

Abstract
Tumor necrosis factor (TNF-α) underlies pathological processes and functional disturbances in acute and chronic neurological disease and injury. The neuroimmunomodulatory peptide α-MSH modulates actions and production of proinflammatory cytokines including TNF-α, but there is no prior evidence that it alters TNF-α induced within the brain. To test for this potential influence of the peptide, TNF-α was induced centrally by local injection of bacterial lipopolysaccharide (LPS). α-MSH given once i.c.v. with LPS challenge, twice daily intraperitoneally (i.p.) for 5 d between central LPS injections, or both i.p. and centrally, inhibited production of TNF-α within brain tissue. Inhibition of TNF-α protein formation by α-MSH was confirmed by inhibition of TNF-α mRNA. Plasma TNF-α concentration was elevated markedly after central LPS, indicative of an augmented peripheral host response induced by the CNS signal. The increase was inhibited by α-MSH treatments, in relation to inhibition of central TNF-α. Presence within normal mouse brain of mRNA for the α-MSH receptor MC-1 suggests that the inhibitory effects of α-MSH on brain and plasma TNF-α might be mediated by this receptor subtype. The inhibitory effect of α-MSH on brain TNF-α did not depend on circulating factors because the effect also occurred in brain tissuein vitro. This indicates that α-MSH can act directly on brain cells to inhibit their production of TNF-α. If central TNF-α contributes to pathology in CNS disease and injury, and promotes inflammation in the periphery, agents that act on brain α-MSH receptors should decrease the pathological TNF-α reaction and promote tissue survival.
Keywords: α-MSH, TNF-α, modulation of CNS inflammation, neurodegenerative disease, LPS, neuroimmunomodulation, anti-inflammatory, anticytokine
With improvement in understanding of cytokine mediators of inflammation and with recent identification of these mediators in certain CNS diseases, it is likely that the importance of inflammatory cytokines and processes in all CNS injuries and disorders will eventually be recognized. Although several inflammatory cytokines likely contribute to CNS inflammation, TNF-α may be especially important in CNS disease. TNF-α has been linked to multiple sclerosis, HIV infection of the CNS, Alzheimer’s disease, meningitis, and acute brain injury as a result of ischemia/reperfusion or of trauma.
TNF-α occurs in abundance in lesions of multiple sclerosis, which, with its capacity to promote myelin destruction and to increase adhesion molecule expression, makes it a prime suspect in etiology of the lesions (Raine, 1994). Expression of TNF-α mRNA predominates in perivascular inflammatory cuffs rather than in parenchymal cells, suggesting that circulating inflammatory cells that have entered the brain are the major source of the cytokine in this disease (Woodroofe and Cuzner, 1993). Experimental multiple sclerosis is improved by anti-TNF-α antibodies and by soluble TNF-α receptors (Selmaj and Raine, 1995). Direct infection of neurons by HIV is not responsible for manifestation of neurological disease. Rather, it seems that macrophages and/or microglia are affected and thereby alter the functions of neurons and glia via release of proinflammatory cytokines such as TNF-α (Koka et al., 1995; Tyor et al., 1995). The cytokines promote dysmyelination and inflammation, and neurological symptoms result. It seems that the HIV envelope protein gp120 is toxic to human brain cell cultures through induction of TNF-α and interleukin 6 (Yeung et al., 1995). It has been known for some time that portions of the HIV envelope protein induce TNF-α in primary brain culture (Merrill et al., 1992). Alzheimer’s disease, marked by progressive intellectual failure, is characterized by formation within the brain of aggregates of β-amyloid protein and infiltration of reactive microglia and astrocytes. Recent evidence suggests that in this disorder, microglia-induced neuronal injury stems from a synergistic effect of β-amyloid protein and interferon-γ in triggering production of proinflammatory and toxic TNF-α and nitrogen intermediates (Meda et al., 1995). TNF-α has been observed in CSF of patients with bacterial meningitis (Glimaker et al., 1993; Dulderian et al., 1995) and in experimental bacterial meningitis I (Kimberlin et al., 1995). There is evidence that such increases are a result of elevated production of TNF-α by ependymal cells of the choroid plexus (Tarlow et al., 1993). In experimental brain ischemia/reperfusion, TNF-α mRNA appears early and precedes infiltration of inflammatory cells into the injured zone (Feuerstein et al., 1994). It is found early in neurons, in and around ischemic tissue, and later in macrophages in infarcted tissue. The cytokine concentration is increased in the CSF of victims of traumatic head injury (Ross et al., 1994).
If TNF-α causes or promotes pathology in multiple CNS disorders, agents that inhibit its production should preserve neurological function. A neuroimmunomodulatory peptide, α-MSH, is known to inhibit the in vivo and in vitro actions and production of proinflammatory cytokines (Lipton and Catania, 1991, 1993, 1997), but no tests have been performed on the influence of the peptide on TNF-α production induced within the brain. The main purpose of the present investigations was to determine whether α-MSH inhibits central production of TNF-α in vivo and in vitro.
MATERIALS AND METHODS
Animals. The experiments were approved by the local Internal Review Board for Animal Research. Male BALB/c mice (Simonsen Laboratories, Gilroy CA), ∼20 gm body weight, were housed at 23–25°C in groups not exceeding five animals per cage [28 cm (length) × 18 cm (width) × 13 cm (height)]. Before the experiments, the mice were acclimatized to standard lighting and temperature conditions with food and water freely available.
Influence of α-MSH treatments on brain TNF-α in vivo. α-MSH1–13 obtained from Sigma (St. Louis, MO) was dissolved in saline just before injection. For determinations ofin vivo brain TNF-α protein and mRNA, and for MC-1 mRNA, brains were removed 1 hr after LPS administration. At the time of decapitation, trunk blood was collected in lightly heparinized 1.5 ml tubes. The LPS used to promote TNF-α production was derived fromSalmonella typhosa (Difco, catalog #0901). Saline (20 μl) or LPS (5 μg) in saline was injected into the cerebral ventricles of mice anesthetized with ether using procedures described previously (Lipton et al., 1991; Lipton and Catania, 1993). α-MSH (10 μg) for i.c.v. injection was likewise dissolved in 0.1 μl saline. Intraperitoneal injections of α-MSH (50 μg) or saline vehicle were 0.1 ml in volume.
Influence of α-MSH on TNF-α production by brain tissuein vitro. Brains were dissected intact, immediately sectioned midsagittally, and the halves were weighed and placed in 1 ml medium (DMEM with 10% FBS, 2 mml-glutamine, 200 U/ml penicillin, and 200 μg/ml streptomycin) in individual wells of a 24-well plate. Each sample was minced with a scalpel blade and washed with medium. After 15 min at room temperature, the washing was repeated. LPS (2.5 μg), LPS + α-MSH, or medium control was added to wells containing 500 μl of DMEM, and incubation at 37°C was continued for 1 hr. Ten microliters of supernatant from each well were assayed for TNF-α concentration.
TNF-α determinations. Concentrations of TNF-α in brain and plasma were determined with an L929 cell cytotoxicity assay, with human TNF-α used as control and for production of standard curves. The triplicate samples were incubated overnight with L929 cells in medium containing 100 μg/ml cyclohexamide (Sigma) in 96-well plates. The TNF-α concentrations were determined with reference to intensity at 550 nm in an ELISA plate reader (BT 2000, Fisher-Biotech). Each plate contained standards.
Northern analysis for TNF-α mRNA. RNA was isolated by homogenization in guanidinium thiocyanate followed by phenol/chloroform extraction. Total RNA (15 μg) was fractionated on a formaldehyde-denaturing agarose gel and transferred overnight to a nylon membrane that was subsequently baked at 80°C for 1 hr. The membrane was prehybridized for 6 hr at 42°C with a solution containing 2× Denhardt’s reagent, 100 μg/ml salmon sperm DNA, 40% formamide, 4× SSC, 7 μm Tris, pH 7.4, and 3% SDS. cDNA probe was radiolabeled with [32P]dCTP (3000 Ci/mmol; DuPont NEN) by random primer labeling (Prime-a-Gene Labeling System, Promega). The blot was hybridized overnight with 1–2 × 106 cpm/ml probe for mouse TNF-α at 42°C in the same solution. Blots were then washed three times in 2× SSC and 0.1% SDS at room temperature for 15 min, and then two more times for 30 min at high stringency (0.1× SSC, 0.1% SDS, 68°C). The blot was then exposed to X-ray film with 2 intensifying screens at −70°C for 2–3 d. Ethidium bromide was used to evaluate equivalence of RNA loading.
Determination of melanocortin 1 receptor mRNA. Total RNA (200 μg) was isolated from normal mouse brain by guanidinium thiocyanate, phenol, chloroform extraction and used to generate cDNA. Genomic DNA was digested with DNase in reverse transcriptase (RT) buffer for 30 min at 37°C. The DNase was inactivated by phenol–chloroform extraction. cDNA was produced using Moloney murine leukemia virus RT (BRL, Gaithersburg, MD). In some tubes, the RT was omitted to control for amplification from contaminating cDNA or genomic DNA. Portions of the cDNA were used for PCR with primer pairs specific for the murine MC-1 isoform (529 bp). The forward primer was (5′ to 3′) GTGAGTCTGGTGGAGAATGTGC and the reverse primer was TTTTGTGGAGCTGGGCAATGCC. The primers were chosen in regions of low similarity among known MC receptors. PCR mixtures contained: 1 μm primers, 1.5–2.5 mm MG2+, 200 μm dNTP, 1× reaction buffer, 1 U of Taq DNA polymerase, and 5 μl cDNA in 20 μl. The PCR profile consisted of 35 cycles of 94°C for 45 sec, 60°C for 45 sec, and 72°C for 75 sec, followed by a 5 min final extension at 72°C. The PCR products were size fractionated by agarose gel electrophoresis, transferred to nylon membrane, and hybridized with a 32P end-labeled internal oligo probe specific for the MC-1 receptor (5′ to 3′) CAGCATCGTCTCCAGCACCCTC.
Statistical analysis. Data were analyzed using one-way ANOVA procedures, followed by Dunnet’s test for multiple comparisons of group means.
RESULTS
Brain TNF-α protein and mRNA
Concentrations of TNF-α protein in control brains harvested after i.c.v. administration of LPS were markedly increased (Fig. 1), whereas in experiments on brains of normal untreated mice, TNF-α was low or undetectable (data not shown). All of the α-MSH treatments inhibited TNF-α protein production within the brain. A single central injection of the peptide greatly reduced TNF-α concentration, a similar inhibition was observed in animals given α-MSH intraperitoneally (i.p.) twice daily for 5 d (final α-MSH injection ∼16 hr before the second LPS injection), and the greatest inhibition occurred in mice given α-MSH systemically for 5 days as well as centrally at the time of LPS injection, 1 hr before brains were removed. These results indicate that a single dose of centrally injected peptide acts rapidly to inhibit local TNF-α production. Further, this single injection is as effective as repeated systemic injections of α-MSH. Given the positive effects of central and systemic α-MSH injections, it is not surprising that the greatest inhibition occurred in mice given α-MSH by both routes. The inhibitory influence of α-MSH on central TNF-α was confirmed by Northern analysis of TNF-α mRNA abundance (Fig.2).
Fig. 1.

α-MSH given centrally (10 μg) and/or systemically (50 μg, repeated) inhibited TNF-α induction within the brain by local injection of LPS (5 μg). Scores are means ± SEM. After ANOVA revealed significant differences among means (p < 0.001), Dunnett’s test was used for comparisons of individual means. **p < 0.01, relative to saline (i.c.v.) + saline (i.p.);+p < 0.01 versus other means.
Fig. 2.
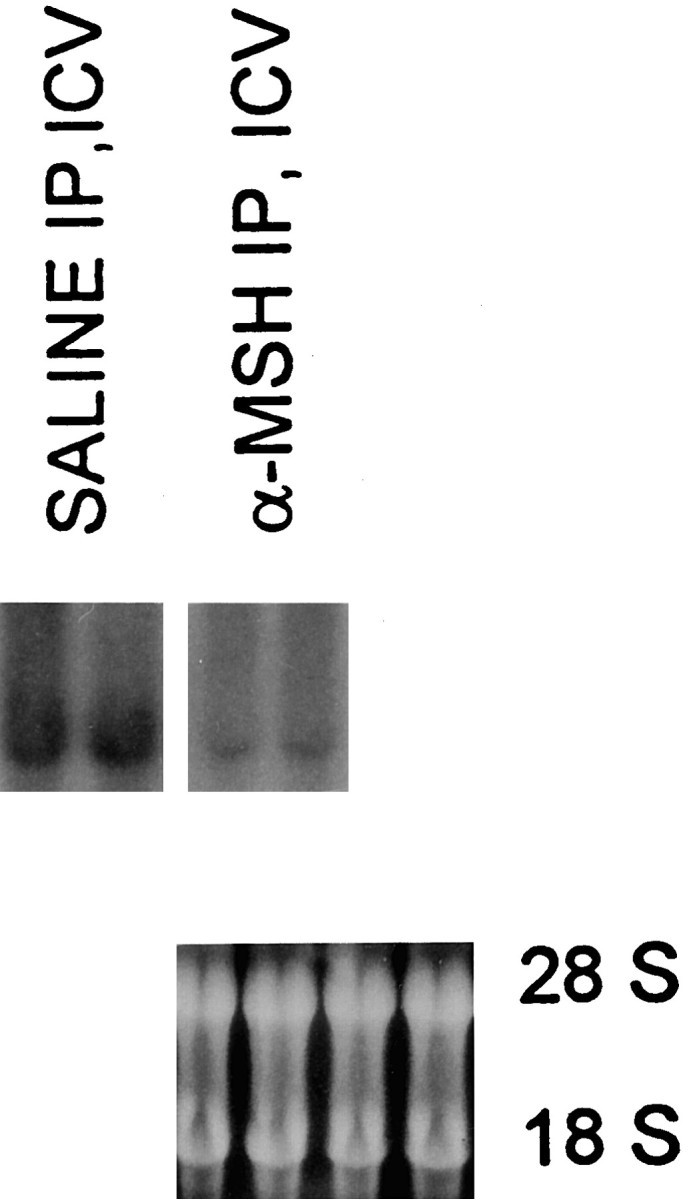
Representative Northern blot of TNF-α mRNA abundance in whole mouse brains. Representative examples from two mice given LPS alone (SALINE IP, ICV) and two others from the group (α-MSH IP, ICV) that showed the greatest inhibitory effect of α-MSH on TNF-α protein. The blot was hybridized with 32P-labeled cDNA probe for mouse TNF-α. Ethidium bromide staining of agarose gel demonstrated equal loading of samples. This is representative of two experiments.
Plasma TNF-α
Central administration of LPS markedly increased circulating TNF-α in animals that did not receive α-MSH treatment (Fig.3). These results indicate that a very small amount of LPS acting within the brain can greatly increase circulating TNF-α. The circulating concentrations of TNF-α after central administration of LPS were five- to sixfold greater than those induced in the brain. Plasma of normal untreated mice had no significant concentrations of TNF-α. Much as for brain TNF-α, plasma TNF-α concentration was reduced by a single injection of α-MSH into the brain, by repeated systemic injections of the peptide, and by combined central and peripheral injections of α-MSH. The marked inhibitory effects of combined central and systemic α-MSH injections suggest that activity of α-MSH receptors in the brain and in the periphery alter systemic TNF-α induced by LPS acting centrally. It is notable that the inhibition of plasma TNF-α paralleled the inhibition of brain TNF-α induced by central α-MSH.
Fig. 3.

α-MSH given centrally and/or peripherally inhibited increases in circulating TNF-α induced by central injection of LPS. Overall, ANOVA was significant (p < 0.001). Dunnett’s test: **p < 0.01 relative to saline (i.c.v.) + saline (i.p.); +p < 0.05 relative to saline (i.c.v.) + α-MSH (i.p.) p< 0.01 relative to α-MSH (i.c.v.) + saline (i.p.).
Evidence of MC-1 receptor expression in the brain
To test the idea that the primary α-MSH receptor MC-1, recently found in murine macrophages and in human macrophages and neutrophils (Star et al., 1995; Rajora et al., 1996; Catania et al., 1996), likewise occurs in brain tissue, normal mouse brain was subjected to RT-PCR and Southern analysis. This analysis revealed a substantial signal at 529 bp, consistent with the size of the cDNA for the murine MC-1 receptor (Fig. 4). Control analysis of other murine tissues (e.g., lung), and of cells of the L929 fibroblast line used for TNF-α determinations, did not show any evidence of MC-1 expression.
Fig. 4.

Representative Southern blot of RT-PCR product for murine MC-1 receptor in normal mouse brain. No bands were observed in lanes containing reaction mixture without tissue (not shown) or without RT (−RT). Lanes containing mouse genomic DNA (1 μg) or MC-1 cDNA also contained the 529 bp product. Approximately 10 μg of cDNA (20 μl) was used in this experiment. Exposure time was 30 min. Washes (all room temperature): first washes = 3 for 5 min, 2× SSC + 0.1% SDS; second washes = 2 for 30 min, 2× SSC + 0.1% SDS.
Effect of α-MSH on TNF-α production by brain tissuein vitro
If, as the results above suggest, α-MSH acts centrally to inhibit TNF-α production, the peptide may reduce production of the cytokine by brain cells per se, without the influence of factors in the blood supply in the live animal. To test this idea, brain samples were incubated with LPS (2.5 μg), and with LPS + concentrations of α-MSH for 1 hr, and samples of medium were analyzed for TNF-α. Marked increases in TNF-α were induced by LPS in control tissue. α-MSH inhibited TNF-α production in a dose-related fashion (Fig.5), which indicates a direct effect of the peptide on brain cells that produce the cytokine. These results demonstrate that no secondary influence of α-MSH via circulating mediators is necessary for the inhibition.
Fig. 5.

Dose-related inhibition by α-MSH of TNF-α protein production by tissue derived from half murine brains incubated with LPS for 1 hr. Control value was the mean TNF-α production (60.1 ± 12.7 U/gm) for all of the opposite hemibrains incubated with LPS but without α-MSH. Scores are means ± SEM of percent inhibition of TNF-α production relative to control derived from three separate experiments.
Brain TNF-α mRNA after a single injection of LPS
In view of reports of CNS tolerance to central LPS administration (Faggioni et al., 1995), the effect of single LPS (5 μg) injections into the brain was tested in otherwise untreated mice. These injections resulted in the greatest abundance of brain TNF-α mRNA observed in this study (Fig. 6). This result indicates that the greatest effect of LPS on TNF-α expression within the brain occurs after the first injection and, with evidence of lesser abundance after a second injection made 5 d later, supports the idea of CNS tolerance to LPS treatment.
Fig. 6.
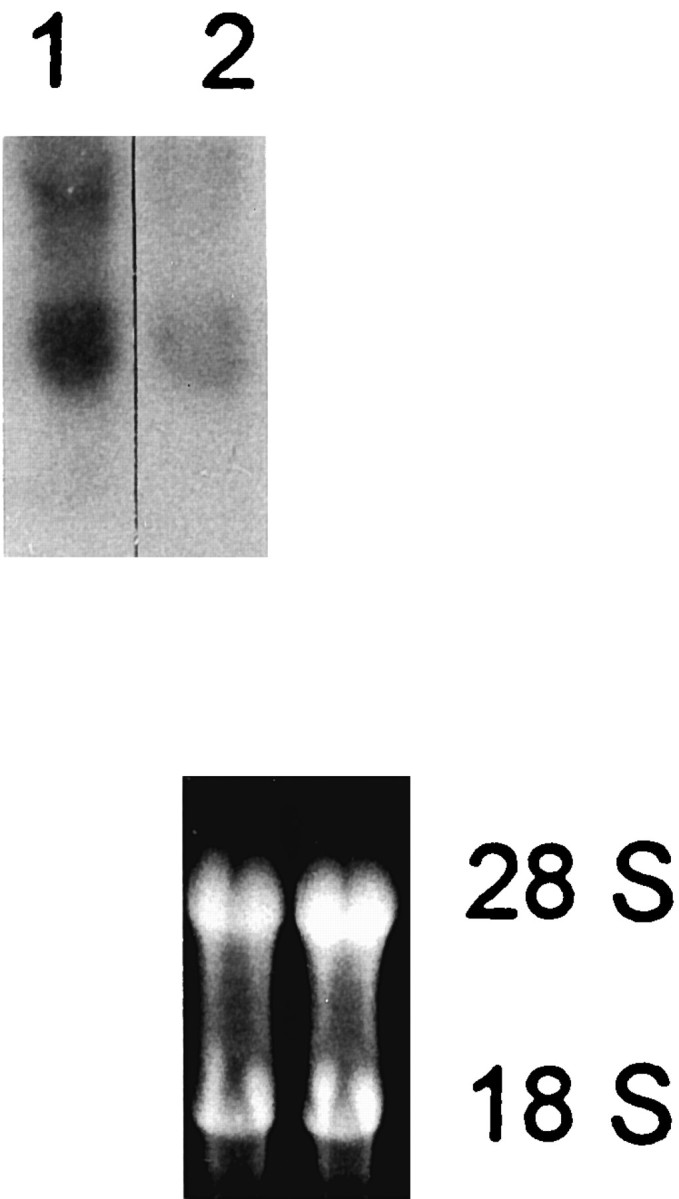
Representative Northern blot showing that single acute i.c.v. injections of LPS (1) had greater effects on brain TNF-α mRNA abundance than injections that were preceded 5 d earlier by a similar i.c.v. injection (2). Ethidium bromide staining of agarose gel demonstrated equal loading of samples. Representative of two experiments: 2 d exposure, −70°C, two intensifying screens.
DISCUSSION
TNF-α within the brain
α-MSH acts locally within the brain to inhibit production of TNF-α both in vivo and in vitro. Central injection of α-MSH inhibited TNF-α production, and expression of TNF-α mRNA, in live mice within 1 hr after local injection of LPS. This result was confirmed in vitro by marked inhibition by α-MSH of TNF-α production by brain tissue. These converging observations indicate that the neuropeptide α-MSH modulates brain TNF-α concentration via actions directly within the parenchyma. The cell source of this rapid increase in TNF-α was not determined; however, it is clear that astrocytes (for example, see Kimberlin et al., 1995) as well as microglia (for example, see Meda et al., 1995) and ependymal cells (for example, see Tarlow et al., 1993) can produce the cytokine.
Systemically administered α-MSH inhibits brain TNF-α production
Repeated i.p. injections of α-MSH likewise inhibited the increase in brain TNF-α caused by central injection of LPS, and the combination of central and systemic α-MSH injections had the greatest inhibitory effect. This observation suggests that repeated systemic administration of α-MSH alters the reactivity of brain cells to LPS stimulation, although the mechanisms underlying this alteration are unknown. It is notable that the greatest inhibitory effect on TNF-α production was caused by repeated administration of α-MSH and of acute central administration of the peptide. It may be that the latter effect results from a combination of influences induced by centrally injected α-MSH acting within the brain, and systemically injected α-MSH acting on peripheral host cells and on brain melanocortin receptors. There is direct evidence that α-MSH administered systemically reaches the brain (Wilson, 1988), albeit in small amounts. Further, systemically administered α-MSH reduces fever in minutes, an antipyretic effect that can occur only through an action of the peptide within the CNS. This observation is very important for potential clinical use of α-MSH to control TNF-α production in animals or humans in whom local brain injections are unsafe or impractical. One of the main questions is whether α-MSH could be administered before neurosurgical procedures and thereby modulate local inflammatory responses to surgery or contamination. In keeping with this idea, in recent research on basilar artery ischemia/reperfusion in dogs (Huh et al., 1997), disturbance of the brainstem auditory evoked potential was less in animals given α-MSH IV, particularly when it was administered during ischemia.
Central LPS markedly increases circulating TNF-α
It is remarkable that a small amount of LPS injected into the brain promotes not only local TNF-α production but circulating TNF-α even moreso. This observation, noted previously (Faggioni et al., 1995), has far-reaching implications. In brief, it may be that the induction of central TNF-α, or a more direct action of LPS within the brain, causes marked increases in production of TNF-α within the circulation, presumably by monocytes. Because the concentrations of TNF-α induced within the brain are substantially less than those induced in the circulation, it is unlikely that the circulating TNF-α originates from the brain. The nature of the signals from brain to the periphery are not clear. TNF-α-producing cells in the brain may invoke neural and/or humoral signals. Nor is the physiological significance of the phenomenon known, although it may be that sensing of central TNF-α promotes circulating TNF-α to counteract challenges to the host. Such an increase in circulating TNF-α after central LPS may explain the increase in peripheral inflammation after central injection of “endogenous pyrogen” (EP, Dulaney et al., 1992). In those experiments, central administration of undiluted EP significantly increased peripheral inflammation in the mouse; dilutions of EP had lesser or no effects. A proinflammatory influence of central TNF-α could be detrimental in organisms with preexisting inflammatory disease in the periphery; for example, central TNF-α might promote peripheral inflammation in rheumatoid arthritis or inflammatory bowel disease by inducing high concentrations of the cytokine in the bloodstream.
Central α-MSH modulates the increase in circulating TNF-α induced by LPS acting solely within the brain
Whatever the basis of the increase in circulating TNF-α, injection of α-MSH into the brain reduced it. The dose of α-MSH that inhibited circulating TNF-α effectively reduced peripheral inflammation after central injection in earlier experiments (Lipton et al., 1991; Ceriani et al., 1994; Macaluso et al., 1994). If circulating TNF-α contributes to the inflammatory response in the periphery, central α-MSH likely inhibits this inflammation in some part by modulating circulating TNF-α. It is clear that descending neurogenic anti-inflammatory pathways in the spinal cord are necessary for the anti-inflammatory actions of central α-MSH (Macaluso et al., 1994). It may be that these pathways are the means through which central α-MSH modulates production of TNF-α by peripheral host cells, although it is likely that modulation of release of peripheral substance P and calcitonin gene-related peptide is also responsible.
Effects of systemic α-MSH
The results indicate that systemically administered α-MSH inhibits circulating TNF-α concentration. There are three potential mechanisms: (1) a direct inhibitory action on TNF-α producing cells (e.g., monocytes) in the circulation, (2) an effect of the α-MSH that penetrates to the brain on central descending anti-inflammatory pathways, or (3) some combination of these two. That α-MSH inhibits release of proinflammatory mediators from monocytic cells is clear; we have noted (Lipton et al., 1997) that TNF-α production by human peripheral blood mononuclear cells is inhibited by α-MSH, which indicates that a direct effect of the peptide on melanocortin receptors on monocytes is possible. As stated above, α-MSH injected systemically is known to reach the brain and may, therefore, inhibit TNF-α production in the circulation via unknown pathways. Finally, it is clear that α-MSH can alter inflammatory processes by acting directly on host cells in the periphery and by acting within the brain. Because systemic α-MSH is available at both sites, it is reasonable to assume that the peptide acts at both these sites.
Evidence for MC-1 receptors within the brain
The inhibitory effects of α-MSH on TNF-α production by brain tissue in vivo and in vitro indicates that receptors for the peptide reside within the brain. Indeed, there is prior evidence for three melanocortin receptor subtypes in brain: MC-3, MC-4, and MC-5 (Hol et al., 1995). Further, our RT-PCR evidence supports observations made in in situ hybridization and immunohistochemistry experiments (Xia et al., 1995) that the MC-1 receptor is likewise expressed in brain tissue. MC-1 may therefore contribute to the anti-TNF-α activity of the peptide. It is not yet possible to determine the precise contribution of the melanocortin receptor subtypes to inhibition of TNF-α production; all four subtypes found within the brain respond to α-MSH and may therefore contribute to its anti-inflammatory effect. However, it should be possible to test the relative contributions of the receptor subtypes to modulation of TNF-α production in whole brain by incubating brain tissue with antibodies to them.
In summary, the results extend to the CNS the observations of the anti-inflammatory effects of the neuropeptide α-MSH. Inflammation in peripheral tissues is modulated by α-MSH acting on its receptors on neutrophils and macrophages and those within the brain. The rapid anti-TNF-α effects of α-MSH within brain tissue per se likely occur via direct actions of the peptide on local brain cells (astrocytes and microglia) that produce TNF-α. Modulation of circulating TNF-α via α-MSH-induced inhibition of central cytokine signals provides a new avenue for control of peripheral inflammatory reactions, via actions on melanocortin receptors within the brain.
Footnotes
This research was supported by National Institutes of Health Grant NS10046 from the National Institute of Neurological Diseases and Stroke, a Comitato Nazionale delle Richerche Grant, VIII Progetto AIDS, Istituto Superiore di Sanita, Italy, Grant PP0406 from the National Multiple Sclerosis Society, and NATO Collaborative Research Grant CGR 950556.
Correspondence should be addressed to Dr. J. M. Lipton, University of Texas Southwestern Medical Center, Dallas TX 75235-9040.
REFERENCES
- 1.Catania A, Rajora N, Capsoni F, Minonzio F, Star RA, Lipton JM. The neuropeptide α-MSH has specific receptors on neutrophils and reduces chemotaxis in vitro. Peptides. 1996;17:675–679. doi: 10.1016/0196-9781(96)00037-x. [DOI] [PubMed] [Google Scholar]
- 2.Ceriani G, Macaluso A, Catania A, Lipton JM. Central neurogenic antiinflammatory action of α-MSH: modulation of peripheral inflammation induced by cytokines and other mediators of inflammation. Neuroendocrinology. 1994;59:138–143. doi: 10.1159/000126650. [DOI] [PubMed] [Google Scholar]
- 3.Dulaney R, Macaluso A, Woerner J, Hiltz ME, Catania A, Lipton JM. Changes in periperal inflammation induced by the central actions of an α-MSH analog and of endogenous pyrogen. Prog Neuroendocrin Immunol. 1992;5:179–186. [Google Scholar]
- 4.Dulderian SJ, Kilpatrick L, Costarino AT, Jr, McCawley L, Fein J, Corcoran L, Zirin S, Harris MC. Cytokine elevations in infants with bacterial and aseptic meningitis. J Pediatr. 1995;125:872–876. doi: 10.1016/s0022-3476(95)70199-0. [DOI] [PubMed] [Google Scholar]
- 5.Faggioni R, Fantuzzi G, Villa P, Buurman W, van Tits LJH, Ghezzi P. Independent down-regulation of central and peripheral tumor necrosis factor production as a result of lipopolysaccharide tolerance in mice. Infect Immunol. 1995;63:1473–1477. doi: 10.1128/iai.63.4.1473-1477.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Feuerstein GZ, Liu T, Barone FC. Cytokines, inflammation, and brain injury: role of tumor necrosis factor. Cerebrovasc Brain Metab Rev. 1994;6:341–360. [PubMed] [Google Scholar]
- 7.Glimaker M, Kragsbjerg P, Forsgren M, Olcen P. Tumor necrosis factor α (TNFα) in cerebrospinal fluid from patients with meningitis of different etiologies: high levels of TNFα indicate bacterial meningitis. J Infect Dis. 1993;167:882–889. doi: 10.1093/infdis/167.4.882. [DOI] [PubMed] [Google Scholar]
- 8.Hol EM, Gispen WH, Bar PR. ACTH-related peptdes: receptors and signal transduction systems involved in their neurotropic and neuroprotective actions. Peptides. 1995;16:979–993. doi: 10.1016/0196-9781(95)00017-e. [DOI] [PubMed] [Google Scholar]
- 9.Huh S-K, Lipton JM, Batjer HHI. The protective effects of α-melanocyte stimulating hormone on canine brainstem ischemia. J Neurosurg. 1997;40:132–139. doi: 10.1097/00006123-199701000-00030. [DOI] [PubMed] [Google Scholar]
- 10.Kimberlin DW, Velasco S, Paris MM, Hickey SM, McCracken GH, Jr, Nisen PD. Modulation of expression of genes involved in the inflammatory response by lipopolysaccharide and temperature in cultured human astroglial cells. Immunol Invest. 1995;24:775–785. doi: 10.3109/08820139509060705. [DOI] [PubMed] [Google Scholar]
- 11.Koka P, He K, Camerini D, Tran T, Yashar SS, Merrill JE. The mapping of HIV-1 gp160 epitopes required for interleukin-1 and tumor necrosis factor production in glial cells. J Neuroimmunol. 1995;57:179–191. doi: 10.1016/0165-5728(94)00184-p. [DOI] [PubMed] [Google Scholar]
- 12.Lipton JM, Catania A. Pyrogenic and inflammatory actions of cytokines and their modulation by neuropeptides: techniques and interpretations. In: DeSouza EB, editor. Neurobiology of cytokines, part B. Academic; Orlando, FL: 1993. pp. 61–79. [Google Scholar]
- 13.Lipton JM, Catania AP (1997) Antiinflammatory actions of the neuroimmunomodulator α-MSH. Immunol Today, in press. [DOI] [PubMed]
- 14.Lipton JM, Macaluso A, Hiltz ME, Catania A. Central administration of the peptide α-MSH inhibits inflammation in the skin. Peptides. 1991;12:795–798. doi: 10.1016/0196-9781(91)90135-c. [DOI] [PubMed] [Google Scholar]
- 15. Lipton JM, Catania A, Rajora N, Ceriani G, Star RA, Boccoli G. Modulation of inflammatory effects of cytokines by the neuropeptide α-MSH. Proinflammatory and antiinflammatory peptides. Lung biology in health and disease. 1997. Dekker; New York: in press. [Google Scholar]
- 16.Macaluso A, McCoy D, Ceriani G, Watanabe T, Biltz J, Catania A, Lipton JM. Antiinflammatory influences of α-MSH molecules: central neurogenic and peripheral actions. J Neurosci. 1994;14:2377–2382. doi: 10.1523/JNEUROSCI.14-04-02377.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Meda L, Cassatella MA, Szendrei GI, Otvos L, Jr, Baron P, Villalba M, Ferrari D, Rossi F. Activation of microglial cells by β-amyloid protein and interferon γ. Nature. 1995;374:647–650. doi: 10.1038/374647a0. [DOI] [PubMed] [Google Scholar]
- 18.Merrill JE, Koyanagi Y, Zack J, Thomas L, Martin F, Chen IS. Induction of interleukin-1 and tumor necrosis factor α in brain cultures by human immunodeficiency virus type I. J Virol. 1992;66:2217–2225. doi: 10.1128/jvi.66.4.2217-2225.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Raine CS. The Dale E. McFarlin Memorial Lecture: the immunology of the multiple sclerosis lesion. Ann Neurol. 1994;36:561–572. doi: 10.1002/ana.410360716. [DOI] [PubMed] [Google Scholar]
- 20.Rajora N, Ceriani G, Catania A, Star RA, Murphy MT, Lipton JM. α-MSH production, receptors, and influence on neopterin, in a human monocyte/macrophage cell line. J Leukocyte Biol. 1996;59:248–253. doi: 10.1002/jlb.59.2.248. [DOI] [PubMed] [Google Scholar]
- 21.Ross SA, Halliday MI, Campbell GC, Byrnes DP, Rowlands BJ. The presence of tumour necrosis factor in CSF and plasma after severe head injury. Br J Neurosurg. 1994;8:419–425. doi: 10.3109/02688699408995109. [DOI] [PubMed] [Google Scholar]
- 22.Selmaj KW, Raine CS. Experimental autoimmune encephalomyelitis: immunotherapy with anti-tumor necrosis factor antibodies and soluble tumor necrosis factor receptors. Neurology. 1995;45:S44–S49. doi: 10.1212/wnl.45.6_suppl_6.s44. [DOI] [PubMed] [Google Scholar]
- 23.Star RA, Rajora N, Huang J, Chavez R, Catania A, Lipton JM. Evidence of autocrine modulation of macrophage nitric oxide synthase by α-MSH. Proc Natl Acad Sci USA. 1995;92:8016–8020. doi: 10.1073/pnas.92.17.8016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tarlow MJ, Jenkins R, Comis SD, Osborne MP, Stephens S, Stanley P, Crocker J. Ependymal cells of the choroid plexus express tumor necrosis factor α. Neuropathol Appl Neurobiol. 1993;19:324–328. doi: 10.1111/j.1365-2990.1993.tb00447.x. [DOI] [PubMed] [Google Scholar]
- 25.Tyor WR, Wesselingh SL, Griffin JW, McArthur JC, Griffin DE. Unifying hypothesis for the pathogenesis of HIV-associated dementia complex, vacuolar myelopathy, and sensory neuropathology. J Acquir Immune Defic Syndr Hum Retrovirol. 1995;9:379–388. [PubMed] [Google Scholar]
- 26.Wilson JF. Low permeability of the blood-brain barier to nanomolar concentrations of immunoreactive α-melanotropin. Psychopharmacology. 1988;96:262–266. doi: 10.1007/BF00177572. [DOI] [PubMed] [Google Scholar]
- 27.Woodroofe MN, Cuzner ML. Cytokine expression in inflammatory multiple sclerosis lesions: detection by non-radioactive in situ hybridization. Cytokine. 1993;5:583–588. doi: 10.1016/s1043-4666(05)80008-0. [DOI] [PubMed] [Google Scholar]
- 28.Xia Y, Wikberg JE, Chhajlani V. Expression of melanocortin 1 receptor in periacqueductal gray matter. NeuroReport. 1995;6:2193–2196. doi: 10.1097/00001756-199511000-00022. [DOI] [PubMed] [Google Scholar]
- 29.Yeung MC, Pulliam L, Lau AS. The HIV envelope protein gp120 is toxic to human brain-cell cultures through the induction of interleukin-6 and tumor necrosis factor α. AIDS. 1995;9:137–143. [PubMed] [Google Scholar]