

Abstract
We have studied the electrophysiological effects of glucose deprivation on morphologically identified striatal neurons recorded from a corticostriatal slice preparation. The large majority of the recorded cells were spiny neurons and responded to aglycemia with a slow membrane depolarization coupled with a reduction of the input resistance. In voltage-clamp experiments aglycemia caused an inward current. This current was associated with a conductance increase and reversed at −40 mV. The aglycemia-induced membrane depolarization was not affected by tetrodotoxin (TTX) or 6-cyano-7-nitroquinoxaline-2,3-dione plus aminophosphonovalerate, antagonists acting respectively on AMPA and NMDA glutamate receptors. Also, the intracellular injection of bis(2-aminophenoxy)ethane-N,N,N′,N′-tetra-acetic acid, a calcium (Ca2+) chelator, and low Ca2+/high Mg2+-containing solutions failed to reduce this phenomenon. Conversely, it was reduced by lowering external sodium (Na+) concentration.
A minority of the recorded cells had the morphological characteristics of large aspiny interneurons and the electrophysiological properties of “long-lasting afterhyperpolarization (LA) cells.” These cells responded to aglycemia with a membrane hyperpolarization/outward current that was coupled with an increased conductance. This current was not altered by TTX, blockers of ATP-dependent potassium (K+) channels, and adenosine A1 receptor antagonists, whereas it was reduced by solutions containing low Ca2+/high Mg2+. This current reversed at −105 mV and was blocked by barium, suggesting the involvement of a K+ conductance. We suggest that the opposite membrane responses of striatal neuronal subtypes to glucose deprivation might account for their differential neuronal vulnerability to aglycemia and ischemia.
Keywords: aglycemia, ischemia, excitatory amino acids, neuronal death, striatum, interneurons
In comparison with the cellular responses reported after anoxia in the CNS, the effects of glucose deprivation on single neuronal populations have been investigated less extensively. It is known that aglycemia hyperpolarizes hippocampal cells (Spuler et al., 1988) and neurons recorded from dorsolateral septal nucleus (Shoji, 1992). Aglycemia-activated K+ channels are present on hippocampal neurons (Tromba et al., 1992); these channels are blocked by the sulfonylurea glibenclamide. In contrast, in human cortical neurons, intracellularly recorded in vitro postsynaptic changes were not observed for periods of glucose depletion of <1 hr (Jiang and Haddad, 1992). Developmental and regional differences in the vulnerability of rat hippocampal slices to lack of glucose have been reported (Crepel et al., 1992).
In the present study we have analyzed the effects produced by glucose deprivation on the membrane properties of striatal neurons intracellularly recorded in a corticostriatal slice preparation. Most of these neurons have been morphologically identified. Moreover, we have investigated the possible involvement of excitatory amino acids in the effects of glucose deprivation and the ionic mechanisms underlying the membrane potential changes induced by aglycemia. We have been encouraged to examine this issue for three main reasons. First, striatum belongs to the selective vulnerable brain regions that are prone to develop neuronal damage in clinical and experimental aglycemia (Auer et al., 1984; Auer and Siesjo, 1988; Kristian et al., 1995; Nakao et al., 1995) and ischemia (Brierley, 1976; Pulsinelli et al., 1982;Hawker and Lang, 1990). Second, subpopulations of striatal neurons show a differential sensitivity to energy deprivation (Beal, 1992). In fact, although spiny neurons are vulnerable to ischemia (Brierley, 1976;Pulsinelli, 1985; Xu, 1995) and excitatory amino acid agonists (Beal et al., 1986; Calabresi et al., 1990a, 1995a), striatal interneurons are more resistant to ischemia (Francis and Pulsinelli, 1982) and excitotoxicity (Koh and Choi, 1988). Third, different subtypes of striatal interneurons have been identified recently on the basis of differential electrophysiological, morphological, and histochemical properties (Wilson et al., 1990; Kawaguchi, 1993; Kawaguchi et al., 1995). Thus, it is of particular interest to investigate the membrane changes induced by aglycemia on spiny neurons and to compare these responses with those observed in interneurons.
MATERIALS AND METHODS
Preparation and maintenance of the slices. Wistar rats (150–250 gm) were used. The preparation and maintenance of coronal slices have been described previously (Calabresi et al., 1990a,b,c, 1991). Briefly, corticostriatal coronal slices (200–300 μm) were prepared from tissue blocks of the brain with the use of a vibratome. A single slice was transferred to a recording chamber and submerged in a continuously flowing Krebs solution (35°C, 2–3 ml/min) gassed with 95% O2/5% CO2. To study glucose metabolism in striatal neurons, we deprived slices of glucose by removing glucose totally from the perfusate and by adding saccharose to balance the osmolarity. In some experiments the osmolarity was balanced by increasing the NaCl concentration (Jiang and Haddad, 1992). Because experiments performed by using these different procedures to replace glucose gave similar results, all the data were pooled. Aglycemic solutions entered the recording chamber no later then 20 sec after a three-way tap was turned. Complete replacement of the medium in the chamber took ∼90 sec, as determined by the speed of diffusion of a colored solution. The composition of the control solution was (in mm): 126 NaCl, 2.5 KCl, 1.2 MgCl2, 1.2 NaH2PO4, 2.4 CaCl2, 11 glucose, 25 NaHCO3. In some experiments, choline chloride was used to replace Na+ chloride. In these experiments, Na+chloride was reduced to 30% (38 mm). In other experiments, low Ca2+ (0.5 mm)/high Mg2+ (10 mm) solutions were used.
Recording technique. In most of the experiments, the intracellular recording electrodes were filled with 2 m KCl (30–60 MΩ). In some experiments, the pipettes were filled with 2m potassium acetate. In other experiments, 200 mm BAPTA was added to the solution of the intracellular pipette. An Axoclamp 2A amplifier was used for recordings in either current-clamp or voltage-clamp mode. In the single-electrode voltage-clamp mode, the switching frequency was 3 kHz. The headstage signal was monitored continuously on a separate oscilloscope. Traces were displayed on an oscilloscope and stored on a digital system. For synaptic stimulation, bipolar electrodes were used. These stimulating electrodes were located either in the cortical areas close to the recording electrode or in the white matter between the cortex and the striatum to activate corticostriatal fibers.
In some experiments, biocytin (Sigma, St. Louis, MO) was used in the intracellular electrode to stain the neurons. In these cases, biocytin at a concentration of 2–4% was added to a 0.5 m KCl pipette solution. Slices containing neurons stained with biocytin were fixed in paraformaldehyde (in 0.1 m phosphate buffer at pH 7.4) overnight and processed according to published protocols (Horikawa and Armstrong, 1988).
In other experiments, the tip of microelectrodes was filled with a solution containing 2 mm fura-2 pentapotassium salt (Molecular Probes, Eugene, OR) and 100 mm KCl, and the shank was filled with 1 m KCl. After intracellular injection of the dye with negative current (0.1–0.5 nA), fluorescence was elicited by epi-illumination with a Xenon arc lamp. Excitation light was filtered at both 340 and 380 nm. Fluorescent emission light was filtered through a long-pass barrier filter and detected by a CCD camera controlled by software (ImproVision) running on Power Macintosh 8100. Video images (see Fig. 1b) are 380 nm images (each of which represents the average of 256 frames).
Fig. 1.
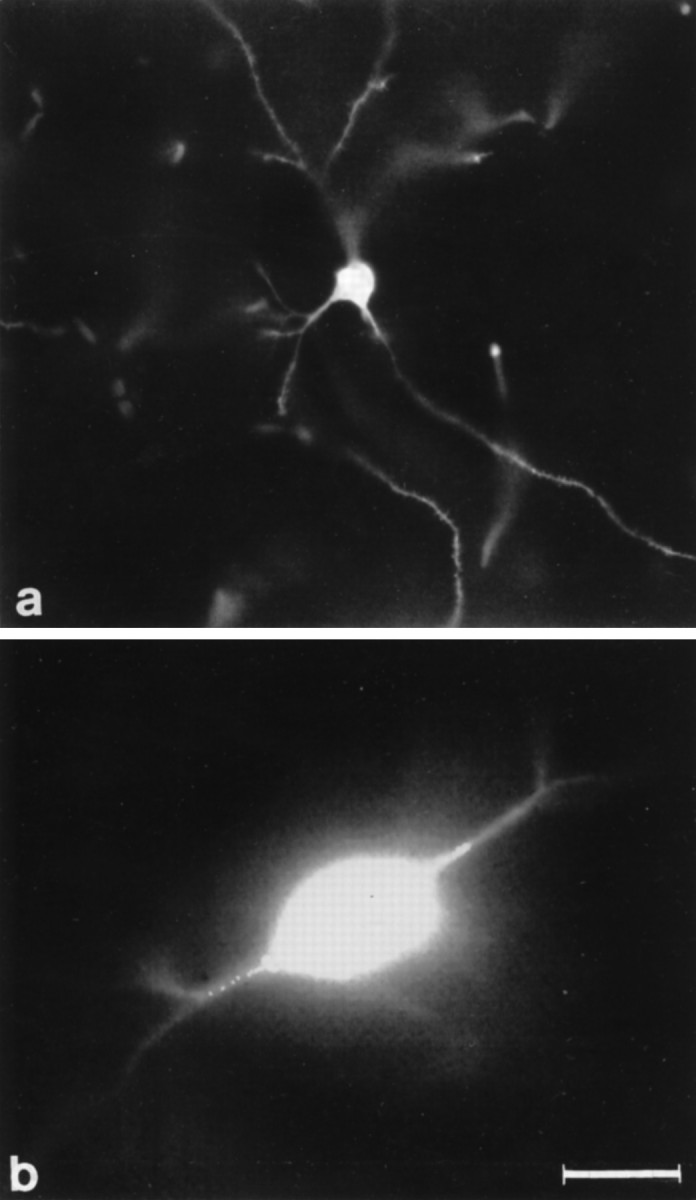
Morphological identification of striatal neurons. The figure represents two striatal neuronal subtypes: a biocytin-injected spiny striatal neuron (a) and a large LA interneuron filled with the dye fura-2 (b) (380 nm image, average of 256 frames) (for details, see Materials and Methods). Scale bar (shown in b): a, 70 μm;b, 30 μm.
Data analysis and drug applications. Values given in the text and in the figures are mean ± SEM of changes in the respective cell populations. Student’s t test (for paired and unpaired observations) was used to compare the means. The characteristics of action potentials and of current–voltage curves (I–V) in different experimental conditions were studied by using a fast chart recorder and a digital system (Nicolet System 400: Benchtop Waveform Acquisition System). Drugs were applied by dissolving them to the desired final concentration in the saline and by switching the perfusion from control saline to drug-containing saline. 6-Cyano-7-nitroquinoxaline-2,3-dione (CNQX) was from Tocris.d-2-amino-5-phosphonovalerate (APV), BAPTA, and tetrodotoxin (TTX) were from Sigma. 8-Cyclopentyl-1,3-dimethylxanthine (CPT) and 1,3-dipropyl-8-cyclopentylxanthine (CPX) were from RBI (Natick, MA). Tolbutamide and glipizide were a gift from Dr. N. B. Mercuri.
RESULTS
Physiological and morphological properties of spiny neurons
We recorded from 197 spiny striatal neurons. In 91 of 197 recorded spiny neurons, the electrophysiological identification was confirmed by a morphological analysis obtained by using biocytin staining (Fig.1a). These cells had a small soma (10–18 μm) and an extensive dendritic tree studded densely with spines. These cells had high resting-membrane potential (−84 ± 5 mV), relatively low apparent input resistance (38 ± 8 MΩ) when measured at the resting potentials from the amplitude of small (<10 mV) hyperpolarizing electrotonic potentials, action potentials of short duration (1.1 ± 0.3 msec), and high amplitude (102 ± 4 mV). These cells were silent at rest and showed membrane rectification and tonic firing activity during depolarizing current pulses (Fig.2C). All of the 40 cells studied with voltage-clamp showed prominent inward rectification in the steady-stateI–V relation. When the currents were evoked by hyperpolarizing command steps, there was no detectable time-dependent component. Some of these properties have been described previously bothin vivo (Preston et al., 1979; Wilson and Groves, 1980;Calabresi et al., 1990c; Onn et al., 1994a; Xu, 1995) and in vitro (Kita et al., 1984; Calabresi et al., 1990b, 1991, 1995b;Jiang and North, 1991; Cepeda et al., 1994) and resemble those reported for intracellularly stained spiny striatal neurons (Preston et al., 1979; Cepeda et al., 1994; Onn et al., 1994b; Calabresi et al., 1995a).
Fig. 2.

Aglycemia depolarizes striatal spiny neurons and decreases their membrane input resistance. A, Thetop part represents selected chart records of the membrane potential changes caused by glucose deprivation. Theblack arrows indicate the onset of hypoglycemia (a), 15 min of aglycemia (b), the onset of the washout (c), and 10 min of washout (d). Interruptions of the traces between two open arrows represent different periods of recording: 13 min betweena and b, 13 min between band c, and 6 min between c andd. The bottom part shows the current trace monitored during the recording. The downward voltage deflections are induced by negative current steps. B, Single sweeps recorded at higher speed at the same times as those indicated inA. Current and voltage calibrations in Aapply also for B. In Bc, thedotted line indicates the original resting membrane potential (RMP = −85 mV). C, The tonic firing discharge (a) induced by a depolarizing pulse (b) in a striatal spiny cell (RMP= −86 mV).
Effects of glucose deprivation on spiny neurons
As shown in Figure 2, periods of glucose deprivation >10 min induced a progressive, slow membrane depolarization. The amplitude of this depolarization was dependent on the duration of aglycemia (Fig.3). The aglycemia-induced membrane depolarization was coupled with a decrease of the apparent input resistance (Fig.2A,B). At 25 min from the onset of the aglycemic solution the input resistance was 55 ± 5% (n = 15) of the control value. This effect persisted also when the membrane was manually clamped to the resting level (n = 7; data not shown). Brief periods (5–10 min) of glucose deprivation did not cause significant changes of membrane potential and input resistance. We also studied the effects of aglycemia in single-microelectrode voltage-clamp experiments. Periods of glucose deprivation >10 min induced inward currents associated with an increase in membrane conductance (Figs. 4, 5A,B). These events had a time course similar to that observed in current-clamp experiments. In most of the experiments, we used a period of glucose deprivation lasting 25 min, which produced reversible membrane depolarization/inward current. In several experiments, a transient overshoot of ∼10 mV was observed (Fig. 2) on restoration of normal glucose concentration. In six experiments, we also studied effects produced by 40 min of aglycemia. This period of glucose deprivation induced irreversible deterioration of the membrane properties in all of the recorded neurons (six of six cells; data not shown). As shown in Figure 5C, the extrapolated reversal potential for the aglycemia-induced inward current was −40 ± 4 mV (n = 7) by using K+ chloride-filled electrodes. This value was obtained in voltage-clamp experiments by applying voltage steps (1–3 sec duration) in both depolarizing and hyperpolarizing directions. In four experiments the intracellular pipette was filled with K+ acetate, and the extrapolated reversal potential for the inward current induced by hypoglycemia was −41 ± 5 mV (data not shown).
Fig. 3.

Time course of the membrane depolarization induced by aglycemia in normal medium and in different experimental conditions. The graphs show the amplitude of the membrane depolarization induced by 25 min of glucose deprivation in control medium (filled circles), in 1 μmTTX (open circles), in 10 μm CNQX plus 50 μm APV (open rhombs), in low Na+ (38 mm, filled rhombs), in the presence of BAPTA-filled electrodes (open squares), in the presence of low calcium (0.5 mm)/high magnesium (10 mm) solutions (filled squares), and in the presence of the adenosine A1 receptor antagonist CPX (300 nm; filled triangles). Each data point represents the mean of at least four single observations.Asterisks indicate significant difference from control values (p < 0.01).
Fig. 4.

Aglycemia induces an inward current and increases the membrane conductance in spiny neurons. A, During a single-microelectrode voltage-clamp experiment, the voltage was monitored (see top part). The glucose deprivation induced an inward current. The black arrows indicate the onset of aglycemia (a), 15 min of aglycemia (b), the onset of the washout after 25 min of aglycemia (c), and 10 min of washout (d). Interruptions of the traces between two open arrowsrepresent different periods of recording: 14 min betweena and b, 7 min between band c, and 8 min between c andd. The downward deflections are induced by negative current steps. B, This part of the figure shows single sweeps recorded at higher speed at the same times as those indicated inA. Current and voltage calibrations in Aapply also for B. The holding potential during the experiment was −85 mV.
Fig. 5.
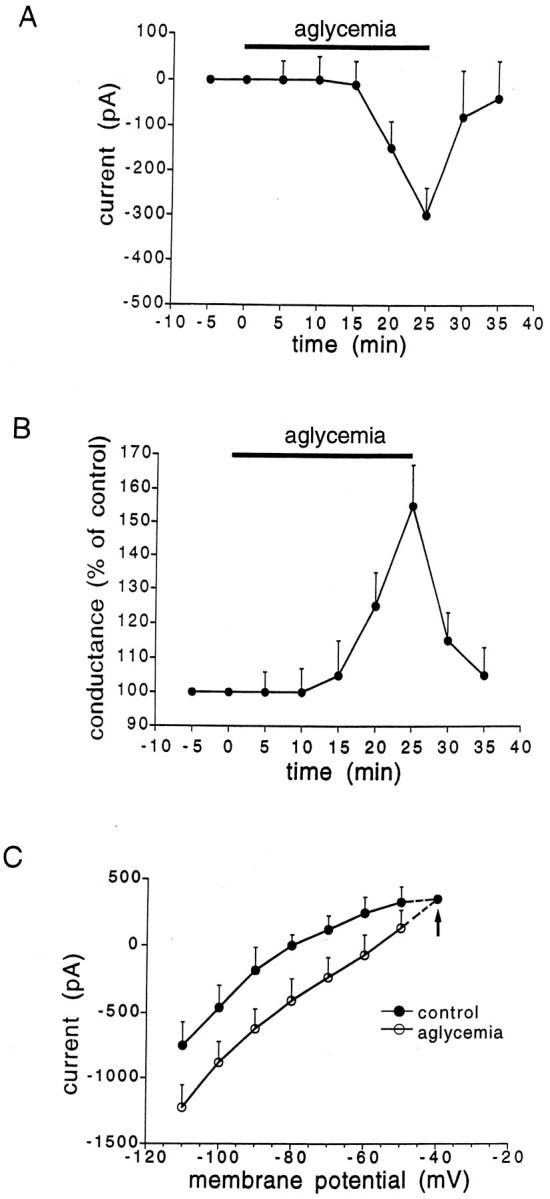
Characteristics of the inward current induced by aglycemia in spiny neurons. A, The graphshows the time course of the inward current induced by 25 min of glucose deprivation. B, The graph indicates the relative membrane conductance changes caused by 25 min of aglycemia.C, I–V relationship showing the extrapolated reversal potential (−40 mV, arrow) of the aglycemia-induced inward current. Long-lasting (1–3 sec) voltage steps were applied in both negative and positive directions before (filled circles) and during (open circles) aglycemia. The holding potential was −80 mV. Each data point represents the mean of at least four single observations.
TTX, low Ca2+/high Mg2+ medium, and glutamate receptor antagonists on aglycemia-induced depolarization in spiny neurons
To analyze whether the aglycemia-induced membrane depolarization observed in spiny cells was caused by a presynaptic mechanism involving an enhanced release of excitatory transmitters, we tested the effect of the preincubation (10 min before the onset of the glucose deprivation) of the slices in 1 μm TTX. As shown in Figure 3, this Na+ channel blocker did not affect the membrane depolarization induced by aglycemia (p > 0.05). To investigate the possible involvement of a TTX-resistant release of excitatory neurotransmitters, we studied the effect of low Ca2+ (0.5 mm)/high Mg2+ (10 mm) medium on the membrane depolarization induced by aglycemia. Incubation of the slices in low Ca2+/high Mg2+ solutions did not reduce the aglycemia-induced membrane depolarization (p > 0.05) (Fig. 3). We also tested whether the direct blockade of postsynaptic glutamate receptors could affect the aglycemia-induced membrane depolarization. We incubated (10 min before the onset of aglycemia) the slices in 10 μm CNQX, an antagonist of AMPA glutamate receptors, plus 50 μm APV, an antagonist of NMDA glutamate receptor. Even in this experimental condition, aglycemia caused membrane depolarizations whose amplitude and duration were similar to those observed in control medium (p > 0.05) (Fig. 3). These concentrations of antagonists fully blocked excitatory postsynaptic potentials evoked by activation of glutamatergic corticostriatal fibers in brain slice preparations (Calabresi et al., 1995b, 1996). CPT (1 μm, data not shown;n = 5) and CPX (300 nm; n = 6) (Fig. 3), antagonists of A1 adenosine receptors, did not affect the membrane depolarization caused by glucose deprivation (p > 0.05).
External low Na+ and intracellular BAPTA on aglycemia-induced membrane depolarization in spiny neurons
To test the possible involvement of a TTX-resistant Na+ influx in the membrane depolarization/inward current observed in spiny neurons after glucose deprivation, we studied the effects of aglycemia in low Na+-containing solutions (38 mm; see Materials and Methods). The lowering of the extracellular Na+ concentration significantly reduced the aglycemia-induced membrane depolarizations (p < 0.01; n = 6) (Fig. 3) and inward currents (n = 3;, data not shown). In low Na+-containing solutions, the reversal potential for the aglycemia-induced inward current was −68 mV ± 4 (n = 3).
The dependence of the aglycemia-induced membrane depolarization/inward current on intracellular Ca2+ was studied with microelectrodes filled with the Ca2+ chelating agent BAPTA (200 mm). As reported previously (Calabresi et al., 1994), 20 min after the intracellular BAPTA injection (performed by applying negative pulses 1 nA in amplitude, 1 sec in duration, 0.3 Hz) the long-term depression (LTD) of excitatory synaptic transmission observed after tetanic stimulation of the corticostriatal fibers was blocked. This test was performed before the glucose deprivation to confirm that the injection of intracellular BAPTA had prevented the rise in internal Ca2+ required to trigger the LTD. In fact, striatal spiny neurons have neither prominent slow afterhyperpolarization nor firing accommodation (Calabresi et al., 1990b,c); thus the blockade of these events by BAPTA could not be used to detect the efficacy of this Ca2+ chelator. In BAPTA-injected neurons the aglycemia-induced membrane depolarization was not altered (p > 0.05; n = 6) (Fig. 3). Moreover, in the presence of intracellular BAPTA the inward current induced by glucose deprivation was unaffected (p> 0.05; n = 3; data not shown).
Physiological and morphological properties of large aspiny interneurons
Twenty-nine recorded cells had electrophysiological characteristics different from those described for spiny neurons and showed properties that have been attributed previously to large aspiny interneurons (Wilson et al., 1990; Jiang and North, 1991; Kawaguchi, 1992, 1993; Kawaguchi et al., 1995). In 11 of these 29 LA interneurons the electrophysiological identification was confirmed by a morphological analysis. This analysis was performed by using either biocytin or fura-2 staining (Fig. 1b). The soma of LA neurons was larger (range, 25–49 μm) than those of spiny cells. The LA interneurons had polygonal or fusiform cell bodies, and their dendrites did not show spines. These cells had low membrane potential (−60 ± 3 mV) and high input resistance (195 ± 55 MΩ). Spontaneous firing occurred in 5 of these 29 cells. As shown in Figure6C, in these neurons depolarizing current pulses of small amplitude (200–400 pA) and short duration (5–30 msec) elicited a single action potential followed by a long-lasting afterhyperpolarization (amplitude 8.9 ± 9 mV, duration 350 ± 130 msec). The amplitude of the action potential was 70.5 ± 3 mV, and the duration of spike at half-amplitude was 0.71 ± 0.05 msec. The hyperpolarizing electrotonic potential declined after about the first 100 msec (Fig. 6). For this reason, the input resistance values were calculated either from the peak amplitude of the electrotonic potential evoked by a 300–400 pA current pulse or at the steady-state. The decline in the electrotonic hyperpolarizing potential was blocked by 2 mm cesium in the external medium (n = 4; data not shown). This finding suggests that it might be attributed to the activation of an Ihcurrent, as has been suggested previously (Jiang and North, 1991).
Fig. 6.

Aglycemia hyperpolarizes LA striatal interneurons and decreases their membrane input resistance. A, Thetop part represents selected chart records of the membrane potential changes caused by glucose deprivation. Theblack arrows indicate the onset of aglycemia (a), 10 min of aglycemia (b), the onset of the washout (c), and 10 min of washout (d). Interruptions of the traces between two open arrows represent different periods of recording: 8 min betweena and b, 3 min between band c, and 7 min between c andd. The bottom part shows the current trace monitored during the recording. The downward voltage deflections are induced by negative current steps. B, Single sweeps recorded at higher speed at the same times as indicated inA. In Ac and Bc, thedotted line indicates the original resting membrane potential (−59 mV). C, A single action potential followed by a long-lasting hyperpolarization (a) is induced by a depolarizing current pulse (b) in an LA striatal interneuron. The dotted line represents the resting membrane potential of the neuron (−59 mV).
Effects of aglycemia on large aspiny interneurons
Unlike spiny neurons, the large aspiny interneurons (LA cells) were hyperpolarized by glucose deprivation (n = 22) (Fig. 6). This hyperpolarization started about 10 min after the onset of the glucose-free solution, and it reversed completely after 10–15 min of washout. As shown in Figure 6, the aglycemia-induced hyperpolarization was coupled with a reduction of the apparent input resistance as detected by the decreased amplitude of long-hyperpolarizing pulses. After 25 min of aglycemia the input resistance measured at the steady-state was 59 ± 5% (n = 9) of the control value. In nine LA interneurons the effect of glucose deprivation was also measured by using the single microelectrode voltage-clamp technique. Under this experimental condition aglycemia produced an outward current associated with an increase of the membrane conductance (Fig. 7). The hyperpolarization generated by glucose deprivation in LA interneurons was blocked by 300 μm barium (p < 0.001; n = 4) but not by 1 mm tolbutamide (p > 0.05; n = 3) or 100 nm glipizide (p > 0.05;n = 3), blockers of ATP-dependent K+channels (Fig. 8A). TTX (1 μm) did not alter the membrane hyperpolarization (p > 0.05; n = 4; data not shown) or the outward current induced by aglycemia in large aspiny neurons (p > 0.05; n = 4; data not shown). Incubation of the slices in low Ca2+ (0.5 mm)/high Mg2+ (10 mm)-containing solutions significantly attenuated the aglycemia-induced membrane hyperpolarization but did not block this phenomenon (p < 0.01; n = 4) (Fig. 8). We also evaluated the possible effect of CPT and CPX, antagonists of adenosine A1 receptors, on the membrane hyperpolarization caused by glucose deprivation. Neither CPT (p > 0.05;n = 3; data not shown) nor CPX (p > 0.05; n = 4) (Fig.8A) altered the time course of the aglycemia-induced membrane hyperpolarization; however, these antagonists blocked the membrane hyperpolarization induced by exogenous adenosine (30 μm; n = 3) in a reversible manner (Fig.8C). The reversal potential of the aglycemia-induced outward current was −105 ± 5 mV (n = 4) (Fig.8B). This value was calculated by measuring the steady-state currents generated by long-lasting (1–3 sec) voltage steps of progressively increasing amplitude delivered from the resting membrane potential in hyperpolarizing direction before and during the application of glucose-free medium.
Fig. 7.

Aglycemia induces an outward current and increases the membrane conductance in LA striatal interneurons.A, During a single-microelectrode voltage-clamp experiment, aglycemia caused an outward current (toppart); during the experiment the voltage was monitored (seebottom part). The black arrows indicate the onset of aglycemia (a), the onset of the washout after 15 min of aglycemia (b), and 10 min after the washout (c). The interruption of the traces betweentwo open arrows represents a period of recording of 12 min. The downward deflections are induced by negative current steps.B, Single sweeps recorded at higher speed at the same times as indicated in A. The holding potential during the experiment was −60 mV.
Fig. 8.

The aglycemia-induced hyperpolarization/outward current in LA interneurons is mediated by a K+ conductance.A, Time course of the membrane changes induced by 25 min of glucose deprivation in different experimental conditions: control (n = 9; filled circles), 300 μm barium (n = 4; open circles), 1 mm tolbutamide (n = 3; open rhombs), 100 nm glipizide (n = 3; filled squares), 300 nm CPX (n = 4; open squares), and low calcium (0.5 mm)/high magnesium (10 mm) solutions (n = 4; filled rhombs). Asterisks indicate significant difference from control values (p < 0.01).B, In an LA interneuron, bath application of 30 μm adenosine induced a membrane hyperpolarization (a); this membrane hyperpolarization was fully blocked by 300 nm CPX, an adenosine A1 receptor antagonist (b); after the washout of this antagonist, the hyperpolarizing action of adenosine was restored (c).C, The reversal potential of the aglycemia-induced outward current is indicated by the arrow (−105 mV;n = 4). This value was calculated by measuring the steady-state currents generated by long-lasting (1–3 sec) voltage steps of progressively increasing amplitude from the holding potential (−60 mV; n = 4) before (filled circles) and during glucose deprivation (25 min, open circles).
DISCUSSION
The main finding of the present study is that glucose deprivation causes opposite membrane potential changes in striatal spiny neurons and in large aspiny interneurons. Spiny neurons are depolarized by aglycemia, whereas large aspiny interneurons are hyperpolarized by glucose deprivation. These different electrophysiological responses may account for the differential neuronal vulnerability observed in these neuronal subtypes after aglycemia and ischemia.
Spiny neurons are depolarized by glucose deprivation
The large majority of striatal neurons is represented by spiny projecting cells whose anatomical, physiological, and pharmacological properties have been widely characterized (Graybiel, 1990; Smith and Bolam, 1990; Surmeier and Kitai, 1994; Calabresi et al., 1996). These small GABAergic cells are highly vulnerable to transient forebrain ischemia (Brierley, 1976; Pulsinelli et al., 1982; Xu, 1995), energy metabolism failure (Beal, 1992, 1995), and aglycemic coma (Auer et al., 1984; Kristian et al., 1995). Accordingly we have found that glucose deprivation depolarizes spiny neurons. Periods of aglycemia <30 min cause reversible membrane potential changes; however, when aglycemia lasts >30 min, the membrane depolarization is irreversible. The aglycemia-induced membrane depolarization is coupled with a decreased input resistance. In voltage-clamp mode, aglycemia generates an inward current associated with an increased conductance. TTX does not alter the amplitude or the time course of the aglycemia-induced depolarization. Low-Na+-containing solutions significantly reduce the amplitude of the depolarization caused by glucose deprivation. A TTX-resistant Ca2+-activated nonspecific cationic current has been reported to act as a depolarizing driving force in neurons of invertebrates (Hofmeier and Lux, 1981) and in rat hippocampal neurons (Crepel et al., 1994). The finding that the aglycemia-induced depolarization is not altered by the intracellular injection of BAPTA, however, does not support the involvement of a Ca2+-activated conductance in the aglycemia-induced effects. There seems to be a discrepancy between the equilibrium potential for Na+ ions and the reversal potential of the aglycemia-induced inward current in spiny neurons that is inconsistent with the involvement of a conductance generated exclusively by Na+ ions. On the basis of an extrapolated I–Vplot (Fig. 5C), this current apparently reverses at approximately −40 mV. Given the ionic composition of the medium, the equilibrium potential for Na+ ions should be approximately +50 mV, whereas the equilibrium potential for K+ ions should be approximately −105 mV (assuming [K]i = 160 mm). Therefore, a reversal potential of approximately −40 mV might indicate a nonselective cation conductance or the combination of independent Na+ and K+ conductance. In either case, the reduction of the extracellular Na+concentration to 38 mm (30% of the control) will reduce the equilibrium potential for Na+ ions to approximately +34 mV and thereby change the reversal potential of a mixed cation conductance to approximately −70 mV. This potential is very close to the value that we obtained in our experiments by using low Na+-containing medium (−68 mV). It has to be stressed that on the restoration of normal glucose concentration a transient membrane potential overshoot of ∼10 mV was observed. This phenomenon may reveal that several compensatory currents are at play that have different rates of recovery. This evidence might also support the idea that the inconsistency between the reversal potential of the aglycemia-induced current and the equilibrium potential for Na+ ions is explained by a second, opposing K+current. Interestingly, an increase of the extracellular K+concentration associated with a decreased intracellular concentration of this ion has been reported in some neuronal subtypes during energy deprivation (Jiang and Haddad, 1991; for review, see Martin et al., 1994). Conversely, the participation of chloride ions in the aglycemia-induced inward current is ruled out, because the reversal potential of this current is not changed by using microelectrodes filled with K+ acetate instead of K+chloride.
The evidence that low Ca2+/high Mg2+ solutions did not reduce the aglycemia-induced membrane depolarization rules out the possible involvement of presynaptic mechanisms in this electrophysiological phenomenon. Moreover, our experiments showing the lack of effect of CNQX and APV on the aglycemia-induced electrophysiological actions clearly indicate that activation of ionotropic glutamate receptors is not involved in the aglycemia-induced depolarization/inward current. It has already been shown that glutamate receptor antagonists fail to alter anoxia-induced membrane depolarization in striatal spiny neurons (Calabresi et al., 1995b) and in brainstem neurons (Haddad and Jiang, 1993). It is possible, however, that although excitatory amino acids are not involved in the “acute” effects of glucose deprivation, they may play a role in the “delayed neuronal death” (Choi, 1990; Choi and Rothmann, 1990). Accordingly, lesions of the glutamatergic corticostriatal projections in the rat ameliorate aglycemic brain damage in the striatum (Wieloch et al., 1985), and the blockade of NMDA receptors prevents the aglycemia-induced neuronal damage (Wieloch, 1985).
We have observed recently that brief periods (5–10 min) of aglycemia cause presynaptic inhibition at corticostriatal synapses. This effect is mediated by the release of endogenous adenosine acting on A1 receptors located on corticostriatal glutamatergic terminals (our unpublished observations). Endogenous adenosine, however, does not seem to be involved in the postsynaptic changes observed in spiny neurons during aglycemia, because A1 receptor antagonists affected neither the amplitude nor the time course of the membrane depolarization caused by glucose deprivation.
Large aspiny interneurons are hyperpolarized by glucose deprivation
The development of cytochemical approaches to identification and classification of interneurons has recently been combined with the physiological analysis of the basic cellular properties of the various subtypes of interneurons (for review, see Kawaguchi et al., 1995). We have studied the membrane responses to glucose deprivation of large aspiny interneurons that have the electrophysiological properties of LA cells (Jiang and North, 1991; Kawaguchi, 1992, 1993). These cells are considered cholinergic interneurons. They receive glutamatergic and dopaminergic inputs and provide an intrinsic cholinergic innervation to spiny neurons. The response of LA interneurons to glucose deprivation is qualitatively different from the effects observed in spiny neurons. In fact, LA cells respond to aglycemia with a membrane hyperpolarization coupled with a decrease of the input resistance. The voltage-clamp analysis has revealed that the hyperpolarization induced by glucose deprivation in these cells is caused by an outward current that reversed at the K+ equilibrium potential. The finding that this outward current is blocked by barium suggests further that it is caused by the activation of a K+-mediated conductance. Moreover, the lack of effect of TTX on the membrane hyperpolarization/outward current-generated aglycemia suggests that this electrophysiological effect is not dependent on a TTX-sensitive release of transmitters. We found that low Ca2+/high Mg2+-containing solutions significantly reduced but did not block the aglycemia-induced membrane hyperpolarization. This finding might have two possible explanations. 1) A component of the K+ conductance generating the aglycemia-induced membrane hyperpolarization is Ca2+-dependent, and 2) a Ca2+-dependent, TTX-insensitive release of transmitters plays a role in this membrane hyperpolarization. This observation, however, demonstrates clearly that at least part of the aglycemia-induced membrane hyperpolarization is postsynaptically mediated. Interestingly, tolbutamide and glipizide fail to block the membrane hyperpolarization/outward current induced by glucose deprivation in LA cells, suggesting that at least in our experimental condition the activation of ATP-sensitive K+ channels does not play a prominent role in the electrophysiological effects induced by glucose deprivation. Membrane potential changes observed in LA interneurons strongly resemble those observed in dorsolateral septal nucleus neurons during aglycemia (Shoji, 1992), in which glucose deprivation causes K+-dependent membrane hyperpolarizations that are not affected by inhibitors of ATP-dependent K+channels.
Extracellular levels of adenosine increase during energy deprivation caused by either anoxia or aglycemia (Martin et al., 1994). Because adenosine has been reported to hyperpolarize various neuronal subtypes by activating K+ channels (Greene and Haas, 1991), we have studied the possible involvement of endogenous adenosine in the aglycemia-induced membrane hyperpolarization. Antagonists of adenosine A1 receptors fully blocked the membrane hyperpolarization induced by exogenous adenosine, but they failed to affect the hyperpolarizing action of aglycemia, suggesting that endogenous adenosine does not play a prominent role in the membrane potential changes observed during glucose deprivation.
The membrane hyperpolarization observed during glucose deprivation in large LA interneurons might represent a protective mechanism activated when energy metabolism fails. This hypothesis can also explain previous morphological results showing that although spiny neurons are highly vulnerable to transient forebrain ischemia (Brierley, 1976; Pulsinelli et al., 1982), energy metabolism failure (Beal, 1992, 1995), and aglycemic coma (Auer et al., 1984; Kristian et al., 1995), large cholinergic interneurons are spared during these pathological events (Francis and Pulsinelli, 1982; Pulsinelli, 1985; Chesselet et al., 1990). Our observation, however, does not exclude the possibility that a reduced expression of excitatory amino acid receptor subtypes or an enhanced activity of protective trophic factors or both might also account for the relative viability of large cholinergic interneurons to hypoxic–ischemic insults (Burke and Kenyon, 1991; Nakao et al., 1995).
Footnotes
This work was supported by Consiglio Nazionale delle Ricerche grants to P.C. and G.B. We thank G. Gattoni and M. Tolu for excellent technical assistance.
Correspondence should be addressed to Paolo Calabresi, Clinica Neurologica, Dip. Sanitá, Universitá di Roma Tor Vergata, Via O. Raimondo 8, 00173 Rome, Italy.
Dr. C. Magarinos Ascone’s present address: Hopital Ramon Y Cajal, Servicio de Neurologia Experimental, Madrid, Spain.
REFERENCES
- 1.Auer RN, Siesjo BK. Biological differences between ischemia, hypoglycemia and epilepsy. Ann Neurol. 1988;24:699–707. doi: 10.1002/ana.410240602. [DOI] [PubMed] [Google Scholar]
- 2.Auer RN, Wieloch T, Olsson Y, Siesjo BK. The distribution of hypoglycemic brain damage. Acta Neuropathol. 1984;64:177–191. doi: 10.1007/BF00688108. [DOI] [PubMed] [Google Scholar]
- 3.Beal MF. Does impairment of energy metabolism result in excitotoxic neuronal death in neurodegenerative illnesses? Ann Neurol. 1992;31:119–30. doi: 10.1002/ana.410310202. [DOI] [PubMed] [Google Scholar]
- 4.Beal MF. Aging, energy and oxidative stress in neurodegenerative diseases. Ann Neurol. 1995;38:357–366. doi: 10.1002/ana.410380304. [DOI] [PubMed] [Google Scholar]
- 5.Beal MF, Kowall NW, Ellison DW, Mazurek MF, Swartz KJ, Martin JB. Replication of the neurochemical characteristics of Huntington’s disease by quinolinic acid. Nature. 1986;321:168–171. doi: 10.1038/321168a0. [DOI] [PubMed] [Google Scholar]
- 6.Brierley JB. Cerebral hypoxia. In: Blackwood W, Corsellis A, editors. Greenfield’s neuropathology. Arnold; London: 1976. pp. 43–85. [Google Scholar]
- 7.Burke RE, Kenyon N. The effect of neonatal hypoxia-ischemia on striatal cholinergic neuropil: a quantitative morphologic analysis. Exp Neurol. 1991;113:63–73. doi: 10.1016/0014-4886(91)90147-5. [DOI] [PubMed] [Google Scholar]
- 8.Calabresi P, De Murtas M, Mercuri NB, Bernardi G. Kainic acid on neostriatal neurons intracellularly recorded in vitro: electrophysiological evidence for differential neuronal sensitivity. J Neurosci. 1990a;10:3960–3969. doi: 10.1523/JNEUROSCI.10-12-03960.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Calabresi P, Mercuri NB, Bernardi G. Synaptic and intrinsic control of membrane excitability of neostriatal neurons. II. An in vitro analysis. J Neurophysiol. 1990b;63:663–675. doi: 10.1152/jn.1990.63.4.663. [DOI] [PubMed] [Google Scholar]
- 10.Calabresi P, Mercuri NB, Stefani A, Bernardi G. Synaptic and intrinsic control of membrane excitability of neostriatal neurons. I. An in vivo analysis. J Neurophysiol. 1990c;63:651–662. doi: 10.1152/jn.1990.63.4.651. [DOI] [PubMed] [Google Scholar]
- 11.Calabresi P, Mercuri NB, De Murtas M, Bernardi G. Involvement of GABA system in the feedback regulation of glutamate- and GABA-mediated synaptic potentials in rat neostriatum. J Physiol (Lond) 1991;440:581–599. doi: 10.1113/jphysiol.1991.sp018726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Calabresi P, Pisani A, Mercuri NB, Bernardi G. Postreceptor mechanisms underlying striatal long-term depression. J Neurosci. 1994;14:4871–4881. doi: 10.1523/JNEUROSCI.14-08-04871.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Calabresi P, De Murtas M, Pisani A, Stefani A, Sancesario G, Mercuri NB, Bernardi G. Vulnerability of medium spiny striatal neurons to glutamate: role of Na+/K+ ATPase. Eur J Neurosci. 1995a;7:1674–1683. doi: 10.1111/j.1460-9568.1995.tb00689.x. [DOI] [PubMed] [Google Scholar]
- 14.Calabresi P, Pisani A, Mercuri NB, Bernardi G. On the mechanisms underlying hypoxia-induced membrane depolarization in striatal neurons. Brain. 1995b;118:1027–1038. doi: 10.1093/brain/118.4.1027. [DOI] [PubMed] [Google Scholar]
- 15.Calabresi P, Pisani A, Mercuri NB, Bernardi G. The corticostriatal projection: from synaptic plasticity to dysfunctions of the basal ganglia. Trends Neurosci. 1996;19:19–24. doi: 10.1016/0166-2236(96)81862-5. [DOI] [PubMed] [Google Scholar]
- 16.Cepeda C, Walsh JP, Peacock W, Buckwald NA, Levine MS. Neurophysiological, pharmacological and morphological properties of human caudate neurons recorded in vitro. Neuroscience. 1994;59:89–103. doi: 10.1016/0306-4522(94)90101-5. [DOI] [PubMed] [Google Scholar]
- 17.Chesselet M-F, Gonzales C, Lin CS, Polsky K, Jin BK. Ischemic damage in the striatum of adult gerbils: relative sparing of somatostatinergic and cholinergic interneurons contrasts with loss of efferent neurons. Exp Neurol. 1990;110:209–218. doi: 10.1016/0014-4886(90)90032-n. [DOI] [PubMed] [Google Scholar]
- 18.Choi DW. Cerebral hypoxia: some new approaches and unanswered questions. J Neurosci. 1990;10:2493–2501. doi: 10.1523/JNEUROSCI.10-08-02493.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Choi DW, Rothmann SM. The role of glutamate neurotoxicity in hypoxic-ischemic neuronal death. Annu Rev Neurosci. 1990;13:171–182. doi: 10.1146/annurev.ne.13.030190.001131. [DOI] [PubMed] [Google Scholar]
- 20.Crepel R, Krnjevic K, Ben-Ari Y. Developmental and regional differences in the vulnerability of rat hippocampal slices to lack of glucose. Neuroscience. 1992;47:579–589. doi: 10.1016/0306-4522(92)90167-z. [DOI] [PubMed] [Google Scholar]
- 21.Crepel V, Aniksztein L, Ben-Ari Y, Hammond C. Glutamate metabotropic receptors increase a Ca2+-activated nonspecific cationic current in CA1 hippocampal neurons. J Neurophysiol. 1994;72:1561–1569. doi: 10.1152/jn.1994.72.4.1561. [DOI] [PubMed] [Google Scholar]
- 22.Francis A, Pulsinelli W. The response of GABAergic and cholinergic neurons to transient cerebral ischemia. Brain Res. 1982;243:271–278. doi: 10.1016/0006-8993(82)90250-5. [DOI] [PubMed] [Google Scholar]
- 23.Graybiel AM. Neurotransmitters and neuromodulators in the basal ganglia. Trends Neurosci. 1990;13:133–254. doi: 10.1016/0166-2236(90)90104-i. [DOI] [PubMed] [Google Scholar]
- 24.Greene RW, Haas HL. The electrophysiology of adenosine in the mammalian central nervous system. Prog Neurobiol. 1991;36:329–341. doi: 10.1016/0301-0082(91)90005-l. [DOI] [PubMed] [Google Scholar]
- 25.Haddad GG, Jiang C. Mechanisms of anoxia-induced depolarization in brainstem neurons: in vitro intracellular studies and voltage clamp studies in the adult rat. Brain Res. 1993;625:261–268. doi: 10.1016/0006-8993(93)91067-3. [DOI] [PubMed] [Google Scholar]
- 26.Hawker K, Lang AE. Hypoxic-ischemic damage of the basal ganglia: case reports and a review of the literature. Mov Disord. 1990;5:219–224. doi: 10.1002/mds.870050306. [DOI] [PubMed] [Google Scholar]
- 27.Hofmeier G, Lux HD. The time courses of intracellular free calcium and related electrical effects after injection of CaCl2 into neurons of the snail, Helix Pomatia. Pflügers Arch. 1981;391:242–251. doi: 10.1007/BF00596178. [DOI] [PubMed] [Google Scholar]
- 28.Horikawa H, Armstrong WE. A versatile means of intracellular labelling: injection of biocytin and its detection with avidin conjugates. J Neurosci Methods. 1988;25:1–11. doi: 10.1016/0165-0270(88)90114-8. [DOI] [PubMed] [Google Scholar]
- 29.Jiang C, Haddad GG. Effect of anoxia on intracellular and extracellular potassium activity in hypoglossal neurons in vitro. J Neurophysiol. 1991;66:103–111. doi: 10.1152/jn.1991.66.1.103. [DOI] [PubMed] [Google Scholar]
- 30.Jiang C, Haddad GG. Differential responses of neocortical neurons to glucose and/or O2 deprivation in human and rat. J Neurophysiol. 1992;68:2165–2173. doi: 10.1152/jn.1992.68.6.2165. [DOI] [PubMed] [Google Scholar]
- 31.Jiang Z-C, North RA. Membrane properties and synaptic responses of rat striatal neurones in vitro. J Physiol (London) 1991;443:533–553. doi: 10.1113/jphysiol.1991.sp018850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kawaguchi Y. Large aspiny cells in the matrix of the rat neostriatum in vitro: physiological identification, relation to the compartments and excitatory postsynaptic currents. J Neurophysiol. 1992;67:1669–1682. doi: 10.1152/jn.1992.67.6.1669. [DOI] [PubMed] [Google Scholar]
- 33.Kawaguchi Y. Physiological, morphological and histochemical characterization of three classes of interneurons in the rat neostriatum. J Neurosci. 1993;13:4908–4923. doi: 10.1523/JNEUROSCI.13-11-04908.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kawaguchi Y, Wilson CJ, Augood SJ, Emson PC. Striatal interneurons: chemical, physiological and morphological characterization. Trends Neurosci. 1995;18:527–535. doi: 10.1016/0166-2236(95)98374-8. [DOI] [PubMed] [Google Scholar]
- 35.Kita T, Kita H, Kitai ST. Passive electrical membrane properties of rat neostriatal neurons in an in vitro slice preparation. Brain Res. 1984;300:129–139. doi: 10.1016/0006-8993(84)91347-7. [DOI] [PubMed] [Google Scholar]
- 36.Koh J-Y, Choi DW. Cultured striatal neurons containing NADPH-diaphorase or acetylcholinesterase are selectively resistant to injury by NMDA receptor agonists. Brain Res. 1988;446:374–378. doi: 10.1016/0006-8993(88)90897-9. [DOI] [PubMed] [Google Scholar]
- 37.Kristian T, Gido G, Siesjo BK. The influence of acidosis on hypoglycemic brain damage. J Cereb Blood Flow. 1995;15:78–87. doi: 10.1038/jcbfm.1995.9. [DOI] [PubMed] [Google Scholar]
- 38.Martin RL, Lloyd HGE, Cowan AI. The early events of oxygen and glucose deprivation: setting the scene for neuronal death? Trends Neurosci. 1994;17:251–257. doi: 10.1016/0166-2236(94)90008-6. [DOI] [PubMed] [Google Scholar]
- 39.Nakao N, Kokaia Z, Odin P, Lindvall O. Protective effects of BDNF and NT-3 but not PDGF against hypoglycemic injury to cultured striatal neurons. Exp Neurol. 1995;131:1–10. doi: 10.1016/0014-4886(95)90002-0. [DOI] [PubMed] [Google Scholar]
- 40.Onn S, Berger TW, Grace A. Identification and characterization of striatal cell subtypes using in vivo intracellular recordings in rats. I. Basic physiology and responses to corticostriatal fiber stimulation. Synapse. 1994a;16:161–180. doi: 10.1002/syn.890160302. [DOI] [PubMed] [Google Scholar]
- 41.Onn S, Berger TW, Grace A. Identification and characterization of striatal cell subtypes using in vivo intracellular recordings in rats. III. Morphological correlates and compartmental localization. Synapse. 1994b;16:231–254. doi: 10.1002/syn.890160308. [DOI] [PubMed] [Google Scholar]
- 42.Preston RJ, Bishop GA, Kitai ST. Medium spiny neuron projection from the rat striatum: an intracellular horse-radish peroxidase study. Brain Res. 1979;183:253–258. doi: 10.1016/0006-8993(80)90462-x. [DOI] [PubMed] [Google Scholar]
- 43.Pulsinelli WA. Selective neuronal vulnerability: morphological and molecular characteristics. Prog Brain Res. 1985;63:29–37. doi: 10.1016/S0079-6123(08)61973-1. [DOI] [PubMed] [Google Scholar]
- 44.Pulsinelli WA, Brierley JB, Plum F. Temporal profile of neuronal damage in a model of transient forebrain ischemia. Ann Neurol. 1982;11:491–498. doi: 10.1002/ana.410110509. [DOI] [PubMed] [Google Scholar]
- 45.Shoji S. Glucose regulation of synaptic transmission in the dorsolateral septal nucleus of the rat. Synapse. 1992;2:322–332. doi: 10.1002/syn.890120409. [DOI] [PubMed] [Google Scholar]
- 46.Smith AD, Bolam JP. The neuronal network of the basal ganglia as revealed by the study of the synaptic connections of identified neurones. Trends Neurosci. 1990;13:259–265. doi: 10.1016/0166-2236(90)90106-k. [DOI] [PubMed] [Google Scholar]
- 47.Spuler A, Endres W, Grafe P. Glucose depletion hyperpolarizes guinea pig hippocampal neurons by an increase in potassium conductance. Exp Neurol. 1988;100:248–252. doi: 10.1016/0014-4886(88)90217-8. [DOI] [PubMed] [Google Scholar]
- 48.Surmeier DJ, Kitai ST. Dopaminergic regulation of striatal efferent pathways. Curr Opin Neurosci. 1994;4:915–919. doi: 10.1016/0959-4388(94)90142-2. [DOI] [PubMed] [Google Scholar]
- 49.Tromba C, Salvalaggio A, Racagni G, Volterra A. Hypoglycemia-activated K+ channels in hippocampal neurons. Neurosci Lett. 1992;143:185–189. doi: 10.1016/0304-3940(92)90262-6. [DOI] [PubMed] [Google Scholar]
- 50.Wieloch T. Hypoglycemia-induced neuronal damage prevented by an N-methyl-d-aspartate antagonist. Science. 1985;230:681–683. doi: 10.1126/science.2996146. [DOI] [PubMed] [Google Scholar]
- 51.Wieloch T, Engelsen B, Westerberg E, Auer R. Lesions of the glutamatergic cortico-striatal projections in the rat ameliorate hypoglycemic brain damage in the striatum. Neurosci Lett. 1985;58:25–30. doi: 10.1016/0304-3940(85)90323-4. [DOI] [PubMed] [Google Scholar]
- 52.Wilson CJ, Groves PM. Fine structure and synaptic connection of the common spiny neuron of the rat neostriatum: a study employing intracellular injection of horseradish peroxidase. J Comp Neurol. 1980;194:599–615. doi: 10.1002/cne.901940308. [DOI] [PubMed] [Google Scholar]
- 53.Wilson CJ, Chang HT, Kitai ST. Firing pattern and synaptic potentials of identified giant aspiny interneurons in the rat neostriatum. J Neurosci. 1990;10:508–519. doi: 10.1523/JNEUROSCI.10-02-00508.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Xu ZC. Neurophysiological changes of spiny neurons in rat neostriatum after transient forebrain ischemia: an in vivo intracellular recording and staining study. Neuroscience. 1995;67:823–836. doi: 10.1016/0306-4522(95)00096-2. [DOI] [PubMed] [Google Scholar]