

Abstract
Activation of cysteine aspartases (caspases) seems to be a required element of apoptotic death in many paradigms. We have shown previously that general inhibitors of cysteine aspartases block apoptosis of PC12 cells and sympathetic neurons evoked by either trophic factor (nerve growth factor and/or serum) deprivation or superoxide dismutase (SOD1) downregulation. Moreover, activation of a caspase family member similar or equivalent to the interleukin-1β-converting enzyme (ICE) was implicated for death caused by SOD1 downregulation, but not withdrawal of trophic support. The experiments presented here demonstrate that diminished expression of the cysteine aspartase Nedd2 in PC12 cells and sympathetic neurons induced by an appropriate vector peptide-linked antisense oligonucleotide rescues them from death caused by trophic factor deprivation without inhibiting apoptosis in the same cell types evoked by SOD1 downregulation. Neither the level (as revealed by Western immunoblotting) nor the cellular distribution (as revealed immunohistochemically) of Nedd2 was altered demonstrably by trophic factor deprivation. However, evidence for proteolytic processing of Nedd2 (consistent with commencement of activation) was observed in PC12 cells after withdrawal of trophic support. These findings indicate that neuronal death triggered by different initial causes may be mediated by distinct members of the cysteine aspartase family.
Keywords: Nedd2, cysteine aspartases, apoptosis, neuronal cell death, oxidative stress, trophic factor deprivation
Neuronal death by apoptosis is a normal feature of development in which it appears that the death program is triggered by the failure of a given neuron to compete for limiting supplies of target-derived neurotrophic factors. Neurons also undergo apoptotic death in the postdevelopmental period when deprived of appropriate trophic factors or when subjected to any of a variety of stresses and injuries. Apoptosis also accounts for at least a portion of cellular loss in degenerative neurological diseases, including Alzheimer’s and amyotrophic lateral sclerosis (Coyle and Puttfarcken, 1993; Brown, 1995; Schapira, 1995; Williams, 1995).
Neuronal apoptotic death may be precipitated by widely different initiating causes. In the rat pheochromocytoma PC12 line, a commonly used model for neuronal differentiation and cell death, apoptosis may be triggered by either trophic factor/nerve growth factor (NGF) withdrawal (Greene, 1978; Batistatou and Greene, 1991; Rukenstein et al., 1991; Mesner et al., 1992; Pittman et al., 1993; Lindenboim et al., 1995) or oxidative stress induced by downregulation of Cu2+/Zn2+ superoxide dismutase (SOD1) (Troy and Shelanski, 1994; Troy et al., 1996a–c). The initiating mechanisms of death seem to be distinct in each instance. Apoptosis triggered by NGF deprivation is blocked by cAMP analogs (Rydel and Greene, 1988;Rukenstein et al., 1991) and high concentrations ofN-acetylcysteine (Ferrari et al., 1995), whereas these agents do not inhibit death induced by downregulation of SOD1 (Troy et al., 1996a,c). In contrast, the latter is blocked by vitamin E (Troy and Shelanski, 1994) and inhibitors of nitric oxide (NO) synthase (Troy et al., 1996a), which have no effect on apoptosis evoked by NGF withdrawal (Ferrari et al., 1995; Farinelli et al., 1996). Despite these initial mechanistic differences, there is evidence for common or similar downstream elements in the pathways that lead to death in both paradigms. In particular, inhibition studies implicate cysteine aspartases as obligate elements of the cell death mechanism in both initiating causes of death (Troy et al., 1996b). However, even at the level of cysteine aspartases, our findings have suggested the presence of parallel pathways. Apoptosis after SOD1 downregulation is suppressed by the peptide YVAD, a potent inhibitor of the interleukin-1-converting enzyme (ICE), by blocking antibodies to IL-1β, and by the IL-1 receptor antagonist IL-1Ra, although such agents have little or no effect on death caused by trophic factor/NGF withdrawal. These findings suggest that ICE itself, or another enzyme with pro-IL-1β cleaving activity, is required for death in the SOD1 downregulation paradigm, whereas a different cysteine aspartase is required for death in the case of trophic factor deprivation (Troy et al., 1996b).
In light of the above, the objective of the present study was to identify a specific cysteine aspartase that is required for neuronal apoptosis triggered by trophic factor deprivation. The cysteine aspartase Nedd2 is the rodent homolog of the human Ich-1/NEDD2 ICE family member and is highly expressed in neurons and PC12 cells (Kumar et al., 1994; Wang et al., 1994). Overexpression of Nedd2/Ich-1 causes apoptosis in fibroblasts and neuroblastoma cells (Kumar et al., 1994), and expression of a NEDD2 antisense construct protects a hematopoietic-derived cell line from death evoked by cytokine deprivation (Kumar, 1995). In the experiments reported here, we used a novel vector-linked antisense oligonucleotide to suppress Nedd2 expression in cultured PC12 cells and sympathetic neurons. Our findings indicate that Nedd2 plays a required role in neuronal apoptosis caused by loss of trophic support. In contrast, it does not seem to be required for death caused by SOD1 downregulation; thus, distinct cysteine aspartases mediate neuronal apoptosis triggered by different causes in the same cell.
MATERIALS AND METHODS
Cell culture
PC12 cells. PC12 cells were grown as previously described (Greene and Tischler, 1976) on rat-tail collagen-coated dishes in RPMI 1640 medium containing 5% fetal calf serum and 10% heat-inactivated horse serum (complete medium). NGF-primed (neuronally differentiated) PC12 cells were grown for at least 7 d in RPMI 1640 medium plus 1% horse serum and NGF (100 ng/ml). For cell survival assays involving trophic factor deprivation, cells (either naive or NGF-pretreated) were washed extensively in serum-free RPMI 1640 medium and replated on fresh collagen-coated 24-well dishes, as previously described (Rukenstein et al., 1991), in RPMI 1640 medium lacking serum or NGF. For SOD1 downregulation survival assays, cells were replated in complete medium with V-ASOD1 (vector-linked antisense oligonucleotide to SOD1, 50 nm), as previously described (Troy et al., 1996a). Various concentrations of V-ANedd (vector-linked antisense oligonucleotide to Nedd2) were included in the medium as indicated. Numbers of viable cells per culture were determined by quantifying intact nuclei, as previously described (Rukenstein et al., 1991). Counts were performed in triplicate and reported as mean ± SEM.
Sympathetic neurons. Sympathetic neuron cultures were prepared from 2-d-old rat pups, as previously described (Ferrari et al., 1995). Cultures were grown in 24-well collagen-coated dishes in RPMI 1640 medium plus 10% horse serum with mouse NGF (100 ng/ml). One day after plating, uridine and 5-fluorodeoxyuridine (10 μm each) were added to the cultures and left for 3 d to eliminate non-neuronal cells. On the sixth day after plating, NGF was removed by washing the cultures three times with RPMI 1640 medium plus 10% horse serum, followed by the addition of medium containing anti-mouse NGF (1:200, Sigma, St. Louis, MO) with or without V-ANedd. Each culture was scored, as previously described (Rydel and Greene, 1988), as numbers of living, phase-bright neurons at various times. Three replicate cultures were assessed for each condition, and data were normalized to numbers of neurons present in each culture at the time of NGF withdrawal and reported as mean ± SEM.
Synthesis of V-ANedd
Oligonucleotides bearing an SH group at their 5′ end and an NH group at their 3′ end were purchased from Operon (Alameda, CA). As previously described (Troy et al., 1996a), oligonucleotides were resuspended in deionized water, an equimolar ratio of Penetratin 1 (Oncor, Gaithersburg, MD) was added, and the mixture was incubated at 37°C for 1 hr. The yield of the reaction, estimated by SDS-PAGE followed by Coomassie blue staining, was routinely above 50%. A scrambled sequence of the antisense oligonucleotide (same base composition, different order), defined as V-SNedd, was synthesized for use as a control.
Antibody preparation
Anti-N-Nedd2, a polyclonal rabbit antiserum, was produced for us by Multiple Peptide Systems (San Diego, CA) using a 16-amino-acid synthetic peptide homologous to the N terminus (amino acids 1–16) as the antigen. The antiserum was affinity-purified with peptide bound to Sulfo-Link gel. Antiserum against a C-terminal peptide of Nedd2 (Nedd2 p12 C20) was purchased from Santa Cruz Biotechnology (Santa Cruz, CA).
Immunofluorescence
PC12 cells were plated on coverslips or on eight-well multichamber slides (LabTek, VWR Scientific Products) coated with Matrigel. After growth overnight, cells were fixed in ice-cold methanol and then immunostained as described (Troy et al., 1990). The primary antibody was either affinity-purified antibody Anti-N-Nedd2 or Nedd2 p12 C20 (Santa Cruz Biotechnology) at a dilution of 1:200. The secondary antibody was fluorescein isothiocyanate-conjugated goat anti-rabbit (Cappel, Durham, NC) at 1:100. For visualization with a Nikon fluorescence microscope, slides were coverslipped with Aqua-mount. Confocal microscopy was done on a Bio-Rad (Richmond, CA) 600 confocal microscope.
Western blotting
PC12 cells grown with or without V-ANedd or V-SNedd were harvested in SDS containing sample buffer and immediately boiled. Equal amounts of protein were separated by 10% PAGE, transferred to nitrocellulose, and immunostained as described (Troy et al., 1992). The affinity-purified anti-N-Nedd2 was used at a dilution of 1:500. The commercial antiserum Nedd2 p12 C20 (Santa Cruz Biotechnology) was used at a dilution of 1:350. Visualization was with ECL, using goat anti-rabbit peroxidase at 1:1000. The relative intensity of the protein bands was quantified with Scion Image 1.55 software, and samples were normalized by stripping and reprobing the blots with anti-peripherin antibody.
Assay of IL-1β
IL-1β was quantified by ELISA with the Intertest-1βX kit (Genzyme, Cambridge, MA), as previously described (Troy et al., 1996b). PC12 cells were grown as described above, on 24-well plates, in 500 μl of medium. After 1 d of incubation, medium was removed and IL-1β was measured following the manufacturer’s instructions; the number of viable cells in each well was quantified.
RESULTS
A vector-linked Nedd2 antisense oligonucleotide (V-ANedd) downregulates Nedd2 protein
To suppress expression of Nedd2 in neuronal cells, we designed an antisense oligonucleotide corresponding to the last 12 bases in the 5′ UTR and the first 9 bases in the coding region of the Nedd2 transcript (Kumar et al., 1994). The antisense oligonucleotide (ANedd; GCTCGGCGCCGCCATTTCCAG) is not homologous to any other reported mRNA sequence, including those of the other known cysteine aspartases. The oligonucleotide was linked to the vector peptide Penetratin 1 (V-) (Theodore et al., 1995; Troy et al., 1996a) to enhance its uptake by cells. The control-scrambled oligonucleotide (SNedd; CCGTAGCGTAGCTCCGCCTGC) also was linked to vector peptide. This vector-linked strategy significantly enhances the potency of antisense oligonucleotides and permits their use in the presence of serum (Troy et al., 1996a).
Using an affinity-purified anti-peptide antiserum (anti-N-Nedd2) generated against a synthetic N-terminal Nedd2 peptide, we examined the expression of Nedd2 in naive and neuronal PC12 cells before and after exposure to V-ANedd. As revealed by immunohistochemistry, in control cells the anti-N-Nedd2 staining was primarily cytoplasmic. This decreased to almost undetectable levels when the cells were pretreated for 24 hr with 400 nm V-ANedd (Fig. 1). In contrast, no change in staining was observed after exposure to 400 nmV-SNedd. Comparable results were found with a commercial antibody generated to a C-terminal peptide of Nedd2 (anti-C-Nedd2; data not shown). Confocal microscopy with either the N-terminal (Fig. 2) or the C-terminal (data not shown) antiserum demonstrated that the Nedd2 staining pattern does not change substantially after 20 hr of trophic factor deprivation (Fig. 2) in either naive or neuronal PC12 cells or after SOD1 downregulation (data not shown). In all cases staining was mainly cytoplasmic with one to two foci of staining seen in many nuclei. In the case of anti-N-Nedd2, all staining was abolished by preincubation with the immunizing peptide.
Fig. 1.

Downregulation of Nedd2 in PC12 cells by Penetratin 1-linked antisense oligonucleotide to Nedd2 (V-ANedd). Naive and neuronally differentiated PC12 cells (pretreated for at least 7 d with NGF) were plated on Matrigel-coated multichamber slides. V-ANedd (400 nm) was added to the indicated cultures after plating. Naive cultures were grown in RPMI 1640 medium with 5% FCS/10% horse serum; neuronal cultures were grown in RPMI 1640 medium with 1% horse serum and NGF (100 ng/ml). After overnight maintenance, cells were fixed in ice-cold methanol and then immunostained with anti-N-Nedd (see Materials and Methods). Cells were observed with a Nikon fluorescence microscope, 120× magnification.
Fig. 2.

Cellular localization of Nedd2 before and after trophic factor deprivation. Naive and neuronally differentiated cells were grown with serum and serum and NGF, respectively. Then the cells were plated (as described in Materials and Methods) in serum-free RPMI 1640 medium for 20 hr with (+NGF) or without (−NGF) NGF, as indicated. Cultures were stained with anti-N-Nedd and were observed with a Bio-Rad MRC600 confocal microscope, 1600× magnification.
By Western blot analysis anti-N-Nedd2 recognizes a major band at 53 kDa in whole PC12 cell lysates (Fig. 3A). The same major band was identified with the commercial C-Nedd antibody (Fig.3B). This apparent molecular weight is in agreement with that calculated from the predicted sequence of the Nedd2 protein (51 kDa). There are also three bands of lesser intensity seen with both antibodies at 70, 60, and 45 kDa. An identical pattern was seen with neuronally differentiated PC12 cells and a similar pattern with cultured sympathetic neurons. Specificity was assessed by absorption of the antiserum with the peptide to which it was generated and showed loss of signal by each of the above species (data not shown). A minor band at 19 kDa was seen on occasion at varying intensity when the N-terminal antibody was used. The major band and the additional molecular weight minor bands were downregulated by 60–70% (n = 4) after 18–22 hr treatment with V-ANedd (Fig.3). In contrast, there was no downregulation of CPP32 on blots of the same samples probed with anti-CPP32 (data not shown), indicating specificity of V-ANedd treatment for Nedd2. V-SNedd, the control oligonucleotide, did not downregulate any of the bands. None of the bands detected by Western blot appeared to be up- or downregulated to a substantial degree in response to either trophic factor withdrawal or short (2–24 hr) or long-term (10–14 d) NGF treatment. However, after withdrawal of trophic support from naive or primed PC12 cells, a cleavage product of ∼36 kDa was detectable by immunoblotting with the N-terminal antiserum before onset of cell death (data not shown).
Fig. 3.

Regulation of Nedd2 by V-ANedd. Naive PC12 cells were grown for 24 hr in serum-free RPMI 1640 medium in the presence or absence of NGF (100 ng/ml) and with or withoutV-ANedd (400 nm) or V-SNedd(400 nm), as indicated. The cells were extracted in sample buffer, the extracts were boiled, and equal amounts of protein were resolved by 10% SDS-PAGE and transferred to nitrocellulose. Blots were probed with (A) anti-N-Nedd at 1:500 or (B) anti-C-Nedd at 1:330, and staining was visualized with ECL. Bands were quantified with Scion Image software and normalized against peripherin levels. The level of downregulation is representative of that obtained in five independent experiments.
Differential effects of V-ANedd on PC12 cell death
To evoke apoptotic neuronal death by trophic factor deprivation, we withdrew NGF and/or serum from cultures of PC12 cells (either naive or neuronally differentiated by NGF pretreatment; Greene and Tischler, 1976) and neonatal rat sympathetic neurons, as previously described (Rydel and Greene, 1988; Ferrari et al., 1995; Troy et al., 1996a). Oxidative stress was induced by exposing cultures to the V-linked copper/zinc SOD1 antisense construct V-ASOD1, which downregulates SOD1 and induces apoptosis in PC12 cells (Troy and Shelanski, 1994; Troy et al., 1996a–c). In each of these paradigms, ∼40–60% of the cells underwent apoptosis within 24 hr.
V-ANedd protected naive PC12 cells from death caused by serum deprivation, with maximal protection at 400 nm when added at the same time as serum withdrawal (Figs. 4, 7). In this and all subsequent experiments, the scrambled V-SNedd construct had no effect on survival or death. Pretreatment of cultures for 4 hr with 50 nm V-ANedd shifted the dose–response curve to the left so that maximal survival was obtained with 100 nm V-ANedd. In contrast, there was no protection from SOD1 downregulation, even at 800 nm (Fig. 4), and pretreatment with V-ANedd was without effect. However, V-ANedd did downregulate Nedd2 in the presence of V-ASOD1, precluding competition by the two vector-linked constructs for cell entry (data not shown). The same concentrations of V-ANedd also protected neuronally differentiated PC12 cells from apoptosis caused by NGF withdrawal (Fig. 5A), but, again, not from downregulation of SOD1 (Fig. 5B). Two successive additions of V-ANedd, at the time of NGF deprivation and 1 d later, maintained survival of >75% of the cells through 4 d (Fig.5C). Although V-ANedd maintained survival, it did not mimic the actions of NGF in promoting either rapid flattening of naive PC12 cells or neurite outgrowth from neuronally differentiated cells (Fig.7).
Fig. 4.

V-ANedd rescues PC12 cells from serum deprivation, but not from SOD1 downregulation. A, V-ANedd differentially protects PC12 cells from serum deprivation. For serum deprivation (4 left-hand bars) cells were washed extensively, as described in Materials and Methods, and the indicated additives (100 ng/ml NGF, 400 nmV-ANedd, or 200 nmV-ICEinh) were added at the time of plating in serum-free RPMI 1640 medium. For SOD1 downregulation (4 right-hand bars), PC12 cells were replated on fresh collagen-coated 24-well dishes in complete medium (RPMI 1640 medium with 10% horse serum/5% fetal bovine serum) with 50 nmV-ASOD1 (vector-linked antisense oligonucleotide to SOD1). Additives (800 nmV-ANedd and 25 nmV-ICEinh) were included as indicated. Control cells were in complete medium. Cultures were incubated for 24 hr and lysed, and the number of intact nuclei was counted. The numbers of surviving cells are expressed relative to the number in the control cultures (designated as 100). Here, as in past studies (Greene and Tischler, 1976; Rukenstein et al., 1991; Troy et al., 1996a,b), NGF or complete medium promotes survival of all cells initially plated. Experiments were performed in triplicate wells, and data are expressed as mean ± SEM. B, Dose–response curve for protection from serum deprivation by V-ANedd. PC12 cells were washed for trophic factor deprivation and plated in serum-free medium with the indicated concentrations of V-ANedd. Cell survival relative to the number present with the addition of NGF was measured at 1 d. C, V-ANedd does not block the V-ASOD1-induced increase of IL-1β production. PC12 cells were plated with the indicated additives (50 nmV-ASOD1, 25 nmV-ICEinh, and 800 nmV-ANedd). Controls contained complete medium. After 20 hr, media were removed, and IL-1β was measured by ELISA with the Intertest-1βX kit. Data are expressed as mean ± SEM (n = 3).
Fig. 7.
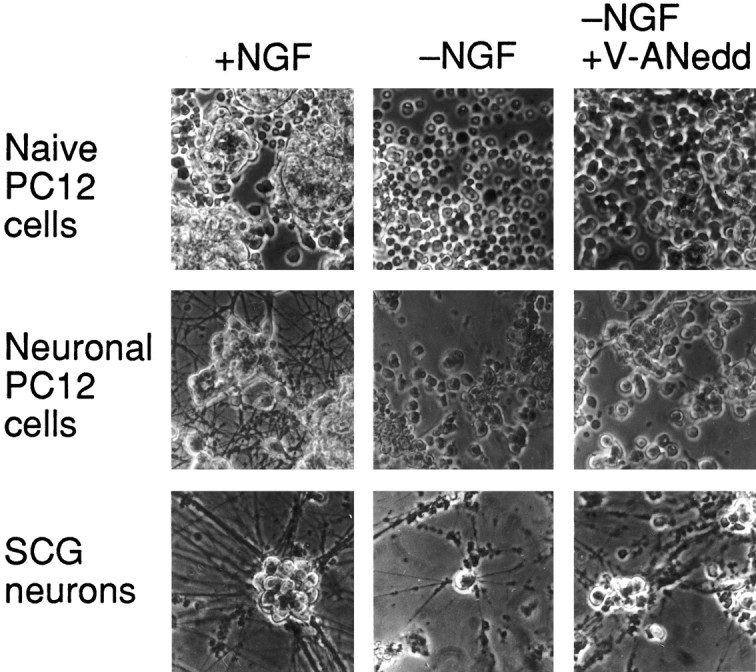
Morphology of cells rescued by V-ANedd. Photomicrographs of cells treated as described in the preceding figures. Top row, Naive PC12 cells:left, in serum-free RPMI 1640 medium with 100 ng/ml NGF (24 hr); middle, in RPMI 1640 medium alone (24 hr);right, in serum-free RPMI 1640 medium with 400 nm V-ANedd (24 hr). Middle row,Neuronal PC12 cells: left, replated in serum-free RPMI 1640 medium with 100 ng/ml NGF (24 hr);middle, replated in serum-free RPMI 1640 medium without NGF (24 hr); right, replated in serum-free RPMI 1640 medium with 400 nm V-ANedd (24 hr). Bottom row, SCG (sympathetic) neurons:left, cultured with 100 ng/ml NGF (3 d);middle, cultured in NGF-free medium with anti-NGF (3 d);right, cultured in NGF-free medium with anti-NGF plus 400 nm V-ANedd (3 d). Phase contrast optics, 80× magnification.
Fig. 5.

Neuronally differentiated PC12 cells are rescued from NGF deprivation, but not from SOD1 downregulation, by V-ANedd.A, V-ANedd differentially protects neuronally differentiated PC12 cells from apoptosis caused by NGF withdrawal. PC12 cells were neuronally differentiated by exposure to NGF (100 ng/ml) for at least 7 d in RPMI 1640 medium plus 1% horse serum. Cells were deprived of serum and NGF and replated as described in Figure 4. Additives present at the time of plating included 400 nmV-ANedd, 400 nmV-ICEinh, or 100 ng/mlNGF (4 left-hand bars). For SOD1 downregulation (4 right-hand bars) neuronally differentiated PC12 cells were plated in RPMI 1640 medium plus 1% horse serum, with 100 ng/ml NGF. At the time of plating, cultures were incubated, as indicated, with 50 nmV-ASOD1and with the indicated additives (800 nmV-ANedd and 50 nmV-ICEinh). Cell survival was determined after 1 d and expressed as in Figure 4.B, Dose–response curve for protection from NGF deprivation by V-ANedd. Neuronally differentiated PC12 cells were washed as above for NGF deprivation and plated in serum-free medium with the indicated concentrations of V-ANedd. Cell survival relative to the number present with the addition of NGF was quantified at 1 d.C, V-ANedd provides long-term protection against NGF deprivation. Neuronally differentiated PC12 cells were deprived of NGF and serum and plated as described in A. V-ANedd (400 nm) was included at the time of NGF deprivation and replenished 1 d later. Cell survival was determined at the indicated times as in Figure 4.
Death of PC12 cells evoked by SOD1 downregulation, but not by withdrawal of trophic support, is associated with enhanced release of IL-1β, and this is blocked by the general inhibitor of cysteine aspartase activity V-IQACRG (V-ICEinh) (Troy et al., 1996b). As illustrated in Figure 4C, V-ANedd did not affect IL-1β release after exposure to ASOD1. This indicates that V-ANedd does not affect processing of pro-IL-1β and that this is not the mechanism by which it blocks death caused by trophic factor deprivation. The data in Figures 4 and 5 also show that, as expected, V-ICEinh protects cells from both trophic factor deprivation and SOD1 downregulation.
V-ANedd protects sympathetic neurons from NGF deprivation, but not from oxidative stress
Parallel results were obtained with sympathetic neurons subjected to NGF deprivation. A single addition of V-ANedd at the time of NGF withdrawal resulted in >60% survival after 4 d and 25% survival at 8 d; at these times all neurons in control cultures were dead (Fig. 6). Although V-ANedd promoted survival, it did not maintain the neurites of NGF-deprived neurons (Fig. 7). Readdition of NGF to such cultures resulted in the reappearance of healthy neurites and maintenance of cell number (data not shown), thereby confirming neuronal survival and function in the presence of V-ANedd.
Fig. 6.

V-ANedd protects sympathetic neurons from NGF withdrawal, but not from oxidative stress. A, V-ANedd protects sympathetic neurons from NGF withdrawal. At the time of NGF deprivation, V-ANedd (400 nm) was added to the cultures, as indicated. Numbers of surviving neurons were determined at the indicated times, as described in Materials and Methods, and are reported as relative to the number present in each culture at the time of NGF withdrawal. B, V-ANedd does not protect sympathetic neurons from death induced by SOD1 downregulation and nitric oxide generation. Sympathetic neurons, after 6 d in culture, were maintained with NGF (100 ng/ml) and mixtures of the following additives as indicated: V-ASOD1 (50 nm), SNAP (100 μm), andV-ANedd (400 nm). Numbers of surviving neurons were determined at the indicated times, as above.
Exposure of cultured sympathetic neurons to antisense SOD1 alone has proved insufficient to produce death although, as for PC12 cells, this treatment reduces SOD1 levels by 50%. In PC12 cell cultures, death caused by SOD1 downregulation requires endogenous NO synthase activity and appears because of generation of peroxynitrite (Troy et al., 1996a). Consistent with this, when V-ASOD1 and the NO generator SNAP (S-nitrosopenicillamine) were added simultaneously to cultured sympathetic neurons, even in the presence of NGF, ∼50% of the cells underwent apoptotic death within 24 hr. Treatment with the NO generator in the absence of SOD1 downregulation did not produce death of either sympathetic neurons or PC12 cells (Farinelli et al., 1996). As in our previous study with PC12 cells (Troy et al., 1996b), the general inhibitor of cysteine aspartase activity, V-IQACRG (V-ICEinh), prevented sympathetic neuron death evoked by V-ASOD1+SNAP (Fig. 6). In contrast, V-ANedd was without effect in this paradigm (Fig. 6B).
DISCUSSION
In the present studies, we used an antisense construct to downregulate the cysteine aspartase Nedd2 in neuronal cells and found that this inhibited death caused by withdrawal of trophic support, but not by oxidative stress. Multiple aspects of our studies support the specificity and utility of our reagents. The major species recognized by both our N-terminal Nedd2 antiserum and a commercial Nedd2 C-terminal antiserum on Western blots migrated at an apparentMr of 53 kDa. This corresponds closely to the predicted Mr of the Nedd2 protein, based on the sequence of the nedd2 transcript from mouse (Kumar, 1995) as well as rat (H. Qi and L. Stefanis, unpublished data). Recognition of this species by anti-N-Nedd2 was abolished in the presence of excess immunizing peptide. Both antisera also provided similar patterns of cellular staining, which, in the case of anti-N-Nedd2, was eliminated by preincubation with the immunizing peptide. Exposure to theV-ANedd antisense construct yielded significant downregulation of Nedd2 protein as assessed by Western blotting and immunostaining with the two different antisera. To assess the specificity of the antisense construct, we also tested V-SNedd, a scrambled version of V-ANedd, and observed that it did not affect Nedd2 protein levels, staining of cells with anti-Nedd2, or cell death. Moreover, the observation that V-ANedd does not promote survival of neuronal cells after SOD1 downregulation seems to rule out nonspecific antiapoptotic actions of this construct. Finally, V-ANedd effectively suppressed death of serum-deprived naive PC12 cells. In such cultures, apoptosis does not require de novo protein translation (Rukenstein et al., 1991), and thus this finding seems to exclude potential nonspecific effects of the antisense construct on synthesis of proteins required for death.
The results of these experiments argue for the existence of at least two distinct parallel pathways to apoptotic cell death in the same neuron. The choice of one or the other pathway is a function of the initial insult to the cell. When SOD1 in PC12 cells is downregulated to ∼40% of its control levels, apoptosis occurs (Troy and Shelanski, 1994). This process seems to be mediated by peroxynitrite (Troy et al., 1996a), although the critical target of peroxynitrite in this model has not been identified. Cultured rat sympathetic neurons survive the downregulation of SOD1 itself but die rapidly when this treatment is coupled with the generation of nitric oxide. Downregulation of SOD1 in PC12 cells is accompanied by an increase in the release of IL-1β, suggesting the activation of an ICE-like enzyme (Troy et al., 1996b). In this case, death can be blocked by addition of anti-IL-1β or the IL-l receptor antagonist (IL-1Rα) to the medium. Death of both PC12 and sympathetic neurons caused by SOD1 downregulation also can be blocked with a variety of inhibitors of the ICE family of proteases (Troy et al., 1996b), but interestingly not by the downregulation of Nedd2. V-ANedd does not alter the release of IL-1β from V-ASOD1-treated cells. These data point strongly to the involvement of ICE itself or an ICE-like activity in this model of free-radical-induced cell death and seem to exclude an obligatory role of Nedd2.
In contrast to the SOD1 downregulation paradigm, antibodies to IL-1β do not rescue PC12 cells and sympathetic neurons from serum and/or trophic factor withdrawal. Moreover, the ICE antagonist peptide ZYVAD-CMK, which effectively rescues the cells from downregulation of SOD1, has negligible effects on death provoked by loss of trophic support (Troy et al., 1996b). However, downregulation of Nedd2 in serum-deprived naive PC12 cells and in NGF-deprived primed PC12 cells and sympathetic neurons rescues them from apoptotic death, pointing to a requisite role of Nedd2 in this process.
Our extracts show a major band at 53 kDa, agreeing with the predicted molecular weight of Nedd2 (Kumar et al., 1994). There are also three minor bands that are detected by both antibodies, two of which are higher than the calculated molecular weight for Nedd2. Although the original report on Nedd2 reported that translation of the construct resulted in a major band of 53 kDa and several minor bands of 45 and 19 kDa (Kumar et al., 1994), the detection of higher molecular weight bands by antibodies against both the C and N termini of Nedd2 and their specific downregulation by V-ANedd strongly suggests that they are Nedd2 products. These bands also are seen after in vitrotranscription translation of rat Nedd2 (Qi and Stefanis, unpublished data).
Previous studies have shown that overexpression of Nedd2 can induce apoptotic death and that an antisense construct can rescue cells from apoptosis (Kumar et al., 1994; Kumar, 1995). The observations presented here extend these findings by demonstrating directly that Nedd2 protein levels are downregulated in neuronal cells by antisense treatment and, more significantly, that Nedd2 is required for neuronal cell death resulting from trophic factor withdrawal and not required when neuronal death is induced by SOD1 downregulation. In addition, we observed that Nedd2 is processed to a 36 kDa cleavage product on withdrawal of trophic support. Cleavage of Nedd2 also has been reported in another death paradigm (Srinivasan et al., 1996). ICE is processed proteolytically to an intermediate 35 kDa peptide that is cleaved further to generate the active form, p20 (Thornberry et al., 1992;Yamin et al., 1996). The 36 kDa Nedd2 cleavage product most likely represents such an intermediate form.
Our results argue against the existence of a single “final common pathway” leading to apoptotic cell death. In the two paradigms presented here, trophic factor deprivation and SOD1 downregulation, the general scheme is similar in that each pathway requires a cysteine aspartase but shows marked selectivity in the specific enzyme required. The differential association of specific cysteine aspartases with apoptosis evoked by different means may account for the proliferation of this family in vertebrates. The use of distinct cysteine aspartases by the same cells to promote death from different initiating stimuli raises the possibility that this selectivity can be exploited for the treatment of specific neurodegenerative disorders.
Footnotes
This work was supported by Javits Neuroscience Investigator Awards from the National Institute of Neurological Disorders and Stroke (NINDS) (M.L.S. and L.A.G.); the Muscular Dystrophy Association of America (C.M.T.); NINDS, National Institute on Aging, the Blanchette Rockefeller Foundation, the Amyotrophic Lateral Sclerosis Association, and the March of Dimes (L.A.G.); and by the Lucille P. Markey Trust (L.S.). We thank Dr. Adriana Rukenstein for excellent assistance with cell culture, Dr. Richik Ghosh for confocal microscopy, and Dan Clayton for technical assistance.
Correspondence should be addressed to Dr. Carol M. Troy, Department of Pathology, College of Physicians and Surgeons, Columbia University, 630 West 168th Street, New York, NY 10032.
REFERENCES
- 1.Batistatou A, Greene LA. Aurintricarboxylic acid rescues PC12 cells and sympathetic neurons from cell death caused by nerve growth factor deprivation: correlation with suppression of endonuclease activity. J Cell Biol. 1991;115:461–471. doi: 10.1083/jcb.115.2.461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brown RH., Jr Amyotrophic lateral sclerosis: recent insights from genetics and transgenic mice. Cell. 1995;80:687–692. doi: 10.1016/0092-8674(95)90346-1. [DOI] [PubMed] [Google Scholar]
- 3.Coyle JT, Puttfarcken P. Oxidative stress, glutamate, and neurodegenerative disorders. Science. 1993;262:689–695. doi: 10.1126/science.7901908. [DOI] [PubMed] [Google Scholar]
- 4.Farinelli SE, Park DS, Greene LA. Nitric oxide delays the death of trophic factor-deprived PC12 cells and sympathetic neurons by a cGMP-mediated mechanism. J Neurosci. 1996;16:2325–2334. doi: 10.1523/JNEUROSCI.16-07-02325.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ferrari G, Yan CYI, Greene LA. N-acetylcysteine (d- and l-stereoisomers) prevents apoptotic death of neuronal cells. J Neurosci. 1995;15:2857–2866. doi: 10.1523/JNEUROSCI.15-04-02857.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Greene LA. Nerve growth factor prevents the death and stimulates the neuronal differentiation of clonal PC12 pheochromocytoma cells in serum-free medium. J Cell Biol. 1978;78:747–755. doi: 10.1083/jcb.78.3.747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Greene LA, Tischler AS. Establishment of a noradrenergic clonal line of rat adrenal pheochromocytoma cells which respond to nerve growth factor. Proc Natl Acad Sci USA. 1976;73:2424–2428. doi: 10.1073/pnas.73.7.2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kumar S. Inhibition of apoptosis by the expression of antisense Nedd2. FEBS Lett. 1995;368:69–72. doi: 10.1016/0014-5793(95)00602-6. [DOI] [PubMed] [Google Scholar]
- 9.Kumar S, Knioshita M, Noda M, Copeland NG, Jenkins NA. Induction of apoptosis by the mouse Nedd2 gene, which encodes a protein similar to the product of the Caenorhabditis elegans cell death gene ced-3 and the mammalian IL-1β-converting enzyme. Genes Dev. 1994;8:1613–1626. doi: 10.1101/gad.8.14.1613. [DOI] [PubMed] [Google Scholar]
- 10.Lindenboim L, Haviv R, Stein R. Inhibition of drug-induced apoptosis by survival factors in PC12 cells. J Neurochem. 1995;64:1054–1063. doi: 10.1046/j.1471-4159.1995.64031054.x. [DOI] [PubMed] [Google Scholar]
- 11.Mesner PW, Winters TR, Green SH. Nerve growth factor withdrawal-induced cell death in neuronal PC12 cells resembles that in sympathetic neurons. J Cell Biol. 1992;119:1669–1680. doi: 10.1083/jcb.119.6.1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pittman RN, Wang S, DiBenedetto AJ, Mills JC. A system for characterizing cellular and molecular events in programmed neuronal cell death. J Neurosci. 1993;13:3669–3680. doi: 10.1523/JNEUROSCI.13-09-03669.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rukenstein A, Rydel RE, Greene LA. Multiple agents rescue PC12 cells from serum-free cell death by translation- and transcription-independent mechanisms. J Neurosci. 1991;11:2552–2563. doi: 10.1523/JNEUROSCI.11-08-02552.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rydel RE, Greene LA. cAMP analogs promote survival and neurite outgrowth in cultures of rat sympathetic and sensory neurons independently of nerve growth factor. Proc Natl Acad Sci USA. 1988;85:1257–1261. doi: 10.1073/pnas.85.4.1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schapira AHV. Oxidative stress in Parkinson’s disease. Neuropathol Appl Neurobiol. 1995;21:3–9. doi: 10.1111/j.1365-2990.1995.tb01022.x. [DOI] [PubMed] [Google Scholar]
- 16.Srinivasan A, Foster LM, Testa M-P, Ord T, Keane RW, Bredesen DE, Kayalar C. Bcl-2 expression in neural cells blocks activation of ICE/CED-3 family proteases during apoptosis. J Neurosci. 1996;16:5654–5660. doi: 10.1523/JNEUROSCI.16-18-05654.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stefanis L, Park DS, Yan CYI, Farinelli SE, Troy CM, Shelanski ML, Greene LA. Induction of CPP32-like activity in PC12 cells by withdrawal of trophic support: dissociation from apoptosis. J Biol Chem. 1996;271:30663–30671. doi: 10.1074/jbc.271.48.30663. [DOI] [PubMed] [Google Scholar]
- 18.Theodore L, Derossi D, Chassaing G, Llirbat B, Kubes M, Jordan P, Chneilweiss H, Godement P, Prochiantz A. Intraneuronal delivery of protein kinase C pseudosubstrate leads to growth cone collapse. J Neurosci. 1995;15:7158–7167. doi: 10.1523/JNEUROSCI.15-11-07158.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Thornberry NA, Bull HG, Calaycay JR, Chapman KT, Howard AD, Kostura MJ, Miller DK, Molineaux SM, Weidner JR, Aunins J, Elliston KO, Ayala JM, Casano FJ, Chin J, Ding GJ-F, Egger LA, Gaffney EP, Limjuco G, Palyha OC, Raju SM, Rolando AM, Salley JP, Yamin T-T, Lee TD, Shively JE, MacCross M, Mumford RA, Schmidt JA, Tocci MJ. A novel heterodimeric cysteine protease is required for interleukin-1 beta processing in monocytes. Nature. 1992;356:768–774. doi: 10.1038/356768a0. [DOI] [PubMed] [Google Scholar]
- 20.Troy CM, Shelanski ML. Down-regulation of copper/zinc superoxide dismutase (SOD1) causes neuronal cell death. Proc Natl Acad Sci USA. 1994;91:6384–6387. doi: 10.1073/pnas.91.14.6384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Troy CM, Brown K, Greene LA, Shelanski ML. Ontogeny of the neuronal intermediate filament protein, peripherin, in the mouse embryo. Neuroscience. 1990;36:217–237. doi: 10.1016/0306-4522(90)90364-a. [DOI] [PubMed] [Google Scholar]
- 22.Troy CM, Greene LA, Shelanski ML. Neurite outgrowth in peripherin-depleted PC12 cells. J Cell Biol. 1992;117:1085–1092. doi: 10.1083/jcb.117.5.1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Troy CM, Derossi D, Prochiantz A, Greene LA, Shelanski ML. Downregulation of Cu/Zn SOD1 leads to cell death via the nitric oxide–peroxynitrite pathway. J Neurosci. 1996a;16:253–261. doi: 10.1523/JNEUROSCI.16-01-00253.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Troy CM, Stefanis L, Prochiantz A, Greene LA, Shelanski ML. The contrasting roles of ICE family proteases and interleukin-1β in apoptosis induced by trophic factor withdrawal and by SOD1 downregulation. Proc Natl Acad Sci USA. 1996b;93:5635–5640. doi: 10.1073/pnas.93.11.5635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Troy CM, Stefanis L, Greene LA, Shelanski ML. Mechanisms of neuronal degeneration: a final common pathway? In: Seil F, editor. Neuronal regeneration, reorganization, and repair. Raven; New York: 1996c. pp. 103–112. [PubMed] [Google Scholar]
- 26.Wang L, Miura M, Bergeron L, Zhu H, Yuan J. Ich-1, an ICE/ced-3-related gene, encodes both positive and negative regulators of programmed cell death. Cell. 1994;78:739–750. doi: 10.1016/s0092-8674(94)90422-7. [DOI] [PubMed] [Google Scholar]
- 27.Williams LR. Oxidative stress, age-related neurodegeneration, and the potential for neurotrophic. Cerebrovasc Brain Metab Rev. 1995;7:55–73. [PubMed] [Google Scholar]
- 28.Yamin T-T, Ayala JM, Miller DK. Activation of the native 45 kDa precursor form of interleukin-1-converting enzyme. J Biol Chem. 1996;271:13273–13282. doi: 10.1074/jbc.271.22.13273. [DOI] [PubMed] [Google Scholar]