

Abstract
Soluble N-ethylmaleimide-sensitive factor attached protein (SNAP) receptor (SNARE) mechanisms are thought to be involved in two important processes in axonal growth cones: (1) membrane expansion for axonal growth and (2) vesicular membrane fusion for mature synaptic transmission. We investigated the localization and interactions among the proteins involved in SNARE complex formation in isolated growth cone particles (GCP) from forebrain. We demonstrated that the SNARE complex is present in GCPs morphologically without synaptic vesicles (SVs) and associated with growth cone vesicles. However, the apparently SV-free GCP was lacking in the regulatory mechanisms inhibiting SNARE complex formation proposed in SV fusion, i.e., the association of synaptotagmin with the SNARE complex, and vesicle-associated membrane protein (VAMP)-synaptophysin complex formation. The core components of the SNARE complex (syntaxin, SNAP-25, and VAMP) accumulated for several days before postnatal day 7, when SVs first appeared, and preceded the accumulation of marker proteins such as synaptophysin, SV2, and V-ATPase. Our present results suggest that the SNARE mechanism for vesicular transmitter release is not fully functional in growth cones before the appearance of SVs, but the SNARE mechanism is working for membrane expansion in growth cones, which supports our recent report. We concluded that the regulation of the SNARE complex in growth cones is different from that in mature presynaptic terminals and that this switching may be one of the key steps in development from the growth cone to the presynaptic terminal.
Keywords: growth cone, SNARE complex, SNARE hypothesis, presynaptic terminal, synaptotagmin, cytoskeleton, growth cone vesicle
A recent breakthrough, the solubleN-ethylmaleimide-sensitive factor attached protein (SNAP) receptor (SNARE) hypothesis proposed by Rothman (1994), has brought rapid progress to understanding the protein machinery underlying neurotransmitter release from the presynaptic terminal (Bajjalieh and Scheller, 1995; Augustine et al., 1996) by explaining Ca2+-regulated exocytosis by a series of interactions among these proteins. The main point of the SNARE hypothesis is that vesicular targeting depends on a protein complex formation between vesicular membrane proteins (vesicular SNAREs) and target membrane proteins (target SNAREs). Among the presynaptic proteins, VAMP/synaptobrevin, SNAP-25, and syntaxin/HPC-1 are thought to be SNAREs (Söllner et al., 1993b). Several other proteins, such as synaptotagmin or synaptophysin, are thought to be regulators of the SNARE complex formation (Bajjalieh and Scheller, 1995). We must note that the framework of the SNARE mechanism is currently applicable to other intracellular vesicular trafficking events, as well as to neurotransmitter release (Rothman, 1994; Söllner, 1995)
The growth cone, formed at the tip of an immature axon, is responsible for axonal guidance and elongation (Jessell and Kandel, 1993). An examination of whether the SNARE mechanism is working in growth cones is important for two reasons. First, the growth cone is regarded as the precursor of the presynaptic terminal (Hall and Sanes, 1993). Because the growth cone gradually changes to the presynaptic terminal after reaching and recognizing its appropriate target, the SNARE mechanism for transmission must be prepared in the growth cone. Second, several reports (including ours) have determined that the SNARE mechanism in growth cones is involved in membrane expansion for axonal growth (Osen-Sand et al., 1993, 1996; Igarashi et al., 1996). One of the important features differentiating a growth cone from a presynaptic terminal is that the growth cone is originally lacking in synaptic vesicles (SVs). Instead, a growth cone has a cluster of vesicles, so-called “growth cone vesicles” (GCV), which are distinct from SVs (Pfenninger et al., 1992; Pfenninger and Friedman, 1993). The hypothesis that these GCVs are added to the plasmalemma in an exocytotic manner in the growth cone is currently the most reliable explanation for membrane expansion, although the details of molecular mechanisms still remain unknown (Osen-Sand et al., 1993; Futerman and Banker, 1996).
In this study we used subcellular fractionation of growth cone particles (GCPs) (Pfenninger et al., 1983) to analyze the localization of the proteins involved in the SNARE mechanisms and interactions among these proteins in isolated growth cones. We demonstrate the presence of the SNARE complex in apparently SV-free GCPs and the association of the SNARE complex (7S) and one larger than 7S with GCV, and the lack of several regulatory protein–protein interactions for the SNARE complex proposed in SV fusion, and the accumulation of these proteins in GCPs. Based on our previous (Igarashi et al., 1996) and present results, we conclude that the SNARE mechanism is functional in apparently SV-free GCPs, but in a manner different from the way the SNARE mechanism in SV fusion works.
Some of these results have been published previously in abstract form (Igarashi et al., 1995).
MATERIALS AND METHODS
Antibodies. Antibodies used in the present study are as follows (mAb indicates monoclonal antibody and the others are rabbit polyclonal antibodies): anti-syntaxin 1 (mAb 10H5), anti-synaptotagmin I (mAb 1D12), anti-VAMP-2, anti-SNAP-25, anti-rab3A, anti-β-SNAP antibodies (M. Takahashi, Mitsubishi Kasei Institute of Life Sciences, Machida, Japan; El Far et al., 1993); anti-SV2 (K. M. Buckley, Harvard Medical School, Boston, MA; Buckley and Kelly, 1985); anti-vacuolar ATPase (V-ATPase) A subunit (Y. Moriyama, Hiroshima University, Hiroshima, Japan; Nakamura et al., 1994); anti-Munc-18/n-sec1/rbsec1 (P. De Camilli, Yale University School of Medicine, New Haven, CT;Garcia et al., 1994); anti-N-ethylmaleimide-sensitive factor (NSF) (Tagaya et al., 1993); and anti-synapsin I (E. Miyamoto, Kumamoto University School of Medicine, Kumamoto, Japan; Fukunaga et al., 1992). Anti-synaptophysin and anti-GAP-43 mAbs were purchased from Boehringer Mannheim (Mannheim, Germany).
Subcellular fractionation. Subcellular fractionation of growth cones from developing rat forebrains of embryonic day 17 (E17), E20, postnatal day 2 (P2), P5, P7, P10, and P14 was as described byPfenninger et al. (1983) or Meiri and Gordon-Weeks (1990), with slight modification (Igarashi et al., 1990; Saito et al., 1992). Perinuclear fraction was prepared from the developing rat brain by the method ofSbaschnig-Agler et al. (1988). In modified Pfenninger’s methods, we collected the interface between 0.32 m/0.83 minterface as the GCP fraction, and 0.83 m/1.0 minterface as the fraction B (Hemlke and Pfenninger, 1995). Synaptosomes from the adult rat cerebral cortex were prepared as described byWhittaker and Barker (1972). The surface area density of the GCPs in each electron micrograph (EM) was calculated as described by Pfenninger et al. (1983), except that we used a microcomputer imaging device (MCID) image analyzer (Image Research). The SV-containing GCPs were also scored in EM, and their percentage of the total number of GCPs were calculated.
The GCV-enriched fraction was fractionated as follows. After trituration (Wood et al., 1992), the GCP was completely lysed by the hypotonic treatment using 6 mm Tris-HCl, pH 8.1 (Ellis et al., 1985), and stirred for 45 min at 4°C. The samples were loaded onto 0.6 m and 0.8 m sucrose and ultracentrifuged at 25,000 rpm (SW 28 rotor, Hitachi Koki Ltd., Tokyo, Japan) for 90 min. The interface between 0.6 m and 0.8m sucrose was collected as GCV, and that between 0.8m and 1.0 m as the plasma membrane fraction of the growth cone [growth cone membranes (GCM)] (Ellis et al., 1985). For electron microscopy, the GCV fraction was fixed using 2.5% glutaraldehyde/2% paraformaldehyde in 0.1 m cacodylate buffer, pH 7.4. After the sample was rinsed with 10% sucrose in the same buffer, it was post-fixed by OsO4, dehydrated, embedded, and thin-sectioned. The volume density of each structure in EMs was determined by measuring the surface area density (Weibel and Bolender, 1973) using the MCID image analyzer.
To obtain cytoskeletal subfraction, the GCP fraction from P2 rat forebrain was extracted by 20 mm HEPES, pH 7.3, 0.3m sucrose, 3 mm MgCl2, 1 mm EGTA, 0.2 μm leupeptin, and 10 μg/ml aprotinin containing 1% Triton X-100 and 0.01% saponin (CSK buffer) (Hemlke and Pfenninger, 1995) for 1 hr at 4°C by stirring, and it was centrifuged for 1 hr at 40,000 rpm. The pellet and the supernatant were collected as the GCP-cytoskeletal and Triton X-soluble subfractions, respectively.
Detection of protein complex formation. Identification of the native SNARE complex present in GCPs was performed as described previously (Hayashi et al., 1994). Briefly, the GCP fraction was mixed with SDS-PAGE sample buffer containing 1% SDS, without boiling. The sample was analyzed by SDS-PAGE and by immunoblotting using anti-syntaxin, anti-SNAP-25, or anti-VAMP antibodies. To detect the core complex formation among the SNAREs, the GCV proteins were solubilized by HKA buffer (10 mm HEPES, pH 7.5, 140 mm potassium acetate, 1 mm MgCl2, 0.1 mm EGTA) containing 2% Triton X-100, as described byPevsner et al. (1994). The sample was loaded onto the continuous 10–35% glycerol gradient, centrifuged at 41,000 rpm for 17 hr using an SW41 rotor (Beckman, Fullerton, CA), and collected in 1 ml samples from fraction 1 (top) to fraction 10 (bottom). Fractionation of SV from adult rat cortex was performed essentially as described (Pevsner et al., 1994), and proteins from the SV were extracted similarly to those from the GCV (as described above). The immunoprecipitation using 10H5 mAb was performed as described previously (Bennett et al., 1992). For the experiment investigating whether the larger complex is formed in growth cones, the GCV proteins were solubilized by HKD buffer [20 mm HEPES, pH 7.0, 0.1 m KCl, 1 mmdithiothreitol (DTT), 2 μm(p-amidinophenyl)methanesulfonyl fluoride] containing 1% Triton X-100. The solubilized membrane proteins were incubated with 0.5 mm ATPγS, 2 mmMgCl2, NSF (15 μg), and His6-α-SNAP (15 μg) for 30 min at 4°C (Pevsner et al., 1994). Then, the sample was loaded onto continuous glycerol gradient, ultracentrifuged, fractionated and collected as described above (Pevsner et al., 1994). BSA (4.6S), catalase (11.3S), and α2-macroglobulin (20S) (Söllner et al., 1993a, 1993b; Pevsner et al., 1994) were used as sedimentation markers.
The immunoprecipitation of the 80 kDa complex from GCP was as described in El Far et al. (1993). Briefly, the proteins of the GCP fraction and of the postnuclear membranes from adult rat brain were solubilized by a buffer (5% glycerol, 0.16 m sucrose, and 25 mmHEPES-Tris, pH 7.4, containing 1% CHAPS and several protease inhibitors). The solubilized proteins were incubated with anti-syntaxin, anti-SNAP-25, anti-VAMP, or the control IgG for 2 hr on ice. Protein G- or protein A-agarose was then added to the antigen–antibody mixture and was incubated for 2 hr at 4°C with gentle agitation, in the presence of 1% BSA. The agarose-bound complex was rinsed several times using the same buffer containing 0.5% CHAPS, and the pellet was collected and solubilized by the sample buffer for SDS-PAGE.
To examine whether NSF is released from GCV in the presence of Mg2+ and ATP, Mg2+-ATPase treatment was carried out as described previously (Hong et al., 1994). The GCV was incubated with 25 mm HEPES, pH 7.2, containing 1 mm DTT, 0.1 m KCl, 0.3 m sucrose, 2% polyethylene glycol, and 5 mm MgCl2/0.5 mm ATP or 2 mm EDTA/0.5 mm ATP on ice for 5 min. The mixtures were centrifuged at 40,000 rpm for 30 min. The supernatant and the pellet were subjected to SDS-PAGE and analyzed by immunoblotting using anti-NSF and anti-β-SNAP antibodies. The SVs from adult rat cerebral cortex and the Golgi apparatus from fetal brain were prepared as described previously (Whittaker and Barker, 1972; Pfenninger et al., 1983; Tagaya et al., 1996).
Immunoblotting. Each fraction was solubilized by the sample buffer for SDS-PAGE containing 1% SDS. To detect the 80 kDa complex (Hayashi et al., 1994), the sample was heated at 60°C for 10 min; otherwise, it was boiled for 10 min. In each lane, 50 μg of the proteins was loaded for SDS-PAGE. The proteins were electroblotted for 1 hr and then identified by immunodetection using the streptavidin–biotin system (Amersham, Buckinghamshire, UK). Quantitation of each immunoblot was performed by densitometric assay using IMAGEQUANT (Molecular Dynamics, Sunnyvale, CA). We checked the linearity of the relationship between the intensity of each protein band and its amount by confirming that each band intensity was twice as large as that of its one-half amount. To examine whether VAMP-synaptophysin complex can be formed in GCP, the cross-linking using disuccinimyl suberate (DSS) in P2-GCP or adult synaptosomes was carried out as described by Edelmann et al. (1994), and detection of the complex was performed by Washbourne et al. (1995). Fresh GCPs or synaptosomes in Krebs’-Ringer’s solution buffer were incubated with 5 mm DSS for 45 min at room temperature and with 0.1m Tris-glycine buffer, pH 7.4, as a quencher was added. After they were centrifuged several times, the supernatants were analyzed by SDS-PAGE and immunoblotting.
Immunohistochemistry. E18 rat hippocampal neurons or dorsal root ganglia neurons were cultured on the laminin-coated chamber slides as described previously (Ozawa and Yuzaki, 1984; Bandtlow et al., 1993). The cultured neurons were fixed for 24 hr by 4% paraformaldehyde at room temperature and permeabilized by 0.25% Triton X-100 for 5 min. After blocking by BSA, the neurons were incubated with the primary antibodies specific to VAMP, synaptotagmin, or syntaxin diluted to 1:250, at 37°C for 2 hr. After they were rinsed, the samples were incubated with fluorescein isothiocyanate (FITC)-labeled secondary antibodies (Amersham). The morphometrical analysis for the distribution of each antigen was performed using MCID. The data of each immunohistochemical micrograph were incorporated into the MCID system through a CCD camera. The fluorescent intensity density (D), representing the amount of the antigen concentration, and the area (S) were scanned separately in the peripheral (P-) domain (actin filament-rich region) and the central (C-) domain (vesicle-rich region) (Dailey and Bridgman, 1993) of the growth cone. P-domain was designated by the staining using rhodamine-labeled phalloidin (Fan et al., 1993). The product between D and S(D × S) in each domain was calculated separately, and the ratio between the value of P-domain and that of the total growth cone area was calculated.
RESULTS
VAMP–SNAP-25–syntaxin complex in GCPs
The core components of SNARE complex, i.e., syntaxin, SNAP-25, and VAMP, were enriched in the growth cone, compared with the C-fraction containing axonal shafts (Pfenninger et al., 1983) (Fig.1A). The amounts of syntaxin, SNAP-25, and VAMP in the GCP fraction were 2.8, 9.1, and 2.7×, respectively, that in the C-fraction (the mean of four independent experiments). The 80 kDa complex, i.e., VAMP–SNAP-25–syntaxin, is the core protein complex of SNARE interaction (Söllner et al., 1993a; Hayashi et al., 1994; Pevsner et al., 1994) and is reported as SDS-resistant and heat-labile (Hayashi et al., 1994). The 80 kDa complex was detected in P2-GCP using the antibodies specific to VAMP, SNAP-25, and syntaxin (Fig. 1B). The amount of the 80 kDa complex, however, was at a lower level than that in adult synaptosomes (Fig.1B). For example, the bound syntaxin in P2-GCP was 9.7 ± 1.0% of that in synaptosomes (four independent experiments). The amount of unbound syntaxin in the GCP was similar to that in synaptosomes (Fig. 1C).
Fig. 1.

A, Enrichment of the SNARE complex components in the GCP fraction (G) compared with the C-fraction (C) containing axonal shafts. Immunoblots of proteins from P2-GCP and P2-C fraction (containing axonal shafts) using anti-VAMP, anti-SNAP-25, and anti-syntaxin antibodies, respectively, are shown. B, Detection of an 80 kDa complex composed of VAMP–SNAP-25–syntaxin in both the P2-GCP fraction (G) and the adult synaptosomes (S). The SDS-resistant 80 kDa complex was recognized by anti-VAMP, anti-SNAP-25, and anti-syntaxin antibodies, respectively. Anti-synaptophysin antibody did not recognize this complex. To detect the 80 kDa complex (Hayashi et al., 1994), the sample was heated at 60°C for 10 min. Arrows indicate the position of 80 kDa. C, Unbound syntaxin was also detected as a 35 kDa protein band in both P2-GCP (G) and adult synaptosomes (S). Note that there is a lower amount of bound (80 kDa) syntaxin in G than inS, but that the amount of the unbound syntaxin inG is almost equal to that inS.
The GCP contains many clear vesicles corresponding to GCV (Fig.2A) (Pfenninger et al., 1983; Igarashi et al., 1990); however, these vesicles are not always found in each GCP in EM, probably because of the spatially nonuniform distribution of the GCVs in a GCP. Subcellular fractionation enriched the GCV (Fig.2B, Table 1). Synaptophysin and SV2, both vesicular marker proteins, were also enriched in this fraction. Their amounts in the GCV fraction were 4.5 and 2.9× those in the GCM (four independent experiments). By solubilization of the proteins bound to the GCV by 2% Triton X-100 and the subsequent fractionation of the proteins using 10–35% glycerol gradient, the three core components of the SNARE complex migrated and were present together in fraction 4, corresponding to 7S position (Fig. 2C). This position was almost identical to that of the 7S complex derived from the adult SV shown in previous reports (Söllner et al., 1993b;Pevsner et al., 1994). The proteins immunoprecipitated by anti-syntaxin mAb also contained SNAP-25 and VAMP but not synaptophysin (Fig. 2D). An endogenous complex containing NSF larger than 7S was present in fraction 8 (Fig. 2C).
Fig. 2.
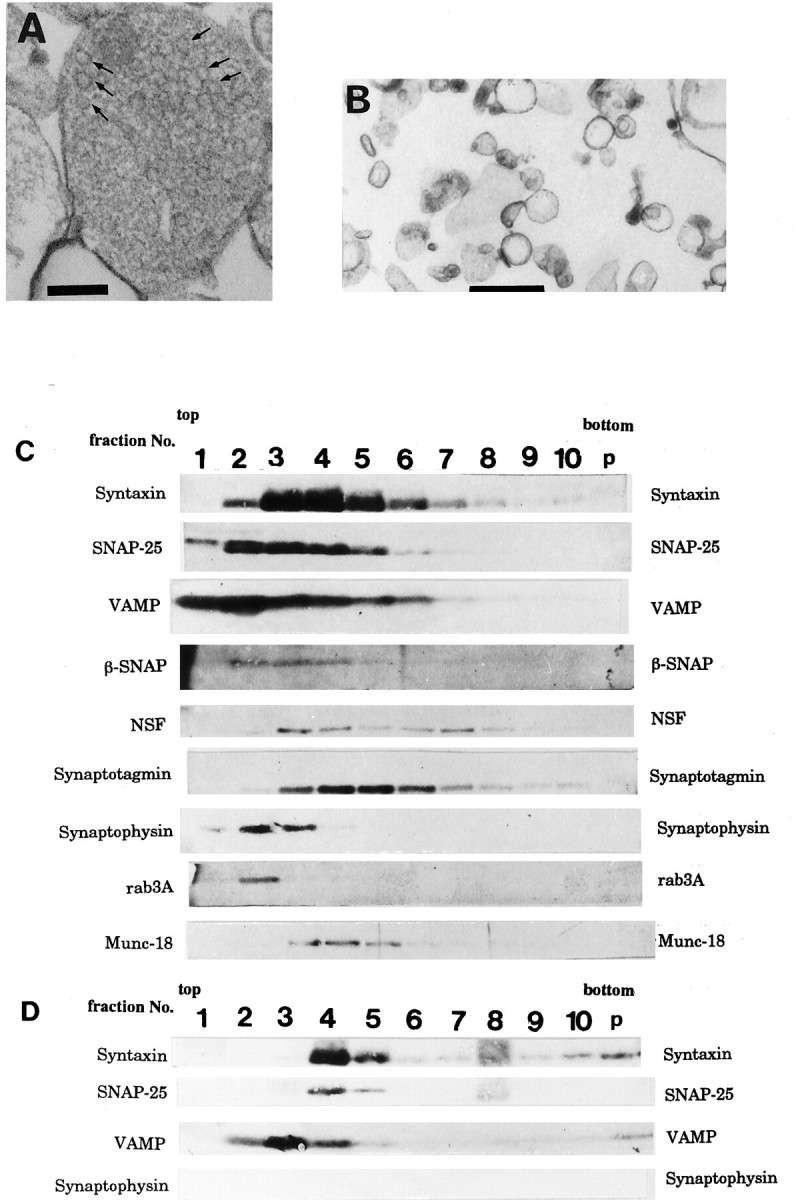
Electron micrographs of (A) P2-GCP and (B) GCV derived from P2-GCP. GCP fractions were prepared and fixed by the methods of Pfenninger et al. (1983), with slight modification (Igarashi et al., 1990). Note that GCVs are seen in a GCP in A (arrows) (see Pfenninger et al., 1983; Igarashi et al., 1990). Scale bars: A, B, 0.5 μm. SNARE complex is associated with P2-GCV (C andD). C, Fractionation of GCV proteins extracted by 1% Triton X-100, using 10–35% continuous glycerol gradient. Each fraction was collected as 1 ml from the top. Each protein was visualized by immunoblotting. Note that fraction 4 (7S position) shows the peak of syntaxin and that SNAP-25 and VAMP are present together with syntaxin there. As sedimentation markers, BSA (4.6S), catalase (11.3S), and α2-macroglobulin (20S) were used (Söllner et al., 1993a,b; Pevsner et al., 1994). D, The immunoprecipitated proteins from GCV using anti-syntaxin antibody after fractionation by glycerol gradient. In fraction 4, VAMP and SNAP-25 were co-precipitated by anti-syntaxin antibody; however, synaptophysin was not. The immunoprecipitation of each fraction inC, using 10H5 mAb (anti-syntaxin antibody), was carried out as described previously (Bennett et al., 1992; Pevsner et al., 1994), and each protein band was detected by immunoblotting using the specific antibodies (see Materials and Methods).
Table 1.
Membrane compartments of the P2-GCV fraction
| Organelles | Relative surface density | |
|---|---|---|
| Raw % ± SEM | Adjusted % | |
| a. Growth cone vesicles | 66.5 ± 5.3 | 75 |
| b. Mitochondria | 4.9 ± 1.9 | 7 |
| c. Dense granules | 4.6 ± 2.6 | 6 |
| d. Lysosomal structures | 0.8 ± 0.2 | 1 |
| e. Plasma membrane | 11.5 ± 0.5 | 14 |
| f. Unidentified membrane | 11.7 ± 0.6 | |
The relative membrane areas were determined by morphometrical analysis using the MCID, a computerized image scanner. The analysis was performed on four random EMs. Adjusted percentage was calculated from the raw percentages, assuming that all elements (a–e) contribute equally to the unidentified category (f). Organelle categories: a, large clear vesicles with diameters of 150–200 nm; b, mitochondria in GCPs, c, small electron-dense granules; d, multivesicular bodies and lysosomes; e, GCP-derived plasma membrane fragments; and f, unclassified membrane fragments. The surface area of each structure was scanned by the MCID system, and its percentage was calculated using that of the total membrane-surrounded structures.
Protein–protein interactions regulatory for the SNARE complex in GCP
Although the SNARE complex was detected in apparently SV-free GCP, as shown in Figures 1B and 2C, the regulatory mechanisms for the complex must be examined to discover whether the SNARE mechanism is fully working in the growth cone. Using the apparently SV-free GCP, we examined several protein–protein interactions regulating the SNARE complex formation in Golgi apparatus or in synaptosomes, as follows.
NSF and β-SNAP release from membrane by Mg2+-ATP (Rothman, 1994)
The GCV-attached NSF was not released by Mg2+-ATP, as well as in SV, but was unlike that in the Golgi apparatus (Fig.3A) (Hong et al., 1994; Tagaya et al., 1996). The distribution of β-SNAP showed a pattern similar to that of NSF (Fig. 3B).
Fig. 3.

Distribution of (A) NSF and (B) β-SNAP in P2-GCV. A, NSF was still bound to the GCVs after the Mg2+-ATP treatment (5 mm MgCl2/0.5 mm ATP on ice for 5 min; see Hong et al., 1994) (lanes 1 and2), similar to the case in SVs (lanes 3and 4) but not the Golgi apparatus (lanes5 and 6). After treatment, the sample was centrifuged at 41,000 rpm for 30 min. Lanes1, 3, and 5, pellet; lanes2, 4, and 6, supernatant. The supernatant and the pellet were subjected to SDS-PAGE and analyzed by immunoblotting using anti-NSF antibody. B, β-SNAP was not released from the membranes in GCV (lanes 1 and2) or in SV (lanes 3 and4), even in the presence of Mg2+-ATP (see above). Under the same conditions, however, β-SNAP was released from membrane in the Golgi apparatus (lanes 5 and6). Lanes 1, 3, and5, pellet; lanes 2, 4, and6, supernatant. The supernatant and the pellet were analyzed by immunoblotting using anti-β-SNAP antibody. Each protein amount was measured by densitometric assay of the immunostained bands.
Recruitment of NSF and β-SNAP to the SNARE complex (Söllner et al., 1993a,b; Rothman, 1994)
In apparently SV-free GCPs, exogenously added NSF and α-SNAP did not cause 20S complex formation, even in the presence of Mg2+ and ATPγS (Fig. 4A; compare Fig. 2C). In contrast, the addition of these exogenous proteins induced a 20S complex formation in the adult synaptosomes under the same conditions as in the case of the GCP, as described previously (Fig. 4B) (Pevsner et al., 1994).
Fig. 4.

Failure to induce formation of 20S complex in P2-GCP by addition of exogenous NSF and α-SNAP (A), unlike in adult SV (B). A, After endogenous NSF and α-SNAP were dissociated from the GCVs by salt treatment and NSF and α-SNAP were exogenously added in the presence of Mg2+-ATPγS, the extracted proteins were incubated, fractionated, electrophoresed, and immunostained after electroblotting. Note that the protein complex larger than 7S in fraction 9(20S position) is not observed compared (A) with Figure2C. B, The same treatment of SVs as inA causes a 20S complex formation. Before (top) and after (bottom) addition of NSF and α-SNAP in the presence of Mg2+-ATPγS, proteins collected into fraction 9 (20S position) were analyzed by immunoblotting. Note that syntaxin, SNAP-25, and VAMP were not detected before addition of the exogenous NSF and α-SNAP.
Association of synaptotagmin with the SNARE complex (Söllner et al., 1993a)
Both antibodies specific to SNAP-25 or to VAMP immunoprecipitated the VAMP–SNAP-25–syntaxin complex from P2-GCP as well as from adult rat synaptosomes, except that synaptotagmin was barely precipitated in the GCV (Fig. 5).
Fig. 5.

The immunoprecipitated proteins from P2-GCP (A) and adult rat synaptosomes (B). In both A and B, each lane represents the immunoprecipitation using the following antibodies: lane1, anti-VAMP; lane 2, anti-SNAP-25; and lane 3, anti-syntaxin antibodies. Lane 4represents the preimmune serum that was used as the control. Note that synaptotagmin was not immunoprecipitated in the GCP. The immunoprecipitation was as described in El Far et al. (1993). The solubilized proteins were incubated with anti-syntaxin, anti-SNAP-25, anti-VAMP, or the control IgG for 2 hr on ice, and then protein G- or protein A-agarose was added to the antigen–antibody mixture and incubated for 2 hr at 4°C with gentle agitation. The agarose-bound complex was rinsed several times using the same buffer containing 0.5% CHAPS, and the pellet was collected and solubilized by the sample buffer for SDS-PAGE. Each protein was visualized by immunoblotting using specific antibodies.
VAMP–synaptophysin complex (Calakos and Scheller, 1994; Edelmann et al., 1995; Washbourne et al., 1995)
The 58 kDa synaptophysin–VAMP complex was not detected in P2-GCP in the presence or absence of DSS, unlike in adult synaptosomes (Fig.6A,B).
Fig. 6.

A cross-linker DSS treatment fails to detect VAMP-synaptophysin complex in the P2-GCP (G), unlike in the adult synaptosome (S). Lane 1, without DSS; lane 2, with DSS. Neither anti-synaptophysin (A) nor anti-VAMP antibodies (B) detected a 58 kDa VAMP-synaptophysin complex. In contrast, DSS treatment made the complex detectable in the adult synaptosome. Fresh GCPs in Krebs’-Ringer’s solution buffer were incubated with 5 mm DSS for 45 min at room temperature, and 0.1 m Tris-glycine buffer, pH 7.4, was added as quencher. After they were centrifuged several times, the supernatants were analyzed by SDS-PAGE and immunoblotting. 58 K andarrows indicate the position of 58 kDa.
Localization of VAMP and synaptotagmin in the cytoskeletal subfraction of the growth cone and filopodia
Because failure to detect interactions among the regulatory proteins in GCP may be attributable to the unique roles of regulatory proteins in growth cones other than vesicular membrane fusion, we examined their localization in growth cones. The transmembrane proteins VAMP and synaptotagmin were also distributed to the Triton-insoluble, i.e., cytoskeletal subfraction of P2-GCP, whereas other transmembrane proteins (syntaxin and synaptophysin) were not (Fig. 7). Compared with the total, 37.0 ± 2.7% of VAMP and 45.1 ± 4.0% of synaptotagmin in the P2-GCP were present in the GCP-cytoskeletal subfraction (six independent experiments). The immunoreactivity of VAMP and that of synaptotagmin were observed not only in the body of the growth cone but also in the filopodia of growth cones of the cultured neurons (Fig.8A,B). In contrast, syntaxin was localized in the C-domain of growth cones and observed minimally in the filopodial region (Fig. 8C). The relative intensity of the antigen localized in the P-domain is shown in Figure8D; those of synaptotagmin and VAMP were significantly larger than that of syntaxin. We show the results using the DRG neurons here (Fig. 8), because their filopodial development is better than that of hippocampal ones, although the results were also quite similar in the hippocampal growth cones.
Fig. 7.
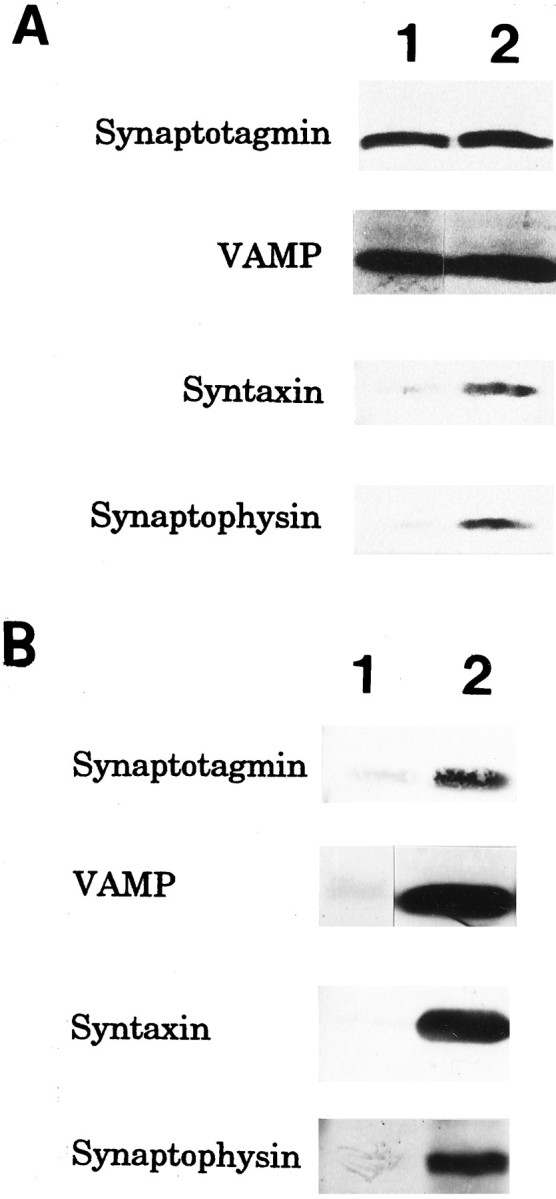
Distribution of the transmembrane proteins (VAMP, synaptotagmin, syntaxin, and synaptophysin) in the Triton-soluble (lane1) and cytoskeletal (lane 2) subfractions of P2-GCP (A) and adult synaptosomes (B). The GCP fraction from P2 rat forebrain was extracted by CSK buffer (see Materials and Methods) containing 1% Triton X-100 and 0.01% saponin for 1 hr at 4°C by stirring, and centrifuged for 1 hr at 40,000 rpm. The pellet and the supernatant were collected as the GCP-cytoskeletal and Triton X-soluble subfractions. After SDS-PAGE, the proteins from each subfraction were electrically transferred to nitrocellulose membrane. VAMP, synaptotagmin, syntaxin, and synaptophysin in each subfraction were visualized by immunostaining, using streptavidin-conjugated alkaline phosphatase.
Fig. 8.
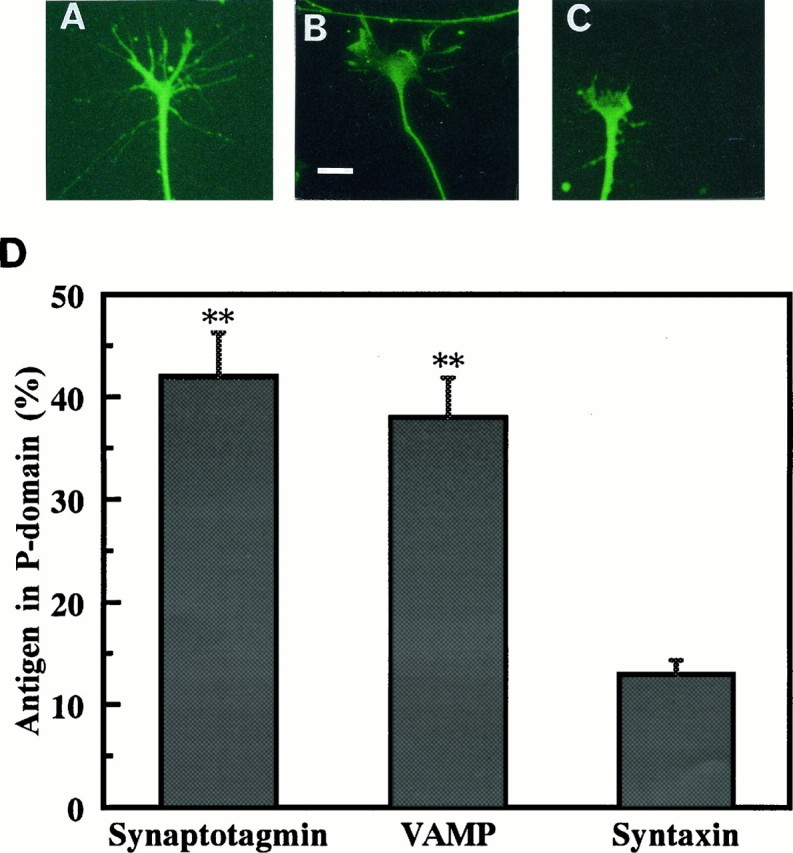
Immunohistochemical localization of VAMP (A), synaptotagmin (B), and syntaxin (C) in growth cones. The DRG neurons were cultured for 24 hr and then fixed by 4% paraformaldehyde. After permeabilization by 0.25% Triton X-100 for 5 min, the neurons were incubated with each primary antibody (diluted to 1:250) for 2 hr at 37°C. FITC-conjugated secondary antibodies were used for fluorescent staining. Note that not only the center of the growth cone but also its filopodia were stained in A and B but not in C. We show the results using the DRG neurons here because their filopodial development is better than that of hippocampal ones, although the results were also quite similar in the hippocampal growth cones. Scale bar, 10 μm. D, The relative distribution of synaptotagmin, VAMP, and syntaxin in the P-domain of the growth cones compared with that of the total growth cone area. The intensity is represented as the product between the area and the fluorescent intensity density, measured using the MCID. Data were the mean ± SEM (n = 10). **p < 0.01.
Developmental patterns of proteins involved in SNARE mechanisms in GCP
The isolated growth cones have characteristics of typical GCPs at every age examined (Fig. 9) (Pfenninger et al., 1983;Taylor and Gordon-Weeks, 1989). Briefly, their diameters were 1–2 μm, and the central area was filled with typical, branching, smooth endoplasmic reticulum. The surface area density showed >70% of the purity of the GCP fractions (see Table 9) in each age. After P7, the particles with SV clustering were found in the GCP fraction (Table2). As a biochemical marker of the GCP fraction, the relative amount of GAP-43, a protein enriched in the growth cone (Meiri et al., 1986), was quantified (Table 2). At every age, GAP-43 was significantly more concentrated in the GCP fraction than in the corresponding B-fraction (0.83 m/1.0 m sucrose interface). It was ∼200% greater in its relative amount of the GCP compared with B-fraction, although the value gradually decreased after P5 (Table 2). After P5, the ratio of the GAP-43 amount between GCP- and B-fractions decreased gradually, probably because of the increase in the synaptosomes with a higher density than the GCPs, which were collected in the B-fraction and contained a certain amount of GAP-43.
Fig. 9.
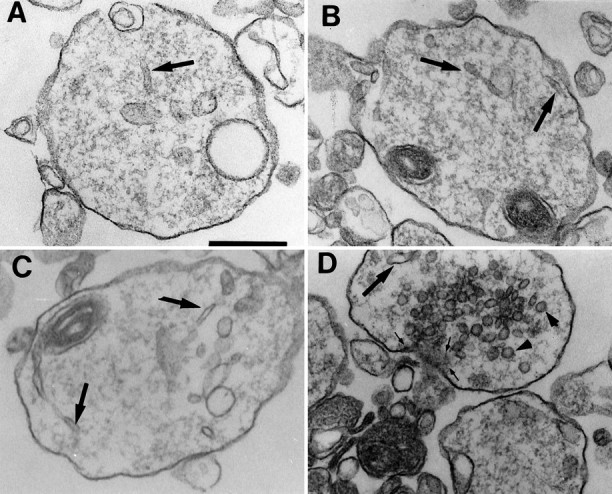
Morphological characterization of the GCP fractions. Typical isolated growth cones of (A) P2, (B) P5, and (C) P10 were shown. Smooth endoplasmic reticuli were observed in every stage of GCPs (thick arrows). Note that typical GCPs are found in 0.32m/0.83 m sucrose interface fraction at every stage. In D, we also found growth cones in P10 with SVs (arrowheads) and an active zone (thin arrow). Scale bar (shown in A): 0.5 μm.
Table 2.
Characterization of GCPs from different ages
| Age | Surface density of GCPs (%) | SV-containing GCPs (%) | GAP-43 (%) |
|---|---|---|---|
| E17 | 74.6 ± 5.0 | 0 | 220 ± 1 |
| P2 | 74.3 ± 2.0 | 0 | 234 ± 18 |
| P5 | 72.6 ± 4.6 | 0 | 254 ± 8 |
| P7 | 70.1 ± 5.6 | 12.5 ± 3.5 | 204 ± 22 |
| P10 | 70.6 ± 4.8 | 21.4 ± 4.8 | 186 ± 2 |
Surface area density of the GCPs in each age was calculated from the EMs as described previously (Pfenninger et al., 1983), except that we used the MCID system. The surface area density of the total membrane-surrounded structures in an EM is shown as 100%. The data regarding surface area density was represented as mean ± SEM of six different preparations. Compared to the total number of GCPs (100%), the number of SV-containing GCPs was scored. The data are shown as the mean ± SEM (six independent experiments). The amount of GAP-43 was determined by densitometric assay for immunoblots, and its relative amount of GCPs was compared with that of the corresponding fraction B (the interface between 0.83 m/1.0 m sucrose). The data are shown as the mean ± SEM (four independent experiments).
Three groups were designated on the basis of the developmental accumulation pattern of the proteins in GCP. Group A represents the proteins accumulated several days before P7 (Fig.10A). This group consists of syntaxin, SNAP-25, VAMP (Fig. 10A), synaptotagmin, Munc-18, rab3A, NSF, and β-SNAP (data not shown) (Table3). These are the components of the SNARE–NSF–SNAP complex, or the regulators (Söllner et al., 1993a,b; Horikawa et al., 1993). Group B represents the group of proteins accumulated in the growth cone on P7 (Fig. 10B); synaptophysin, SV2, and V-ATPase belong to this group. These are also typical markers of SV. The amount of synapsin I did not change in the growth cones at the different developmental ages examined (Fig. 10C), and it was not considered to belong to group A or B. Synapsin I was thus defined as group C.
Fig. 10.
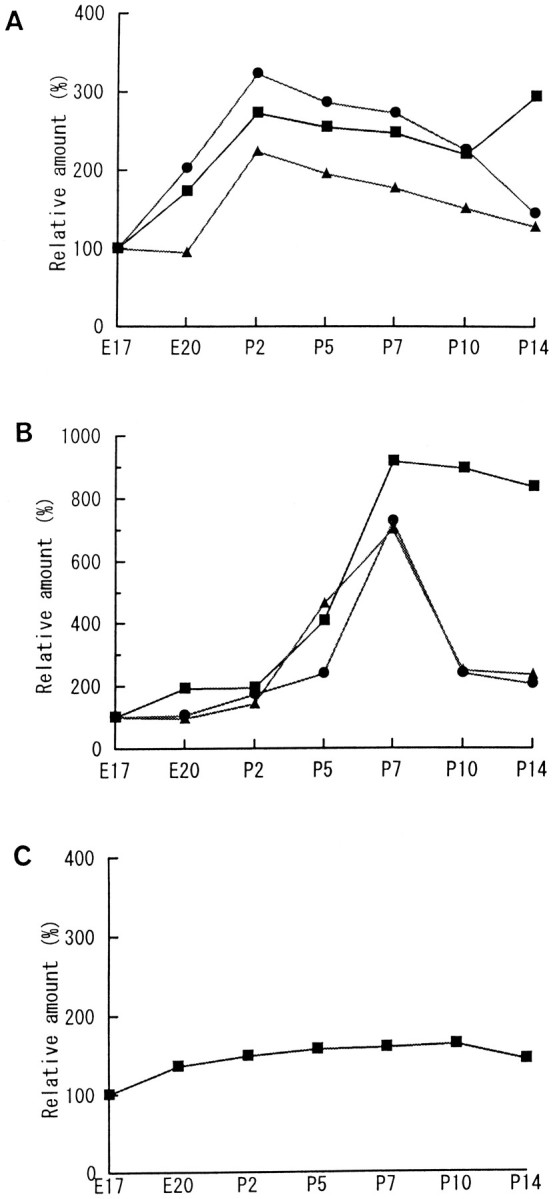
Accumulation patterns of “presynaptic proteins” in the growth cones. These proteins are classified into three groups according to when each protein reaches the peak amount in the GCPs. A, Group A consists of the proteins accumulated before P7. The core components of SNARE complex, namely, SNAP-25 (square), syntaxin (circle), and VAMP (triangle) are shown here, and several other proteins belong to this group (see Table 1). B, Group B are the proteins accumulated only on P7. Synaptophysin (square), SV2 (circle), and V-ATPase (triangle) are classified in this group. Note that P7 is the time when SV appears in the growth cone of rat forebrain (Taylor and Gordon-Weeks, 1989). C, Synapsin I did not change significantly in amount during the different stages of the growth cones. It was thus classified as group C. Each protein amount was measured by densitometric assay of the immunostained bands. The relative amount was calculated with the value of E17 as 100%. Data are shown as the mean of the four independent experiments.
Table 3.
Relative peak amount of the group A proteins (%)
| Synaptotagmin | 211 |
| Munc-18 | 170 |
| rab3A | 215 |
| NSF | 354 |
| β-SNAP | 300 |
In the CGP, these proteins reach the peak on P2 as do VAMP, SNAP-25, and syntaxin (Fig. 10). Each relative amount of the peak value is calculated with the value of E17-GCP as 100% and shown as the mean of four independent experiments.
Group A proteins increased in amount only 200%, at most, in synaptosomes compared with the peak value in the growth cones (Fig.11), whereas groups B and C increased in amount by severalfold (Fig. 11).
Fig. 11.

The relative amount of each protein in adult synaptosome compared with that in growth cones at the peak. The amount of each group A protein in synaptosome is, at most, 2× its peak amount in the growth cone, whereas group B and C proteins increased in amount more than fivefold. Each protein amount was measured by densitometric assay of the immunostained bands. The relative amount was calculated with the value of E17 as 100%. The data are shown as the mean ± SEM (n = 4).
DISCUSSION
Because axonal growth cones convert to presynaptic terminals, the SNARE mechanism for synaptic transmission should develop accompanied by the maturation of axon terminals. Regarding axon terminal development, we have classified axon terminals into three stages: stage I, the growth cone stage, without SV;stage II, the transition stage from a growth cone to a presynaptic terminal; and stage III, the mature presynaptic terminal stage, free of growth cone properties. We summarized our present results in Table 4 according to the above definition. The SNARE mechanism is accepted not only in SV fusion but also in the mechanism explaining general intracellular vesicular fusion (Rothman, 1994). As an example of the latter case, we have recently demonstrated that the SNARE mechanism is involved in membrane expansion for axonal growth (Igarashi et al., 1996). Our results demonstrated that growth cones at stage I have the SNARE complex but several regulatory mechanisms are deficient, indicating that the SNARE mechanism working at stage I may be sufficient for axonal growth but is not fully functional in SV fusion.
Table 4.
Developmental stages of the axon terminal in rat forebrain
| Stage I (growth cone stage) | Stage II (transition stage) | Stage III (mature presynaptic terminal stage) | |
|---|---|---|---|
| Age | Before P7 | P7–P14 | After P14–adult |
| GCV | ++ | +4-a | − |
| SV | − | +4-a | +++ |
| Group A | ++ | ++ | +++ |
| Group B | + | ++ | ++++ |
| Group C | + | + | ++++ |
| Ca2+-dependent release | +4-a | +4-a | +++ |
| Membrane expansion | ++ | +? | − |
| Association of synaptotagmin4-b | − | +? | ++ |
Cited from Taylor and Gordon-Weeks (1989).
Association of synaptotagmin with endogenous SNARE complex.
SNARE complex is associated with GCV at stage I similar to its association with SV at stage III
We demonstrated that the core of the SNARE complex, i.e., VAMP–SNAP-25–syntaxin complex, is already formed at stage I, although the amount is approximately one-tenth that of the synaptic SNARE complex (Fig. 1B,C). These proteins were enriched in P2-GCP (Fig. 1A), and the core complex was associated with GCV (Fig. 2C,D). Two vesicle-associated proteins, SV2 and synaptophysin, were enriched in GCVs (see Results). Because neither P2-GCP nor P2-GCV contains SV (Tables 1, 2), the SNARE complex detected in this study at stage I is associated with GCV and not with SV. We found that a small amount of protein complex larger than the core complex exists endogenously (e.g., fraction 8 in Fig. 2C) associated with GCV, probably corresponding to the SNARE–NSF–SNAP complex (cf. Pevsner et al., 1994). This did not migrate at the 20S position, probably because synaptotagmin is absent from this complex (Fig. 5; see below). Because the 20S complex is formed by recruitment of NSF and α/β-SNAP to the SNARE complex (Söllner et al., 1993a,b; Rothman, 1994), this suggests that we succeeded in detecting biochemical evidence for the working of the SNARE mechanism in apparently SV-free GCP. This supports the theory that the SNARE complex is involved in the vesicle docking process of growth cones for membrane expansion (Osen-Sand et al., 1993; Pfenninger and Friedman, 1993; Bark and Wilson, 1994; Catsicas et al., 1994; Schulze et al., 1995; Igarashi et al., 1996).
Several protein–protein interactions proposed to regulate SNARE complex formation are deficient at stage I: low-stringent regulation in growth cones versus high-stringent regulation in mature presynaptic terminals
Although the SNARE mechanisms are widely accepted in intracellular vesicular fusion systems, such as in presynaptic terminals, in Golgi apparatus, and in endosomes, the protein–protein interactions regulatory for SNARE complex association and dissociation in one vesicular docking and fusion system differ from those in others, probably because the SNARE mechanisms are functionally modulated in each fusion system (Rothman, 1994; Söllner, 1995; Südhof, 1995; Tagaya et al., 1996). To better characterize the SNARE mechanism properties, we examined the biochemical reactions in growth cones.
NSF bound to GCV could not be easily released from GCV membrane by Mg2+-ATP alone (Fig. 3A). This property of NSF is similar to that in SV or in endosome but distinct from that in the Golgi apparatus (Fig. 3A) (Hong et al., 1994; Colombo et al., 1996; Tagaya et al., 1996). However, the exogenously added NSF and α-SNAP did not induce formation of a larger complex, such as 20S, despite our detection of an endogenous, larger complex associated with GCVs (Fig. 4A), unlike in the adult synaptosomes and in Golgi apparatus (Fig. 4B). We concluded that recruitment of NSF and SNAPs to the SNARE complex in growth cones is considerably different from that in Golgi apparatus where the original SNARE hypothesis was postulated (Hong et al., 1994; Rothman, 1994;Moriyama et al., 1995; Colombo et al., 1996), and that the SNARE mechanism at stage I is similar to but not the same as that at stage III.
We also demonstrated that two protein–protein interactions, proposed as inhibitory to SNARE complex formation in presynaptic terminals, are lacking at stage I: recruitment of synaptotagmin to the SNARE complex, and VAMP-synaptophysin binding (Figs. 5, 6). These results suggest that inhibition of the SNARE complex formation by these mechanisms is not working at stage I. At this stage, the growth cone uses the SNARE mechanism for axonal growth (Osen-Sand et al., 1993; Igarashi et al., 1996; Williamson et al., 1996). In this case, low-stringent regulation of the membrane fusion is favorable for continuous axon growth. From the aspect of preparation for neurotransmitter release, we revealed that the two inhibitory mechanisms for vesicular fusion that assure high-stringent regulation for fusion in transmitter release were absent at stage I, indicating that they are added in the course of axon terminal development. Our results also suggest that release regulated by synaptotagmin I, a Ca2+ sensor ubiquitously distributed in the CNS presynaptic terminals (Yoshida et al., 1992; Südhof and Rizo, 1996), is not working at this stage (Fig. 5), although another synaptotagmin isoform might be localized (Südhof and Rizo, 1996). Our observation is consistent with a previous report that GABA release from the growth cones of rat forebrain is not Ca2+-dependent until P7 (Taylor and Gordon-Weeks, 1989). High-stringent regulation of vesicular fusion by synaptophysin (Calakos and Scheller, 1994; Edelmann et al., 1995) starts after its accumulation there (Fig. 10). We showed that synaptotagmin and VAMP are cytoskeletal components in growth cones as well as participants in membrane fusion regulators (Figs. 5, 6). Such different localization of these proteins from that in presynaptic terminals might explain that the two inhibitory mechanisms involving these two proteins were not detected at stage I.
Presynaptic proteins do not accumulate simultaneously in growth cones
More than 10 presynaptic proteins have been well characterized, and their roles in synaptic transmission have been partly elucidated (Südhof, 1995). Because most of them are involved in the SNARE mechanism, we investigated molecular aspects of the SNARE mechanism development in growth cones using different developmental stages of GCPs, by immunoquantitation of each component.
Morphological quantitation of volume density in EM (Fig. 9 and Table 2) and biochemical quantitation of GAP-43 (Table 2) in GCP fractions from the different ages indicated similar characteristics (Taylor and Gordon-Weeks, 1989).
According to the developmental accumulation time course of the presynaptic proteins in the growth cones, we have classified them into the following three groups (Figs. 10, 11).
Group A contains VAMP, SNAP-25, syntaxin, synaptotagmin, Munc-18, rab3A, NSF, and β-SNAP (Fig. 10A, Table 3); all members are proteins involved in vesicular fusion systems (Südhof, 1995). These components accumulate before morphological SV appearance (Fig. 10A, Table 3) (Lou and Bixby, 1995), suggesting that they are also important to growth cone functions such as membrane expansion (Igarashi et al., 1996), but that they are insufficient to cause transition from growth cones to mature presynaptic terminals.
Group B consists of several typical SV marker proteins (Fig.10B). Our previous report failed to detect synaptophysin in P5-GCP (Saito et al., 1992), probably because of the lower amount of synaptophysin in GCPs before P7 than that in synaptosomes (Fig. 11). The accumulation time of these proteins in growth cones is consistent with the time when SV first appears, at stage II (Taylor and Gordon-Weeks, 1989). Because the mature SV is synthesized in axon terminals (Mundigl and De Camilli, 1994; Okada et al., 1995), the SV probably does not appear until group B components accumulate and are associated with the preexisting group A proteins, such as synaptotagmin, VAMP, and rab3A.
Because the amount of synapsin I remains constant in GCPs through several different stages, synapsin I was classified as group C (Fig.10C). Its synaptosomal amount reaches ∼700% of the peak amount of GCP (P10-GCP; Fig. 11); thus, synapsin I dramatically accumulates at the last stage of synaptogenesis, i.e., stage III, to accomplish rapid transmitter release in the mature terminal (Südhof, 1995).
How are the molecular events correlated with conversion from a growth cone to a presynaptic terminal?
The SNARE mechanisms are involved in two processes in growth cones: (1) axonal growth and (2) synaptic maturation. Our results suggest that group A components are sufficient to accomplish the former function, but groups B and C, in addition to group A, are necessary for the latter. Because axonal growth terminates after target recognition but conversion to the mature terminal continues, and because groups B and C accumulate in the axon terminals after group A accumulation (Fig.10), the switching of axonal growth to mature transmission must occur through the appearance of several protein–protein interactions. Which protein–protein interaction is necessary to regulate SNARE complex formation at stage I and how such a conversion occurs in axon terminals are currently unknown. Identifying the proteins involved in these processes is an important next step.
Footnotes
This work was supported in part by scientific grants from the Ministry of Education, Science, Sports, and Culture to M.I., M.T., and Y.K, and from Naito Memorial Foundation to M.I. We thank the researchers who kindly provided specific antibodies used in this study (see Materials and Methods). We are also grateful to T. Hijikata, X. Sun, and K. Sudo for their help in taking electron micrographs. Thanks are also due to T. Tashiro and M. Takahashi for their advice on immunoprecipitation. We are indebted to Richard A. Cripe Jr. for English editing of this manuscript.
Correspondence should be addressed to Michihiro Igarashi, M.D., Ph.D., Department of Molecular and Cellular Neurobiology, Gunma University School of Medicine, 3-39-22 Showa-machi, Maebashi, Gunma 371, Japan.
REFERENCES
- 1.Augustine GJ, Burns ME, DeBello WM, Petit DL, Schweizer FE. Exocytosis: proteins and perturbations. Annu Rev Pharmacol Toxicol. 1996;36:659–701. doi: 10.1146/annurev.pa.36.040196.003303. [DOI] [PubMed] [Google Scholar]
- 2.Bajjalieh SM, Scheller RH. The biochemistry of neurotransmitter secretion. J Biol Chem. 1995;270:1971–1974. doi: 10.1074/jbc.270.5.1971. [DOI] [PubMed] [Google Scholar]
- 3.Bandtlow CE, Schmidt MF, Hassinger TD, Schwab ME, Kater SB. Role of intracellular calcium in NI-35-evoked collapse of neuronal growth cones. Science. 1993;259:80–83. doi: 10.1126/science.8418499. [DOI] [PubMed] [Google Scholar]
- 4.Bark IC, Wilson MC. Regulated vesicular fusion in neurons: snapping together the details. Proc Natl Acad Sci USA. 1994;91:4621–4624. doi: 10.1073/pnas.91.11.4621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bennett MK, Calakos N, Kreiner T, Scheller RH. Synaptic vesicle membrane proteins interact to form a multimeric complex. J Cell Biol. 1992;116:761–775. doi: 10.1083/jcb.116.3.761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Buckley KM, Kelly RB. Identification of a transmembrane glycoprotein specific for secretory vesicles of neurons and endocrine cells. J Cell Biol. 1985;100:1284–1294. doi: 10.1083/jcb.100.4.1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Calakos N, Scheller RH. Vesicle-associated membrane protein and synaptophysin are associated on the synaptic vesicle. J Biol Chem. 1994;269:24534–24537. [PubMed] [Google Scholar]
- 8.Catsicas S, Grenningloh G, Pich EM. Nerve-terminal proteins: to fuse to learn. Trends Neurosci. 1994;17:368–373. doi: 10.1016/0166-2236(94)90045-0. [DOI] [PubMed] [Google Scholar]
- 9.Colombo MI, Taddese M, Whiteheart SW, Stahl PD. A possible predocking attachment site for N-ethylmaleimide-sensitive fusion protein: insights from in vitro endosome fusion. J Biol Chem. 1996;268:18810–18816. doi: 10.1074/jbc.271.31.18810. [DOI] [PubMed] [Google Scholar]
- 10.Dailey ME, Bridgman PC. Vacuole dynamics in growth cones: correlated EM and video observation. J Neurosci. 1993;13:3375–3393. doi: 10.1523/JNEUROSCI.13-08-03375.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Edelmann L, Hanson PI, Chapman ER, Jahn R. Synaptobrevin binding to synaptophysin: a potential mechanism for controlling the exocytotic fusion machine. EMBO J. 1995;14:224–231. doi: 10.1002/j.1460-2075.1995.tb06995.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.El Far O, Charvin N, Leveque C, Martin-Moutot N, Takahashi M, Seager MJ. Interaction of a synaptobrevin (VAMP)-syntaxin complex with presynaptic calcium channels. FEBS Lett. 1993;361:101–105. doi: 10.1016/0014-5793(95)00156-4. [DOI] [PubMed] [Google Scholar]
- 13.Ellis L, Wallis I, Abreu E, Pfenninger KH. Nerve growth cones isolated from fetal rat brain. IV. Preparation of a membrane subfraction and identification of a membrane glycoprotein expressed on sprouting neurons. J Cell Biol. 1985;101:1977–1989. doi: 10.1083/jcb.101.5.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fan J, Mansfield G, Redmon T, Gordon-Weeks PR, Raper JA. The organization of F-actin and microtubules in growth cones exposed to a brain-derived collapsing factor. J Cell Biol. 1993;121:867–878. doi: 10.1083/jcb.121.4.867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fukunaga K, Soderling TR, Miyamoto E. Activation of Ca2+/calmodulin-dependent protein kinase II and protein kinase C by glutamate in cultured rat hippocampal neurons. J Biol Chem. 1992;267:25527–22533. [PubMed] [Google Scholar]
- 16.Futerman AH, Banker GA. The economics of neurite outgrowth: the addition of new membrane to growing axon. Trends Neurosci. 1996;19:144–149. doi: 10.1016/s0166-2236(96)80025-7. [DOI] [PubMed] [Google Scholar]
- 17.Garcia EP, Gatti E, Butler M, Burton J, De Camilli P. A rat brain Sec1 homologue related to Rop and UNC18 interacts with syntaxin. Proc Natl Acad Sci USA. 1994;91:2003–2007. doi: 10.1073/pnas.91.6.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hall ZW, Sanes JR. Synaptic structure and development: the neuromuscular junction. Neuron. 1993;10:99–121. doi: 10.1016/s0092-8674(05)80031-5. [DOI] [PubMed] [Google Scholar]
- 19.Hayashi T, McMahon H, Yamasaki S, Binz T, Hata Y, Südhof TC, Niemann H. Synaptic vesicle membrane fusion complex: action of clostridial neurotoxins on assembly. EMBO J. 1994;13:5051–5061. doi: 10.1002/j.1460-2075.1994.tb06834.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hemlke S, Pfenninger KH. Growth cone enrichment and cytoskeletal association of non-receptor tyrosine kinases. Cell Motil Cytoskeleton. 1995;30:194–207. doi: 10.1002/cm.970300304. [DOI] [PubMed] [Google Scholar]
- 21.Hong R-M, Mori H, Fukui T, Moriyama Y, Futai M, Yamamoto A, Tashiro Y, Tagaya M. Association of N-ethylmaleimide-sensitive factor with synaptic vesicles. FEBS Lett. 1994;350:253–257. doi: 10.1016/0014-5793(94)00778-0. [DOI] [PubMed] [Google Scholar]
- 22.Horikawa HPM, Saisu H, Ishizuka T, Sekine Y, Tsugita A, Odani S, Abe T. A complex of rab3A, SNAP-25, VAMP/synaptobrevin-2 and syntaxins in brain presynaptic terminals. FEBS Lett. 1993;330:236–240. doi: 10.1016/0014-5793(93)80281-x. [DOI] [PubMed] [Google Scholar]
- 23.Igarashi M, Waki H, Hirota M, Hirabayashi Y, Obata K, Ando S. Differences in the lipid composition between isolated growth cones from the forebrain and those from the brainstem in the fetal rat. Dev Brain Res. 1990;51:1–9. doi: 10.1016/0165-3806(90)90252-t. [DOI] [PubMed] [Google Scholar]
- 24.Igarashi M, Kozaki S, Terakawa S, Komiya Y. Presynaptic proteins in growth cones. Cell Struct Funct [Suppl] 1995;20:566. [Google Scholar]
- 25.Igarashi M, Kozaki S, Terakawa S, Kawano S, Ide C, Komiya Y. Growth cone collapse and inhibition of neurite growth induced by botulinum neurotoxin C1: a t-SNARE is involved in axonal growth. J Cell Biol. 1996;134:205–215. doi: 10.1083/jcb.134.1.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jessell TM, Kandel ER. Synaptic transmission: a bidirectional and self-modifiable form of cell-cell communication. Neuron. 1993;10:1–30. doi: 10.1016/s0092-8674(05)80025-x. [DOI] [PubMed] [Google Scholar]
- 27.Lou X, Bixby JL. Patterns of presynaptic gene expression define two stages of synaptic differentiation. Mol Cell Neurosci. 1995;6:252–262. doi: 10.1006/mcne.1995.1020. [DOI] [PubMed] [Google Scholar]
- 28.Meiri KF, Gordon-Weeks PR. GAP-43 in growth cones is associated with areas of membrane that are tightly bound to substrate and is a component of a membrane skeleton subcellular fraction. J Neurosci. 1990;10:255–265. doi: 10.1523/JNEUROSCI.10-01-00256.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Meiri KF, Pfenninger KH, Willard MB. Growth-associated protein, GAP-43, a polypeptide that is induced when neurons extend axons, is a component of growth cones and corresponds to pp46, a major polypeptide of a subcellular fraction enriched in growth cones. Proc Natl Acad Sci USA. 1986;83:3537–3541. doi: 10.1073/pnas.83.10.3537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moriyama Y, Yamamoto A, Tagaya M, Tashiro Y, Michibata H. Localization of N-ethylmaleimide-sensitive fusion protein in pinealocytes. NeuroReport. 1995;6:1757–1760. doi: 10.1097/00001756-199509000-00012. [DOI] [PubMed] [Google Scholar]
- 31.Mundigl O, De Camilli P. Formation of synaptic vesicles. Curr Opin Cell Biol. 1994;6:561–567. doi: 10.1016/0955-0674(94)90077-9. [DOI] [PubMed] [Google Scholar]
- 32.Nakamura H, Moriyama Y, Futai M, Ozawa H. Immunohistochemical localization of vacuolar H+-ATPase in osteoclasts of rat tibiae. Arch Histol Cytol. 1994;57:535–539. doi: 10.1679/aohc.57.535. [DOI] [PubMed] [Google Scholar]
- 33.Okada Y, Yamazaki H, Sekine-Aizawa Y, Hirokawa N. The neuron-specific kinesin superfamily protein KIF1A is a unique monomeric motor for anterograde axonal transport of synaptic vesicle precursors. Cell. 1995;81:769–780. doi: 10.1016/0092-8674(95)90538-3. [DOI] [PubMed] [Google Scholar]
- 34.Osen-Sand A, Catsicas M, Staple JK, Jones KA, Ayala G, Knowles J, Grenningloh G, Catsicas S. Inhibition of axonal growth by SNAP-25 antisense oligonucleotides in vitro and in vivo. Nature. 1993;364:445–448. doi: 10.1038/364445a0. [DOI] [PubMed] [Google Scholar]
- 35.Osen-Sand A, Staple JK, Naldi E, Schiavo G, Rossetto O, Petitpierre S, Malgaroli A, Montecucco C, Catsicaas S. Common and distinct fusion proteins in axonal growth and transmitter release. J Comp Neurol. 1996;367:222–234. doi: 10.1002/(SICI)1096-9861(19960401)367:2<222::AID-CNE5>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 36.Ozawa S, Yuzaki M. Patch-clamp studies of chloride channels activated by gamma-aminobutyric acid in cultured hippocampal neurones of the rat. Neurosci Res. 1984;1:275–293. doi: 10.1016/0168-0102(84)90034-8. [DOI] [PubMed] [Google Scholar]
- 37.Pevsner J, Hsu S-C, Braun JEA, Calakos N, Ting AE, Bennett MK, Scheller RH. Specificity and regulation of a synaptic vesicle docking complex. Neuron. 1994;13:353–364. doi: 10.1016/0896-6273(94)90352-2. [DOI] [PubMed] [Google Scholar]
- 38.Pfenninger KH, Friedman LB. Sites of plasmalemmal expansion in growth cones. Dev Brain Res. 1993;71:181–192. doi: 10.1016/0165-3806(93)90170-f. [DOI] [PubMed] [Google Scholar]
- 39.Pfenninger KH, Ellis L, Johnson MP, Friedman LB, Somlo S. Nerve growth cone isolated from fetal brain: subcellular fractionation and characterization. Cell. 1983;35:573–584. doi: 10.1016/0092-8674(83)90191-5. [DOI] [PubMed] [Google Scholar]
- 40.Pfenninger KH, de la Houssaye BA, Frame L, Hemlke S, Lockerbie RO, Lohse K, Miller V, Negre-Aminou P, Wood MR. Biochemical dissection of plasmalemmal expansion at the growth cone. In: Letourneau PC, Kater SB, Macagno ER, editors. The nerve growth cone. Raven; New York: 1992. pp. 111–123. [Google Scholar]
- 41.Rothman JE. Mechanisms of intracellular protein transport. Nature. 1994;372:55–63. doi: 10.1038/372055a0. [DOI] [PubMed] [Google Scholar]
- 42.Saito S, Fujita T, Komiya Y, Igarashi M. Biochemical characterization of nerve growth cones isolated from both fetal and neonatal rat forebrains: the growth cone particle fraction mainly consists of axonal growth cones in both stages. Dev Brain Res. 1992;65:179–184. doi: 10.1016/0165-3806(92)90177-x. [DOI] [PubMed] [Google Scholar]
- 43.Sbaschnig-Agler M, Pfenninger KH, Ledeen RW. Gangliosides and other lipids of the growth cone membranes. J Neurochem. 1988;51:212–220. doi: 10.1111/j.1471-4159.1988.tb04858.x. [DOI] [PubMed] [Google Scholar]
- 44.Schiavo G, Gmachi MJS, Stenbeck G, Söllner TH, Rothman JE. A possible docking and fusion particle for synaptic transmission. Nature. 1995;378:733–736. doi: 10.1038/378733a0. [DOI] [PubMed] [Google Scholar]
- 45.Schulze KL, Broadie K, Perin MS, Bellen HJ. Genetic and electrophysiological studies of Drosophila syntaxin-1A demonstrate its role in nonneuronal secretion and neurotransmission. Cell. 1995;80:311–320. doi: 10.1016/0092-8674(95)90414-x. [DOI] [PubMed] [Google Scholar]
- 46.Söllner T. SNAREs and targeted membrane fusion. FEBS Lett. 1995;389:80–83. doi: 10.1016/0014-5793(95)00594-y. [DOI] [PubMed] [Google Scholar]
- 47.Söllner T, Bennett MK, Whiteheart SW, Scheller RH, Rothman JE. A protein assembly-disassembly pathway in vitro that may correspond to sequential steps of synaptic vesicle docking, activation, and fusion. Cell. 1993a;75:409–418. doi: 10.1016/0092-8674(93)90376-2. [DOI] [PubMed] [Google Scholar]
- 48.Söllner T, Whiteheart W, Brunner M, Erdjument-Bromage H, Geromanos S, Tempst P, Rothamn JE. SNAP receptors implicated in vesicle targeting and fusion. Nature. 1993b;362:318–324. doi: 10.1038/362318a0. [DOI] [PubMed] [Google Scholar]
- 49.Südhof TC. The synaptic vesicle cycle: a cascade of protein-protein interactions. Nature. 1995;375:645–653. doi: 10.1038/375645a0. [DOI] [PubMed] [Google Scholar]
- 50.Südhof TC, Rizo J. Synaptotagmins: C2-domain proteins that regulate membrane traffic. Neuron. 1996;17:379–388. doi: 10.1016/s0896-6273(00)80171-3. [DOI] [PubMed] [Google Scholar]
- 51.Tagaya M, Wilson DW, Brunner M, Arango N, Rothman JE. Domain structure of an N-ethylmaleimide-sensitive fusion protein involved in vesicular transport. J Biol Chem. 1993;268:2662–2666. [PubMed] [Google Scholar]
- 52.Tagaya M, Furuno A, Mizushima S. SNAP prevents Mg2+-ATP-induced release of N-ethylmaleimide-sensitive factor from the Golgi apparatus in digitonin-permeabilized PC12 cells. J Biol Chem. 1996;271:466–470. doi: 10.1074/jbc.271.1.466. [DOI] [PubMed] [Google Scholar]
- 53.Taylor J, Gordon-Weeks PR. Developmental changes in the calcium dependency of γ-aminobutyric acid release from isolated growth cones: correlation with growth cone morphology. J Neurochem. 1989;53:834–843. doi: 10.1111/j.1471-4159.1989.tb11780.x. [DOI] [PubMed] [Google Scholar]
- 54.Washbourne P, Schiavo G, Montecucco C. Vesicle-associated membrane protein-2 (synaptobrevin-2) forms a complex with synaptophysin. Biochem J. 1995;305:721–724. doi: 10.1042/bj3050721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Weibel ER, Bolender RP. Stereological techniques for electron microscopic morphometry. In: Hayat MA, editor. Principles and techniques of electron microscopy: biological applications, Vol 3. Van Nostrand-Reinhold; New York: 1973. pp. 239–291. [Google Scholar]
- 56.Whittaker VP, Barker LA. The subcellular fractionation of brain tissue with special references to the preparation of synaptosomes and their component organelles. In: Fried R, editor. Methods for neurochemistry, Vol 2. Marcel Dekker; New York: 1972. pp. 1–52. [Google Scholar]
- 57.Williamson LC, Halpern JL, Montecucco C, Brown JE, Naele EA. Clostridial neurotoxins and substrate proteolysis in intact neurons: botulinum neurotoxin C acts on synaptosomal-associated protein of 25 kDa. J Biol Chem. 1996;271:7694–7699. doi: 10.1074/jbc.271.13.7694. [DOI] [PubMed] [Google Scholar]
- 58.Wood MR, DeBin J, Strichartz GR, Pfenninger KH. Plasmalemmal insertion and modification of sodium channels at the nerve growth cone. J Neurosci. 1992;12:2948–2959. doi: 10.1523/JNEUROSCI.12-08-02948.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yoshida A, Oho C, Omori A, Kuwahara R, Ito T, Takahashi M. HPC-I is associated with synaptotagmin and ω-conotoxin receptor. J Biol Chem. 1992;267:24925–24928. [PubMed] [Google Scholar]