

Abstract
Spiral ganglion neurons (SGNs) require both pre- and postsynaptic contacts to maintain viability. BDNF, NT-3, chlorphenylthio-cAMP, and depolarization (veratridine or elevated [K+]o) all promote survival of SGNs in vitro, depolarization being the most effective. Combining different trophic stimuli increases survival in an additive manner. Neurotrophins and depolarization maintain comparable soma size and neurite extension, but SGNs are shrunken in cAMP. Elevated [K+]o has a biphasic effect on SGN survival; survival improves as [K+]o is raised to 30 mm (30K) and falls as [K+]o is further increased; SGN survival in 80 mm[K+]o (80K) is poor relative to survival in 30K. These responses to elevated [K+]o are potentiated by an L-type channel agonist, whereas L-type Ca2+ channel blockers antagonize the trophic effect of depolarization. Four hours after depolarization, steady-state [Ca2+]i is elevated in SGNs in 30K and further elevated in SGNs in 80K. At 22 hr after depolarization, by which time death of neurons in 80K has begun, elevated [Ca2+]i levels in surviving neurons in 80K are not higher than those in neurons in 30K (∼150–450 nm), suggesting that neurons with high [Ca2+]i are preferentially lost. Veratridine causes oscillatory increases in [Ca2+]i to 250–350 nm. Thus, [Ca2+]i is predictive of cell survival; [Ca2+]i elevated to 100–500 nm in a sustained or oscillatory manner permits SGN survival independent of exogenous neurotrophic factors. Higher [Ca2+]i is associated with cell death.
Keywords: depolarization, calcium, neurotrophic factor, neurotrophin, spiral ganglion neuron, cell survival
Neuronal survival is regulated in a complex environment in which neurons are exposed simultaneously to a variety of stimuli, including peptide neurotrophic factors and depolarization. Integration of multiple distinct trophic stimuli may be required for neuronal survival, to achieve specific combinations of signals or to reach a threshold level of trophic signaling. Trophic support derived from both pre- and postsynaptic cells may be required. Presynaptic cells can provide trophic support by releasing neurotrophic factors or neurotransmitters or by depolarization attributable to synaptic activity. Target-derived neurotrophic support has been extensively investigated, but neurotrophic support by presynaptic cells has received less attention.
Spiral ganglion neurons (SGNs) provide a favorable system for experimental investigation of neurotrophic support by presynaptic cells. SGNs are bipolar neurons; a central axon projects to the cochlear nucleus and a peripheral axon to the organ of Corti. Hair cells, the auditory receptors, are the sole presynaptic input to the SGNs. Both pre- and postsynaptic partners are necessary for SGN survival in vivo; neither is sufficient alone. Hair cells can be selectively killed in vivo with aminoglycoside antibiotics, thus deafferenting the SGNs. This results in the eventual death of the SGNs (Spoendlin, 1975; Webster and Webster, 1981; Koitchev et al., 1982; Bichler et al., 1983), even though the central projection remains intact. This suggests that different sources of neurotrophic stimuli are summed to a threshold to support survival. One aim of this study is to test this possibility using cultured SGNs. Our data indicate that different types of neurotrophic stimuli are indeed additive.
SGNs express TrkB and TrkC (Ylikoski et al., 1993; Schecterson and Bothwell, 1994) and are supported by BDNF, NT-4, and NT-3 in vitro (Avila et al., 1993; Vazquez et al., 1994; Zheng et al., 1995). SGNs can receive target-derived neurotrophic support by either BDNF or NT-3 in vivo, because both are expressed in the cochlear nucleus (Lefebvre et al., 1994). Type I SGNs can receive presynaptic neurotrophic support from NT-3, expressed in inner hair cells (Ylikoski et al., 1993; Schecterson and Bothwell, 1994).
Electrical activity is another potential source of presynaptic neurotrophic support and has been implicated in neuronal survivalin vivo. Blockade of afferent input increases death in vivo in central (Catsicas et al., 1992; Galli-Resta et al., 1993) and peripheral neurons (Wright, 1981; Furber et al., 1987; Meriney et al., 1987; Maderdrut et al., 1988). In the avian auditory system, removal of the cochlea causes rapid atrophic changes, culminating in a 25–30% loss of neurons in the target, nucleus magnocellularis (Born and Rubel, 1985; Steward and Rubel, 1985; Sie and Rubel, 1992); blockade of electrical activity results in rapid atrophic changes and loss of magnocellularis neurons comparable with those after complete cochlear ablation (Born and Rubel, 1988; Pasic and Rubel, 1989; Rubel et al., 1990; Sie and Rubel, 1992).
Depolarization is also a neurotrophic stimulus in vitro for central and peripheral neurons (Scott and Fisher, 1970; Bennett and White, 1979; Chalazonitis and Fischbach, 1980; Wakade et al., 1983;Gallo et al., 1987); conversely, blockade of electrical activityin vitro reduces neuronal survival (Lipton, 1986; Ruitjer et al., 1991). The neurotrophic effect of depolarization is apparently a consequence of a sustained rise in cytosolic Ca2+, entering through L-type Ca2+ channels (Gallo et al., 1987; Collins and Lile, 1989; Koike et al., 1989; Collins et al., 1991). This is in spite of the critical role of cytosolic Ca2+ in mediating neuronal degeneration (Choi, 1988; Siesjo et al., 1989). A hypothesis unifying these observations proposes that [Ca2+]i must rise to a particular “setpoint” to promote survival in the absence of neurotrophic factor; degeneration is the result of very high [Ca2+]i (Collins et al., 1991; Koike and Tanaka, 1991; Franklin and Johnson, 1992). Thus, survival occurs within a range of elevated [Ca2+]i, with the lower end of the range determined by the Ca2+ setpoint for that neuron and the upper end by that neuron’s sensitivity to Ca2+-mediated neurotoxicity. Although this “Ca2+ setpoint” hypothesis can account for neuronal survival in varying levels of sustained depolarization and after exposure to excitotoxins, a recent study of sympathetic neurons (Franklin et al., 1995) found no significant correlation between survival and [Ca2+]i, inconsistent with the hypothesis.
Investigating the mechanisms of presynaptic neurotrophic support is the second aim of this study. We find that depolarization is a potent source of neurotrophic support for SGNs, more effective than neurotrophins or a permeant cAMP analog. Ca2+ entry via L-type channels is necessary for trophic support by depolarization, and the steady-state level of cytosolic Ca2+ is indeed predictive of cell survival. Our data support a “Ca2+setpoint” hypothesis and can explain observations (Franklin et al., 1995) not apparently consistent with it.
MATERIALS AND METHODS
Cell culture and trophic factor deprivation.Dissociated spiral ganglion cell cultures were prepared from postnatal day 5 (P5) Sprague Dawley (Sasco) rat cochleae and maintained using a procedure modified from that described by Lefebvre et al. (1991). The protocol was approved by the University of Iowa Animal Care and Use Committee. Cochleae were aseptically removed from the temporal bone and placed in ice-cold PBS. The bony cochlear capsule was removed, followed by the spiral ligament. The organ of Corti was then removed, transecting the outer radial fibers, leaving the SGNs within the modiolus. Modiolar bone was removed and surrounding connective tissue incompletely removed. Ganglia were collected in ice-cold HBSS. Enzymatic dissociation was then performed in Ca2+/Mg2+-free HBSS with 0.1% collagenase, 0.1% trypsin, and 0.01% DNase I (Boehringer Mannheim, Indianapolis, IN) in a gently shaking 37°C water bath for 25 min. FCS (Life Technologies, Gaithersburg, MD) was added to 10% to inhibit enzymatic activity, followed by three washes in serum-free DMEM and one wash in culture medium (see below). The ganglia were dissociated mechanically using two fire-polished reduced-orifice glass Pasteur pipettes, the second considerably more narrow than the first. The ganglia were gently triturated approximately 15 times with each pipette and diluted with culture medium (8–10 ganglia/2 ml). Cells were allowed to adhere for 4 hr before the addition of trophic factors or other changes made to the culture medium.
Dissociated spiral ganglion cell cultures were maintained in a serum-free culture medium consisting of high-glucose (4.5 mg/ml) DMEM with 0.1 mg/ml penicillin and 0.1 mg/ml streptomycin, in addition to a serum-free supplementation, a modification of the N2 formulation (Bottenstein and Sato, 1979). Our supplementation consisted of human apo-transferrin (100 μg/ml), putrescine (100 μm), progesterone (20 nm), selenium (30 nm), crystalline BSA (20 μg/ml), and d-glucose (1.5 mg/ml; to a final glucose concentration of 6 mg/ml in the culture medium). Stocks (100 ×) were prepared and kept at −80°C. Fresh insulin (10 μg/ml) was added to the supplemental medium on the day of culture. All of these medium supplements were from Sigma (St. Louis, MO). In all chronic elevated [K+]o depolarization experiments, Na+ was replaced by equimolar K+to maintain osmolarity.
Cells were cultured in 96-well tissue culture dishes (Falcon) that had been coated sequentially with poly-ornithine (0.1 mg/ml in 10 mm borate buffer, pH 8.4) for 1 hr at 20°C followed by laminin (mouse EHS, Boehringer Mannheim and Life Technologies, 20 μg/ml in HBSS) overnight at 4°C. Cells were grown in 100 μl medium per well at 37°C in a 6.5% CO2 incubator.
Neuronal survival and neurite growth assay. Spiral ganglion cells were cultured for 48 hr in control or experimental media, then fixed with freshly prepared 4% paraformaldehyde and 5% sucrose in 0.1m phosphate buffer. Fixation was performed at 4°C for 30 min, and the cultures were then washed with PBS. The cells were treated with 0.1% H2O2 in 100% methanol for 30 min at 20°C to reduce endogenous peroxidase activity and then washed three times with PBS. The cells were permeabilized using 0.2% Triton X-100 (Fisher, Houston, TX) in PBS for 30 min at 20°C. A “blocking buffer” (2% BSA, 10% goat serum, and 0.2% Triton X-100 in PBS) was then added to reduce nonspecific antibody binding. After 45 min at 37°C, the blocking buffer was removed and rabbit anti-neuron specific enolase (anti-NSE, Zymed, San Francisco, CA), diluted 1:100 in blocking buffer, was added. Anti-NSE specifically labels neurons and in experiments not shown here labels the same cells as does anti-68 kDa neurofilament antibody. The cells were incubated overnight at 4°C in anti-NSE then washed three times with PBS. HRP-conjugated goat anti-rabbit-HRP (Zymed), diluted 1:100 in blocking buffer, was added. After 30 min at 20°C, the antibody was removed, and the cultures were washed twice in PBS and once in 0.1 m acetate buffer, pH 5.2, before adding the chromogen 3-amino-9-ethylcarbazole (Sigma). The AEC (1.25 mm in 0.1 m acetate buffer + 0.03% H2O2) was allowed to react at room temperature, with the end-point (typically ∼5 min) determined by monitoring under an inverted microscope, and the reaction terminated by washing with PBS.
SGNs were counted and photographed using a Nikon Diaphot inverted microscope. Counting was done by two individuals. Criteria used to determine neuronal viability were (1) NSE positivity, with a visible nucleus, and (2) absence of nuclear pyknosis. Separate tallies were made of surviving neurons with neurites ≥3 cell diameters in length and those without such neurites. Each trophic factor test was performed in triplicate and repeated on at least three different occasions.
Calcium imaging experiments. Cells were plated in the center of a 25 mm glass coverslip (Fisher), sequentially coated with poly-ornithine and laminin (as described above), within a small well made with an 8 × 8 mm glass cloning cylinder (Bellco) and silicone caulk. The cells were cultured for 2 hr in control medium to allow attachment, then the medium was switched to elevated [K+]o or maintained as control for an additional 4 or 22 hr. At 6 or 24 hr after plating, the cells were loaded with 1 μm fura-2-acetoxymethyl ester and 0.025% pluronic acid (Molecular Probes, Eugene, OR) in 0.035% DMSO at 37°C for 25 min. The cells were then washed three times with N2 media (containing factors to be studied). The coverslip was clamped into a chamber and then placed in a temperature-controlled stage (Medical Systems, Greenvale, NY), held at 37°C, and superfused with 7% CO2/93% air. Viable cells were identified using Hoffman optics and evidence of fura-loading. Neurons were identified by morphology under Hoffman optics. Measurements were taken randomly throughout the culture, obtaining >100 cells per condition.
Microfluorimetry was performed on a Nikon Diaphot microscope with excitation provided by two pulsed lasers (337 and 380 nm, 3 nsec pulse duration, Laser Science, Newton, MA) coupled via a fused quartz light guide to the epi-illumination. The diffused and collimated light was reflected by a dichroic mirror (Omega, 400 DCLP) into the objective (Nikon UV-Fluor, 40×). Fluorescence emitted by the cell was transmitted through a 510 nm edge-pass filter (Omega 510 WB 40) onto the faceplate of an intensified CCD camera (ICCD 2525FS, Video Scope, Washington, DC). Images were acquired by alternating the firing of the two lasers every 90 msec. Intracellular Ca2+ concentrations were estimated using the ratio R of the fluorescent intensity at 337 nm to that at 380 nm in the equation of Grynkiewicz et al. (1985):
where KD is the dissociation constant of fura-2, Rmin is the ratio with 0 calcium, Rmax is the ratio with saturating calcium, and Sf2/Sb2 is the ratio of the intensity of fluorescence with 380 nm excitation for a solution with no calcium divided by that for a solution with saturating calcium. In vitro calibration was performed using a set of buffered Ca2+ concentrations (Molecular Probes). The average intensity of fluorescence over a cell was determined using a rectangular region of interest in the program Transform (Spyglass, Champaign, IL).
Supplies. NT-3 was the generous gift of Genentech (San Francisco, CA). BDNF was purchased from Promega (Madison, WI). BAY K 8644, nifedipine, verapamil, and ω-conotoxin GVIA were obtained from Calbiochem (La Jolla, CA). Chlorphenylthio-cAMP was obtained from Boehringer Mannheim. All culture media were prepared by the University of Iowa Diabetes and Endocrinology Research Center Cell Culture Core, with the exception of the Ca2+-free DMEM, which was obtained from Life Technologies. All other chemicals were obtained from Sigma. Lipid-soluble chemicals were maintained as stocks at 10,000× working concentration in DMSO, except that BAY K 8644 was dissolved in ethanol. These vehicles alone had no effect on survival at the dilutions used.
RESULTS
BDNF, NT-3, depolarization, and cAMP rescue SGNs from cell deathin vitro
A single experiment uses dissociated spiral ganglia from five P5 rats plated in 36 wells of a 96-well plate. This yields 800–900 neurons per well, a plating efficiency of ∼13% given that there are ∼24,000 SGNs in each cochlea of a P5 rat (Rueda et al., 1987).
Neuronal survival was determined by counting NSE-positive cells as described in Materials and Methods. Because NSE is a neuron-specific soluble cytosolic enzyme that should diffuse away from cell debris, we reason that cells positive for NSE immunoreactivity are neurons that must have been alive and intact at the the time of fixation. After 48 hr in culture in N2 medium with no additional trophic agents (control medium), fewer than 10 neurons remained alive in each well. As shown in Figure 1A, the neurotrophins NT-3 and BDNF promoted survival in a dose-dependent manner, with the effect saturating at 100 ng/ml, as has been shown previously (Lefebvre et al., 1994; Pirvola et al., 1994; Zheng et al., 1995). The permeant cAMP analog chlorphenylthio-cAMP (cpt-cAMP) promoted survival, with a maximal effect (at 1 mm), quantitatively comparable with the effect of the neurotrophins.
Fig. 1.

A, Trophic support of SGNs by neurotrophins, chlorphenylthio-cAMP, and depolarization. Spiral ganglion cultures were fixed and immunostained with anti-NSE, as described in Materials and Methods, after culturing for 48 hr in control DMEM/N2 with no additives (Con), with BDNF or NT-3 at the indicated concentrations, with 1 mmchlorphenylthio-cAMP (cA), with 30 mm[K+]o (30K), or with 1.5 μm veratridine (Vrt). Neuronal viability in the experimental conditions is expressed as a percentage of average number of NSE-positive cells in triplicate parallel cultures maintained in 30K (arbitrarily defined as 100%). The mean value is shown in the figure. All NSE-positive cells were included in these counts, regardless of neurite length. The number of determinations for each condition is shown adjacent to eachbar. Each such determination was performed using three separate culture wells. Error bars in this and all subsequent figures indicate SD. B, Trophic support of SGNs by combinations of neurotrophins, chlorphenylthio-cAMP, and depolarization. Spiral ganglion cultures were prepared, maintained, and scored for neuronal survival as in A, except that trophic agents are added in combinations, as shown, instead of singly. Agents used are 30 mm [K+]o(30K), 100 ng/ml BDNF (B), 100 ng/ml NT-3 (N), and 1 mmchlorphenylthio-cAMP (cA).
Cells are depolarized by culturing in medium containing 30 mm K+ or by adding 1.5 μmveratridine (Na+ channel agonist). (External K+concentration is denoted here as [K+]o.) Remarkably, depolarization is a much more effective trophic stimulus than is NT-3, BDNF, or cpt-cAMP. Whereas the neurotrophins and cpt-cAMP increase SGN survival by 10- to 20-fold over the N2 control, depolarization with 30 mm [K+]o(30K) or 1.5 μm veratridine increases survival by ∼40-fold over control (Fig. 1A). Nerve gowth factor, EGF, and FGF have no trophic effect when added at concentrations up to 100 ng/ml (data not shown).
These data show that at least four stimuli, BDNF, NT-3, a cAMP analog, and depolarization (mediated by 30K or veratridine), are trophic to SGNs. To determine whether the trophic effects are additive, combinations of factors were added to SGNs. Each factor in the combination was present at a level at which it provided its maximal trophic effect. Addition of factors in combination resulted in an increase in SGN survival approximating the arithmetic sum of the survival effect of each factor alone (Fig. 1B), consistent with an additivity of the trophic effects of the different factors. A possible exception to this apparent additivity is that cAMP and neurotrophins appear to interact synergistically: addition of cpt-cAMP in combination with BDNF and NT-3 leads to better SGN survival than might be expected by assuming strict additivity. The survival in these conditions exceeds that in 30K with BDNF and NT-3, even though 30K alone promotes survival better than does cpt-cAMP alone. However, any such synergistic effect appears to be small relative to the variability among the samples.
SGN soma size and neurites are supported by neurotrophins and depolarization in vitro but are reduced by cpt-cAMP
SGN appearance was examined in cells cultured under the same conditions as used for the cell counts described above; the cells were plated on polyornithine-laminin in serum-free N2-based medium. The agents shown above to be capable of supporting cell survival were added in various combinations. The cultures were fixed 48 hr after plating and immunostained with anti-NSE, and representative fields photographed under bright-field illumination.
Figure 2 shows examples of the appearance of SGNs cultured under these conditions. SGNs in the presence of any trophic agent generally produced a single neuritic process in vitro, whereas these neurons are bipolar in vivo. Those few neurons that survived in the absence of added neurotrophic agents typically had neurites, although these neurites were relatively short and the cell bodies small. Neurotrophins or depolarization maintained soma size and promoted neurite extension. SGNs depolarized with 30K, or with veratridine (data not shown), exhibited soma size comparable with that of cells treated with neurotrophins. Although depolarization was more efficacious than neurotrophins at promoting survival, neurite outgrowth in depolarizing conditions was markedly less than in neurotrophins (Fig. 2, Table 1). SGNs treated with combinations of depolarization and neurotrophins show somatic size and neuritic growth comparable with SGNs treated with neurotrophins alone (Fig. 2), in contrast to the significant increment in cell survival observed after combining these agents (Fig. 1B).
Fig. 2.
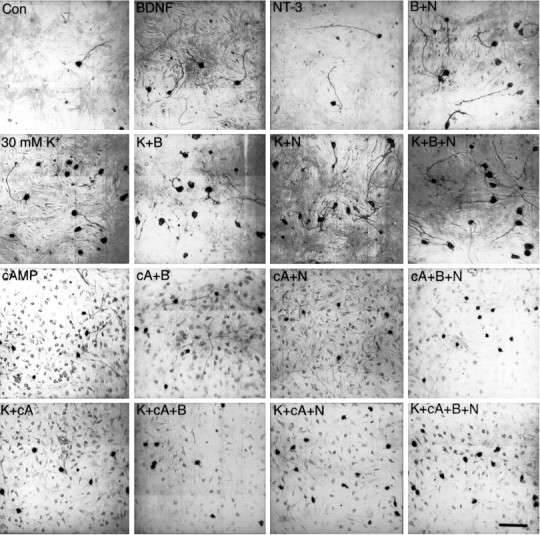
Morphology of SGNs cultured in neurotrophins, chlorphenylthio-cAMP, and 30K. Spiral ganglion cultures were fixed and immunostained with anti-NSE, as described in Materials and Methods, after culturing for 48 hr in control DMEM/N2 with no additives (Con), or in DMEM/N2 with 100 ng/ml BDNF (B), 100 ng/ml NT-3 (N), 30 mm [K+]o(K), or 1 mm chlorphenylthio-cAMP (cA). The cells were viewed and photographed in a Nikon Diaphot microscope and photographed under bright-field illumination. The photographs were made at different times with different methods and thus vary in background and contrast. Scale bar (all photographs), 100 μm.
Table 1.
Percentage of neurite-bearing cells among SGNs surviving in various conditions
| Conditions | SGNs counted | Neurites ≥3 cell diameter |
|---|---|---|
| Control | 1664 | 81.3 ± 18.6% (N = 104) |
| BDNF (100 ng/ml) | 9310 | 88.8 ± 6.10% (N = 42) |
| NT-3 (100 ng/ml) | 6603 | 88.9 ± 6.60% (N = 54) |
| 30 mM [K+]o | 44,892 | 58.1 ± 13.3% (N = 90) |
| Cpt-cAMP (1 mM) | 9545 | 80.7 ± 11.0% (N = 45) |
| 30K + BDNF | 15,063 | 78.3 ± 6.80% (N = 15) |
| 30K + NT-3 | 14,238 | 79.6 ± 5.90% (N = 15) |
| 30K + BDNF + NT-3 | 16,849 | 77.1 ± 4.20% (N = 15) |
| Cpt-cAMP + BDNF | 10,581 | 79.5 ± 7.20% (N = 18) |
| Cpt-cAMP + NT-3 | 9168 | 81.1 ± 8.10% (N = 18) |
| Cpt-cAMP + BDNF + NT-3 | 8005 | 68.8 ± 24.2% (N = 12) |
| 30K + cpt-cAMP | 19,489 | 14.3 ± 6.70% (N = 30) |
| 30K + cpt-cAMP + BDNF | 7045 | 20.1 ± 13.0% (N = 9) |
| 30K + cpt-cAMP + NT-3 | 6992 | 24.9 ± 15.1% (N = 9) |
| 30K + cpt-cAMP + BDNF + NT-3 | 13,762 | 18.1 ± 7.00% (N = 18) |
Spiral ganglion cultures are fixed and immunostained with anti-NSE, as described in Materials and Methods, after culturing for 48 hr in control DMEM/N2 with no additives (Control) or in DMEM/N2 with 100 ng/mL BDNF (BDNF), 100 ng/mL NT-3 (NT-3), 30 mM [K+]o (30K), 1 mM chlorphenylthio-cAMP, or various combinations of these factors. The cells are viewed in a Nikon Diaphot microscope and separate counts made of SGNs with neurites ≥3 cell diameters and those with shorter neurites. Shown is the total number of cells scored for this purpose and the percentage of these cells with neurites ≥3 cell diameters. The number N used to calculate means and SD values is the number of individual wells used for each determination. The relatively small number of cells per well in the control condition reflects the poor viability of cells in this condition.
The permeant cAMP analog cpt-cAMP maintained SGN survival but, in contrast to depolarized SGNs or SGNs treated with neurotrophins, SGNs treated with cpt-cAMP had a shrunken appearance with small cell bodies and thin neurites (Fig. 2). The neurites were nearly as long as those of SGNs in neurotrophins (Fig. 2, Table 1). The addition of cpt-cAMP or 30K to neurotrophins resulted in a small reduction in neurite outgrowth relative to that in neurotrophins alone (Table 1). However, the combination of cpt-cAMP and 30K strongly reduced SGN neurite outgrowth (Table 1). The reduced size of SGNs treated with cpt-cAMP was not simply attributable to a deficiency of trophic support by cpt-cAMP. SGNs treated with cpt-cAMP in combination with neurotrophins and/or depolarization still exhibited shrunken somata and thin neurites. Therefore, the cAMP analog antagonizes SGN growth regardless of the presence of growth-promoting neurotrophic stimuli.
The trophic effects of depolarization are mediated by Ca2+ entry through L-type Ca2+ channels
SGNs depolarized with 30K or veratridine in the absence of extracellular Ca2+ did not display any increase in survival relative to nondepolarized SGNs (Fig. 3). This suggests that Ca2+ entry through voltage-gated Ca2+channels, open as a result of the depolarization, is responsible for the trophic effect of depolarization. Nifedipine (5 μm) and verapamil (10 μm) were used to block dihydropyridine-sensitive L-type Ca2+ channels. Figure 3shows that these compounds were effective in blocking the trophic effect of depolarization caused by 30K, veratridine, or both. Nifedipine and verapamil had no effect on SGN survival in neurotrophins.
Fig. 3.

The effect of Ca2+ channel blockers and removal of [Ca2+]o on depolarization-dependent survival. Spiral ganglion cultures were prepared, maintained, and scored for neuronal survival after 48 hr in culture, as in Figure 1. The cells were cultured in control DMEM/N2 medium (white bars), 30K depolarizing medium (black bars), or 1.5 μm veratridine depolarizing medium (gray bars). In addition, the medium either lacked Ca2+ (−Ca); included Ca2+-channel blockers 5 μm nifedipine (Nif), 10 μm verapamil (Vpl), or 1 μm Ω-conotoxin GVIA (Ctx); or had no additional modifications (Con). Neuronal viability in the experimental conditions is expressed as a percentage of average number of NSE-positive cells in triplicate parallel cultures maintained in 30K depolarizing medium (or veratridine depolarizing medium for veratridine-containing conditions) arbitrarily defined as 100%. The number of determinations for each condition is shown adjacent to eachbar.
Previous studies of cultured guinea pig SGNs have shown that inward Ca2+ currents caused by depolarization are slowly inactivating (Hisashi et al., 1995) and that the increase in [Ca2+]i caused by depolarization is completely blocked by nifedipine or verapamil but is enhanced by Bay K 8644 (Han et al., 1994). These data make it unlikely that depolarization could cause significant Ca2+ influx into SGNs through channels other than L-type. Nonetheless, because N-type channels are expressed on many neurons, we also assessed the effect of the N-type channel blocker ω-conotoxin GVIA (1 μm) on cell survival. As shown in Figure 3, ω-conotoxin GVIA had no effect on the ability of 30K or veratridine to promote survival. We conclude that Ca2+ influx through L-type Ca2+ channels, specifically, is necessary for the trophic effect of depolarization.
Elevated [K+]o has a biphasic effect on SGN survival
The survival of SGNs cultured in medium with different K+ concentrations was assessed by counting cells as above and is presented in Figure 4A. All media used were iso-osmolar with control DMEM/N2. Cell survival increased with increasing [K+]o above control (5.4 mm [K+]o), reaching a maximum at 30 mm [K+]o. Above 30 mm [K+]o, survival was reduced; 80 mm [K+]o (the highest concentration that can practically be achieved with the culture media we use) had only a marginal effect on SGN survival.
Fig. 4.

Spiral ganglion neuronal survival as a function of [K+]o. Spiral ganglion cultures were prepared, maintained, and scored for neuronal survival after 48 hr in culture, as in Figure 1, with survival in 30 mm[K+]o (maximal survival) defined as 100%. The culture medium was DMEM/N2 with Na+ replaced with equimolar K+ to achieve the indicated values of [K+]o. Each of the four individualplots represents a separate experiment, and eachpoint represents the mean of triplicate wells counted.B, Spiral ganglion neuronal survival as a function of [K+]o in Bay K 8644. SGN viability in different [K+]o values was assessed as inA, except that the L-type Ca2+ channel agonist Bay K 8644 (1 μm) was added (solid symbols). Neuronal viability in the experimental conditions is expressed as in A, except that survival in 15 mm [K+]o (the condition at which survival was maximal with 1 μm Bay K 8644) was defined as 100%. Survival determinations in control medium without Bay K 8644 are represented by open symbols. Each of the three individual plots represents a separate experiment, and each point represents the mean of triplicate wells counted.
Our data implicate Ca2+ entry through L-type Ca2+ channels in the trophic effect of depolarization on SGNs. This was tested further by using the dihydropyridine L-type Ca2+ channel agonist Bay K 8644 (1 μm). Bay K 8644 potentiated both the survival-promoting and survival-inhibiting effects of elevated [K+]o (Fig.4B), consistent with a role for the L-type Ca2+ channel. In effect, the relationship of survival to [K+]o was qualitatively similar but shifted to lower values of [K+]o when Bay K 8644 is present. In the presence of Bay K 8644, increased SGN survival was observed in 5.4 mm [K+]o, the concentration normally present in culture medium. Maximal survival in the presence of Bay K 8644 was at 15 mm[K+]o instead of 30 mm[K+]o, and survival declined with [K+]o >15 mm. However, maximal survival was much less in the presence of Bay K 8644 than in its absence. These data suggest that elevated Ca2+ plays a role in both survival-promoting and survival-inhibiting effects of elevated [K+]o, with the effect shifting from survival to toxicity as Ca2+ influx is increased.
Direct measurement of [Ca2+]i in depolarized SGNs show a correlation between [Ca2+]i and cell survival
The data above show a biphasic dependence of SGN survival on [K+]o. If the effects of elevated [K+]o are attributable to an elevated cytosolic Ca2+ concentration (cytosolic Ca2+concentration is denoted here as [Ca2+]i), then cell survival should be correlated with [Ca2+]i. Previous studies have found such a correlation (Collins et al., 1991; Koike and Tanaka, 1991; Franklin and Johnson, 1992); specifically, increasing [Ca2+]i over the level in nondepolarized cells is associated with cell survival, but very high [Ca2+]i is associated with reduced viability. However, Franklin et al. (1995) recently found that increased [K+]o and consequent depolarized membrane potential was associated with increased survival of sympathetic neurons but that this was not correlated with [Ca2+]imeasured 24 hr after initiating depolarization. Rather, the relationship between survival and [Ca2+]i was not the same at all values of membrane potential. We measured [Ca2+]i in nondepolarized SGNs and in SGNs depolarized with either 30 mm[K+]o (30K), which has maximal trophic effect, or 80 mm [K+]o (80K), in which SGN viability is poor. Measurement of [Ca2+]i was performed 4 hr after elevating [K+]o, at which time SGNs in all conditions exhibit comparable viability (and [Ca2+]i has already reached steady-state level), and 22 hr after elevating [K+]o, at which time neurons in 80K exhibit diminished viability relative to those in 30K.
SGNs respond to depolarization with a biphasic increase in [Ca2+]i; there is an initial steep increase in [Ca2+]i followed by gradual decline to a steady-state level, which is maintained thereafter (Han et al., 1994). This plateau value (∼0.5 μm) is comparable with the mean [Ca2+]i level that we find in SGNs depolarized for 4 hr and therefore can be considered a “steady-state” value.
SGNs cultured for 2 hr in control medium (5.4 mm[K+]o) and then for 4 hr in control medium, in 30K or 80K, exhibited comparable viability. SGNs cultured for 6 hr in control medium (5.4 mm [K+]o) had a mean [Ca2+]i = 110 nm(SD = 66 nm, n = 134) (Fig.5). SGNs exposed to elevated [K+]o for 4 hr exhibited [Ca2+]i greatly elevated over control (Fig.5). The histograms show that there were very few SGNs in either 30K or 80K, with [Ca2+]i levels comparable with those of SGNs in control medium. In either 30K or 80K, the elevated [Ca2+]i levels were very widely distributed. In 30K, the average [Ca2+]i was 571 nm with an SD of 233 nm (n = 110) and in 80K, the average [Ca2+]i was 922 nm with an SD of 393 nm (n = 101), but the range of [Ca2+]i values was so great that the average value has little significance. However, the histograms in Figure 5 show that neuronal [Ca2+]i levels are higher in 80K than in 30K; most of the SGNs in 80K have higher [Ca2+]ithan most of the neurons in 30K, although some overlap exists. These data are consistent with the hypothesis that [Ca2+]i is predictive of cell fate, with either very low [Ca2+]i (i.e., <100–200 nm) or very high [Ca2+]i (i.e., >600–700 nm), incompatible with neuronal viability. The cytotoxicity of high [Ca2+]i could account for poor survival in 80K.
Fig. 5.

[Ca2+]i histograms after 6 hr in 5.4 (control), 30, or 80 mm[K+]o. Spiral ganglia were dissociated and the cells plated on polyornithine/laminin-coated glass coverslips as described in Materials and Methods. Elevated [K+]o medium was added 2 hr after plating. [Ca2+]i was determined from the ratio of Fura-2 fluorescence at 337 and 380 nm, as described in Materials and Methods. The images captured were from fields randomly distributed on the coverslip. Ratios (256) of each field, taken at 90 msec intervals, were averaged. A sufficient number of fields were imaged to to allow assay of [Ca2+]i in >100 neurons for each condition. All of the images were captured within an interval of 30 min. Two to three coverslips were used for each condition, assayed on different days. The calculated [Ca2+]ivalues, grouped in 50 nm bins, are plotted in thehistograms shown.
The levels of [Ca2+]i in SGNs continuously depolarized for 22 hr in 80K were significantly different from those in cells depolarized for 4 hr (Fig. 6). SGNs in control medium (5.4 mm [K+]o) had an average [Ca2+]i = 39 nm (SD = 30 nm, n = 138), whereas for SGNs in 30K, the average [Ca2+]i was 316 nmwith an SD of 164 nm (n = 138), and in 80K, the average [Ca2+]i was 285 nmwith an SD of 202 nm (n = 190). As was the case after 4 hr of depolarization, the levels of [Ca2+]i were very widely distributed. After 22 hr in 80K, >30% of the SGNs have already died (M. Hansen and S. Green, unpublished observations) and >80% die within 48 hr (Fig. 4A). The distribution of [Ca2+]i levels in the surviving SGNs after 22 hr in 80K is similar to that in SGNs in 30K. Relatively few cells exhibited very high [Ca2+]i, as was the case after 4 hr in 80K. This suggests that the cells lost are expressly those with high [Ca2+]i or that SGNs alter their regulation of [Ca2+]i with time in culture under these conditions.
Fig. 6.

[Ca2+]i histograms after 24 hr in 5.4 (control), 30, or 80 mm[K+]o. [Ca2+]ilevels in dissociated SGNs were determined as in Figure 5. The calculated [Ca2+]i values, grouped in 50 nm bins, are plotted in the histogramsshown. To facilitate comparison, the abscissae are at the same scale as in the histograms in Figure 5.
A population of SGNs with low [Ca2+]i was observed in cultures depolarized with 80K, but not with 30K (Fig. 6). This may reflect long-term changes in Ca2+ influx, transport, or buffering in these cells.
Cells cultured with 1 μm Bay K 8644 survived best in 15 mm [K+]o (Fig.4B). [Ca2+]i levels in SGNs cultured in 1 μm Bay K 8644 + 15 mm[K+]o for 22 hr are shown in Figure7. SGNs so cultured had [Ca2+]i levels comparable with SGNs treated with 30K (compare Fig. 6). In 1 μm Bay K 8644 + 15 mm [K+]o, the average [Ca2+]i was 371 nm, with an SD of 183 nm (n = 83). The addition of 1 μm Bay K 8644 to control medium (5.4 mm[K+]o) resulted in oscillations in [Ca2+]i from control levels to 150–200 nm (data not shown). These oscillatory increases in [Ca2+]i may account for the small enhancement of SGN survival observed in 1 μm Bay K 8644 (Fig.4B). The behavior of SGNs in 1 μm Bay K 8644 was thus unlike the behavior of SGNs depolarized by 30K, 80K, or 1 μm Bay K 8644 + 15 mm[K+]o, which consistently exhibited stable nonoscillating [Ca2+]i regardless of the variability in the level of steady-state [Ca2+]i among the SGNs.
Fig. 7.

[Ca2+]i histograms after 24 hr in 15 mm [K+]o + 1 μm Bay K 8644. [Ca2+]i levels in dissociated SGN were determined as in Figure 5. The calculated [Ca2+]i values, grouped in 50 nmbins, are plotted in the histograms shown. To facilitate comparison, the abscissae are at the same scale as in the histograms in Figures 5 and 6. m
The Na+ channel agonist veratridine (1.5 μm) was approximately equal to 30K in the ability to promote SGN survival (Fig. 1). However, veratridine caused oscillatory increases in [Ca2+]i, an oscillation that began at application of the drug (data not shown) and persisted for at least 24 hr. Four representative examples of SGNs cultured for 24 hr in medium containing 1.5 μm veratridine are shown in Figure8. The oscillatory increases in [Ca2+]i are up to 250–350 nm[Ca2+]i, greater than the peak [Ca2+]i levels observed in SGNs in 1 μm Bay K 8644 + 5.4 mm[K+]o.
Fig. 8.

Veratridine-induced [Ca2+]i oscillations in SGN. [Ca2+]i levels in dissociated SGNs were determined as in Figure 5 and plotted as a function of time. Ratios were acquired every 240 msec for rectangular areas of interest overlying the neurons. Records from four different neurons are shown here.
From these data, it appears that the initial steady-state level of [Ca2+]i is predictive of SGN survival. The level seen after 24 hr reflects that in those SGNs selected for ability to survive in the particular depolarizing condition. Apparently, these are the neurons having [Ca2+]i in the range displayed in the histograms in Figures 7 and 8, ∼150–450 nm, which can be considered the range permissive for survival in vitro in the absence of exogenous neurotrophic factors.
DISCUSSION
Trophic support by neuronal afferents can involve neurotrophic factors, cAMP, and membrane electrical activity
SGNs die after deafferentation in vivo and after isolation in culture. In either case, the death is apparently apoptotic (J. Hegarty and S. Green, unpublished observations), characteristic of neurons that have lost trophic support (Johnson and Deckworth, 1993). Studies of auditory system reveal that pre- and postsynaptic sources of neurotrophic support cooperate to prevent programmed cell death. Type I SGNs, which constitute >90% of the population, receive presynaptic input from the inner hair cells and are dependent on them for survival (Spoendlin, 1975; Webster and Webster, 1981; Koitchev et al., 1982;Bichler et al., 1983). As with other cells in the vicinity of the neuron and its terminals, presynaptic cells can contribute to neuronal survival by releasing neurotrophic factors. In addition, presynaptic cells can provide neurotrophic support by causing depolarization through synaptic activity or by releasing neurotransmitters that activate trophic second messenger systems. The cochlea is well suited as a system for study of the means by which presynaptic input contributes to neuronal survival, because the SGNs are relatively accessible to in vivo experimental manipulations, including deafferentation and direct electrical stimulation (Wong-Riley et al., 1981; Lousteau, 1987; Hartshorn et al., 1991; Leake et al., 1991, 1992;Lustig et al., 1994).
Support by neurotrophins
The embryonic rat auditory sensory epithelium expresses BDNF and NT-3 (Wheeler et al., 1994). However, the SGNs, which express both TrkB and TrkC (Ylikoski et al., 1993; Pirvola et al., 1994; Schecterson and Bothwell, 1994), require only NT-3 for survival. Mice homozygous for NT-3 knockout are born lacking >85% of the SGNs (Fariñas et al., 1994; Ernfors et al., 1995), whereas mice homozygous for BDNF knockout show only a small loss of SGNs at birth (Conover et al., 1995;Ernfors et al., 1995). Nonetheless, BDNF, like NT-3, supports survival of cultured embryonic and postnatal SGNs (Avila et al., 1993; Lefebvre et al., 1994; Pirvola et al., 1994; Vazquez et al., 1994; Zheng et al., 1995). It is not clear why only NT-3, and not BDNF, is necessary for prenatal trophic support of SGNs; possibly, SGNs lack access to sufficient BDNF at this time.
We show here that BDNF and NT-3 are trophic to P5 rat SGNs in vitro, consistent with previous studies that showed that BDNF, NT-3, and NT-4 are trophic to postnatal SGNs (Lefebvre et al., 1994;Vazquez et al., 1994; Zheng et al., 1995). At P5, NT-3 expression is low in auditory hair cells, and BDNF is not expressed (Pirvola et al., 1994; Wheeler et al., 1994), concomitant with a postnatal reorganization of afferent SGN synapses and period of SGN death during which ∼20% of SGNs are lost (Rueda et al., 1987). In mature organ of Corti, NT-3 is expressed in the inner hair cells, and BDNF is not expressed (Ylikoski et al., 1993; Pirvola et al., 1994; Schecterson and Bothwell, 1994; Wheeler et al., 1994). Thus, if a hair cell-derived neurotrophin is necessary for support of mature type I SGNs, it is NT-3. BDNF is produced in the cochlear nucleus (Lefebvre et al., 1994) and can supply postsynaptic trophic support.
Support by nerve membrane electrical activity
Direct electrical stimulation is sufficient to promote survival of deafferented SGNs in vivo (Wong-Riley et al., 1981;Lousteau, 1987; Hartshorn et al., 1991; Leake et al., 1991, 1992;Lustig et al., 1994). The present study, using an in vitromodel, support the hypothesis that membrane electrical activity attributable to presynaptic input is a crucial source of trophic support for SGNs. Membrane depolarization is a significant neurotrophic stimulus for cultured SGNs, as it is for other neurons. Indeed, SGNs exhibit markedly greater survival in depolarizing conditions than in neurotrophins.
Although trophic to sympathetic neurons, depolarization did not support somatic or neuritic growth (Franklin et al., 1995). This does not appear to be a general property of neurons; SGNs maintained in depolarizing media displayed somatic and neuritic growth comparable with that of SGNs maintained in neurotrophins. This difference may be attributable to differences in culture conditions (the SGNs were plated on laminin and the sympathetic neurons on other substrates) or to differences between the cells. For instance, membrane electrical activity promotes neurotrophin synthesis in SGNs (M. Hansen and S. Green, unpublished observations) possibly allowing support of neurite outgrowth by an autocrine effect. In contrast, Franklin et al. (1995)could not detect a depolarization-induced neurotrophin autocrine mechanism in sympathetic neurons.
Support by cAMP analogs
Permeant cAMP analogs have been shown previously to promote sympathetic neuronal survival (Rydel and Greene, 1988). This may be relevant to support by afferent input in vivo, because neurotransmitters can stimulate adenylate cyclase activity. We show here that cpt-cAMP promoted SGN survival and neurite length comparable with that in neurotrophins, but cell somata and neurites appeared shrunken and thin, unlike their appearance in 30K or neurotrophins.Rydel and Greene (1988) found that NGF promoted sympathetic neuron somatic hypertrophy and neurite growth to a much greater degree than did cAMP analogs. However, in sympathetic neurons, cAMP analogs did not antagonize somatic or neuritic growth promoted by NGF nor antagonize growth in neurons plated on laminin, whereas SGNs maintained with cpt-cAMP have shrunken somata and thin neurites, even in the presence of laminin and neurotrophins. Apparently, a growth-inhibiting activity of cAMP is antagonized by exogenous growth-promoting factors, such as NGF or laminin, in sympathetic neurons, but not in SGNs. In PC12 cells, cAMP antagonizes neurite outgrowth in the presence of NGF (Greene et al., 1986), an even stronger inhibitory effect than what is observed SGNs. A strong inhibition of neurite outgrowth in cultured SGNs is observed in the presence of both cAMP and depolarization, possibly attributable to summation of inhibitory stimuli that separately have only a small effect on SGNs.
Additivity can account for the requirement for multiple sources of trophic support by SGNs in vivo
Our observation that depolarization in vitro is a more potent trophic stimulus than neurotrophins or cAMP analogs is consistent with the possibility that electrical stimulation is the primary trophic stimulus provided by afferent neurons in vivo. However, the observation of additivity and, possibly, some synergism among the various neurotrophic agents that we used suggests that, in vivo, it is the combination of trophic stimuli received from all pre- and postsynaptic sources that is necessary to fully promote survival. The observed additivity in neurotrophic support by various stimuli could reflect the existence of different subpopulations of SGNs, each supported by a different stimulus. Because additional increments of trophic support were provided by each of four different stimuli, there would have to be at least four different SGN subpopulations to account for our results. Although formally possible, this is an unlikely possibility.
Additivity or synergism among different neurotrophic stimuli is likely to be attributable to interactions among the intracellular signal pathways that they use. There are at least two types of interactions. First, summation of neurotrophic effects may result from increased magnitude or duration in a single intracellular signal pathway. This can account for the additivity reported here or for possible synergism (R. Davis, personal communication) in the neurotrophic effects of BDNF and NT-3. Both signal through Trk family protein-tyrosine kinases and thus are likely to activate the same intracellular signaling pathways to promote survival. Second, summation may be attributable to activation of different intracellular signaling pathways by different neurotrophic stimuli. Ca2+ and cAMP function in intracellular pathways that are distinct from each other and from the pathway used by neurotrophin signal transduction. Trophic support of sympathetic or sensory neurons by NGF does not require cAMP signaling (Rydel and Greene, 1988), nor does trophic support of cerebellar granule cells by peptide growth factors of cAMP require increased [Ca2+]i (Galli et al., 1995). Additivity among depolarization, peptide neurotrophic factor, and substratum in the trophic support of cultured ciliary ganglion neurons (Schmidt and Kater, 1993) appears to depend on the recruitment of different intracellular signals by laminin and depolarization (Schmidt and Kater, 1995). Moreover, our observation here of dissimilar effects of different neurotrophic stimuli on SGN morphology argues that they act in different intracellular signal pathways. A critical question is whether these different signaling pathways ultimately converge on a single molecular regulator of cell survival or whether the cell death machinery is independently regulated at different points by these various intracellular trophic signaling pathways.
Neurotrophic support by depolarization requires cytosolic Ca2+ to be within a set concentration range
Data presented here indicate that an increase in cytosolic Ca2+ concentration ([Ca2+]i) occurs in response to depolarization of SGNs. Regardless of the means used to depolarize the SGNs, the cellular levels of [Ca2+]i are very broadly distributed. This heterogeneity may reflect differing Ca2+ buffering capacities among the cells or differing densities of Ca2+channels or pumps, perhaps because of differences in frequency-tuning or maturity among the cells.
Ca2+ influx through L-type channels is specifically required for the trophic effect of depolarization on SGNs, as has been shown previously for several types of neurons (Gallo et al., 1987;Collins and Lile, 1989; Koike et al., 1989; Collins et al., 1991). The increase in Ca2+ need not be constant to promote SGN survival; veratridine depolarization supports survival as effectively as depolarization by 30 mm [K+]o, even though, unlike the latter, veratridine depolarization does not appear to cause a stable increase in Ca2+ but rather an oscillation of [Ca2+]i with a periodicity of several minutes. The mechanism underlying this oscillation is unknown.
As with depolarization by 30K, depolarization by veratridine causes [Ca2+]i to exceed 200 nm, but not 350 nm, in most SGNs. This range of [Ca2+]i appears to be critical for survival. Depolarization by 80K causes a sustained increase in [Ca2+]i greater than that caused by depolarization with veratridine or 30 mm[K+]o. In most SGNs cultured in 80K, [Ca2+]i exceeds 400 nm. A large fraction of SGNs in 80K die within 24 hr, and this seems to be related to the high [Ca2+]i levels; because the surviving SGNs are those with lower [Ca2+]ilevels, most have [Ca2+]i < 350 nm.
These results are comparable with those reported previously for sympathetic neurons (Koike et al., 1989; Koike and Tanaka, 1991), although maximal SGN survival appears to occur at a slightly lower [Ca2+]i than does maximal sympathetic neuronal survival (Franklin et al., 1995). Most important, our data are consistent with the “calcium homeostasis” or “setpoint” hypothesis for neuronal survival proposed by these authors (Koike et al., 1989; Koike and Tanaka, 1991), which is comparable with one proposed earlier for growth cone stability (Kater et al., 1988). Specifically, maintenance of [Ca2+]i within a particular range is sufficient for neuronal survival. With [Ca2+]i below this range, neurons survive only in the presence of exogenous neurotrophic factors. [Ca2+]i exceeding this range results in neuronal death, presumably because of Ca2+-mediated excitotoxicity (Choi, 1988; Siesjo et al., 1989). This is of particular consequence for auditory neurons that have high levels of spontaneous and even higher levels of evoked activity, which could compromise survival through increased Ca2+ influx. Cochlear input to nucleus magnocellularis neurons attenuates Ca2+ influx by a metabotropic glutaminergic postsynaptic mechanism distinct from the glutaminergic excitatory stimulation (Lachica et al., 1995; Zirpel et al., 1995). Other compensatory mechanisms presumably exist in other auditory or highly active neurons.
Although the calcium homeostasis hypothesis can successfully account for the survival or death of neurons in various depolarizing conditions or after exposure to excitatory amino acids (Collins et al., 1991;Koike and Tanaka, 1991; Franklin and Johnson, 1992), Franklin et al. (1995) have suggested recently that the relationship between [Ca2+]i and survival may be more complex. In their study of sympathetic neurons, they found that at high levels of depolarization, in which neuronal survival was poor, the [Ca2+]i level was comparable with the level at lower depolarization, in which survival was good. In that study, [Ca2+]i was assessed at 24 hr after shift to depolarizing conditions. Our data also seem to show that [Ca2+]i levels are comparable after 24 hr in 30K, in which survival is good, and in 80K, in which survival is poor. However, this is the result of comparing only that selected subpopulation of neurons that survives for 24 hr in 80K with neurons cultured for 24 hr in 30K, which are most of the neurons plated. Only this surviving population has comparable [Ca2+]i; most of the neurons in 80K had much higher [Ca2+]i levels when assessed at 6 hr after depolarization, when all were still alive. Thus, the results of Franklin et al. do not require reassessment of the calcium homeostasis hypothesis. In that study, neurons that did not exhibit high [Ca2+]i after 24 hr in high [K+]o may well be from a population that experienced very high [Ca2+]i levels earlier. Possibly, cells present after 24 hr in high [K+]o are those that initially had lower [Ca2+]i levels. Alternatively, neurons in culture may change their means of regulating [Ca2+]i and could vary in their ability to do so. Additional studies are required to identify those differences among neurons that result in varying survival in high [K+]o and to determine the relationship between regulation of [Ca2+]i and neuronal survival.
A variety of intracellular signals that critically dependents on [Ca2+]i are potential mediators of the trophic effect of depolarization. The robust trophic response of SGNs to depolarization in vitro suggests that they will of value in investigation of these mechanisms. Moreover, the in vivorelevance of mechanisms identified in cultured SGNs can be addressed because of the ease of deafferentation and electrical stimulation of the spiral ganglion in vivo.
Footnotes
This study was supported by an American Otological Society Research grant, a University of Iowa CIFRE award, a Carver Scientific Research Initiative grant, and a University of Iowa Diabetes and Endocrinology Research Core Seed grant; and funded by National Institutes of Health (NIH) Grant DK25295 (S.H.G.), NIH Grant NS30654, and a grant from the Office of Naval Research (A.R.K.), and a grant from the Academy of Otolaryngology (J.L.H.). J.L.H. was supported by NIH Training Grant DC00040. We thank Drs. Lloyd Greene and Robin Davis and members of the Green lab for comments on this manuscript, Mr. Matt Cardoni for assisting with the cell counts, and Genentech for graciously providing NT-3.
Correspondence should be addressed to Dr. Steven H. Green, Department of Biological Sciences, University of Iowa, 138 Biology Building, Iowa City, IA 52242-1324.
REFERENCES
- 1.Avila MA, Varela-Nieto I, Romero G, Mato JM, Giraldez F, Van De Water TR, Represa J. Brain-derived neurotrophic factor and neurotrophin-3 support the survival and neuritogenesis response of developing cochleovestibular ganglion cells. Dev Biol. 1993;159:266–275. doi: 10.1006/dbio.1993.1239. [DOI] [PubMed] [Google Scholar]
- 2.Bennett MR, White W. The survival and development of cholinergic neurons in potassium-enriched media. Brain Res. 1979;173:549–553. doi: 10.1016/0006-8993(79)90250-6. [DOI] [PubMed] [Google Scholar]
- 3.Bichler E, Spoendlin H, Rauchegger H. Degeneration of cochlear neurons after amikacin intoxication in the rat. Arch Otorhinolaryngol. 1983;237:201–208. doi: 10.1007/BF00453725. [DOI] [PubMed] [Google Scholar]
- 4.Born DE, Rubel EW. Afferent influences on brain stem auditory nuclei of the chicken: neuron number and size following cochlea removal. J Comp Neurol. 1985;231:435–445. doi: 10.1002/cne.902310403. [DOI] [PubMed] [Google Scholar]
- 5.Born DE, Rubel EW. Afferent influences on brain stem auditory nuclei of the chicken: presynaptic action potentials regulate protein synthesis in nucleus magnocellularis neurons. J Neurosci. 1988;8:901–919. doi: 10.1523/JNEUROSCI.08-03-00901.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bottenstein JE, Sato G. Growth of a rat neuroblastoma cell line in serum-free supplemented medium. Proc Natl Acad Sci USA. 1979;76:514–517. doi: 10.1073/pnas.76.1.514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Catsicas M, Péquinot Y, Clarke PGH. Rapid onset of neuronal death induced by blockade of either axoplasmic transport or action potentials in afferent fibers during brain development. J Neurosci. 1992;12:4642–4650. doi: 10.1523/JNEUROSCI.12-12-04642.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chalazonitis A, Fischbach GD. Elevated potassium induces morphological differentiation of dorsal root ganglionic neurons in dissociated cell culture. Dev Biol. 1980;78:172–183. doi: 10.1016/0012-1606(80)90327-9. [DOI] [PubMed] [Google Scholar]
- 9.Choi DW. Calcium-mediated neurotoxicity: relationship to specific channel types and role in ischemic damage. Trends Neurosci. 1988;11:465–467. doi: 10.1016/0166-2236(88)90200-7. [DOI] [PubMed] [Google Scholar]
- 10.Collins F, Lile JD. The role of dihydropyridine-sensitive voltage gated calcium channels in potassium mediated neuronal survival. Brain Res. 1989;502:99–108. doi: 10.1016/0006-8993(89)90465-4. [DOI] [PubMed] [Google Scholar]
- 11.Collins F, Schmidt MF, Guthrie PB, Kater SB. Sustained increase in intracellular calcium promotes neuronal survival. J Neurosci. 1991;11:2582–2587. doi: 10.1523/JNEUROSCI.11-08-02582.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Conover JC, Erickson JT, Katz DM, Bianchi LM, Poueymirou WT, McClain J, Pan L, Helgren M, Ip NY, Boland P, Friedman B, Wiegand S, Vejsada R, Kato AC, DeChiara TM, Yancopoulos GD. Neuronal deficits, not involving motor neurons, in mice lacking BDNF and/or NT4. Nature. 1995;375:235–238. doi: 10.1038/375235a0. [DOI] [PubMed] [Google Scholar]
- 13.Ernfors P, Van De Water T, Loring J, Jaenisch R. Complementary roles of BDNF and NT-3 in vestibular and auditory development. Neuron. 1995;14:1153–1164. doi: 10.1016/0896-6273(95)90263-5. [DOI] [PubMed] [Google Scholar]
- 14.Fariñas I, Jones KR, Backus C, Wang X-Y, Reichardt LF. Severe sensory and sympathetic deficits in mice lacking neurotrophin-3. Nature. 1994;369:658–661. doi: 10.1038/369658a0. [DOI] [PubMed] [Google Scholar]
- 15.Franklin JL, Johnson EM., Jr Suppression of programmed neuronal death by sustained elevation of cytoplasmic calcium. Trends Neurosci. 1992;15:501–508. doi: 10.1016/0166-2236(92)90103-f. [DOI] [PubMed] [Google Scholar]
- 16.Franklin JL, Sanz-Rodriguez C, Juhasz A, Deckwerth TL, Johnson EM., Jr Chronic depolarization prevents programmed death of sympathetic neurons in vitro but does not support growth: requirement for Ca2+ influx but not Trk activation. J Neurosci. 1995;15:643–664. doi: 10.1523/JNEUROSCI.15-01-00643.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Furber S, Oppenheim RW, Prevette D. Naturally-occurring neuron death in the ciliary ganglion of the chick embryo following removal of preganglionic input: evidence for the role of afferents in ganglion cell survival. J Neurosci. 1987;7:1816–1832. doi: 10.1523/JNEUROSCI.07-06-01816.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Galli C, Meucci O, Scorziello A, Werge TM, Calissano P, Schettini G. Apoptosis in cerebellar granule cells is blocked by high KCl, forskolin, and IGF-1 through distinct mechanisms of action: the involvement of intracellular calcium and RNA synthesis. J Neurosci. 1995;15:1172–1179. doi: 10.1523/JNEUROSCI.15-02-01172.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Galli-Resta L, Ensini M, Fusco E, Gravina A, Margheritti B. Afferent spontaneous electrical activity promotes the survival of target cells in the developing retinotectal system of the rat. J Neurosci. 1993;13:243–250. doi: 10.1523/JNEUROSCI.13-01-00243.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gallo V, Kingsbury A, Balazs R, Jorgensen OS. The role of depolarization in the survival and differentiation of cerebellar granule cells in culture. J Neurosci. 1987;7:2203–2213. doi: 10.1523/JNEUROSCI.07-07-02203.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Greene LA, Drexler SA, Connolly JL, Rukenstein A, Green SH. Selective inhibition of responses to nerve growth factor and of microtubule-associated protein phosphorylation by activators of adenylate cyclase. J Cell Biol. 1986;103:1967–1978. doi: 10.1083/jcb.103.5.1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- 23.Han D-Y, Harada N, Tomoda K, Yamashita T. Characterization of the calcium influx induced by depolarization of guinea pig cochlear spiral ganglion cells. ORL J Otorhinolaryngol Relat Spec. 1994;56:125–129. doi: 10.1159/000276626. [DOI] [PubMed] [Google Scholar]
- 24.Hartshorn DO, Miller JM, Altschuler RA. Protective effect of electrical stimulation in the deafened guinea pig cochlea. Otolaryngol Head Neck Surg. 1991;104:311–319. doi: 10.1177/019459989110400305. [DOI] [PubMed] [Google Scholar]
- 25.Hisashi K, Nakagawa T, Yasuda T, Kimitsuki T, Komune S, Komiyama S. Voltage dependent Ca2+ channels in the spiral ganglion cells of guinea pig cochlea. Hearing Res. 1995;91:196–201. doi: 10.1016/0378-5955(95)00191-3. [DOI] [PubMed] [Google Scholar]
- 26.Johnson EM, Deckworth TL. Molecular mechanisms of developmental neuronal death. Annu Rev Neurosci. 1993;16:31–46. doi: 10.1146/annurev.ne.16.030193.000335. [DOI] [PubMed] [Google Scholar]
- 27.Kater SB, Mattson MP, Cohan C, Connor J. Calcium regulation of the neuronal growth cone. Trends Neurosci. 1988;11:315–321. doi: 10.1016/0166-2236(88)90094-x. [DOI] [PubMed] [Google Scholar]
- 28.Koike T, Tanaka S. Evidence that nerve growth factor dependence of sympathetic neurons for survival in vitro may be determined by levels of cytoplasmic free Ca2+. Proc Natl Acad Sci USA. 1991;88:3892–3896. doi: 10.1073/pnas.88.9.3892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Koike T, Martin DP, Johnson EM., Jr Role of Ca2+ channels in the ability of membrane depolarization to prevent neuronal death induced by trophic-factor deprivation: evidence that levels of internal Ca2+ determine nerve growth factor dependence of sympathetic ganglion cells. Proc Natl Acad Sci USA. 1989;86:6421–6425. doi: 10.1073/pnas.86.16.6421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Koitchev K, Guilhaume A, Cazals Y, Aran J-M. Spiral ganglion changes after massive aminoglycoside treatment in the guinea pig. Counts and ultrastructure. Acta Otolaryngol. 1982;94:431–438. doi: 10.3109/00016488209128931. [DOI] [PubMed] [Google Scholar]
- 31.Lachica EA, Rubsamen R, Zirpel L, Rubel EW. Glutamatergic inhibition of voltage-operated calcium channels in the avian cochlear nucleus. J Neurosci. 1995;15:1724–1734. doi: 10.1523/JNEUROSCI.15-03-01724.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Leake PA, Hradek GT, Rebscher SJ, Snyder RL. Chronic intracochlear electrical stimulation induces selective survival of spiral ganglion neurons in neonatally deafened cats. Hearing Res. 1991;54:251–271. doi: 10.1016/0378-5955(91)90120-x. [DOI] [PubMed] [Google Scholar]
- 33.Leake PA, Snyder RL, Hradek GT, Rebscher SJ. Chronic intracochlear electrical stimulation in neonatally deafened cats: effects of intensity and stimulating electrode location. Hearing Res. 1992;64:99–117. doi: 10.1016/0378-5955(92)90172-j. [DOI] [PubMed] [Google Scholar]
- 34.Lefebvre PP, Van de Water TR, Weber T, Rogister B, Moonen G. Growth factor interactions in cultures of dissociated adult acoustic ganglia: neuronotrophic effects. Brain Res. 1991;567:306–312. doi: 10.1016/0006-8993(91)90809-a. [DOI] [PubMed] [Google Scholar]
- 35.Lefebvre PP, Malgrange B, Staecker H, Moghadass M, Van De Water TR, Moonen G. Neurotrophins affect survival and neuritogenesis by adult injured auditory neurons in vitro. NeuroReport. 1994;5:865–868. doi: 10.1097/00001756-199404000-00003. [DOI] [PubMed] [Google Scholar]
- 36.Lipton SA. Blockade of electrical activity promotes the death of mammalian retinal ganglion cells in culture. Proc Natl Acad Sci USA. 1986;83:9774–9778. doi: 10.1073/pnas.83.24.9774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lousteau RJ. Increased spiral ganglion cell survival in electrically stimulated deafened guinea pig cochleae. Laryngoscope. 1987;97:836–842. [PubMed] [Google Scholar]
- 38.Lustig LR, Leake PA, Snyder RL, Rebscher SJ. Changes in the cat cochlear nucleus following neonatal deafening and chronic intracochlear electrical stimulation. Hearing Res. 1994;74:29–37. doi: 10.1016/0378-5955(94)90173-2. [DOI] [PubMed] [Google Scholar]
- 39.Maderdrut JL, Oppenheim RW, Prevette D. Enhancement of naturally-occurring cell death in the sympathetic and parasympathetic ganglia of the chicken embryo following blockade of ganglionic transmission. Brain Res. 1988;444:189–194. doi: 10.1016/0006-8993(88)90928-6. [DOI] [PubMed] [Google Scholar]
- 40.Meriney SD, Pilar G, Ogawa M, Nunez R. Differential neuronal survival in the avian ciliary ganglion after chronic acetylcholine receptor blockade. J Neurosci. 1987;7:3840–3849. doi: 10.1523/JNEUROSCI.07-12-03840.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pasic TR, Rubel EW. Rapid changes in cochlear nucleus cell size following blockade of auditory nerve electrical activity in gerbils. J Comp Neurol. 1989;283:474–480. doi: 10.1002/cne.902830403. [DOI] [PubMed] [Google Scholar]
- 42.Pirvola U, Arumae U, Moshnyakov M, Palgi J, Saarma M, Ylikoski J. Coordinated expression and function of neurotrophins and their receptors in the rat inner ear during target innervation. Hearing Res. 1994;75:131–144. doi: 10.1016/0378-5955(94)90064-7. [DOI] [PubMed] [Google Scholar]
- 43.Rubel EW, Hyson RL, Durham D. Afferent regulation of neurons in the brain stem auditory system. J Neurobiol. 1990;21:169–196. doi: 10.1002/neu.480210112. [DOI] [PubMed] [Google Scholar]
- 44.Rueda J, De La Sen C, Juiz JM, Merchán JA. Neuronal loss in the spiral ganglion of young rats. Acta Otolaryngol. 1987;104:417–421. doi: 10.3109/00016488709128269. [DOI] [PubMed] [Google Scholar]
- 45.Ruitjer JM, Baker RE, De Jong BM, Romijn HJ. Chronic blockade of bioelectric activity in neonatal rat cortex grown in vitro. Morphological effects. Int J Dev Neurosci. 1991;9:331–338. doi: 10.1016/0736-5748(91)90054-p. [DOI] [PubMed] [Google Scholar]
- 46.Rydel RE, Greene LA. cAMP analogs promote survival and neurite outgrowth in cultures of rat sympathetic and sensory neurons independently of nerve growth factor. Proc Natl Acad Sci USA. 1988;85:1257–1261. doi: 10.1073/pnas.85.4.1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schecterson LC, Bothwell M. Neurotrophin and neurotrophin receptor mRNA expression in developing inner ear. Hearing Res. 1994;73:92–100. doi: 10.1016/0378-5955(94)90286-0. [DOI] [PubMed] [Google Scholar]
- 48.Schmidt MF, Kater SB. Fibroblast growth factors, depolarization, and substratum interact in a combinatorial way to promote neuronal survival. Dev Biol. 1993;158:228–237. doi: 10.1006/dbio.1993.1181. [DOI] [PubMed] [Google Scholar]
- 49.Schmidt MF, Kater SB. Depolarization and laminin independently enable bFGF to promote neuronal survival through different second messenger pathways. Dev Biol. 1995;168:235–246. doi: 10.1006/dbio.1995.1076. [DOI] [PubMed] [Google Scholar]
- 50.Scott BS, Fisher KC. Potassium concentration and number of neurons in cultures of dissociated ganglia. Exp Neurol. 1970;27:16–22. doi: 10.1016/0014-4886(70)90197-4. [DOI] [PubMed] [Google Scholar]
- 51.Sie KC, Rubel EW. Rapid changes in protein synthesis and cell size in the cochlear nucleus following eighth nerve activity blockade or cochlea ablation. J Comp Neurol. 1992;320:501–508. doi: 10.1002/cne.903200407. [DOI] [PubMed] [Google Scholar]
- 52.Siesjo BK, Bengtsson F, Grampp W, Theander S. Calcium, excitotoxins, and neuronal death in the brain. Ann NY Acad Sci. 1989;568:234–251. doi: 10.1111/j.1749-6632.1989.tb12513.x. [DOI] [PubMed] [Google Scholar]
- 53.Spoendlin H. Retrograde degeneration of the cochlear nerve. Acta Otolaryngol. 1975;79:266–275. doi: 10.3109/00016487509124683. [DOI] [PubMed] [Google Scholar]
- 54.Steward O, Rubel EW. Afferent influences on brain stem auditory nuclei of the chicken: cessation of amino acid incorporation as an antecedent to age-dependent transneuronal degeneration. J Comp Neurol. 1985;231:385–395. doi: 10.1002/cne.902310308. [DOI] [PubMed] [Google Scholar]
- 55.Vazquez E, Van de Water TR, Del Valle M, Vega JA, Staecker H, Giráldez F, Represa J. Pattern of trkB protein-like immunoreactivity in vivo and the in vitro effects of brain-derived neurotrophic factor (BDNF) on developing cochlear and vestibular neurons. Anat Embryol. 1994;189:157–167. doi: 10.1007/BF00185774. [DOI] [PubMed] [Google Scholar]
- 56.Wakade AR, Edgar D, Thoenen H. Both nerve growth factor and high K+ concentrations support the survival of chick embryo sympathetic neurons. Evidence for a common mechanism of action. Exp Cell Res. 1983;144:377–384. doi: 10.1016/0014-4827(83)90417-2. [DOI] [PubMed] [Google Scholar]
- 57.Webster M, Webster DB. Spiral ganglion neuron loss following organ of Corti loss: a quantitative study. Brain Res. 1981;212:17–30. doi: 10.1016/0006-8993(81)90028-7. [DOI] [PubMed] [Google Scholar]
- 58.Wheeler EF, Bothwell M, Schecterson LC, von Bartheld CS. Expression of BDNF and NT-3 mRNA in hair cells of the organ of Corti: quantitative analysis in developing rats. Hear Res. 1994;73:46–56. doi: 10.1016/0378-5955(94)90281-x. [DOI] [PubMed] [Google Scholar]
- 59.Wong-Riley MTT, Walsh SM, Leake-Jones PA. Maintenance of neuronal activity by electrical stimulation of unilaterally deafened cats demonstrable with cytochrome oxidase technique. Ann Otol Rhinol Laryngol. 1981;90:30–32. doi: 10.1177/00034894810902s211. [DOI] [PubMed] [Google Scholar]
- 60.Wright L. Cell survival in chick embryo ciliary ganglion is reduced by chronic ganglionic blockade. Dev Brain Res. 1981;1:283–286. doi: 10.1016/0165-3806(81)90114-0. [DOI] [PubMed] [Google Scholar]
- 61.Ylikoski J, Pirvola U, Moshnyakov M, Palgi J, Arumäe U, Saarma M. Expression patterns of neurotrophin and their receptor mRNAs in the rat inner ear. Hearing Res. 1993;65:69–78. doi: 10.1016/0378-5955(93)90202-c. [DOI] [PubMed] [Google Scholar]
- 62.Zheng JL, Stewart RR, Gao W-Q. Neurotrophin-4/5 enhances survival of cultured spiral ganglion neurons and protects them from cisplatin neurotoxicity. J Neurosci. 1995;15:5079–5087. doi: 10.1523/JNEUROSCI.15-07-05079.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zirpel L, Lachica EA, Lippe WR. Deafferentation increases the intracellular calcium of cochlear nucleus neurons in the embryonic chick. J Neurophysiol. 1995;74:1355–1357. doi: 10.1152/jn.1995.74.3.1355. [DOI] [PubMed] [Google Scholar]