

Abstract
We have shown previously that a selective metabotropic glutamate receptor (mGluR) agonist, 1S,3R-1-aminocyclo-pentane-1,3-dicarboxylate (1S,3R-ACPD), evokes an inward current in CA1 pyramidal neurons of rat hippocampal slices in the presence of K+ channel blockers (Crépel et al., 1994). This current has been characterized as a Ca2+-activated nonselective cationic (CAN) current. Using whole-cell patch-clamp recordings and intracellular dialysis, we now have identified the mGluR subtype and the mechanisms underlying the CAN current (ICAN) and report for the first time the presence of a synaptic ICANin the mammalian CNS. First, we have shown pharmacologically that activation of ICAN by 1S,3R-ACPD involves the group I mGluRs (and not the groups II and III) and a G-protein-dependent process. We also report that ICAN is modulated by the divalent cations (Mg2+, Cd2+, and Zn2+). Second, we have isolated a slow synaptic inward current evoked by a high-frequency stimulation in the presence of K+ channel blockers, ionotropic glutamate, and GABAA receptor antagonists. This current shows similar properties to the exogenously evoked ICAN: its reversal potential is close to the reversal potential of the 1S,3R-ACPD-evokedICAN, and it is G-protein- and Ca2+-dependent. Because the amplitude and duration of ICAN increased in the presence of a glutamate uptake blocker, we suggest that this synaptic current is generated via the activation of mGluRs. We propose that the synapticICAN, activated by a brief tetanic stimulation and leading to a long-lasting inward current, may be involved in neuronal plasticity and synchronized network-driven oscillations.
Keywords: Ca2+-activated nonselective cationic current, slow synaptic inward current, postsynaptic mGluRs, intracellular perfusion, whole-cell voltage clamp, CA1 pyramidal neurons, hippocampus
Metabotropic glutamate receptors (mGluRs; Sugiyama et al., 1987) form a heterogenous family of G-protein-coupled receptors, which play a crucial role in controlling the cell excitability in the CNS. Multiple physiological roles for mGluRs have been identified, including neuronal excitation (increase of cell excitability, potentiation of glutamate release, coinduction, or facilitation of LTP) as well as neuronal inhibition (hyperpolarization, presynaptic inhibition of glutamate and GABA release, coinduction, or facilitation of LTD), and a possible implication in glutamate-induced neurotoxicity (for review, see Schoepp and Conn, 1993; Gallagher et al., 1994; Ben-Ari and Aniksztejn, 1995; Pin and Duvoisin, 1995). The mGluRs are coupled to a large variety of second messenger systems [including activation of phospholipase C (PLC) and inhibition of adenylyl cyclase] and modulate several ligand-gated ionic channels (for review, see Schoepp and Conn, 1993; Pin and Duvoisin, 1995).
In a previous report, using intracellular recordings, we showed that, in the presence of K+ channel blockers, ionotropic glutamate, and GABAA receptor antagonists, the selective mGluR agonist 1S,3R-1-aminocyclopentane-1,3-dicarboxylate (1S,3R-ACPD) evoked an inward current in CA1 pyramidal neurons of the rat hippocampus (Crépel et al., 1994). This current was characterized as a Ca2+-activated nonselective cationic current (CAN current) on the basis of its ionic properties and its blockade by intracellular injection of the Ca2+ chelating agent BAPTA. However, several questions remain unanswered. (1) Does the link between mGluRs and CAN current involve G-proteins? (2) What types of mGluRs are implicated in the activation of this CAN current (and consequently, which second messenger pathway)? (3) Can this current be activated synaptically?
We now have used whole-cell patch-clamp recordings and intracellular dialysis to study the properties of ICAN. Our observations show that (1) ICAN is activated by group I mGluRs via a G-protein- and Ca2+-dependent pathway, (2) ICAN is modulated by divalent cations, and (3) a high-frequency stimulation (HFS) synaptically generates a slow inward current through mGluRs, once glutamate and GABA ionotropic receptors have been blocked. This synaptic current demonstrates features specific to the CAN current; in particular, it is calcium-dependent (blocked by BAPTA) and G-protein-dependent (blocked by G-proteins inhibitors). Therefore, in CA1 pyramidal neurons, in addition to the well characterized fast excitatory currents resulting from the activation of ionotropic AMPA and NMDA receptors, brief tetani generate, via the activation of mGluRs, a long-lasting CAN current. Because of its unique features ICAN may play a major role in neuronal plasticity and network-driven oscillations in neurons of the mammalians CNS.
MATERIALS AND METHODS
Hippocampal slices preparation. Experiments were performed in CA1 hippocampal neurons in slices obtained from 100–150 gm male Wistar rats (20–40 d old). Rats were anesthetized with ether and decapitated. After decapitation, the brain was removed quickly from the skull, and hippocampi were dissected free on ice in a 0–5°C oxygenated artificial cerebrospinal fluid (ACSF) containing (in mm): 126 NaCl, 3.5 KCl, 1 CaCl2, 2 MgCl2, 1.2 NaH2PO4, 25 NaHCO3, and 11 glucose, equilibrated with 95% O2/5% CO2, pH 7.4. Transverse slices (500 μm) were prepared with a MacIlwain tissue chopper and were incubated in ACSF at room temperature, as previously described (Cherubini et al., 1987). After a 2 hr recovery period, hippocampal slices were transferred one at a time to a submerged recording chamber and superfused continuously (2.5–3 ml/min at 28–30°C) with a phosphate-free ACSF (P-free ACSF). The superfusing ACSF did not contain NaH2PO4 to avoid precipitation in the following conditions: (1) in the presence of the K+ channel blockers TEA (5–25 mm) and 4-AP (5 mm), (2) when the external Mg2+concentration was raised (from 2 to 10 mm), or (3) when Cd2+ (200 μm) or Zn2+ (200 μm) was added.
Whole-cell patch-clamp recordings. Whole-cell patch-clamp recordings were obtained from CA1 pyramidal neurons by using the “blind” patch-clamp technique. Membrane currents were recorded in the voltage-clamp mode with an Axopatch 200A amplifier (Axon Instruments, Foster City, CA). Patch electrodes were pulled from borosilicate thin-wall glass capillaries (GC150F-15, Clarck Electromedical Instruments, Pangbourne, UK) with a vertical puller (Glass Microelectrodes Puller PP83, Narishige, London, UK). Broad taper pipettes were used to optimize the solution exchange at their tips. These pipettes had a resistance of 5–7 MΩ when filled with a Cs gluconate (CsGlu) internal solution that contained (in mm): 120 CsGlu, 10 CsCl, 10 NaCl, 1 CaCl2, 2 MgATP, 0.5 GTP, 10 EGTA, and 10 HEPES, pH 7.25 (intracellular-free Ca2+ ≈ 100 nm). In some experiments CsGlu was substituted for equimolar Cs chloride (CsCl). Resting membrane potential was estimated from the potential observed on withdrawal of the electrode from the cell. Holding potential was maintained at −60 mV for all cells recorded. Membrane input conductance was monitored by application of hyperpolarizing voltage steps of 10 mV (0.03 Hz) throughout the experiments. Cells showing changes in intrinsic membrane input resistance during the experiments were discarded.
In all experiments, except during synaptic recordings (see below), 1 μm tetrodotoxin (TTX) and 50 μmmethoxyverapamil (D600), 10 μm bicuculline, and 1 mm kynurenate were present in the P-free ACSF to inhibit Na+ and voltage-dependent Ca2+currents, respectively, and to antagonize GABAA, AMPA, and NMDA receptor-mediated currents, respectively. In these experiments K+ currents were suppressed by concomitant intracellular perfusion of CsGlu-containing pipette solution and by the addition of 6 mm CsCl (to depressIQ), 5–25 mmtetraethylammonium chloride (TEACl; to depressIM, IK, and the fast Ca2+-dependent K+current IC), and 5 mm4-aminopyridine (4-AP; to block the early fast-inactivatingIA and the slow-inactivatingID currents) to the P-free ACSF (Segal and Barker, 1984; Storm, 1988, 1990). This superfusing medium was called medium A. The glutamatergic [200 μm1S,3R-ACPD, 200 μm3,5-dihydroxyphenylglycine (DHPG), 10 μm(2S,1′R,2′R,3′R)-2-(2,3-dicarboxycyclopropyl) glycine (DCG IV), and 1 mml-2-amino-4-phosphonobutyrate (l-AP4)] or muscarinic (60–120 μm carbachol) metabotropic receptor agonists, the glutamatergic [1 mm(S)-α-methyl-4-carboxyphenylglycine (S)-MCPG] or muscarinic (10 μmatropine) metabotropic receptor antagonists, and the activator of adenylyl cyclase (50 μm forskolin) or the glutamate uptake inhibitor dihydrokainic acid (DHK; 250–500 μm) were dissolved in this medium A and were bath-applied. The voltage dependence of these metabotropic receptor-induced currents was studied using (+50 to −100 mV, 8 sec) ramp voltage commands: the membrane potential was stepped from VH to +50 mV, held at +50 mV for 500 msec, and then ramped to −100 mV in 7.5 sec (ramp A).
Synaptic recordings were performed with recording pipettes containing 2 mm of N-(2,6-dimethylphenylcarbamoylmethyl) triethylammonium bromide (QX 314; diluted into the CsGlu solution) to block Na+ voltage-dependent channels. In these experiments the superfusing medium contained 20 μmbicuculline, 40 μm CNQX, 200 μmdl-2-amino-5-phosphono-valeric acid (dl-APV) (to block GABAA, AMPA, and NMDA receptor-mediated currents, respectively), K+ channel blockers (5–25 mm TEA, 5 mm 4-AP, and 6 mmCs+), and 1 mm Mg2+(medium B). The voltage dependence of the synaptically evoked current was studied using (−20 mV to −100 mV, 1 sec) ramp voltage commands: the membrane potential was stepped from VH to −20 mV, held at −20 mV for 100 msec, and then ramped to −100 mV in 1 sec (ramp B).
Pipette–whole-cell perfusion. Experiments requiring intracellular perfusion were performed with a 2PK+pipette–whole-cell perfusion kit combined with an MRC-6 multireservoir carrousel (ALA Scientific Instruments, Rockville Center, NY). The reservoirs contained various intracellular solutions (drugs were diluted to their final concentration in the CsGlu pipette solution) and were connected to a common pressure vessel. The outlet from the vessel was fed into a valve, and a thin polyethylene tube (PE 10) connected to the output of this valve was inserted into the recording pipette through the perfusion port of a specific pipette holder. The polyethylene tube ended by a connection to a polyimide-coated quartz microperfusion capillary, thinned down at the other end by a high flame pulling (cleaned and cut for a final tip size of 30–50 μm), extending close to the tip of the recording pipette in a position in which its orifice was approximately one-half of the internal diameter of the pipette. The inlet to the pressure vessel was fed into a valve and connected to a pressure/vacuum generator. Pipette–whole-cell perfusion was achieved by two successive 5 min periods of active perfusion (application of a well defined positive pressure on the inlet to the positive pressure vessel and a conversely negative pressure on the suction port of the pipette holder, both simultaneously generated by the pressure/vacuum generator), interspersed by 5 min of passive diffusion.
Fluorescence measurements. Fluorescence measurements were performed as previously described (Leinekugel et al., 1995) on neurons loaded with the Ca2+-sensitive dye Fluo-3 in the impermeant form (whole-cell configuration CsGlu pipette solution containing 0.01 mm Fluo-3), using a confocal laser scanning microscope (MRC Bio-Rad 600, Munich, Germany) equipped with an argon–krypton laser and photomultiplier. Excitation was delivered at 488 nm, and emission intensity was measured at wavelengths >500 nm. Images were acquired every 10 sec with the program SOM (Bio-Rad) and analyzed off-line with the program Fluo (Imstar, Paris, France). All results were expressed as ΔF/F0, with Fequal to fluorescence from the defined portion of the image corresponding to the cell under investigation andF0 equal to the mean base line fluorescence in the selected area from at least five consecutive images. Because Fluo-3 is a single wavelength chromophore and fluorescence is a function of the concentration of Ca2+ and dye (Kao et al., 1989), we have used this dye only for an approximate estimation of [Ca2+]i and included for analysis only experiments in which the fluorescence level recovered to control value after cell excitation. As described earlier (Leinekugel et al., 1995), individual neurons were selected by using the optics of an Axioscope Karl Zeiss microscope (water immersion objective 40×) that allows recognition of pyramidal neurons in slices.
Materials. DCG IV was generously supplied by Dr. K. Shimamoto (Suntori Institute for Bio-organic Research, Osaka, Japan). DHPG, l-AP4, [(S)-MCPG], 1S,3R-ACPD, bicuculline, 6-cyano-7-nitroquin-oxaline-2,3-dione (CNQX), anddl-APV were purchased from Tocris Cookson (Bristol, UK). Fluo-3 was purchased from Molecular Probes (Eugene, OR). Kynurenate and all other drugs were purchased from Sigma (St. Louis, MO).
Data analysis. Membrane responses were digitized and displayed simultaneously on a Nicolet digital oscilloscope and on a computer-driven chart recorder. Data were analyzed off-line on computer (programs G, Sadoc, France) and are presented as means ± SEM. Statistical significance (p ≤ 0.05) was assessed by the Student’s t test analysis (paired data).
RESULTS
Experiments were performed on a homogenous population of 78 pyramidal cells of the CA1 hippocampal region. On average, resting membrane potential was −62 ± 0.5 mV, and input resistance was 173 ± 5.7 MΩ.
In the presence of K+ channel blockers, 1S,3R-ACPD induced a Ca2+-activated nonselective cationic current:ICAN
As previously shown with intracellular recordings (Crépel et al., 1994), with continual application of K+ channel blockers (5–25 mm TEA, 5 mm 4-AP, and 6 mm Cs+ diluted in the superfusing medium A; see Materials and Methods), bath application of 1S,3R-ACPD (200 μm, 2 min) evoked a reversible inward current of −64.4 ± 3.4 pA, peak amplitude atVH = −60 mV (IACPD; Fig.1A-a). This current was associated with a significant increase in membrane conductance of 42.9 ± 3.4% (2.2 ± 0.1 and 3.1 ± 0.1 nS in the absence and in the presence of 1S,3R-ACPD, respectively;n = 55, p = 0.0001). The voltage dependence of IACPD was studied by plottingI/V relations derived from hyperpolarizing ramp commands from +50 to −100 mV (ramp A; see Materials and Methods).IACPD had a reversal potential of −17 ± 1.3 mV (n = 55), slightly more negative than that predicted by the Nernst equation (+3.5 mV) at the experimental conditions of [cations]o = 160.5 mm and [cations]i = 140 mm (but see Discussion). The I/V relation ofIACPD, obtained by subtracting the current recorded in the presence of 1S,3R-ACPD from that recorded in the absence of 1S,3R-ACPD, was not linear but displayed an area of reduced slope conductance at voltages more negative than −40 mV (Fig. 1D,E). The conductances calculated for the two representative membrane potential intervals, −100 to −60 mV (which will be referred to as negative conductance) and 0 to +40 mV (which will be referred to as positive conductance) were significantly different, 0.92 ± 0.08 and 2.1 ± 0.1 nS, respectively (n = 55,p = 0.0001), showing a negative rectification of −48 ± 5.7% (n = 55). These current properties were observed similarly in the presence of CsGlu-containing and CsCl-containing pipette solution (data not shown), confirming the chloride gradient independence of the current.
Fig. 1.

In the presence of K+ channel blockers, 1S,3R-ACPD induced a Ca2+-activated nonselective cationic current,ICAN. In control conditions 1S,3R-ACPD activates an inward current (IACPD) with a reversal potential close to that of a nonselective cationic current. Whole-cell perfusion of the calcium chelator BAPTA prevents activation ofIACPD. A-a, Membrane current and conductance changes evoked by 1S,3R-ACPD (200 μm, 2 min,VH = −60 mV) in control conditions recorded (in this and in Figs. 2, 3, 4, 5, 6, 7) in the presence of P-free ACSF containing 1 μm TTX, 10 μm bicuculline, 1 mmkynurenate, 50 μm D600, and K+ channel blockers (5–25 mm TEA, 5 mm 4-AP, and 6 mm CsCl) in patch-clamp whole-cell configuration using CsGlu-containing pipette solution. Hyperpolarizing voltage step of 10 mV (0.03 Hz) was applied constantly during the experiment.A-b, Membrane current and conductance changes evoked in the same cell by 1S,3R-ACPD (200 μm, 2 min, VH = −60 mV) 10 min after pipette–whole-cell perfusion with BAPTA-containing (20 mm) pipette solution. B, C, Individual current–voltage (I/V) relations (ramp command A; see Materials and Methods) obtained at the times indicated by numbers in A-a and A-bin the absence (B) and in the presence (C) of BAPTA. D,I/V relations obtained by subtracting the current traces obtained in the absence (B) and in the presence (C) of BAPTA. E, MeanI/V relation of 1S,3R-ACPD-induced current in the absence and in the presence of intracellular BAPTA (n = 5; paired data). In this and in all following figures, the error bars represent the SEM.
The dependence of IACPD on [Ca2+]i was studied first by recording CA1 pyramidal cells in the whole-cell configuration in the presence of Fluo-3 diluted in the CsGlu pipette solution (see Materials and Methods). Changes of [Ca2+]i were monitored in the soma of pyramidal neurons with a confocal laser scanning microscope simultaneously to the recording ofIACPD (performed in the presence of the superfusing medium A). As previously described in cultured hippocampal neurons and CA1 pyramidal cells (Mayer and Miller, 1990; Frenguelli et al., 1993; Jaffe and Brown, 1994; Petrozzino and Connor, 1994;Shirasaki et al., 1994), 1S,3R-ACPD induced a significant rise of [Ca2+]i of 49 ± 14% (VH = −60 mV; n = 4,p < 0.05) (Fig. 2). The increase of [Ca2+]i was correlated closely to theIACPD simultaneously recorded, and both displayed similar latency (47.5 ± 10.3 and 60 ± 10.8 sec, respectively; n = 4, p = 0.15) and time to peak (127.5 ± 16 and 136.3 ± 9 sec, respectively;n = 4, p = 0.37) (Fig. 2).
Fig. 2.
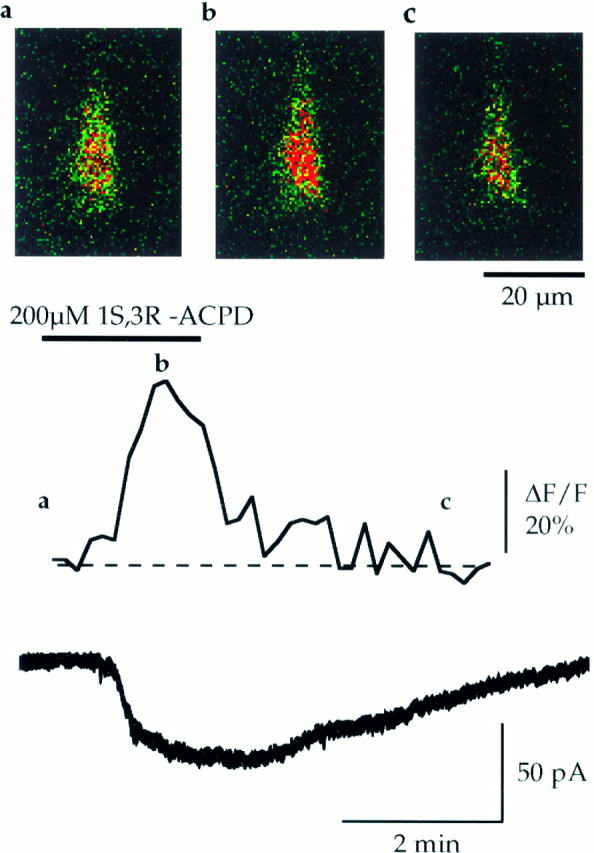
IACPD is associated with an increase in intracellular [Ca2+]. Changes in intracellular [Ca2+] induced by application of 1S,3R-ACPD were recorded from the soma of CA1 pyramidal neurons loaded in whole-cell configuration with the Ca2+-sensitive dye Fluo-3 (0.01 mmdiluted in the CsGlu pipette solution). Fluorescence images were acquired every 10 sec; [Ca2+]i was quantified and correlated to the simultaneously recorded CAN current. Top panels, Successive pseudocolored photomicrographs of the fluorescence collected before (a), during (b), and after (c) application of 1S,3R-ACPD (200 μm, 2 min,VH = −60 mV). Middle trace, Mean changes of the Ca2+-dependent fluorescence, observed in four CA1 pyramidal neurons, during the corresponding electrophysiologically recorded ICAN(bottom trace). Note that the rise in [Ca2+]i is strictly correlated to the rise phase of ICAN.
To show directly that IACPD depends on the rise in [Ca2+]i, we used pipette–whole-cell perfusion with the Ca2+chelating agent BAPTA (20 mm) diluted in the CsGlu pipette solution. Two series of preliminary experiments were performed to confirm the efficacy of the pipette–whole-cell perfusion system. The first consisted of three successive applications of 1S,3R-ACPD, one before and two after a perfusion with the same CsGlu pipette solution. We showed that the perfusion system by itself did not change IACPDsignificantly (see Fig. 3), which could be evoked by up to six consecutive applications of 1S,3R-ACPD (data not shown). The second experiment consisted of testing the effects of whole-cell perfusion of BAPTA on the slow afterhyperpolarization (sAHP) after a depolarizing pulse. As previously described with intracellular recordings (Crépel et al., 1994), 10 min after BAPTA perfusion spike frequency accommodation and sAHP disappeared, confirming adequate BAPTA loading (data not shown). In control conditions (before intracellular perfusion of BAPTA), 1S,3R-ACPD generated a reversible inward current (peak amplitude = −86.2 ± 9 pA; reversal potential = −17.6 ± 4.4; VH = −60 mV;n = 5) (Fig. 1A-a) associated with an increase in membrane conductance of 51.6 ± 13% (n = 5). After intracellular BAPTA perfusion (20 mm added to the CsGlu internal solution), subsequent applications of 1S,3R-ACPD failed to induce inward current (Fig. 1A-b); under this condition the 1S,3R-ACPD-mediated current was nearly abolished (the peak amplitude was reduced by 93.7 ± 2.9%;n = 5, p = 0.0004) and was not associated with a significant increase in membrane conductance (0.8 ± 2.6%; n = 5, p = 0.004). Analysis of the I/V relations (ramp command A) confirm that BAPTA perfusion completely suppressed the 1S,3R-ACPD-mediated current (Fig.1C–E). This showed that IACPD is Ca2+-dependent, as previously reported (Crépel et al., 1994).
Fig. 3.

Intracellular perfusion with control CsGlu pipette solution does not modify ICAN.A, Membrane currents (ΔI) and conductance changes (Δgm) evoked in the same cell by three successive applications of 1S,3R-ACPD (200 μm, 2 min, VH = −60 mV) in control (▪), after 15 min of cell dialysis with the control CsGlu pipette solution (▴), and after 30 min of cell dialysis with the control CsGlu pipette solution (•). Note that the successive applications of 1S,3R-ACPD induced three similar ICAN. B,I/V relations of the CAN currents induced, in the same cell, by three successive 1S,3R-ACPD applications and performed, respectively, before (▪), 15 (▴), and 30 min (•) after pipette–whole-cell perfusion with control CsGlu pipette solution. These I/V relations have been obtained by subtracting the current traces at the time indicated by thenumbers in A.
In conclusion, using whole-cell recordings, we have confirmed that, in the presence of K+ channel blockers, 1S,3R-ACPD induced a current with a reversal potential close to that of a nonselective cationic current, which was triggered by a rise in intracellular Ca2+concentration. This current will be referred to hereafter as a calcium-activated nonselective cationic current (ICAN), as per Crépel et al. (1994); its properties are not modified by whole-cell recordings.
ICAN is mediated via G-proteins
To clarify the transduction system leading to an increase in [Ca2+]i required for the activation of the CAN current, we used intracellular dialysis of the specific G-protein inhibitors GTPγS and GDPβS.
In the first set of experiments we investigated whether 1S,3R-ACPD still could generateICAN when G-proteins were blocked in an activated state. GTPγS (500 μm) was diluted in the GTP-free CsGlu pipette solution to replace GTP at equimolar concentration. In five of five cells, in the presence of GTPγS, the first application of 1S,3R-ACPD (200 μm, 2 min) evoked an irreversibleICAN (peak amplitude = −47 ± 5.5 pA; reversal potential = −19 ± 6.8 mV) (Fig.4A-a); the second application of 1S,3R-ACPD failed to evoke any subsequent current, as illustrated in Figure 4A-b(ICAN was reduced by 98.2 ± 1.8%;n = 5, p = 0.0001). The analysis of theI/V relations (ramp command A) confirm that GTPγS completely suppressed the second 1S,3R-ACPD-mediated current (Fig.4B).
Fig. 4.

In the presence of the nonhydrolyzable analog of GTP, GTPγS, 1S,3R-ACPD irreversibly activates ICAN, because intracellular perfusion with the G-protein inhibitor GDPβS prevents the activation of ICAN. A-a, Membrane current and conductance changes evoked by a first application of 1S,3R-ACPD (200 μm, 2 min,VH = −60 mV) after 20 min of cell dialysis with 500 μm GTPγS. Note thatICAN becomes irreversible in the presence of GTPγS. A-b, Membrane current and conductance changes evoked in the same cell by a second application of 1S,3R-ACPD (200 μm, 2 min,VH = −60 mV) after 40 min of cell dialysis with 500 μm GTPγS. Note that, in the presence of GTPγS, a second application of 1S,3R-ACPD fails to evoke a subsequent response. B, Mean I/V relations of the CAN currents obtained in the same cell by two successive 1S,3R-ACPD applications performed 20 min (•) and 40 min (○), respectively, after cell dialysis with 500 μm GTPγS (n = 5; paired data).C, Mean I/V relations of the CAN currents obtained by two successive 1S,3R-ACPD applications in the same cell before (•) and after (○) pipette–whole-cell perfusion with 500 μm GDPβS (n = 7; paired data).
In the second set of experiments we examined whether 1S,3R-ACPD could induceICAN when G-proteins were blocked in an inactivated state. In this set of experiments GDPβS was dialyzed into the cell via the pipette–whole-cell perfusion system; GDPβS (500 μm) was diluted in the GTP-free CsGlu pipette solution to replace GTP at equimolar concentration. In control conditions, before perfusion of GDPβS, a first application of 1S,3R-ACPD induced the expected fully reversibleICAN (peak amplitude = −79.4 ± 10.9 pA; reversal potential = −16.43 ± 1.9 mV; n= 7). In the presence of GDPβS, a subsequent application of 1S,3R-ACPD failed to induceICAN (Fig. 4C): the second 1S,3R-ACPD-mediated current was nearly abolished (the peak amplitude of ICAN was reduced by 94.7 ± 6.4%; n = 7, p = 0.0007). The analysis of the I/V relations (ramp command A) confirmed that GDPβS completely suppressed the second 1S,3R-ACPD-mediated current (Fig.4C).
Therefore, 1S,3R-ACPD activatesICAN via a G-protein-dependent process.
ICAN is mediated by group I mGluRs
Glutamatergic metabotropic receptors are divided into three groups. Group I (including mGluRs 1 and 5) activates PLC [i.e., the inositol triphosphate (IP3) production pathway]; group II (including mGluRs 2 and 3) and group III (including mGluRs 4, 6, 7, and 8) inhibit adenylyl cyclase (i.e., the cAMP production pathway) (for review, see Schoepp and Conn, 1993; Pin and Duvoisin, 1995). To specify the type of mGluRs involved in the activation ofICAN, we performed pharmacological experiments to test the ability of selective agonists for these three groups to induce ICAN. In each cell in which mGluRs agonists were tested, we subsequently applied 1S,3R-ACPD (200 μm, 2 min) to verify the ability of these cells to generateICAN; only cells that displayedICAN were kept for analysis. These data are summarized in Table 1.
Table 1.
Parameters of the CAN currents evoked in CA1 pyramidal neurons by different metabotropic receptor agonists

Shown are the effects of extracellular divalent cations in the 1S,3R-ACPD-induced CAN current.
Previous immunohistochemical studies have shown that CA1 pyramidal neurons prominently express mGluR5 (Abe et al., 1992; Shigemoto et al., 1993; Luján et al., 1996). Therefore, we first tested the most selective agonist of the group I mGluRs, the DHPG (Ito et al., 1992;Schoepp et al., 1994). With superfusion of medium A (see Materials and Methods), bath application of 200 μm DHPG evoked a current identical to that evoked by 1S,3R-ACPD. The currents evoked by DHPG and 1S,3R-ACPD had similar peak amplitudes, reversal potentials, and slope conductances; they both exhibited an area of reduced slope conductance at potentials more negative than −40 mV (see Fig. 5, Table 1). Second, we tested the ability of specific agonists of the group II (DCG IV; Ishida et al., 1993) and the group III (l-AP4; (Thomsen et al., 1992) mGluRs to induce ICAN. Under perfusion with medium A (see Materials and Methods), DCG IV (10 μm; n = 6) and l-AP4 (1 mm; n = 5) were bath-applied for 2–10 min at concentrations 10–50 times higher than their reported EC50 in hippocampal slices (Gereau and Conn, 1995). We found that DCG IV and l-AP4 failed to evoke any current (Table 1), whereas in the same cells 1S,3R-ACPD evoked ICAN (data not shown).
Fig. 5.

The group I mGluRs selective agonist DHPG (200 μm) evoked a CAN current identical to that evoked by 1S,3R-ACPD. A, Membrane current and conductance changes evoked by DHPG (200 μm, 2 min, VH = −60 mV). B, MeanI/V relations of DHPG-induced (○) and 1S,3R-ACPD-induced (•) currents (n = 5; paired data). Note that I/Vrelations are very similar (reversal potential, slope conductance; but see also Table 1).
Finally, we tested the ability of the competitive nonselective mGluR antagonist (S)-MCPG (Eaton et al., 1993) to antagonize the DHPG-evoked ICAN. (S)-MCPG (1 mm; n = 3) was bath-applied after recording a first DHPG-evokedICAN in a CA1 pyramidal neuron at least 15–30 min before a second application of DHPG (100–200 μm) on the same neuron. (S)-MCPG failed to induce a significant reduction of the DHPG-evokedICAN, which displayed similar peak amplitudes (−62.8 ± 13.6 and −68.6 ± 20.3 pA;n = 3, p = 0.3) and slope conductances (negative conductances: 0.2 ± 0.47 and 0.3 ± 0.7 nS;n = 3, p = 0.35; positive conductances: 2.4 ± 0.4 and 1.9 ± 0.65 nS; n = 3,p = 0.11), respectively, in the absence and in the presence of (S)-MCPG (data not shown).
These results strongly suggest that ICAN was evoked by metabotropic receptors positively linked to the IP3 production pathway (i.e., mGluRs of the group I) and not by metabotropic receptors negatively linked to the adenylyl cyclase pathway (i.e., mGluRs of the group II and III). If so, other types of metabotropic receptors linked to the IP3 production pathway (for review, see Schoepp and Conn, 1993) should activateICAN also. In keeping with this hypothesis and in agreement with previous data (Shen and North, 1992; Colino and Halliwell, 1993; Fraser and MacVicar, 1996), we found that activation of muscarinic receptors induced a current showing similar features to those of ICAN: under perfusion with medium A, bath application of 60–120 μm carbachol (a selective agonist of muscarinic receptors linked to IP3 production pathway) (Fisher et al., 1983, 1984; Dutar and Nicoll, 1988) generated a CAN current that was smaller but otherwise comparable toICAN induced by 1S,3R-ACPD (n = 9; Fig. 6C, Table 1). To demonstrate further that ICAN is not attributable to a change of intracellular cAMP content, we tested the ability of 1S,3R-ACPD to induceICAN in the presence of forskolin, an activator of adenylyl cyclase (which increases the intracellular concentration of cAMP). In this set of experiments 50 μm forskolin (diluted in medium A) was bath-applied before (10 min), during (2 min), and after (5 min) the application of 1S,3R-ACPD (2 min, 200 μm). First, we observed that forskolin itself did not generate any significant current (−0.21 ± 6 pA;n = 8); second, in the presence of forskolin, the current evoked by 1S,3R-ACPD was not significantly different from that evoked in the absence of forskolin (Fig. 6A): neither the peak amplitude (96.5 ± 27% of the control; n = 8, p = 0.15) nor the reversal potential (−8.5 ± 3.3 and −8.3 ± 3.7 mV, respectively, in the presence and in absence of forskolin;n = 8, p = 0.3) nor the I/Vrelation of ICAN was altered by forskolin (Fig.6B).
Fig. 6.

ICAN induced by 1S,3R-ACPD is not regulated by forskolin, an activator of adenylyl cyclase, and also can be induced by carbachol, a selective agonist of muscarinic receptors. A, Membrane current and conductance changes evoked in the same cell by two successive applications of 1S,3R-ACPD (200 μm, 2 min, VH = −60 mV) in the absence (a) and the presence (b) of forskolin (50 μm; added at least 10 min before the second application of 1S,3R-ACPD).B, Mean I/V relations of the forskolin-induced currents (▵) and of 1S,3R-ACPD-induced currents in control (•) and in the presence of forskolin (○) (n = 8; paired data). Note that forskolin itself did not induce any current and that the I/V relations of 1S,3R-ACPD-induced currents in the absence and in the presence of forskolin are very similar (reversal potential, slope conductance; but see also Table 1). C, Mean I/V relations of the carbachol-induced (○; n = 9) and 1S,3R-ACPD-induced currents (•;n = 55). Note that the carbachol- and 1S,3R-ACPD-induced CAN currents are qualitatively similar (same reversal potential and similarI/V relations, despite the smaller amplitude of the carbachol-induced current; but see also Table 1).
Therefore, in CA1 pyramidal cells the CAN current can be activated by group I mGluRs and muscarinic receptors, which are positively linked to PLC pathway. Our results also demonstrate that the transduction system involved in the activation of ICAN does not implicate a change of cAMP production.
ICAN is sensitive to external divalent cations
As shown in Figures 1, 2, 3, 4, 5, 6, the I/V relation ofICAN exhibited an area of reduced slope conductance at potentials more negative than −40 mV. We reasoned that this negative rectification could be attributable to a voltage-dependent block by external divalent cations, as has been demonstrated for NMDA channels (Ascher and Nowak, 1988; Mayer et al., 1989) or cyclic nucleotide-gated nonselective channels (for review, seeZufall et al., 1994).
We thus investigated the possible regulatory role of external Mg2+ on ICAN evoked by 1S,3R-ACPD in seven cells superfused with medium A containing the following external Mg2+concentrations: 0.1, 2, and 10 mm. Attempts to investigate effects of medium A containing zero external Mg2+were abandoned, because we failed to obtain stable long-lasting recordings with such superfusing solution (K+channel blockers, low Ca2+, and 0 Mg2+). The results of these experiments are illustrated in Figure 7A and summarized in Table 1. In the presence of 0.1 mm Mg2+,ICAN exhibited a larger peak amplitude than that previously observed with physiological Mg2+concentration; its negative and positive conductances were nearly identical, and consequently its I/V relation was linear. When the external Mg2+ concentration was raised from 0.1 to 2 mm, we observed a significant decrease of the peak amplitude of ICAN associated with a significant reduction of the negative conductance (−58.8 ± 3.7%;p = 0.0001). The positive conductance was unchanged, leading to a nonlinear I/V curve. The reversal potential ofICAN in 2 mm Mg2+was similar to that observed in 0.1 mmMg2+. When the external Mg2+concentration was raised from 2 to 6–10 mm, the peak amplitude and the negative conductance of ICANwere decreased further, but the positive conductance also was strongly reduced, and the reversal potential was shifted to a more positive value.
Fig. 7.

The divalent cation magnesium induces a voltage-dependent block, whereas divalent cations cadmium and zinc induce a voltage-independent block of CAN currents produced by 1S,3R-ACPD. A,Top, Mean I/V relations of CAN currents produced by 1S,3R-ACPD (200 μm, 2 min, VH = −60 mV) in the presence of three different magnesium concentrations: 0.1 mm (open circles), 2 mm(filled circles), and 10 mm(barred squares) (n = 7; paired data). Bottom, Mean conductances of theI/V relations obtained in A,top, illustrating the decrease of conductance induced at negative potentials by increasing the extracellular magnesium concentration from 0.1 (open column) to 2 mm(filled column) and the decrease of conductance induced at all potentials by increasing magnesium concentration from 2 mm (filled column) to 10 mm (barred column) (n = 7; paired data; see also Table 1). The 2 mm condition was compared with the 0.1 mm and then with the 10 mm condition. In this and the following figures, the statistical significance was assessed by paired t test analysis (*p < 0.05; **p < 0.01). B, Top, Mean I/Vrelations of CAN currents produced by two successive applications of 1S,3R-ACPD (200 μm, 2 min,VH = −60 mV) in the absence (filled circles) and in the presence (open circles) of cadmium in the superfusing medium (200 μm at least 10 min before the second application of 1S,3R-ACPD) (n = 14; paired data). Bottom, Mean conductances of theI/V relations obtained in B,top, illustrating the decrease of conductance induced at all potentials by cadmium (200 μm, n= 14; paired data; see also Table 1). C,Top, Mean I/V relations of CAN currents produced by two successive applications of 1S,3R-ACPD (200 μm, 2 min,VH = −60 mV) in the absence (filled circles) and in the presence (open circles) of zinc in the superfusing medium (200 μm) at least 10 min before the second application of 1S,3R-ACPD (n = 12; paired data). Bottom, Mean conductances of theI/V relations obtained in C,top, illustrating the decrease of conductance induced at all potentials by zinc (200 μm, n = 12; paired data; see also Table 1).
Thus, as for NMDA and cyclic nucleotide-gated currents, there is a voltage-dependent regulation of the CAN current by physiological external Mg2+ concentrations (between 1 and 2 mm) at negative membrane potentials. Higher external Mg2+ concentration also shifted the reversal potential to more positive values and reduced CAN current at positive membrane potentials.
As for NMDA currents and cyclic nucleotide-gated currents (Ascher and Nowak, 1988; Mayer et al., 1989; Zufall et al., 1994), other divalent cations such as Cd2+ and Zn2+also modulate ICAN. In the presence of 200 μm of external Cd2+ the peak amplitude of ICAN was reduced significantly, and its reversal potential shifted to more positive values; both negative and positive conductances were strongly reduced (n = 14; Fig. 7B, Table 1). Similarly, in the presence of 200 μm of external Zn2+ peak amplitude ofICAN was reduced, and its reversal potential shifted to more positive values. As with Cd2+ and high concentrations of Mg2+, both negative and positive conductances were strongly reduced by Zn2+(n = 12; Fig. 7C, Table1).
In conclusion, external divalent cations regulate the CAN current by reducing its slope conductance. Their effects can be classified in two groups: the voltage-dependent blockers, such as Mg2+(at physiological concentrations), and the non- (or weak) voltage-dependent blockers, such as Cd2+ and Zn2+ (or high concentrations of Mg2+).
ICAN can be evoked synaptically
We determined the conditions required for synaptic activation ofICAN to elucidate its possible role in synaptic transmission. In these experiments the CA1 region was isolated surgically from CA3. Synaptic responses were evoked by a bipolar stimulating electrode placed in the stratum radiatum or at the border between stratum radiatum and stratum lacunosum moleculare and recorded in the presence of K+ channel blockers. Low-frequency stimulations (0.05 Hz) evoked postsynaptic responses (Fig. 8A), which were abolished completely by bicuculline, CNQX, and dl-APV (medium B; see Materials and Methods) (Fig. 8A-b,A-c). However, HFS (25–100 Hz, 1 sec) evoked a slow inward current that was maximal at the frequency of 100 Hz (as illustrated in Fig.8B-a). The current evoked by a 100 Hz HFS had a peak amplitude of −42.9 ± 3.5 pA (VH = −60 mV; n = 36) and a total duration of 11 ± 1.3 sec (n = 36). The amplitude of this postsynaptic current was dependent on the frequency of stimulation (Fig.8B-a), the stimulus intensity (Fig.8B-c), and the duration of the HFS (Fig.8B-b), suggesting its dependence on the level of transmitter release. Furthermore, this current was blocked completely by bath application of TTX (1 μm), confirming its synaptic origin (n = 5; Fig. 9A-a,A-c). The voltage dependence of synaptically evoked currents was studied using ramp voltage commands applied at the peak amplitude of the evoked response. To avoid the voltage-dependent activation of calcium channels, we limited the ramp voltage commands to negative potentials (−20 mV to −100 mV, ramp B; see Materials and Methods). The recorded synaptically evoked current showed a reversal potential of −22.8 ± 8.2 mV (close to the reversal potential ofIACPD) and a conductance of 0.7 ± 0.11 nS (calculated between −100 and −60 mV; n = 13) (Fig. 9C). Thus HFS evoked a slow inward postsynaptic current resistant to ionotropic glutamate and GABA receptor antagonists.
Fig. 8.

High-frequency stimulation (HFS) evoked a slow inward postsynaptic current resistant to ionotropic glutamate (and GABAA) receptor antagonists. A, Postsynaptic response evoked in a CA1 pyramidal neuron (VH = −60 mV) by a single shock stimulation of 1.5 and 3 mA (60 μsec) in control conditions (A-a, 1.5 mA) and in the presence of CNQX (40 μm),dl-APV (200 μm), and bicuculline (20 μm) containing superfusion medium B (see Materials and Methods) (A-b, 1.5 mA; A-c, 3 mA). Note that the response to a single shock stimulation is blocked by the ionotropic glutamate and GABAA antagonists.B, Slow inward postsynaptic currents evoked as a function of HFS parameters in the presence of superfusion medium B (as in A-b, A-c; see Materials and Methods).B-a, Slow inward postsynaptic currents evoked by HFS (1 sec, 3 mA) with frequencies of 20, 50, and 100 Hz.Inset, Illustration of the onset of the response to tetanic stimulation at an expanded time scale. B-b, Slow inward postsynaptic currents evoked by HFS (100 Hz, 3 mA) with duration of 100, 500, and 1000 msec. B-c, Slow inward postsynaptic currents evoked by HFS (1 sec, 100 Hz) with stimulus intensities of 2, 3, and 4 mA. Note that the response to a single shock stimulation, even at high intensity (3 mA, 60 μsec), is blocked by ionotropic glutamate and GABAA antagonists, whereas in the same conditions the same stimulus delivered at high frequency (50–100 Hz, 1 sec) evokes a slow inward postsynaptic current.
Fig. 9.

HFS stimuli evoked a nonionotropic glutamate-mediated excitatory postsynaptic current. A, Slow inward postsynaptic currents evoked in the same cell by four successive HFS (1 sec, 100 Hz, 3 mA) and recorded in medium B (see Materials and Methods): in control conditions (a), in the presence of the muscarinic receptor antagonist atropine (b) (10 μm) at least 10 min before the second HFS, in the presence of tetrodotoxin (c) (TTX, 1 μm) after a 10 min wash of atropine and at least 10 min before the third HFS, and back in control conditions after a 30 min wash of TTX (d) (n = 5; paired data). Note that atropine did not change the peak amplitude or the duration of the response, which was blocked completely by TTX and recovered with the wash ofTTX. B, Slow inward postsynaptic currents evoked in the same cell by two successive HFS (1 sec, 100 Hz, 3 mA): in the absence (a) and in the presence of the glutamate uptake inhibitor dihydrokainic acid (b) (DHK, 250 μm) added at least 15 min before the second HFS. Note that DHK significantly increased the duration of the HFS-evoked synaptic current. C, MeanI/V relations of the slow inward postsynaptic currents evoked by HFS (ramp command B; see Materials and Methods) (n = 13). Note that this HFS-induced current and the 1S,3R-ACPD-induced CAN current (see Fig. 1E) have qualitatively similar meanI/V relations (same reversal potential and similar shape, despite the smaller amplitude of the HFS-induced current).
Previous studies have described slow excitatory synaptic responses mediated by muscarinic receptors in CA1 and activated by HFS (Cole and Nicoll, 1984; Madison et al., 1987). Therefore, we tested the ability of the muscarinic receptor antagonist atropine to block the HFS-evoked response. In five of five cells, atropine (up to 20 μm) changed neither the peak amplitude (−2.2 ± 2.2%; paired data;p = 0.2) nor the duration (−8.2 ± 3.4%; paired data; p = 0.09) of the response (Fig.9A-a,A-b).
To study the possible activation of the synaptically evoked inward current by nonionotropic glutamate receptors, we tested its sensitivity to the glutamate uptake inhibitor DHK (Watkins and Evans, 1981). As shown in Figure 9B, bath application of DHK (250 to 500 μm for 15 min) significantly increased the duration of the HFS-evoked synaptic current (by 120 ± 18.5%;n = 5, p = 0.0002; paired data). DHK also slightly increased the peak amplitude of the current in two of the five cells. Thus, in CA1 pyramidal cells HFS evoked a nonionotropic glutamate-mediated excitatory postsynaptic current.
As shown for the 1S,3R-ACPD-evoked CAN current, the synaptically evoked current also involves Ca2+- and G-protein-dependent processes. In the presence of BAPTA, GDPβS, or GTPγS added to the pipette solution, the first responses evoked by the HFS within the 5 min after the passage to whole-cell configuration were similar to the responses evoked in the absence of BAPTA, GDPβS (Fig. 10A-a, B-a, respectively), or GTPγS (data not shown). After 20 min of cell dialysis with BAPTA (20 mm; n = 5; Fig.10A-b) or GDPβS (500 μm;n = 7; Fig. 10B-b), a second HFS failed to evoked a response. With GTPγS, after 20 min of cell dialysis the first response evoked by HFS was significantly longer in four of six cells (29 ± 3.1 vs 11 ± 1.3 sec in control,p = 0.001) and irreversible in the two other cells (Fig. 10C-a); subsequent HFS-evoked responses were nearly abolished (Fig. 10C-b).
Fig. 10.

HFS-generated nonionotropic slow inward currents are triggered by a rise in [Ca2+]i and mediated by G-proteins. A, Slow inward postsynaptic currents evoked in the same cell by two successive HFS (1 sec, 100 Hz, 3 mA) in the presence of the calcium chelator BAPTA (20 mm) in the CsGlu pipette solution within the 5 min after the passage to whole cell (a) and after 20 min of cell dialysis (b) (n = 5; paired data).B, Slow inward postsynaptic currents evoked in the same cell by two successive HFS (1 sec, 100 Hz, 3 mA) in the presence of the G-protein inhibitor GDPβS (500 μm) in the CsGlu pipette solution within the 5 min after the passage to whole cell (a) and after 20 min of cell dialysis (b) (n = 7; paired data). Note that in the presence of BAPTA or GDPβS a second HFS failed to evoked any subsequent response.C, Slow inward postsynaptic currents evoked in the same cell by two successive HFS (1 sec, 100 Hz, 3 mA) in the presence of the nonhydrolyzable analog of GTP, GTPγS (500 μm), in the CsGlu pipette solution after 20 (a) and 40 min (b) of cell dialysis (n = 6; paired data). Note that in the presence of GTPγS the first response evoked by HFS is significantly longer (even irreversible) than in control (see Figs. 6B, 7A-a,B-a) and that subsequent HFS-evoked responses are nearly abolished (b).
Therefore HFS generates a mGluRs-dependent slow inward current that is triggered by a rise in [Ca2+]i, mediated by G-proteins. This current exhibit properties of a synapticICAN.
DISCUSSION
The present report describes a slow nonselective cationic current (ICAN) triggered by a [Ca2+]i rise, generated by glutamate acting on group I mGluRs via a G-protein-dependent process. It provides the first characterization in the mammalian CNS of a synapticICAN generated by HFS. The presence of a mGluR-mediated ICAN in the CNS has several important physiological implications.
Properties of ICAN evoked by 1S,3R-ACPD in CA1 pyramidal neurons
In the CNS several studies have described an inward current associated with an increase in membrane conductance, mediated by exogenous activation of glutamate metabotropic receptors, in the presence of K+ channel blockers (Glaum and Miller, 1992; Staub et al., 1992; Mercuri et al., 1993). A nonselective cationic current induced by 1S,3R-ACPD was, however, first described in CA1 pyramidal neurons of adult rat hippocampal slices by Crépel et al. (1994). In that work the authors characterized a Ca2+-dependent, weakly temperature-sensitive, nonselective cationic current corresponding to the CAN cationic current defined in invertebrate preparations (Swandulla and Partridge, 1990). The 1S,3R-ACPD-induced current described in the present study using whole-cell patch-clamp recordings has similar features to those currents described above, namely in terms of nonselectivity for monovalent cations, reversal potential, and dependence on [Ca2+]i.
CAN channels have been found principally to be permeable to Na+ and K+ (for review, seeSwandulla and Partridge, 1990) and occasionally to Cs+ ions (Yellen, 1982; Lipton, 1986; Simmoneau et al., 1987; Teulon et al., 1987). In our experimental conditions we have observed that the reversal potential of the CAN current was more negative than that expected from the Nernst equation. We suspect that it may be attributable to a better permeability of CAN channels for Cs+, which will tend to displace the reversal potential to more negative values. Using the Goldman–Hodgkin–Katz equation (Goldman, 1943; Hodgkin and Katz, 1949), we estimated, under our experimental conditions, a permeability ratioPCs+/PNa+= 3.
Additionally, hippocampal ICAN is modulated by the external divalent cations Mg2+, Cd2+, and Zn2+ via two pathways: a voltage-dependent block (Mg2+) and a voltage-independent one (Cd2+, Zn2+). These properties, and in particular the voltage-dependent block by external Mg2+, are reminiscent of other types of cationic channels, including NMDA channels (Ascher and Nowak, 1988; Mayer et al., 1989), cyclic nucleotide-gated channels (for review, see Zufall et al., 1994), and a nonselective cationic conductance recently described in CA3 pyramidal cells (Caeser et al., 1993; Guérineau et al., 1995). The voltage-dependent modulation of ICAN suggests that CAN channels are regulated directly by external Mg2+. It is not clear, however, whether this property will enable mGluRs to exert a coincident detection property that typically is associated with NMDARs. Future studies will have to determine the consequences of this feature.
ICAN is generated via a G-protein-dependent process by group I mGluRs
Previous studies have shown clearly that mGluRs are linked to phospholipase C and adenylyl cyclase via G-proteins (for review, seeSchoepp and Conn, 1993; Pin and Duvoisin, 1995). As expected, we found that activation of ICAN by 1S,3R-ACPD involved a G-protein-dependent process. In the presence of GTPγS, which blocks the G-protein in its activated state, 1S,3R-ACPD irreversibly activated ICAN, whereas in the presence of GDPβS, which blocks the G-protein in its inactivated state, 1S,3R-ACPD did not evoke any current. These two complementary results clearly demonstrate the role of a G-protein-dependent process in the 1S,3R-ACPD-induced ICANand further confirm that this current is activated by mGluRs via a metabotropic pathway (Sladeczek et al., 1985; Sugiyama et al., 1987).
Previous immunohistological studies have shown that group I mGluRs are expressed prominently in the CA1 region of hippocampus (Abe et al., 1992; Shigemoto et al., 1993; Luján et al., 1996), whereas group II mGluRs are not (Ohishi et al., 1994). In keeping with this (also seeGereau and Conn, 1995), in CA1 pyramidal neuronsICAN is mediated by group I mGluRs, which are known to be positively linked to the IP3 production pathway and do not implicate a change of adenylyl cyclase activity, because (1) DHPG, a selective agonist of group I mGluRs (mGluR 1 and 5) (Ito et al., 1992; Schoepp et al., 1994), generates a current identical to that evoked by 1S,3R-ACPD; (2) DCG IV andl-AP4, selective agonists of group II and III mGluRs, respectively, which are both negatively linked to the adenylyl cyclase pathway (Tanabe et al., 1992, 1993), do not evokeICAN; and (3) forskolin, an activator of adenylyl cyclase, does not modify ICAN.
Because mGluR5s are expressed prominently in CA1 pyramidal neurons (Abe et al., 1992; Shigemoto et al., 1993; Luján et al., 1996), whereas mGluR1 subtypes are localized in CA1 interneurons (Martin et al., 1992; Baude et al., 1993; Luján et al., 1996), the CAN current described in the present report probably is mediated by mGluR5. In addition, the absence of effect of MCPG provides further evidence that ICAN involved the activation of mGluR5 and not mGluR1, because MCPG antagonizes mGluR1-mediated, but not mGluR5-mediated, responses (Joly et al., 1995) and has no effect on CA1 pyramidal neurons (Chinestra et al., 1993; Izumi and Zorumski, 1994;Manzoni et al., 1994; Selig et al., 1995) (but see Bashir et al., 1993;Brown et al., 1994). Direct evidence in support of this must await the development of a selective mGluR5 antagonist.
Our pharmacological study shows that ICAN is evoked by group I mGluRs, which are known to be positively linked to the IP3 production pathway. We then suggest thatICAN can be activated by other receptors linked to the same messenger pathway. In keeping with this and in agreement with previous data (Shen and North, 1992; Colino and Halliwell, 1993;Fraser and MacVicar, 1996), we found that activation of muscarinic receptors, linked to the IP3 production pathway in the CA1 area (Fisher et al., 1983; Dutar and Nicoll, 1988), also activates a current with the same feature as ICAN.
Properties and functional significance of a synapticICAN generated by mGluRs
Several lines of evidence show that the current activated synaptically by a brief HFS of afferent fibers in the stratum lacunosum moleculare–radiatum area was similar to theICAN evoked by exogenous application of 1S,3R-ACPD. HFS induced an excitatory postsynaptic response composed of a fast ionotropic component and a slow metabotropic component that persist in the presence of ionotropic glutamate and GABAA receptor antagonists. This slow inward current increases in amplitude and duration in the presence of glutamate uptake inhibitor and is not blocked by muscarinic receptor antagonists. In addition, it is blocked by the Ca2+chelating agent BAPTA and by G-protein inhibitors (GTPγS and GDPβS), and it displayed a reversal potential similar to that of the 1S,3R-ACPD-activated ICAN. Taken together, these data strongly suggest that this slow synaptic inward current is generated, as IACPD, through mGluRs via G-protein- and Ca2+-dependent activation of an excitatory postsynaptic nonselective cationic currentICAN. Although synaptic currents evoked in the presence of ionotropic glutamate receptor antagonists have been reported in earlier studies (Charpak and Gähwiler, 1991; Glaum and Miller, 1992; Batchelor et al., 1994; Pozzo Miller et al., 1995;Batchelor and Garthwaite, 1997), to the best of our knowledge the present report is the first description of a synaptically triggered CAN current in mammalian CNS neurons. This synapticICAN exhibited a slow kinetic and may mediate a long-lasting excitation outlasting the action of most voltage-dependent ionic currents (as NMDA, for instance). This property is in keeping with the specific slow kinetics of the CAN single channel observed in several invertebrate preparations (for review, see Swandulla and Partridge, 1990). In the present study the synapticICAN required relatively strong HFS to be generated. This is likely the consequence of the perisynaptic location of mGluR5 in CA1 pyramidal neurons (Baude et al., 1993; Luján et al., 1996). An accumulation of glutamate in the synaptic cleft and a “spillover” may be required to reach the perisynaptic receptors and trigger a mGluR-mediated response. Therefore, at least in CA1 pyramidal neurons ICAN probably does not participate in ongoing spontaneous activity; a different situation may prevail in interneurons (see Miles and Poncer, 1993).
The demonstration of a synaptically activated CAN current mediated by mGluRs adds a novel facet to the pleiotropic repertoire of modulations exerted by glutamate metabotropic receptors in the CNS. In the hippocampus, activation of cationic currents through metabotropic receptors generates long-lasting plateau potentials (Fraser and MacVicar, 1996) and provides the basis for synchronized activities (Yaari and Jensen, 1989; Miles and Poncer, 1993; Bianchi and Wong, 1995). The synaptically activated ICAN described in the present report may play an important role in synaptic integration, notably as frequency sensors, in regard to its strong frequency dependence. Interestingly, Batchelor and Garthwaite (1997)recently have described, in the cerebellum, an inward current generated through mGluRs that integrates temporally dispersed signals from two different inputs. It will be important to establish whether this synaptic response is mediated by a CAN current.
We suggest that CAN currents activated in the hippocampus, presumably as a consequence of an important accumulation of glutamate, may contribute not only to the normal physiological functions, such as the neuronal plasticity, but also to the long-lasting excitotoxic depolarizations observed in pathological conditions, such as epilepsy or ischemia.
Footnotes
Financial support from Institut National de la Santé et de la Recherche Médicale, Centre National pour la Recherche Scientifique, and Direction des Recherches, Etudes et Techniques, is acknowledged. We are grateful to Dr. K. Schimamoto for her generous gift of (2S,1′R,2′R,3′R)-2-(2,3-dicarboxycyclopropyl) glycine and to Drs. H. McLean, C. Hammond, S. Williams, and D. D. Fraser for their helpful comments and discussions.
Correspondence should be addressed to Dr. Patrice Congar, Institut National de la Santé et de la Recherche Médicale Unité 29, 123 Boulevard de Port Royal, 75674 Paris Cedex 14, France.
REFERENCES
- 1.Abe T, Sugihara H, Nawa H, Shigemoto R, Mizuno N, Nakanishi S. Molecular characterization of a novel metabotropic glutamate receptor mGluR5 coupled to inositol phosphate/Ca2+ signal transduction. J Biol Chem. 1992;19:13361–13368. [PubMed] [Google Scholar]
- 2.Ascher P, Nowak L. The role of divalent cations in the N-methyl-d-aspartate responses of mouse central neurones in culture. J Physiol (Lond) 1988;399:247–266. doi: 10.1113/jphysiol.1988.sp017078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bashir ZI, Bortolotto ZA, Davies CH, Beretta N, Irving AJ, Seal AJ, Henley JM, Jane DE, Watkins JC, Collingridge GL. Induction of LTP in the hippocampus needs synaptic activation of glutamate receptors. Nature. 1993;363:347–350. doi: 10.1038/363347a0. [DOI] [PubMed] [Google Scholar]
- 4.Batchelor AM, Garthwaite J. Frequency detection and temporally dispersed synaptic signal association through a metabotropic glutamate receptor pathway. Nature. 1997;385:74–77. doi: 10.1038/385074a0. [DOI] [PubMed] [Google Scholar]
- 5.Batchelor AM, Madge DJ, Garthwaite J. Synaptic activation of metabotropic glutamate receptors in the parallel fibre–Purkinje cell pathway in rat cerebellar slices. Neuroscience. 1994;63:911–915. doi: 10.1016/0306-4522(94)90558-4. [DOI] [PubMed] [Google Scholar]
- 6.Baude A, Nusser Z, Roberts JDB, Mulvihill E, McIlhinney RAJ, Somogyi P. The metabotropic glutamate receptor (mGluR1α) is concentrated at perisynaptic membrane of neuronal subpopulations as detected by immunogold reaction. Neuron. 1993;11:771–787. doi: 10.1016/0896-6273(93)90086-7. [DOI] [PubMed] [Google Scholar]
- 7.Ben-Ari Y, Aniksztejn L. Role of glutamate metabotropic receptors in long-term potentiation in the hippocampus. Semin Neurosci. 1995;7:127–135. [Google Scholar]
- 8.Bianchi R, Wong RKS. Excitatory synaptic potentials dependent on metabotropic glutamate receptor activation in guinea-pig hippocampal pyramidal cells. J Physiol (Lond) 1995;487:663–676. doi: 10.1113/jphysiol.1995.sp020908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brown RE, Rabe H, Reymann KG. (R-S)-α-Methyl-4-carboxy-phenylglycine (MCPG) does not block theta burst-induced long-term potentiation in area CA1 of rat hippocampal slices. Neurosci Lett. 1994;170:17–21. doi: 10.1016/0304-3940(94)90228-3. [DOI] [PubMed] [Google Scholar]
- 10.Caeser M, Brown DA, Gähwiler BH, Knöpfel T. Characterization of a calcium-dependent current generating a slow afterdepolarization of CA3 pyramidal cells in rat hippocampal slices cultures. Eur J Neurosci. 1993;5:560–569. doi: 10.1111/j.1460-9568.1993.tb00521.x. [DOI] [PubMed] [Google Scholar]
- 11.Charpak S, Gähwiler BH. Glutamate mediates a slow synaptic response in hippocampal slice cultures. Proc R Soc Lond [Biol] 1991;243:221–226. doi: 10.1098/rspb.1991.0035. [DOI] [PubMed] [Google Scholar]
- 12.Cherubini E, Ben-Ari Y, Gho M. Long-term potentiation of synaptic transmission in the hippocampus induced by a bee venom peptide. Nature. 1987;328:70–73. doi: 10.1038/328070a0. [DOI] [PubMed] [Google Scholar]
- 13.Chinestra P, Aniksztejn L, Diabira D, Ben-Ari Y. (R-S)-α-Methyl-4-carboxyphenylglycine neither prevents induction of LTP nor antagonizes metabotropic glutamate receptors in CA1 hippocampal neurons. J Neurophysiol. 1993;70:2684–2689. doi: 10.1152/jn.1993.70.6.2684. [DOI] [PubMed] [Google Scholar]
- 14.Cole AE, Nicoll RA. Characterization of a slow cholinergic postsynaptic potential recorded in vitro from rat hippocampal pyramidal cells. J Physiol (Lond) 1984;352:173–188. doi: 10.1113/jphysiol.1984.sp015285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Colino A, Halliwell JV. Carbachol potentiates Q current and activates a calcium-dependent nonspecific conductance in rat hippocampus in vitro. Eur J Neurosci. 1993;5:1198–1209. doi: 10.1111/j.1460-9568.1993.tb00974.x. [DOI] [PubMed] [Google Scholar]
- 16.Crépel V, Aniksztejn L, Ben-Ari Y, Hammond C. Glutamate metabotropic receptors increase a Ca2+-activated nonspecific cationic current in CA1 hippocampal neurons. J Neurophysiol. 1994;72:1561–1569. doi: 10.1152/jn.1994.72.4.1561. [DOI] [PubMed] [Google Scholar]
- 17.Dutar P, Nicoll RA. Classification of muscarinic responses in hippocampus in terms of receptors subtypes and second messenger systems: electrophysiological studies in vitro. J Neurosci. 1988;8:4214–4224. doi: 10.1523/JNEUROSCI.08-11-04214.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Eaton SA, Jane DE, Jones PLSJ, Porter RHP, Pook PC-K, Sunter DC, Udvarhelyi PM, Roberts PJ, Salt TE, Watkins JC. Competitive antagonism at metabotropic glutamate receptors by (S)-4-carboxy-phenylglycine and (R-S)-α-methyl-4-carboxyphenylglycine. Eur J Pharmacol. 1993;244:195–197. doi: 10.1016/0922-4106(93)90028-8. [DOI] [PubMed] [Google Scholar]
- 19.Fisher SK, Klinger PD, Agranoff BW. Muscarinic agonist binding and phospholipid turnover in brain. J Biol Chem. 1983;258:7358–7363. [PubMed] [Google Scholar]
- 20.Fisher SK, Figueiredo JC, Bartus RT. Differential stimulation of inositol phospholipid turnover in brain by analogs of oxotremorine. J Neurochem. 1984;43:1171–1179. doi: 10.1111/j.1471-4159.1984.tb12858.x. [DOI] [PubMed] [Google Scholar]
- 21.Fraser DD, MacVicar BA. Cholinergic-dependent plateau potential in hippocampal CA1 pyramidal neurons. J Neurosci. 1996;16:4113–4128. doi: 10.1523/JNEUROSCI.16-13-04113.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Frenguelli BG, Potier B, Slater NT, Alford S, Collingridge GL. Metabotropic glutamate receptors and calcium signaling in dendrites of hippocampal CA1 neurones. Neuropharmacology. 1993;32:1229–1237. doi: 10.1016/0028-3908(93)90017-w. [DOI] [PubMed] [Google Scholar]
- 23.Gallagher JP, Zheng F, Shinnick-Gallhager P. Long-lasting modulation of synaptic transmission by metabotropic glutamate receptors. In: Conn PJ, Patel J, editors. The metabotropic glutamate receptors. Humana; Totowa, NJ: 1994. pp. 173–193. [Google Scholar]
- 24.Gereau RW, Conn PJ. Role of specific metabotropic glutamate receptor subtypes in regulation of hippocampal CA1 pyramidal cell excitability. J Neurophysiol. 1995;74:122–129. doi: 10.1152/jn.1995.74.1.122. [DOI] [PubMed] [Google Scholar]
- 25.Glaum SR, Miller RJ. Metabotropic glutamate receptors mediate excitatory transmission in the nucleus of the solitary tract. J Neurosci. 1992;12:2251–2258. doi: 10.1523/JNEUROSCI.12-06-02251.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Goldman DE. Potential, impedance, and rectification in membranes. J Gen Physiol. 1943;27:37–60. doi: 10.1085/jgp.27.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guérineau NC, Bossu JL, Gähwiler BH, Gerber U. Activation of a nonselective cationic conductance by metabotropic glutamatergic and muscarinic agonists in CA3 pyramidal neurons of the rat hippocampus. J Neurosci. 1995;15:4395–4407. doi: 10.1523/JNEUROSCI.15-06-04395.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hodgkin AL, Katz B. The effect of sodium ions on the electrical activity of the giant axon of the squid. J Physiol (Lond) 1949;108:37–77. doi: 10.1113/jphysiol.1949.sp004310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ishida M, Saitoh T, Shimamoto K, Ohfune Y, Shinozaki H. A novel metabotropic glutamate receptor agonist: marked depression of monosynaptic excitation in the newborn rat isolated spinal cord. Br J Pharmacol. 1993;109:1169–1177. doi: 10.1111/j.1476-5381.1993.tb13745.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ito I, Kohda A, Tanabe S, Hirose E, Hayashi M, Mitsunaga S, Sugiyama H. 3,5-Dihydrophenylglycine: a potent agonist of metabotropic glutamate receptors. NeuroReport. 1992;3:1013–1016. [PubMed] [Google Scholar]
- 31.Izumi Y, Zorumski CF. Developmental changes in the effects of metabotropic glutamate receptor antagonists on CA1 long-term potentiation in rat hippocampal slices. Neurosci Lett. 1994;176:89–92. doi: 10.1016/0304-3940(94)90878-8. [DOI] [PubMed] [Google Scholar]
- 32.Jaffe DB, Brown TH. Metabotropic glutamate receptor activation induces calcium waves within hippocampal dendrites. J Neurophysiol. 1994;72:471–474. doi: 10.1152/jn.1994.72.1.471. [DOI] [PubMed] [Google Scholar]
- 33.Joly C, Gomeza J, Brabet I, Curry K, Bockaert J, Pin J-P. Molecular, functional, and pharmacological characterization of the metabotropic glutamate receptor type 5 splice variants: comparison with mGluR1. J Neurosci. 1995;15:3970–3981. doi: 10.1523/JNEUROSCI.15-05-03970.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kao JPY, Harootunian AT, Tsien RY. Photochemically generated cytosolic calcium pulses and their detection by Fluo-3. J Biol Chem. 1989;264:8179–8184. [PubMed] [Google Scholar]
- 35.Leinekugel X, Tseeb V, Ben-Ari Y, Bregestovski P. Synaptic GABAA activation induces Ca2+ rise in pyramidal cells and interneurons from rat neonatal hippocampal slices. J Physiol (Lond) 1995;487:319–329. doi: 10.1113/jphysiol.1995.sp020882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lipton SA. Antibody activates cationic channels via second messenger Ca2+. Biochim Biophys Acta. 1986;856:59–67. doi: 10.1016/0005-2736(86)90010-6. [DOI] [PubMed] [Google Scholar]
- 37.Luján R, Nusser Z, Roberts JDB, Shigemoto R, Somogyi P. Perisynaptic location of metabotropic glutamate receptors mGluR1 and mGluR5 on dendrites and dendritic spines in the rat hippocampus. Eur J Neurosci. 1996;8:1488–1500. doi: 10.1111/j.1460-9568.1996.tb01611.x. [DOI] [PubMed] [Google Scholar]
- 38.Madison DV, Lancaster B, Nicoll RA. Voltage-clamp analysis of cholinergic action in the hippocampus. J Neurosci. 1987;7:733–741. doi: 10.1523/JNEUROSCI.07-03-00733.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Manzoni OJ, Weisskop MG, Nicoll RA. MCPG antagonizes metabotropic glutamate receptors but not long-term potentiation in the hippocampus. Eur J Neurosci. 1994;6:1050–1054. doi: 10.1111/j.1460-9568.1994.tb00599.x. [DOI] [PubMed] [Google Scholar]
- 40.Martin LJ, Blackstone CD, Huganir RL, Price DL. Cellular localization of a metabotropic glutamate receptor in rat brain. Neuron. 1992;9:259–270. doi: 10.1016/0896-6273(92)90165-a. [DOI] [PubMed] [Google Scholar]
- 41.Mayer ML, Miller R. Excitatory amino acid receptors, second messengers, and regulation of intracellular calcium in mammalian neurones. Trends Pharmacol Sci. 1990;11:254–259. doi: 10.1016/0165-6147(90)90254-6. [DOI] [PubMed] [Google Scholar]
- 42.Mayer ML, Vyklicky L, Jr, Westbrook GL. Modulation of excitatory amino acid receptors by group IIB metal cations in cultured mouse hippocampal neurons. J Physiol (Lond) 1989;415:329–350. doi: 10.1113/jphysiol.1989.sp017724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mercuri NB, Stratta F, Calabresi P, Bernardi G. Activation of metabotropic glutamate receptors induces an inward current in rat dopamine mesencephalic neurons. Neuroscience. 1993;56:399–407. doi: 10.1016/0306-4522(93)90341-c. [DOI] [PubMed] [Google Scholar]
- 44.Miles R, Poncer JC. Metabotropic glutamate receptors mediate a post-tetanic excitation of guinea-pig hippocampal inhibitory neurones. J Physiol (Lond) 1993;463:461–473. doi: 10.1113/jphysiol.1993.sp019605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ohishi H, Ogawa-Meguro R, Shigemoto R, Kaneko T, Nakanishi S, Mizuno N. Immunohistochemical localization of metabotropic glutamate receptors, mGluR2 and mGluR3, in rat cerebellar cortex. Neuron. 1994;13:55–66. doi: 10.1016/0896-6273(94)90459-6. [DOI] [PubMed] [Google Scholar]
- 46.Petrozzino JJ, Connor JA. Dendritic Ca2+ accumulations and metabotropic glutamate receptor activation associated with an N-methyl-d-aspartate receptor-independent long-term potentiation in hippocampal CA1 neurons. Hippocampus. 1994;4:546–558. doi: 10.1002/hipo.450040504. [DOI] [PubMed] [Google Scholar]
- 47.Pin JP, Duvoisin R. Review: neurotransmitter receptors. I. The metabotropic glutamate receptors: structure and functions. Neuropharmacology. 1995;34:1–26. doi: 10.1016/0028-3908(94)00129-g. [DOI] [PubMed] [Google Scholar]
- 48.Pozzo Miller LD, Petrozzino JJ, Connor JA. G-protein-coupled receptors mediate a fast excitatory postsynaptic current in CA3 pyramidal neurons in hippocampal slices. J Neurosci. 1995;15:8320–8330. doi: 10.1523/JNEUROSCI.15-12-08320.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schoepp DD, Conn PJ. Metabotropic glutamate receptors in brain function and pathology. Trends Pharmacol Sci. 1993;14:13–20. doi: 10.1016/0165-6147(93)90107-u. [DOI] [PubMed] [Google Scholar]
- 50.Schoepp DD, Goldsworthy J, Johnson BG, Salhoff CR, Baker SR. 3,5-Dihydrophenylglycine is a highly selective agonist for phospho-inositide-linked metabotropic glutamate receptors in the rat hippocampus. J Neurochem. 1994;63:769–772. doi: 10.1046/j.1471-4159.1994.63020769.x. [DOI] [PubMed] [Google Scholar]
- 51.Segal M, Barker JL. Rat hippocampal neurons in culture: potassium conductances. J Neurophysiol. 1984;51:1409–1433. doi: 10.1152/jn.1984.51.6.1409. [DOI] [PubMed] [Google Scholar]
- 52.Selig DK, Lee H-K, Bear MF, Malenka RC. Reexamination of the effects of MCPG on hippocampal LTP, LTD, and depotentiation. J Neurophysiol. 1995;74:1075–1082. doi: 10.1152/jn.1995.74.3.1075. [DOI] [PubMed] [Google Scholar]
- 53.Shen KS, North RA. Muscarine increases cation conductance and decreases potassium conductance in rat locus coeruleus neurones. J Physiol (Lond) 1992;455:471–485. doi: 10.1113/jphysiol.1992.sp019312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shigemoto R, Nomura S, Ohishi H, Sugihara H, Nakanishi S, Mizuno N. Immunohistochemical localization of a metabotropic glutamate receptor, mGluR5, in the rat brain. Neurosci Lett. 1993;163:53–57. doi: 10.1016/0304-3940(93)90227-c. [DOI] [PubMed] [Google Scholar]
- 55.Shirasaki T, Harata N, Akaike NJ. Metabotropic glutamate response in acutely dissociated hippocampal CA1 pyramidal neurones of the rat. J Physiol (Lond) 1994;475:439–453. doi: 10.1113/jphysiol.1994.sp020084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Simmoneau M, Distasi C, Tauc L, Barbin G. Potassium channels in mouse neonate dorsal root ganglion cells: a patch-clamp study. Brain Res. 1987;412:224–232. doi: 10.1016/0006-8993(87)91128-0. [DOI] [PubMed] [Google Scholar]
- 57.Sladeczek F, Pin JP, Recassens M, Bockaert J, Weiss S. Glutamate stimulates inositol phosphate formation in striatal neurones. Nature. 1985;317:717–719. doi: 10.1038/317717a0. [DOI] [PubMed] [Google Scholar]
- 58.Staub C, Vranesic I, Knöpfel T. Responses to metabotropic glutamate receptor activation in cerebellar Purkinje cells: induction of an inward current. Eur J Neurosci. 1992;4:832–839. doi: 10.1111/j.1460-9568.1992.tb00193.x. [DOI] [PubMed] [Google Scholar]
- 59.Storm JF. Temporal integration by a slowly inactivating K+ current in hippocampal neurons. Nature. 1988;336:379–381. doi: 10.1038/336379a0. [DOI] [PubMed] [Google Scholar]
- 60.Storm JF. Potassium currents in the hippocampal pyramidal cells. Prog Brain Res. 1990;83:161–187. doi: 10.1016/s0079-6123(08)61248-0. [DOI] [PubMed] [Google Scholar]
- 61.Sugiyama H, Ito I, Hirono C. A new type of glutamate receptor linked to inositol phospholipid metabolism. Nature. 1987;325:531–533. doi: 10.1038/325531a0. [DOI] [PubMed] [Google Scholar]
- 62.Swandulla D, Partridge LD. Nonspecific cation channels. In: Cook NS, editor. Potassium channels. Ellis Horwood; Chichester, UK: 1990. pp. 167–180. [Google Scholar]
- 63.Tanabe Y, Masu M, Ishii T, Shigemoto R, Nakanishi S. A family of metabotropic glutamate receptors. Neuron. 1992;8:169–179. doi: 10.1016/0896-6273(92)90118-w. [DOI] [PubMed] [Google Scholar]
- 64.Tanabe Y, Nomura A, Masu M, Shigemoto R, Mizuno N, Nakanishi S. Signal transduction, pharmacological properties, and expression patterns of two rat metabotropic glutamate receptors, mGluR3 and mGluR4. J Neurosci. 1993;13:1372–1378. doi: 10.1523/JNEUROSCI.13-04-01372.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Teulon J, Paulais M, Bouthier M. A Ca2+-activated cation-selective channel in the basolateral membrane of the cortical thick ascending limb of Henle’s loop of the mouse. Biochim Biophys Acta. 1987;905:125–132. doi: 10.1016/0005-2736(87)90016-2. [DOI] [PubMed] [Google Scholar]
- 66.Thomsen C, Kristensen P, Mulvihill E, Haldeman B, Suzdak PD. l-2-Amino-4-phosphonobutyrate (l-AP4) is a agonist at the type IV metabotropic glutamate receptor, which is negatively coupled to adenylate cyclase. Eur J Pharmacol. 1992;227:361–362. doi: 10.1016/0922-4106(92)90018-q. [DOI] [PubMed] [Google Scholar]
- 67.Watkins JC, Evans RH. Excitatory amino acid transmitters. Annu Rev Pharmacol Toxicol. 1981;21:165–204. doi: 10.1146/annurev.pa.21.040181.001121. [DOI] [PubMed] [Google Scholar]
- 68.Yaari Y, Jensen MS. Cholinergic modulation of hippocampal epileptic activity in vitro. In: Frotscher M, Misgeld U, editors. Central cholinergic synaptic transmission. Birkhäuser; Basel: 1989. pp. 150–158. [DOI] [PubMed] [Google Scholar]
- 69.Yellen G. Single Ca2+-activated nonselective cation channels in neuroblastoma. Nature. 1982;296:357–359. doi: 10.1038/296357a0. [DOI] [PubMed] [Google Scholar]
- 70.Zufall F, Firestein S, Shepherd GM. Cyclic nucleotide-gated ion channels and sensory transduction in olfactory receptor neurons. Annu Rev Biophys Biomol Struct. 1994;23:577–607. doi: 10.1146/annurev.bb.23.060194.003045. [DOI] [PubMed] [Google Scholar]