

Abstract
In this study, the distribution and concentration of β1, β2, and α2adrenergic receptors were examined in the frontal cortex, hypothalamus, and cerebellum of Alzheimer’s disease (AD) and age-matched control human brains by receptor autoradiography. The purpose of this study was to detect changes in adrenergic receptor concentrations in key areas of the brain known to affect behavior. For these studies, [125I]iodopindolol ([125I]IPIN) was used to visualize total β adrenergic sites (with ICI-89,406 and ICI-118,551 as subtype-selective antagonists to visualize β2 and β1receptors, respectively). [3H]UK-14,304 was used to localize the α2 sites. Essentially no significant difference in adrenergic receptor concentration was found between total AD cases taken together and control patients. It was found, however, that there were important distinctions within the AD group when cases were subdivided according to the presence or absence of aggression, agitation, and disruptive behavior. Aggressive AD patients had markedly increased (by ∼70%) concentrations of α2 receptors in the cerebellar cortex compared with nonaggressive patients with similar levels of cognitive deficit. The levels of cerebellar α2receptors in aggressive AD patients were slightly above the healthy elderly controls, suggesting that these receptors are preserved and perhaps increased in this subgroup of AD. β1 And β2 adrenergic receptors of the cerebellar cortex showed smaller but significant (∼25%) increases in concentration in aggressive AD subjects versus both nonaggressive AD patients and controls. No significant differences were found in adrenergic receptor concentrations within the frontal cortex or hypothalamus. These results point out the importance of distinguishing behavioral subgroups of AD when looking for specific neurochemical changes. These autoradiographic results may reflect the importance of the cerebellum in behavioral control.
Keywords: autoradiography, dementia, aggression, behavioral disorder, receptor localization, frontal cortex, hypothalamus
Much evidence exists that the adrenergic system has an important role in normal CNS function as well as in brain disease. It has been proposed that the main function of the locus ceruleus (LC) and its projections is to determine the brain’s global orientation concerning events in the external world and within the viscera (Cooper et al., 1991). Central norepinephrine (NE) neurons are suggested to have a role in learning and memory, reinforcement, sleep–wake cycle regulation, affective psychoses, and the regulation of aggression. Drugs effective in the treatment of neuropsychiatric illness, including depression and aggressive or disruptive behavior, are active on adrenergic receptors.
Several studies have been completed to investigate changes in neurotransmitters or their receptors in Alzheimer’s disease (AD) so that the neurological deficits in AD can be better understood and more effective treatments can be developed. The LC, the predominant source of noradrenergic projection neurons in the brain, is significantly damaged in Alzheimer’s disease (Ishino and Otsuki, 1975). Although adrenergic receptors in these projection areas of AD brains have been studied, and some abnormalities have been observed (Kalaria et al., 1989a; Meana et al., 1992), there have been variable results. A possible reason for a lack of significant changes in some receptor studies is the heterogeneity among AD cases. Much evidence exists for subgroups within the AD population. For example, in addition to variations in the severity of the cognitive deficits in AD patients, there are various behavioral abnormalities that appear to be present in subgroups of AD patients (Reisberg et al., 1987; Mega et al., 1996). These behavioral symptoms include apathy, agitation/aggression, dysphoria, and aberrant motor behaviors.
This study has sought to investigate changes in adrenergic receptor concentrations in key areas of the brain known to affect behavior. Limbic regions of the brain, such as the frontal neocortex and hypothalamus, are projection regions of the LC known to contain significant levels of NE and adrenergic receptors, and to function in the modulation of behavior (Weiger and Bear, 1988). The cerebellar cortex is another LC projection area that has been shown to participate in behavioral control, in addition to motor control (Schmahmann, 1991). Adrenergic receptor concentration and distribution were investigated in these brain regions of AD patients and normal elderly controls by way of receptor autoradiography.
MATERIALS AND METHODS
Patient selection and tissue preparation
Brain tissue was obtained from the University of California, Irvine, Alzheimer’s Disease Research Center Repository Core (Table1). Tissue was sectioned into 1-cm-thick coronal slices and frozen on dry ice at autopsy with a postmortem interval of 2–14 hr and stored at −80°C until processed for autoradiography. All tissue was handled in the same manner, and all cases were examined neuropathologically for definitive AD diagnosis. Controls were matched to experimental tissue for age and postmortem delay. Control sections were mounted side by side with AD sections on the same slide. Brain areas studied included the frontal neocortex (examining separately the dorsolateral prefrontal and orbitofrontal cortices; Brodmann areas 10 and 11, respectively), the hypothalamus (ventromedial, dorsomedial, posterior, and lateral nuclei), and the cerebellar cortex (from the cerebellar hemispheres).
Table 1.
Sources of brain tissue
| Case | Source | Age | Sex | PMI | MMSE | Cause of death | Medications |
|---|---|---|---|---|---|---|---|
| Ag-AD | |||||||
| 1 | ADRC clinic | 88 | F | 14 | 8 | Cardiac arrest | Dyazide, haloperidol, ibuprofen, multivitamins |
| 2 | ADRC clinic | 85 | M | 12 | 20, 11 | Cardiopulmonary arrest | Loxapine |
| 3 | ADRC clinic | 90 | M | 8 | 13 | Myocardial infarction | Aspirin |
| 4 | ADRC clinic | 73 | F | 2 | 18 | Cardiopulmonary arrest | Furosemide, potassium, digoxin, enalapril, nifedipine |
| Clorazepic acid, trazodone, tylenol, coumadin | |||||||
| 5 | TRCP | 70 | M | 2.5 | 18 | Cardiopulmonary arrest | Nortriptyline |
| 6 | ADRC clinic | 86 | F | 2.25 | 21, 19 | Respiratory failure | Hydrochlorothiazide, furosemide, naproxen, tacrine |
| meclizine, digoxin, diclofenac sodium, amitriptyline | |||||||
| 7 | ADRC clinic | 74 | M | 6.5 | 23 | Cardiac arrest | Bumetanide, colchicine |
| 8 | ADRC clinic | 73 | F | 9.5 | 2 | Respiratory failure | Digoxin, haloperidol, ranitidine, omperazole, estrogen, prednisone, theophylline |
| Controls | |||||||
| 9 | TRCP | 89 | M | 9 | N/A | Cardiac arrest | None |
| 10 | TRCP | 83 | M | 7.5 | N/A | Aspiration pneumonia, cancer | Vicodin, morphine, triazolam |
| 11 | TRCP | 71 | F | 4 | N/A | Chronic pulmonary disease | Theophylline, metaproterenol, iron, prednisone |
| 12 | TRCP | 65 | M | 4.5 | N/A | Cardiac arrest | Captopril, furosemide, nitroglycerine, iron, fluoxetine, diltiazem, coumadin, aspirin, albuterol, ipra-tropium bromide, triamcinolone acetonide |
| 13 | TRCP | 81 | F | 6 | N/A | Cardiac arrest | Digoxin, prednisone, albuterol, trazodone |
| 14 | TRCP | 77 | M | 6.5 | N/A | Cardiac arrest | Thyroid hormone |
| 15 | TRCP | 76 | F | 4.5 | N/A | Myocardial infarction | Prednisone, lorazepam, theophylline |
| nAg-AD | |||||||
| 16 | ADRC clinic | 79 | M | 12 | 23, 20 | Pneumonia | Dihydroergotamine mesylate, aspirin, florinef, salt tablets, piroxicam |
| 17 | ADRC clinic | 73 | M | 8 | 9 | Cardiopulmonary arrest | Carbidopa, amitriptyline, vitamin E, selegeline |
| 18 | ADRC clinic | 72 | F | 12 | 5 | Brain embolism | Phenytoin, estrogen |
| 19 | ADRC clinic | 73 | M | 6 | 19, 6 | Cardiac arrest | Ergometrinine, aspirin, imipramine |
| 20 | ADRC clinic | 77 | M | 6.5 | 19 | Cardiac arrest | Imipramine, cycloserine (study), quinidine |
| 21 | ADRC clinic | 90 | F | 8.5 | 18 | Cardiac arrest | Captopril, aspirin, multivitamins |
| 22 | ADRC clinic | 80 | F | 3 | 19 | Stroke | Nitrofurantoin, carbidopa, oxybutynin, warfarin, Atenolol, triamterine |
| 23 | ADRC clinic | 95 | F | 9.5 | 14 | Cardiopulmonary arrest | None |
Ag-AD, Aggressive Alzheimer’s disease subgroup; nAg-AD, non-aggressive AD subgroup; ADRC, Alzheimer’s Disease Research Center; TRCP, Tissue Repository Consent Program; MMSE, Mini Mental State Exam scores (two scores reflect two evaluations in consecutive years); PMI, postmortem interval (hours).
Autoradiography
For autoradiographic experiments, the brain tissue was cryostat-sectioned at −15°C (12 μm for α2 assays and 20 μm for β), thaw-mounted onto gelatin-subbed slides over ice, and stored at −20°C for up to 1 week until used in the assays. As described above, sections from the three patient groups were mounted on the same slide to minimize any interslide variability in conditions. Sections for assays were taken in triplicate, and repeats were randomized with respect to position on the slide (top or bottom) and cases with which they were paired.
α2-adrenergic receptors.α2-Adrenoceptors were labeled with the selective agonist [3H]bromoxidine ([3H]UK-14,304, 64.0 Ci/mmol, New England Nuclear, Boston, MA) according to the procedure of Pazos et al. (1988). After a 15 min preincubation at room temperature in Tris-HCl buffer (50 mm, pH 7.7) containing 0.1 mmMnCl2, slide-mounted, 12 μm tissue sections were incubated with 6 nm [3H]UK-14,304 for 90 min under the same conditions. After the incubation, sections were washed for 5 min in ice-cold buffer and dried in a cold air stream. Nonspecific binding was defined as that remaining in the presence of 10 mm phentolamine. Sections were exposed at 4°C for 8 weeks to tritium-sensitive film (Hyperfilm-3H, Amersham, Arlington Heights, IL) before development and analysis.
β-Adrenergic receptors. The general procedure described byRainbow et al. (1984) was used to determine the distribution of total β-adrenergic receptor sites and of the β1 and β2 subtypes. Cryostat sections (20 μm thickness) were used for these experiments. Slides were placed horizontally on trays, and 1 ml of buffer containing [125I]iodopindolol ([125I]IPIN) was layered on the tissue. The slide-mounted tissue sections were incubated for 70 min with 200 pm [125I]IPIN in Tris-saline buffer to determine the distribution of total [125I]IPIN sites. In serial sections, 50 nm of the selective β2 receptor antagonist ICI-118,551 or 70 nmof the selective β1 receptor antagonist ICI-89,406 was included to visualize binding of [125I]IPIN to β1 and β2 subtypes, respectively. Nonspecific binding was determined from sections co-incubated in the presence of 100 mm isoproterenol, a nonselective agonist. Sections were then washed twice for 15 min each in the incubation buffer at 4°C, rinsed quickly in cold water to remove buffer salts, dried under a stream of cool air, and stored with desiccant at 4°C overnight to remove any remaining moisture.
To prepare autoradiographs, the incubated slides were loaded into cassettes and apposed to tritium-sensitive film (Hyperfilm-3H, Amersham) for 12–18 hr at room temperature before the film was developed.
Analysis of autoradiograms. Receptor densities as reflected in autoradiograms were analyzed by computer-assisted densitometry. The illuminated image of each autoradiograph was collected by a camera connected to an IBM computer with an MCID (St. Catherines, Ontario, Canada) image processing system. Autoradiographic images were calibrated relative to [125I]- or [3H]-labeled standards exposed together with the tissue to the film. Areas (7 × 0.2 mm2) were randomly selected from each cell layer or nucleus in each of three sections per case to determine silver grain density. Differences in [125I]IPIN or [3H]UK-14,304 binding between cases were determined by paired Student’s ttests.
RESULTS
α2-Adrenergic receptor binding
[3H]UK-14,304 binding to human brain tissue sections was saturable and of high affinity, with distributions and pharmacological competition profiles corresponding to α2receptors.
Cerebellum
In the human cerebellar cortex, high levels of α2-adrenergic receptor binding were observed in both molecular and granule cell layers, with no labeling in the subcortical white matter (Fig. 1). Overall, the concentration of α2 receptor labeling in the AD cases was quite variable compared with normal, healthy controls, and when averaged, appeared to be slightly lower, or essentially equal (difference nonsignificant; Fig. 2A). We next sought to investigate whether subdivision of the AD cases by a specific behavioral derangement would result in more homogeneity within each group. The AD cases examined were divided according to the presence or absence of agitated and aggressive behaviors during the patients’ disease course. This was determined by reviewing the results of the California ADDTC Behavior Questionnaire (Mungas et al., 1993), a caregiver-completed, ordinally scaled instrument with 62 questions rating the frequency of a broad range of behaviors (including agitation, aggression, depression, insomnia, and psychosis) and 19 items rating the severity of emotional symptoms. For all cases examined, the questionnaire was administered by the same trained registered nurse. The aggressive AD cases were matched to AD patients with no history of aggression by degree of cognitive impairment, as determined by the Mini Mental State Exam (Folstein et al., 1975). Now with three patient groups examined, the α2 receptor levels were strikingly highest in the agitated, aggressive subgroup of AD patients, with lowest levels seen in the nonagitated AD group (a 70% increase in agitated vs nonagitated subgroups) (Fig.2B). Elderly control patients without AD diagnoses showed α2 receptor concentrations slightly lower than those of the agitated AD subgroup (difference not statistically significant).
Fig. 1.
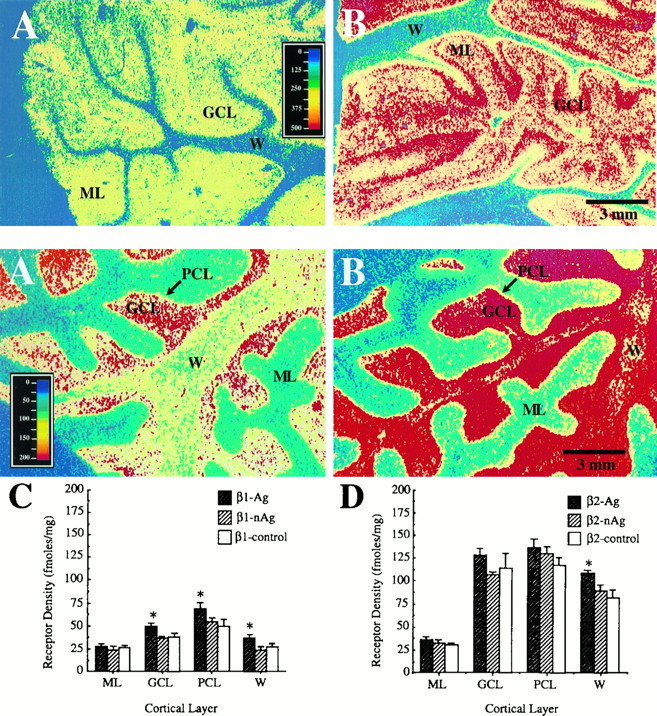
Top. Pseudocolor images of α2adrenergic receptor distribution in human cerebellar cortex.A and B are photomicrographs of the autoradiographic distribution of [3H]UK-14,304 binding sites in the cerebella of nonaggressive and aggressive AD subjects, respectively. Note the higher level of α2receptor concentration in the granule cell and molecular layers of this cerebellar cortical section from an aggressive AD patient (B) compared with a patient from the nonaggressive AD subgroup (A). W, White matter; GCL, granule cell layer;ML, molecular layer. Approximate pseudocolor scale is in femtomoles/mg protein. Scale bar, 3 mm.
Fig. 2.
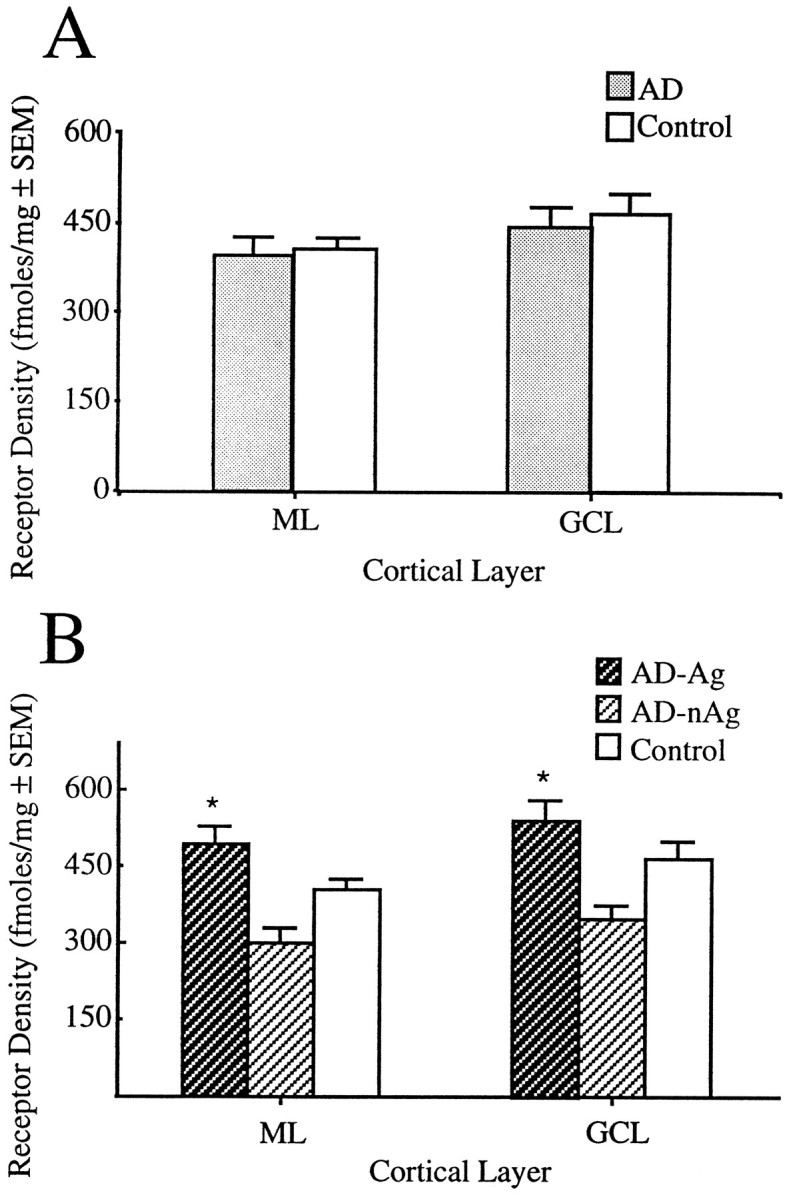
α2 receptor density in the cerebellar cortex. A, Density of α2adrenergic receptor ([3H]UK-14,304) binding in the cerebellar cortex of AD (all patients combined) and normal elderly controls. B, Density of α2 adrenergic receptors in the cerebellar cortices of AD patients with (AD-Ag) and without (AD-nAg) a history of aggression and in normal age-matched controls. Note the lack of significant difference in receptor density when all AD patients are combined and the striking separation in receptor densities between the two subgroups of AD patients. The aggressive subgroup shows an ∼70% increase in density over the nonagitated patients, whose level of α2 receptor concentration is lower than that of controls.ML, Molecular layer; GCL, granule cell layer; * p < 0.005 (for difference between AD-Ag and AD-nAg).
Frontal cortex
Highest levels of α2-adrenergic receptor binding in the orbitofrontal cortex were observed in layer I, with intermediate levels in layer III, and relatively low levels in layers II and IV–VI. There was no labeling observed in the subcortical white matter. In the dorsolateral prefrontal cortex, highest binding levels were evident in layers I and III, with intermediate levels in layers V/VI, and low levels in layers II and IV. No significant differences were found in α2-adrenergic receptor distribution or densities between AD patients and age-matched controls in either of these cortical areas, nor were differences observed between the agitated and nonagitated subgroups of AD patients. For illustrative purposes, the results from the orbitofrontal cortex are shown in Table 2 (the dorsolateral prefrontal cortex also showed no significant differences between the three groups).
Table 2.
α2-Adrenergic receptor concentration in the hypothalamus and orbitofrontal cortex
| Hypothalamus | ||||||
|---|---|---|---|---|---|---|
| Nuclei | n | Ag | n | nAg | n | Control |
| DM | 5 | 263.10 ± 23.07 | 5 | 261.66 ± 19.53 | 0 | |
| VM | 4 | 290.04 ± 26.34 | 3 | 305.01 ± 24.15 | 1 | 249.6 ± 0.00 |
| LAT | 10 | 363.93 ± 51.12 | 5 | 328.50 ± 26.01 | 3 | 360.81 ± 95.76 |
| POST | 8 | 172.53 ± 27.75 | 4 | 120.75 ± 9.30 | 3 | 181.41 ± 30.06 |
| Orbitofrontal cortex | ||||||
| Layer | n | Ag | n | nAg | n | Control |
| I | 4 | 630.45 ± 44.46 | 4 | 680.16 ± 89.76 | 4 | 775.20 ± 69.93 |
| III | 4 | 472.44 ± 26.22 | 4 | 453.36 ± 40.59 | 4 | 488.25 ± 32.88 |
| IV | 4 | 382.35 ± 24.78 | 4 | 390.99 ± 36.27 | 4 | 392.70 ± 30.15 |
| V–VI | 4 | 358.95 ± 20.22 | 4 | 351.15 ± 28.14 | 4 | 362.25 ± 36.33 |
Densities of α2-adrenergic receptors in orbitofrontal cortex and in nuclei of the hypothalamus of Alzheimer’s disease patients with (AD-Ag) and without (AD-nAg) a history of aggression, and in normal age-matched controls, in femtomoles/mg protein ± SEM. No significant differences were found in receptor densities among the three groups of patients either in orbitofrontal (chosen for illustration) or dorsolateral prefrontal cortices or in the hypothalamus. DM, Dorsomedial nucleus; VM, ventromedial nucleus; LAT, lateral nucleus; POST, posterior nucleus.
Hypothalamus
The highest levels of α2 receptor binding in the human hypothalamus were present in the lateral nucleus, with intermediate levels in the dorsomedial nucleus, and low levels in the ventromedial and posterior nuclei. Receptor binding densities in these four hypothalamic nuclei were compared between AD and control and between the two AD subgroups, and no significant differences were found in α2 receptor concentrations (Table 2).
β-Adrenergic receptor binding
Binding of [125I]IPIN to human brain tissue sections was of high affinity, saturable, and with a pharmacological competition profile corresponding to β-adrenergic receptors. Displacement by the selective β1 antagonist ICI-89,406 or the selective β2 antagonist ICI-118,551 was used to define binding to the β2 and β1 receptors, respectively. All human brain regions examined contained both receptor subtypes, with the relative ratio of β1/β2 ranging from 70:30 in certain layers of the prefrontal cortex to 20:80 in the cerebellar cortex. In all regions, the sum of the densities of the two receptors was approximately equal to the total binding for [125I]IPIN.
Cerebellum
The overall ratio of β1- to β2-adrenergic receptor binding in this brain area was ∼20:80. Highest levels of β receptors were observed in the granule and Purkinje cell layers, with low levels in the molecular layer, and intermediate levels over the subcortical white matter (Fig.3). This agrees with β-adrenergic autoradiographic distributions reported previously in human brain (Reznikoff et al., 1986). There were no significant differences in receptor densities in the cerebellar cortices of all AD patients taken together versus controls (data not shown). Cerebella of aggressive demented patients showed small but significant increases in total β-adrenergic receptor density versus both healthy controls and nonaggressive AD patients (Fig. 3A,B). The β1subtype showed small but significant increases in density in the granule cell layer and Purkinje layer as well as in white matter (Fig.3C), whereas the β2 subtype showed significant increases in the subcortical white matter only (Fig. 3D). These results suggest that the observed β-adrenergic receptor binding increases are specific to AD with aggression/agitation.
Fig. 3.
Bottom. β adrenergic receptor binding in cerebellar cortex. A, B, Pseudocolor images of total β adrenergic receptor distribution in human cerebellar cortex. A and B are photomicrographs of the autoradiographic distribution of [125I]IPIN binding sites in the cerebella of nonaggressive (A) and aggressive (B) AD subjects. C andD are histograms of receptor density (in femtomole/milligram protein) for the two subtypes of β adrenergic receptors. C shows the levels of β1receptor binding in the different layers of the cerebellar cortex of two subgroups of AD patients (agitated and nonagitated) and normal elderly controls. Note the moderate but significant increases in β1 receptor concentration in the granule cell layer, Purkinje cell layer, and subcortical white matter of aggressive AD patients over both nonaggressive AD patients and controls.D Displays the levels of β2 adrenergic receptors in these groups. Significant increases in concentration for this β2 receptor subtype of agitated AD patients over both the nonagitated subgroup and the controls are detected in subcortical white matter only. W, White matter;GCL, granule cell layer; PCL, Purkinje cell layer; ML, molecular layer. Approximate pseudocolor scale is in femtomoles/mg protein. Scale bar, 3 mm.
Because receptor concentration increases are evident in the white matter as well as in cerebellar cortical layers, it is possible that the receptors observed in this study may include β-adrenergic receptors on cerebral microvessels and/or glia. Adrenergic receptors (primarily β), innervated by noradrenergic LC neurons, are known to exist in brain microvessels (Kobayashi et al., 1982; Kalaria et al., 1989c). In fact, β2 receptors in cerebral microvessel fractions from human brain have been found to be increased significantly in AD (Kalaria and Harik, 1989). Both normal and reactive astrocytes are known to express β-adrenergic receptors in adult rat brain (Sutin and Shao, 1992). In the visual cortex of the adult cat, ∼50% of cells expressing β-adrenergic receptors have been shown to be astrocytes (Liu et al., 1992). This includes cells both within the cortical layers and within the subcortical white matter.
Frontal cortex
In the human orbitofrontal cortex, high levels of β1receptors were observed in layers I and II, with low levels in layers III–V, and intermediate levels in layer VI. There was no labeling in subcortical white matter. β2 Receptors were observed in a more uniform distribution throughout cortical layers (including the subcortical white matter), with levels at ∼35% of total β-adrenergic receptors. In the dorsolateral prefrontal cortex, high levels of β1 receptors were visible in layers I, II, and VI, with low levels in layers III–V. As in the orbitofrontal cortex, β2 receptors were distributed uniformly throughout the dorsolateral prefrontal cortex at relatively low levels (∼35% of total). No statistically significant differences were found in β-adrenergic receptor distribution or density between AD patients and age-matched controls in either of these cortical areas, nor were differences observed between the agitated and nonagitated subgroups of AD patients. Table 3 shows the results for β1 receptors in the orbitofrontal cortex.
Table 3.
Densities of β-adrenergic receptors in the nuclei of the hypothalamus and in layers of the orbitofrontal cortex
| Hypothalamus—total β-adrenergic receptors | ||||||
|---|---|---|---|---|---|---|
| Nuclei | n | Ag | n | nAg | n | Control |
| DM | 5 | 74.694 ± 9.922 | 5 | 50.066 ± 3.564 | 1 | 64.630 ± 0.00 |
| VM | 4 | 62.200 ± 7.591 | 2 | 40.170 ± 2.420 | 2 | 64.255 ± 22.575 |
| LAT | 9 | 47.023 ± 5.557 | 5 | 45.090 ± 11.795 | 3 | 55.027 ± 5.466 |
| POST | 7 | 51.409 ± 5.182 | 5 | 39.882 ± 9.831 | 3 | 41.200 ± 1.511 |
| Orbitofrontal cortex—β 1-adrenergic receptors | ||||||
| Layers | n | Ag | n | nAg | n | Control |
| I–II | 4 | 22.312 ± 2.520 | 7 | 21.526 ± 1.791 | 4 | 14.442 ± 3.699 |
| III–IV | 3 | 17.907 ± 1.611 | 7 | 16.410 ± 1.077 | 2 | 13.760 ± 4.720 |
| VI | 4 | 18.703 ± 1.338 | 1 | 19.130 ± 1.486 | 1 | 17.640 ± 0.000 |
Data are expressed in femtomole/milligram ± SEM. No statistically significant differences were found among the patient groups examined in any layers of either orbitofrontal (chosen for illustration) or dorsolateral prefrontal cortex, or in the hypothalamus. DM, Dorsomedial nucleus; VM, ventromedial nucleus; LAT, lateral nucleus; POST, posterior nucleus.
Hypothalamus
In the hypothalamus, the overall ratio of β1- to β2-adrenergic receptor binding was ∼30:70. Of the areas examined, the highest levels of β2 binding were present in the dorsomedial and ventromedial nuclei, with intermediate levels in the lateral and posterior nuclei. Among the three patient groups, somewhat higher levels of β-adrenergic receptors (particularly the β2 subtype) appeared to exist in the aggressive subgroup of AD patients and in the controls than in the nonaggressive AD subgroup (∼30%). These differences, however, were not statistically significant (Table 3).
DISCUSSION
Our studies have demonstrated that abnormally low levels of cerebellar α2 adrenergic receptors are restricted to a subgroup of Alzheimer’s patients showing no symptoms of aggression or agitation. In the aggressive subgroup of AD patients, we found receptor levels that were at least as high as (or slightly higher than) controls, representing an ∼70% increase over the nonaggressive AD subgroup. A similar, but less pronounced, increase was observed for β-adrenergic receptors in the same aggressive AD population (vs both nonaggressive AD and control patients).
Target areas of the LC, a region known to be damaged in many AD patients, include the cerebral cortices, specific areas within the limbic system (such as thalamic and hypothalamic nuclei and the hippocampus), and the cerebellum (Cooper et al., 1991). However, many studies investigating the distribution and concentration of adrenergic receptors in these areas in the brains of AD patients have been equivocal; either no significant differences from controls are found, or the changes are small. In the majority of these studies, the AD population has not been divided into behavioral subpopulations, and the examination of specific brain areas has been conducted primarily in homogenates. For example, Meana et al. (1992) showed a reduction in the concentration of α2 receptors in the AD cerebellum, frontal cortex, and hypothalamus (∼30%). Kalaria et al. (1989a) also showed a decrease in α2 receptors in the prefrontal cortex (∼50%), but not in the cerebellum. Total β-adrenergic receptor content showed no change in the AD prefrontal cortex, but when separate subtypes were examined, there was a slight decrease in β1 and a larger (∼35%) increase in β2receptors (Kalaria et al., 1989b). No significant changes have been reported in cerebellar β-receptor concentration in AD. In one autoradiographic study (Vogt et al., 1991), differences in β-receptor concentrations in the cingulate cortex between AD and control brains were found to be nonuniform. Anatomical subclasses were found within the AD group that appeared to vary in neuronal losses within the specific layers of the cingulate cortex. Certain subclasses showed no change in β-receptor concentration, and others showed substantial increases (as much as 65%). It was speculated that clinical factors may correlate with the differences among subclasses. It is of interest that if the levels from all AD patients in our studies were averaged, only a slight decrease in cerebellar α2 receptors would be evident compared with the normal elderly patient population [similar to findings by Meana et al. (1992)], and no change would be evident in cerebellar β receptors. Thus, our results underscore the importance of distinguishing subgroups within a disease population and a particular need for attention to behavioral symptoms that could benefit from specific treatments.
Aggression, irritability, and agitation are among the most common and problematic symptoms of Alzheimer’s disease. During the course of the illness, as many as 48% of AD patients develop these behavioral symptoms, forcing them out of outpatient care settings into institutions with close supervision (Reisberg et al., 1987; Chandler and Chandler, 1988). In the normal brain, several specific cortical and subcortical regions that contain high levels of NE (Pifl et al., 1991) are thought to be associated with the modulation and control of aggression. These include the hypothalamus, amygdala/medial temporal lobe, and frontal neocortex (Weiger and Bear, 1988). Elevated NE levels have been associated with increases in aggressive behavior, and inhibitors of NE function decrease aggression. For example, agents that enhance central NE function, such as tricyclic/monoamine oxidase inhibitor antidepressants (Eichelman and Barchas, 1975) and presynaptic α2 antagonists (Haller, 1995), have been shown to increase fighting in rodents. Lithium, which decreases NE availability and increases central tryptophan uptake, reduces shock-induced fighting in rodents (Eichelman et al., 1973). In humans, β-adrenergic blockade with propranolol has been successful in managing violent behavior in neuropsychiatric syndromes (Yudofsky et al., 1981; Sorgi et al., 1986), and there is emerging clinical evidence that a subgroup of AD patients obtain significant relief from aggressive symptoms through treatment with low-dose propranolol (Weiler et al., 1988; Pauszek, 1991; Shankle et al., 1995). It has been suggested that lithium can have a clinically useful effect on impulsive aggressive behavior in humans when the behavior is not associated with psychosis (Sheard et al., 1976). Clonidine has shown promise in controlling aggression in children (Kemph et al., 1993) and adults with the neuropsychiatric syndrome autism (Koshes and Rock, 1994).
In addition to cortical and subcortical limbic regions, the cerebellum also appears to be involved in behavioral control (Schmahmann, 1991). NE fibers from the LC project via the superior cerebellar peduncle to the cerebellar cortex (Pickel et al., 1974), where the Purkinje cell appears to be the primary target. These fiber afferents make contact with tertiary or secondary Purkinje cell dendrites in the molecular layer (Bloom, 1971). In addition, NE-containing fibers are also found in the superficial region of the granule cell layer, particularly around the glomeruli, making close contact with granule cell dendrites (Kimoto et al., 1981). The axons of the Purkinje cells (after synapsing with deep cerebellar nuclei) provide the major output of the cerebellar cortex, projecting through the thalamus to the prefrontal cortex (Asanuma et al., 1983) and other association areas, such as the posterior parietal cortex (Kasdon and Jacobson, 1978) and the upper bank of the superior temporal sulcus (Yeterian and Pandya, 1989). Therefore, in addition to its well known function in the modulatory control of limb movements and other motor-associated behaviors, the cerebellar Purkinje cell output may serve in the modulation of affective and defensive/aggressive behavior, possibly by influencing circuits in the prefrontal cortex and other association areas.
Human postmortem study, particularly in an elderly patient population, can be potentially complicated by the existence of multiple medications in the patients’ histories. Evidence exists that neuroleptic medication treatments can influence the levels of adrenergic receptors in mammalian brain. Because several of the patients included in this study had received neuroleptics during their history (Table 1), it is important to address the possibility that any of the observed receptor changes could have been brought about by their treatments. Of the eight patients in the aggressive subgroup, three were treated with antipsychotic medication (two with haloperidol and one with loxapine). Only one of these patients (patient 1), however, took this medication within 3 weeks of death, and neuroleptic-induced receptor concentration changes have been shown to be reversed within 7 d of stopping treatment (Wolfe et al., 1978). Several patients in our study had received antidepressant medications during their treatment, but this treatment was fairly equally distributed among the three patient groups (3/8 in the aggressive AD subgroup, 3/8 in the nonaggressive AD subgroup, and 2/7 in the elderly control group). All neuroleptic-induced adrenergic receptor changes, when present, appear to be evident in several regions throughout the brain (Greenberg, 1978;Maggi et al., 1980; Weiss and Greenberg, 1980). The changes in adrenergic receptor concentrations that we have reported were confined to the cerebellum. In summary, multiple lines of evidence suggest that the receptor concentration changes we report are not attributable to neuroleptic medication effects.
Aggression and agitation are often the factors leading to institutionalization of an individual with AD. Therefore, these symptoms account for a large part of the caregiver distress and cost involved in this disease, and it is essential to work toward understanding the neurochemical mechanisms behind these symptoms so that better treatments can be developed. Ours is among the first reports examining neurochemical lesions of specific Alzheimer’s disease subgroups (based on behavioral symptoms), and the first study, to our knowledge, that has specifically investigated neurotransmitter receptor distributions/concentrations in a behavioral subgroup of dementia. A recent study examining the neuropathological correlates of agitation and physical aggression in AD revealed that AD patients with histories of unequivocal interpersonal violence had significantly greater neuron counts in the substantia nigra pars compacta than did nonviolent patients (Victoroff et al., 1996). Therefore, other evidence is appearing that important differences in monamine neurotransmitter function may exist in this aggressive subgroup of AD patients.
Our autoradiographic results raise questions about the possible mechanisms underlying the observed changes in the cerebellar cortex. Are adrenergic receptor increases in the aggressive AD subgroup a result of denervation supersensitivity, or is there a preservation of cerebellar adrenergic inputs? Studies in our laboratory have shown a relative preservation of tyrosine hydroxylase-containing neuronal fibers in the cerebellar cortex of aggressive versus nonaggressive AD patients (our unpublished data). The results reported in this paper suggest that noradrenergic inputs, which appear to decline in the normal aging cerebellum (Jones and Olpe, 1983) and diminish sharply in many AD patients, are preserved in the agitated subgroup of AD patients. We hypothesize that in the agitated AD patients presented in this autoradiographic study, inhibitory influences of NE on Purkinje cells are preserved, in the face of a cortical lesion known to be present in AD. Therefore, in the aggressive subgroup of AD, the inhibitory/modulatory output from the cerebellum is low compared with that in the nonaggressive subgroup, whereas the AD-lesioned cerebral cortex is already impaired in its ability to judge and modulate behavior, resulting in poor behavioral control. In this case, a normal level of cerebellar NE function (i.e., levels of α2adrenergic receptors that approximate normal controls), in the face of cerebral cortical impairment, can actually be abnormally high. Another question raised by our results is why the receptor changes have been observed in the cerebellum only, and not in other brain areas examined that are known to be involved in the mediation and modulation of aggression. One possibility is based on the observation that the cerebellum is one of the last brain regions to develop neuropathology and undergo degeneration in AD. In this case, it is possible that the cerebellum is the only one of these brain areas capable of maintaining/preserving noradrenergic receptors or inputs to the extent suggested.
Footnotes
This work was supported by U.S. Public Health Service Grant MH-02166. We thank Drs. Adrienne Frostholm and Andrej Rotter for valuable comments in review of this manuscript, and Toska J. Zomorodian for assistance in preparing figures and tables.
Correspondence should be addressed to Dr. Amelia Russo-Neustadt, Bio Sci II Room 1305, Institute for Brain Aging and Dementia, University of California, Irvine, CA 92697-4540.
REFERENCES
- 1.Asanuma C, Thach WT, Jones EG. Distribution of cerebellar terminations and their relation to other afferent terminations in the ventral lateral thalamic region of the monkey. Brain Res. 1983;286:237–265. doi: 10.1016/0165-0173(83)90015-2. [DOI] [PubMed] [Google Scholar]
- 2.Bloom FEH, Hoffer BJ, Siggins GR. Studies on norepinephrine-containing afferents to Purkinje cells of art cerebellum. I. Localization of the fibers and their synapses. Brain Res. 1971;25:501–521. doi: 10.1016/0006-8993(71)90457-4. [DOI] [PubMed] [Google Scholar]
- 3.Chandler JD, Chandler JE. The prevalence of neuropsychiatric disorders in a nursing home population. J Geriatr Psychiatry Neurol. 1988;1:71–76. doi: 10.1177/089198878800100203. [DOI] [PubMed] [Google Scholar]
- 4.Cooper JR, Bloom FE, Roth RH. The biochemical basis of neuropharmacology, Ed 6. Oxford UP; New York: 1991. [Google Scholar]
- 5.Eichelman B, Barchas J. Facilitated shock-induced aggression following antidepressive medication in the rat. Pharmacol Biochem Behav. 1975;3:601–604. doi: 10.1016/0091-3057(75)90180-x. [DOI] [PubMed] [Google Scholar]
- 6.Eichelman B, Thoa NB, Perez-Cruet J. Alkali metal cations: effects on aggression and adrenal enzymes. Pharmacol Biochem Behav. 1973;1:121–123. doi: 10.1016/0091-3057(73)90066-x. [DOI] [PubMed] [Google Scholar]
- 7.Folstein MF, Folstein SE, McHugh PR. “Mini-mental state.” A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12:189–198. doi: 10.1016/0022-3956(75)90026-6. [DOI] [PubMed] [Google Scholar]
- 8.Greenberg LH, Weiss B. Beta-adrenergic receptors in the aging rat brain: modifications induced by psychotropic drugs. In: Szabadi E, Bradshaw CM, Bevan P, editors. Recent advances in the pharmacology of adrenoceptors. Elsevier/North Holland Biomedical; New York: 1978. pp. 241–250. [Google Scholar]
- 9.Haller J. Alpha-2 adrenoceptor blockade and the response to intruder aggression in Long–Evans rats. Physiol Behav. 1995;58:101–106. doi: 10.1016/0031-9384(95)00030-m. [DOI] [PubMed] [Google Scholar]
- 10.Ishino H, Otsuki S. Frequency of Alzheimer’s neurofibrillary tangles in the basal ganglia and brain-stem in Alzheimer’s disease, senile dementia and the aged. Folia Psychiatr Neurol Jpn. 1975;29:279–287. doi: 10.1111/j.1440-1819.1975.tb02344.x. [DOI] [PubMed] [Google Scholar]
- 11.Jones RS, Olpe HR. Altered sensitivity of forebrain neurones to iontophoretically applied noradrenaline in aging rats. Neurobiol Aging. 1983;4:97–99. doi: 10.1016/0197-4580(83)90060-x. [DOI] [PubMed] [Google Scholar]
- 12.Kalaria RN, Harik SI. Increased alpha 2- and beta 2-adrenergic receptors in cerebral microvessels in Alzheimer disease. Neurosci Lett. 1989;106:233–238. doi: 10.1016/0304-3940(89)90231-0. [DOI] [PubMed] [Google Scholar]
- 13.Kalaria RN, Andorn AC, Harik SI. Alterations in adrenergic receptors of frontal cortex and cerebral microvessels in Alzheimer’s disease and aging. Prog Clin Biol Res. 1989a;317:367–374. [PubMed] [Google Scholar]
- 14.Kalaria RN, Andorn AC, Tabaton M, Whitehouse PJ, Harik SI, Unnerstall JR. Adrenergic receptors in aging and Alzheimer’s disease: increased beta 2-receptors in prefrontal cortex and hippocampus. J Neurochem. 1989b;53:1772–1781. doi: 10.1111/j.1471-4159.1989.tb09242.x. [DOI] [PubMed] [Google Scholar]
- 15.Kalaria RN, Stockmeier CA, Harik SI. Brain microvessels are innervated by locus ceruleus noradrenergic neurons. Neurosci Lett. 1989c;97:203–208. doi: 10.1016/0304-3940(89)90164-x. [DOI] [PubMed] [Google Scholar]
- 16.Kasdon DL, Jacobson S. The thalamic afferents to the inferior parietal lobule of the rhesus monkey. J Comp Neurol. 1978;177:685–706. doi: 10.1002/cne.901770409. [DOI] [PubMed] [Google Scholar]
- 17.Kemph JP, DeVane CL, Levin GM, Jarecke R, Miller RL. Treatment of aggressive children with clonidine: results of an open pilot study. J Am Acad Child Adolesc Psychiatry. 1993;32:577–581. doi: 10.1097/00004583-199305000-00013. [DOI] [PubMed] [Google Scholar]
- 18.Kimoto Y, Tohyama M, Satoh K, Sakumoto T, Takahashi Y, Shimizu N. Fine structure of rat cerebellar noradrenaline terminals as visualized by potassium permanganate “in situ perfusion” fixation method. Neuroscience. 1981;6:47–58. doi: 10.1016/0306-4522(81)90242-6. [DOI] [PubMed] [Google Scholar]
- 19.Kobayashi H, Frattola L, Ferrarese C, Spano P, Trabucchi M. Characterization of beta-adrenergic receptors on human cerebral microvessels. Neurology. 1982;32:1384–1387. doi: 10.1212/wnl.32.12.1384. [DOI] [PubMed] [Google Scholar]
- 20.Koshes RJ, Rock NL. Use of clonidine for behavioral control in an adult patient with autism. Am J Psychiatry [Letter] 1994;151:1714. doi: 10.1176/ajp.151.11.1714b. [DOI] [PubMed] [Google Scholar]
- 21.Liu Y, Jia WG, Strosberg AD, Cynader M. Morphology and distribution of neurons and glial cells expressing beta-adrenergic receptors in developing kitten visual cortex. Brain Res Dev Brain Res. 1992;65:269–273. doi: 10.1016/0165-3806(92)90188-3. [DOI] [PubMed] [Google Scholar]
- 22.Maggi A, U’Prichard DC, Enna SJ. Differential effects of antidepressant treatment on brain monoaminergic receptors. Eur J Pharmacol. 1980;61:91–98. doi: 10.1016/0014-2999(80)90152-1. [DOI] [PubMed] [Google Scholar]
- 23.Meana JJ, Barturen F, Garro MA, García-Sevilla JA, Fontán A, Zarranz JJ. Decreased density of presynaptic alpha 2-adrenoceptors in postmortem brains of patients with Alzheimer’s disease. J Neurochem. 1992;58:1896–1904. doi: 10.1111/j.1471-4159.1992.tb10067.x. [DOI] [PubMed] [Google Scholar]
- 24.Mega MS, Cummings JL, Fiorello T, Gornbein J. The spectrum of behavioral changes in Alzheimer’s disease. Neurology. 1996;46:130–135. doi: 10.1212/wnl.46.1.130. [DOI] [PubMed] [Google Scholar]
- 25.Mungas D, Jagust W, Reed B (1993) Neuropsychological and behavioral measures for Alzheimer’s disease and other dementias. Presented at the California Alzheimer’s Disease Diagnostic and Treatments Centers Strategic Planning Conference, Lake Arrowhead, CA, April.
- 26.Pauszek ME. Propranolol for treatment of agitation in senile dementia. Indiana Med. 1991;84:16–17. [PubMed] [Google Scholar]
- 27.Pazos A, González AM, Pascual J, Meana JJ, Barturen F, García-Sevilla JA. Alpha 2-adrenoceptors in human forebrain: autoradiographic visualization and biochemical parameters using the agonist [3H]UK-14304. Brain Res. 1988;475:361–365. doi: 10.1016/0006-8993(88)90626-9. [DOI] [PubMed] [Google Scholar]
- 28.Pickel VM, Segal M, Bloom FE. Axanol proliferation following lesions of cerebellar peduncles. A combined fluorescence microscopic and radioautographic study. J Comp Neurol. 1974;155:43–60. doi: 10.1002/cne.901550104. [DOI] [PubMed] [Google Scholar]
- 29.Pifl C, Schingnitz G, Hornykiewicz O. Effect of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine on the regional distribution of brain monoamines in the rhesus monkey. Neuroscience. 1991;44:591–605. doi: 10.1016/0306-4522(91)90080-8. [DOI] [PubMed] [Google Scholar]
- 30.Rainbow TC, Parsons B, Wolfe BB. Quantitative autoradiography of beta 1- and beta 2-adrenergic receptors in rat brain. Proc Natl Acad Sci USA. 1984;81:1585–1589. doi: 10.1073/pnas.81.5.1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Reisberg B, Borenstein J, Salob SP, Ferris SH, Franssen E, Georgotas A. Behavioral symptoms in Alzheimer’s disease: phenomenology and treatment. J Clin Psychiatry [Suppl] 1987;48:9–15. [PubMed] [Google Scholar]
- 32.Reznikoff GA, Manaker S, Rhodes CH, Winokur A, Rainbow TC. Localization and quantification of beta-adrenergic receptors in human brain. Neurology. 1986;36:1067–1073. doi: 10.1212/wnl.36.8.1067. [DOI] [PubMed] [Google Scholar]
- 33.Schmahmann JD. An emerging concept. The cerebellar contribution to higher function [see Comments]. Arch Neurol. 1991;48:1178–1187. doi: 10.1001/archneur.1991.00530230086029. [DOI] [PubMed] [Google Scholar]
- 34.Shankle WR, Nielson KA, Cotman CW. Low-dose propranolol reduces aggression and agitation resembling that associated with orbitofrontal dysfunction in elderly demented patients. Alzheimer Dis Assoc Disord. 1995;9:233–237. [PubMed] [Google Scholar]
- 35.Sheard MH, Marini JL, Bridges CI, Wagner E. The effect of lithium on impulsive aggressive behavior in man. Am J Psychiatry. 1976;133:1409–1413. doi: 10.1176/ajp.133.12.1409. [DOI] [PubMed] [Google Scholar]
- 36.Sorgi PJ, Ratey JJ, Polakoff S. Beta-adrenergic blockers for the control of aggressive behaviors in patients with chronic schizophrenia. Am J Psychiatry. 1986;143:775–776. doi: 10.1176/ajp.143.6.775. [DOI] [PubMed] [Google Scholar]
- 37.Sutin J, Shao Y. Resting and reactive astrocytes express adrenergic receptors in the adult rat brain. Brain Res Bull. 1992;29:277–284. doi: 10.1016/0361-9230(92)90057-5. [DOI] [PubMed] [Google Scholar]
- 38.Victoroff J, Zarow C, Mack WJ, Hsu E, Chui HC. Physical aggression is associated with preservation of substantia nigra pars compacta in Alzheimer disease. Arch Neurol. 1996;53:428–434. doi: 10.1001/archneur.1996.00550050058024. [DOI] [PubMed] [Google Scholar]
- 39.Vogt BA, Crino PB, Volicer L. Laminar alterations in gamma-aminobutyric acid A, muscarinic, and beta adrenoceptors and neuron degeneration in cingulate cortex in Alzheimer’s disease. J Neurochem. 1991;57:282–290. doi: 10.1111/j.1471-4159.1991.tb02126.x. [DOI] [PubMed] [Google Scholar]
- 40.Weiger WA, Bear DM. An approach to the neurology of aggression. J Psychiatr Res. 1988;22:85–98. doi: 10.1016/0022-3956(88)90073-8. [DOI] [PubMed] [Google Scholar]
- 41.Weiler PG, Mungas D, Bernick C. Propranolol for the control of disruptive behavior in senile dementia. J Geriatr Psychiatry Neurol. 1988;1:226–230. doi: 10.1177/089198878800100408. [DOI] [PubMed] [Google Scholar]
- 42.Weiss B, Greenberg LH. Modulation of beta-adrenergic receptors and calmodulin following acute and chronic treatment with neuroleptics. Adv Biochem Psychopharmacol. 1980;24:139–146. [PubMed] [Google Scholar]
- 43.Wolfe BB, Harden TK, Sporn JR, Molinoff PB. Presynaptic modulation of beta adrenergic receptors in rat cerebral cortex after treatment with antidepressants. J Pharmacol Exp Ther. 1978;207:446–457. [PubMed] [Google Scholar]
- 44.Yeterian EH, Pandya DN. Thalamic connections of the cortex of the superior temporal sulcus in the rhesus monkey. J Comp Neurol. 1989;282:80–97. doi: 10.1002/cne.902820107. [DOI] [PubMed] [Google Scholar]
- 45.Yudofsky S, Williams D, Gorman J. Propranolol in the treatment of rage and violent behavior in patients with chronic brain syndromes. Am J Psychiatry. 1981;138:218–220. doi: 10.1176/ajp.138.2.218. [DOI] [PubMed] [Google Scholar]