PubMed] [Google Scholar]
- 12.Curtis DR, Wilson VJ, Malik R. The effect of GABA on the terminations of vestibulospinal neurons in the cat spinal cord. Brain Res. 1984;295:372–375. doi: 10.1016/0006-8993(84)90989-2. [DOI] [PubMed] [Google Scholar]
- 13.Eccles JC, Eccles RM, Magni F. Central inhibitory action attributable to presynaptic depolarization produced by muscle afferent volleys. J Physiol (Lond) 1961;159:147–166. doi: 10.1113/jphysiol.1961.sp006798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Edgley SA, Jankowska E. Field potentials generated by group II muscle afferents in the middle lumbar segments of the cat spinal cord. J Physiol (Lond) 1987a;385:393–413. doi: 10.1113/jphysiol.1987.sp016498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Edgley SA, Jankowska E. An interneuronal relay for group I and II muscle afferents in the mid-lumbar segments of the cat spinal cord. J Physiol (Lond) 1987b;389:647–674. doi: 10.1113/jphysiol.1987.sp016676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gogan P, Gueritaud JP, Tyc-Dumont S. Comparison of antidromic and orthodromic action potentials of identified motor axons in the cat’s brain stem. J Physiol (Lond) 1983;335:205–220. doi: 10.1113/jphysiol.1983.sp014529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Graham B, Redman S. A simulation of action-potentials in synaptic boutons during presynaptic inhibition. J Neurophysiol. 1994;71:538–549. doi: 10.1152/jn.1994.71.2.538. [DOI] [PubMed] [Google Scholar]
- 18.Hultborn H, Meunier S, Pierrot-Deseilligny E, Shindo M. Changes in presynaptic inhibition of Ia fibres at the onset of voluntary contraction in man. J Physiol (Lond) 1987;389:757–772. doi: 10.1113/jphysiol.1987.sp016681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kriz N, Sykova E, Ujec E, Vycklicky L. Changes of extracellular potassium concentration induced by neuronal activity in the spinal cord of the cat. J Physiol (Lond) 1974;238:1–15. doi: 10.1113/jphysiol.1974.sp010507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lipski J. Antidromic activation of neurons as an analytical tool in the study of the central nervous system. J Neurosci Methods. 1982;4:1–32. doi: 10.1016/0165-0270(81)90015-7. [DOI] [PubMed] [Google Scholar]
- 21.Lundberg A, Malmgren K, Schomburg E. Reflex pathways from group II muscle afferents. 1. Distribution and linkage to α-motoneurones. Exp Brain Res. 1987;65:271–281. doi: 10.1007/BF00236299. [DOI] [PubMed] [Google Scholar]
- 22.Luscher H-R, Ruenzel P, Henneman E. Topographic distribution of terminals of Ia and group II fibers in spinal cord, as revealed by postsynaptic population potentials. J Neurophysiol. 1980;49:968–985. doi: 10.1152/jn.1980.43.4.968. [DOI] [PubMed] [Google Scholar]
- 23.Riddell JS, Jankowska E, Huber J. Organization of neuronal systems mediating presynaptic inhibition of group-II muscle afferents in the cat. J Physiol (Lond) 1995;483:443–460. doi: 10.1113/jphysiol.1995.sp020596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rudomin P. Presynaptic control of synaptic effectiveness of muscle spindle and tendon organ afferents in the mammalian spinal cord. In: Binder MD, Mendell LM, editors. The segmental motor system. Oxford UP; New York: 1990. pp. 349–380. [DOI] [PubMed] [Google Scholar]
- 25.Rudomín P, Jankowska E. Presynaptic depolarization of terminals of rubrospinal tract fibers in intermediate nucleus of cat spinal cord. J Neurophysiol. 1981;46:517–531. doi: 10.1152/jn.1981.46.3.517. [DOI] [PubMed] [Google Scholar]
- 26.Rudomín P, Engberg I, Jimenez I. Mechanism involved in presynaptic depolarization of group I and rubrospinal fibers in cat spinal cord. J Neurophysiol. 1981;46:532–551. doi: 10.1152/jn.1981.46.3.532. [DOI] [PubMed] [Google Scholar]
- 27.Rudomín P, Jimenez I, Enriquez M. Effects of stimulation of group-I afferents from flexor muscles on heterosynaptic facilitation of monosynaptic reflexes produced by Ia and descending inputs: a test for presynaptic inhibition. Exp Brain Res. 1991;85:93–102. doi: 10.1007/BF00229990. [DOI] [PubMed] [Google Scholar]
- 28.Schmidt RF. Presynaptic inhibition in the vertebrate central nervous system. Ergeb Physiol. 1971;63:20–101. doi: 10.1007/BFb0047741. [DOI] [PubMed] [Google Scholar]
- 29.Schmied A, Fetz EE. Activity-related changes in electrical thresholds of pyramidal tract axons in the behaving monkey. Exp Brain Res. 1987;65:353–360. doi: 10.1007/BF00236308. [DOI] [PubMed] [Google Scholar]
- 30.Swadlow HA, Kocsis JD, Waxman SG. Modulation of impulse conduction along the axonal tree. Annu Rev Biophys Bioeng. 1980;9:143–179. doi: 10.1146/annurev.bb.09.060180.001043. [DOI] [PubMed] [Google Scholar]
- 31.Wall PD. Excitability changes in afferent fibre terminations and their relation to slow potentials. J Physiol (Lond) 1958;142:1–21. doi: 10.1113/jphysiol.1958.sp005997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wall PD. Control of impulse conduction in long range branches of afferents by increases and decreases of primary afferent depolarization in the rat. Eur J Neurosci. 1994;6:1136–1142. doi: 10.1111/j.1460-9568.1994.tb00611.x. [DOI] [PubMed] [Google Scholar]
- 33.Zhang SLJ, Jackson MB. GABA(A) receptor activation and the excitability of nerve-terminals in the rat posterior pituitary. J Physiol (Lond) 1995;483:583–595. doi: 10.1113/jphysiol.1995.sp020608. [DOI] [PMC free article] [PubMed] [Google Scholar]