

To the Editor: Mitochondrial diseases are a common group of human genetic diseases that can occur at any decade of life. Mitochondrial DNA (mtDNA) mutations are responsible for most adult-onset mitochondrial diseases.[1] There are over 260 known pathogenic mutations in mtDNA.[2] In the last two decades, a large number of pathogenic mutations of mtDNA have been reported, which associate with a wide spectrum of clinical features.[3]
G8363A is a rare mtDNA tRNALys gene mutation that associates with myoclonic epilepsy and ragged red fibers-like syndrome, ataxia and lipomas, and Leigh syndrome. Patients develop signs of encephalomyopathy, sensorineural hearing loss, ataxia, and hyperthyroidism.[4]
Few articles about G8363A mutation have been published. In this report, we describe a patient with a G8363A mutation in combination with parietal and temporal lobe atrophy, which has never been reported before.
The patient was a 54-year-old man who presented with slurred speech, fatigue, hearing loss, and limb weakness. He had slurred speech when he was 5 years old. At the age of 43 years, he started complaining of hearing loss. At the age of 46 years, he developed limb weakness and fatigue. The symptoms were progressive. The patient had no complaints of muscle stiffness, myalgia, or numbness. Physical examination showed obvious muscle atrophy. A slightly shallow nasolabial fold and deviation of the tongue were present. The patient's muscle strength was 4+/5 (Medical Research Council Scale) in the proximal lower limbs, 4/5 in the proximal upper limbs, and almost normal in the distal limbs. Sensory examination was intact with the absence of deep tendon reflexes. Hoffmann or Babinski signs were not observed. In addition, he had ataxia and impaired tandem gait. Serum blood parameters were normal except mildly elevated creatine kinase isoenzymes levels (34.1 U/L, normal 0–25 U/L). Electromyography revealed a myogenic pattern in the muscles of the limbs. The patient's clinical symptoms improved after treatment with vitamins, lipoic acid, and coenzymes. However, he stopped taking drugs 2 months later. The disease did not progress. He did not develop cognitive decline during the follow-up. The patient's family history was negative.
Pathological examination revealed no abnormal alterations. Hematoxylin and eosin staining revealed few rounded or angular atrophic muscle fibers without abnormal material deposits (not shown). Nicotinamide-adenine dinucleotide and succinate dehydrogenase staining showed limited staining of muscle fiber oxidase, which was unevenly distributed [Figure 1A and 1B]. No specific abnormalities were observed on modified Gomori trichrome [Figure 1C], periodic acid-Schiff, and oil red O staining (not shown).
Figure 1.
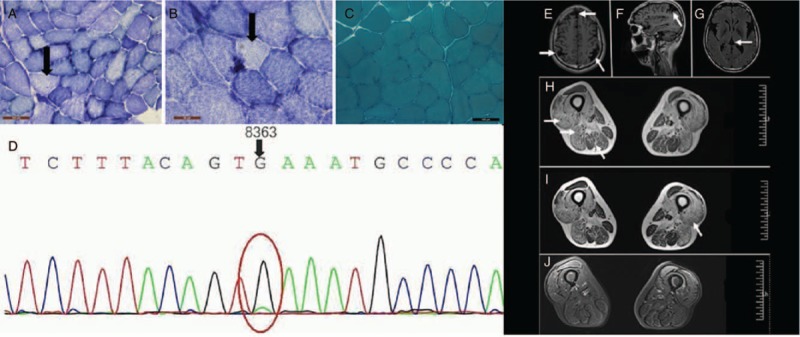
Muscle pathology and MRI of the patient with G8363A mutation. (A and B) SDH staining showed limited staining of muscle fiber oxidase, which was unevenly distributed (original magnification ×200). (C) No specific abnormalities were observed on MGT staining (original magnification ×200). (D) The figure shows the patient's G8363A mutation. (E–G) Brain atrophy can be seen on imaging (E and G: axial; F: sagittal). (H–J) Muscle MRI shows atrophy of the muscle with diffuse infiltration (axial). MGT: Modified Gomori trichrome; MRI: Magnetic resonance imaging; SDH: Succinate dehydrogenase.
Brain magnetic resonance imaging (MRI) showed parietal and temporal lobe atrophy [Figure 1E–1G]. However, the hypophysis, optic chiasm, and auditory nerve trunk were spared. Muscle MRI revealed abnormal high signal infiltrates into the muscles on T1- and T2-weighted images and low signals on fat-saturated T2-weighted images. Severe muscle fatty infiltrations and diffuse atrophy were observed in the vastus lateralis, biceps femoris short head, semitendinosus, and semimembranosus. The rectus femoris and gracilis were not involved. Diffusion-weighted images revealed no abnormal findings [Figure 1H–1J].
Targeted next-generation sequencing identified a novel heterozygous mutation: one single base pair substitution in G8363A, which has previously been reported to be a disease-causing mutation for mitochondrial diseases [Figure 1D].[4] Sanger sequencing confirmed the mutation in the proband.
In this report, we describe a patient who presented with the recognized clinical features in mitochondrial disease: muscle weakness, lactic acidosis, and ophthalmoplegia.[4] Genetic screening demonstrated a causative heterozygous mutation in the 8363 mtDNA site. These findings provide sufficient evidence to establish a diagnosis of mitochondrial disease caused by a G8363A mutation.
Though brain atrophy is a common feature of brain disorders, significant parietal and temporal lobe atrophy in patients with mitochondrial diseases caused by G8363A mutations has not been reported before.
Mutations in mtDNA can impair mitochondrial function and cause the electron transport chain (ETC) complex deficiencies, finally resulting in programmed cell death (apoptosis).[5] Furthermore, the brain is very sensitive to damage. Once neurons are damaged, apoptosis may occur, which causes brain atrophy. The underlying pathogenesis of this process requires further exploration.
Although next-generation sequencing technologies have improved the genetic diagnosis of mitochondrial diseases,[2] there are still patients for whom the diagnosis is challenging. In this report, MRI played an important role in the diagnosis.
Mitochondrial diseases lack effective treatments. The patient's clinical symptoms improved after treatment with vitamins, lipoic acid, and coenzymes. However, treatment therapies can only deal with the complications of mitochondrial diseases. We hope there will be major advances in this area.
In conclusion, we identified parietal and temporal lobe atrophy in a Chinese patient with a G8363A mutation uncommonly. The present case raises the awareness that brain atrophy may be a feature of G8363A mutations.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form, the patient has given his consent for his images and other clinical information to be reported in the journal. The patient understands that his name and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Funding
This work was supported by grants from the Young Scientists Fund of the National Natural Science Foundation of China (No. 81601093), and the Young Scientists Fund of the First Affiliated Hospital of Zhengzhou University (2015, Director: Hong-Liang Xu).
Conflicts of interest
None.
Footnotes
How to cite this article: Xu HL, Lian YJ, Chen X. Brain atrophy in a patient with mitochondrial DNA G8363A mutation. Chin Med J 2019;00:00–00. doi: 10.1097/CM9.0000000000000395
References
- 1.Lightowlers RN, Taylor RW, Turnbull DM. Mutations causing mitochondrial disease: what is new and what challenges remain. Science 2015; 349:1494–1499. doi: 10.1126/science.aac7516. [DOI] [PubMed] [Google Scholar]
- 2.Craven L, Alston CL, Taylor RW, Turnbull DM. Recent advances in mitochondrial disease. Annu Rev Genomics Hum Genet 2017; 18:257–275. doi: 10.1146/annurev-genom-091416-035426. [DOI] [PubMed] [Google Scholar]
- 3.Zeviani M, Carelli V. Mitochondrial disorders. Curr Opin Neurol 2007; 20:564–571. doi: 10.1097/WCO.0b013e3282ef58cd. [DOI] [PubMed] [Google Scholar]
- 4.Virgilio R, Ronchi D, Bordoni A, Fassone E, Bonato S, Donadoni C, et al. Mitochondrial DNA G8363A mutation in the tRNA Lys gene: clinical, biochemical and pathological study. J Neurol Sci 2009; 281:85–92. doi: 10.1016/j.jns.2009.01.025. [DOI] [PubMed] [Google Scholar]
- 5.Rossignol DA, Frye RE. Mitochondrial dysfunction in autism spectrum disorders: a systematic review and meta-analysis. Mol Psychiatry 2012; 17:290–314. doi: 10.1038/mp.2010.136. [DOI] [PMC free article] [PubMed] [Google Scholar]