

Abstract
The mechanism underlying dopamine D1receptor-mediated attenuation of glutamatergic synaptic input to nucleus accumbens (NAcc) neurons was investigated in slices of rat forebrain, using whole-cell patch-clamp recording. The depression by dopamine of EPSCs evoked by single-shock cortical stimulation was stimulus-dependent. Synaptic activation of NMDA-type glutamate receptors was critical for this effect, because dopamine-induced EPSC depressions were blocked by the competitive NMDA receptor antagonistd/l-2-amino-5-phosphonopentanoate (AP5). Application of NMDA also depressed the EPSC, and both this effect and the dopamine depressions were blocked by the A1 receptor antagonist 8-cyclopentyl-1,3-dipropylxanthine (DPCPX), implicating adenosine release in the EPSC depression. A1 receptor agonists also depressed EPSCs by a presynaptic action, causing increased paired-pulse facilitation, but this was insensitive to AP5. Activation of D1 receptors enhanced both postsynaptic inward currents evoked by NMDA application and the isolated NMDA receptor-mediated component of synaptic transmission. The biochemical processes underlying the dopamine-induced EPSC depression did not involve either protein kinase A or the production of cAMP and its metabolites, because this effect was resistant to the protein kinase inhibitors H89 and H7 and the cAMP-specific phosphodiesterase inhibitor rolipram. We conclude that activation of postsynaptic D1receptors enhances the synaptic activation of NMDA receptors in nucleus accumbens neurons, thereby promoting a transsynaptic feedback inhibition of glutamatergic synaptic transmission via release of adenosine. Unusually for D1 receptors, this phenomenon occurs independently of adenylyl cyclase stimulation. This process may contribute to the locomotor stimulant action of dopaminergic agents in the NAcc.
Keywords: Key words nucleus accumbens, whole-cell patch-clamp recording, rat, brain slices, glutamatergic synaptic transmission, presynaptic inhibition, retrograde messenger, dopamine, adenosine, glutamate, dopamine D1 receptors, NMDA receptors, adenosine A1 receptors, adenylyl cyclase, cyclic AMP, protein kinase A
The ventral extension of the striatum, the nucleus accumbens (NAcc), is a major projection field of the mesolimbic dopamine system (Deutch and Cameron, 1992). NAcc neurons also receive excitatory glutamatergic inputs primarily from cortical regions and also from the thalamus, hippocampus, and amygdala (see Pennartz et al., 1994). Because the predominant cell type, the GABA-containing medium spiny projection neurons, are generally quiescent in nature, this excitatory drive is extremely important for generation of outputs from the NAcc (Pennartz et al., 1994; Wilson and Kawaguchi, 1996).
Dopamine within the NAcc has been implicated critically in promoting locomotion (Pijnenberg and Van Rossum, 1973) (see also Pennartz et al., 1994; Iversen, 1995), in motivation, behavioral drive, and reward (Fibiger and Phillips, 1986; Robbins and Everitt, 1996), including the behaviors associated with addictive drugs of abuse (Koob, 1992; Kalivas et al., 1993; Wise, 1996), and also in the cognitive dysfunction of schizophrenia (Iversen, 1995; Wan et al., 1995). Interactions between dopaminergic and glutamatergic processes in the NAcc often have been proposed to contribute to these behaviors (Kalivas et al., 1993;Iversen, 1995; Hyman, 1996; Robbins and Everitt, 1996; Wise, 1996). Two functional cellular models for such processes have emerged recently. First, induction of gene expression in striatal neurons by the psychomotor stimulant drug amphetamine depends on activation of both postsynaptic dopamine D1 and NMDA-type glutamate receptors (Konradi et al., 1996). Second, D1 receptor activation enhances depolarization of striatal neurons resulting from NMDA application (Levine et al., 1996). D1 receptor-stimulated adenylyl cyclase (Stoof and Kebabian, 1981) has been implicated in both of these phenomena (Colwell and Levine, 1995; Konradi et al., 1996).
Electrophysiological studies have demonstrated that activation of D1-like receptors depresses glutamatergic synaptic input to the NAcc (Higashi et al., 1989; Pennartz et al., 1992; Harvey and Lacey, 1996c; Nicola et al., 1996). This is thought to involve a presynaptic mechanism, because dopamine-induced depressions are accompanied by an increase in the degree of paired-pulse facilitation (Pennartz et al., 1992; Nicola et al., 1996) and occur in the absence of any change in the postsynaptic cell membrane potential or conductance (Pennartz et al., 1992; Harvey and Lacey, 1996c; Nicola et al., 1996) (but see Higashi et al., 1989). Furthermore, dopamine also reduces the frequency, but not the amplitude, of miniature EPSCs (Nicola et al., 1996). However, this dopamine-induced depression of EPSCs does not seem to involve adenylyl cyclase stimulation (Harvey and Lacey, 1996c).
In contrast to these latter electrophysiological findings, anatomical studies of the striatum provide little support for presynaptic D1 receptors but demonstrate the existence of both postsynaptic D1 receptors (Hersch et al., 1995) and the messenger RNA encoding their synthesis (Gerfen et al., 1990; Le Moine and Bloch, 1995). We sought to resolve this discrepancy, and here we demonstrate electrophysiologically that a postsynaptic interaction between D1 and NMDA receptors results in liberation of a retrograde messenger, which itself inhibits presynaptically the release of glutamate.
Some of these results have been reported previously in abstract form (Harvey and Lacey, 1996a,b).
MATERIALS AND METHODS
Brain slice preparation and recording techniques.Experiments were performed on horizontal slices of ventral forebrain prepared from male Wistar rats 4–5 weeks of age, as described previously (Harvey and Lacey, 1996c). In brief, after inhalational anesthesia (3% Fluothane) animals were decapitated and their brains removed. Horizontal forebrain slices (350 μm thick) were prepared at 4–10°C using a Vibroslice (Campden Instruments, Loughborough, UK). All slices that were used contained or were within 350 μm of the dorsal or ventral extent of the anterior commissure. Slices were maintained in the recording chamber at 32–33°C and superfused continuously at 2–3 ml/min with artificial cerebrospinal fluid comprising (in mm): NaCl 126, KCl 2.5, NaHCO3 26, NaHPO4 1.2, CaCl2 2.4, MgCl2 1.3, and d-glucose 10, saturated with 95% O2/5% CO2 at pH 7.4.
Recordings were obtained using the “blind” whole-cell patch-clamp recording technique (Blanton et al., 1989). Recording pipettes were positioned in the ventral portion of the nucleus accumbens and had resistances of 5–7 MΩ when filled with electrolyte solution comprising (in mm): potassium gluconate 125, MgCl2 2, NaCl 10, CaCl2 1, HEPES 10, 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid (BAPTA) 10, adenosine trisphosphate (ATP) 2, and guanosine trisphosphate (GTP) 0.3, buffered to pH 7.2–7.3 with KOH. Membrane potential and current were measured by an Axopatch-1D patch-clamp amplifier (Axon Instruments, Foster City, CA). Series resistance was measured in current clamp, and after electrical compensation (by 70–85%) this ranged from 10 to 22 MΩ. Throughout voltage-clamp recordings input resistance and whole-cell access were monitored via voltage steps (+10 mV, 50 msec) delivered every 20 sec. Neurons that displayed >20% change in the shape or size of capacitance transients or input resistance (in the absence of drugs) were excluded from analysis.
Synaptic currents were evoked by delivering single shocks (0.1 msec, 1–5 mV) every 20 sec, using a bipolar stimulating electrode positioned 300–900 μm rostral to the recording pipette, adjacent to the cerebral cortex. To study solely the glutamate receptor-mediated excitatory postsynaptic currents (Harvey and Lacey, 1996c), we performed all experiments in the presence of picrotoxin (50 μm) to block GABAA receptor-mediated synaptic currents. Picrotoxin was applied within 5–10 min of obtaining the whole-cell configuration. In all experiments cells were voltage-clamped at between −80 and −90 mV (close to the resting membrane potential), unless otherwise stated (i.e., when NMDA receptor-mediated excitatory synaptic currents were studied).
Data acquisition and analysis, together with generation of voltage and current pulses and timing of electrical stimulation, were performed by pCLAMP software (Axon Instruments). Synaptic currents were stored as the average of five consecutive records, and the peak amplitude of the averaged EPSCs was measured. The percentage of depression/facilitation induced by an agent was expressed relative to the control EPSC amplitude averaged over the 5 min period immediately before drug additions. Numerical data are expressed as mean ± SEM. Statistical analyses were performed by Student’s paired ttest, and all data were significantly different at p < 0.05 unless otherwise stated.
Drugs were applied directly to the superfusate in known concentrations, reaching the recording chamber within 15 sec of switching a tap in the perfusion line. Drugs used were 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX) and d/l-2-amino-5-phosphonopentanoic acid (AP5) (both from Tocris Cookson, Bristol, UK); picrotoxin, adenosine, dopamine, NMDA, tetrodotoxin (TTX), 2-chloropentyl adenosine (CPA), [R]-N6-(2-phenylisopropyl) adenosine (R-PIA), and l-nitroarginine (l-NARG) (all from Sigma, Aldrich, UK). Rolipram and 8-cyclopentyl-1,3-dipropylxanthine (DPCPX) were obtained from Research Biochemicals (Natick, MA).
RESULTS
Properties of nucleus accumbens neurons
Whole-cell patch-clamp recordings were obtained from a total of 127 neurons in the ventrolateral nucleus accumbens. After the whole-cell configuration was obtained, the membrane potential was voltage-clamped at −90 mV to enable cell stabilization. Five minutes later the pipette access resistance was measured and optimally compensated in current-clamp mode. Under these conditions the resting membrane potential and input resistance of these neurons were −83 ± 1.6 mV and 108 ± 2.4 MΩ, respectively. These characteristics correspond to those of the medium spiny projection neurons of the NAcc (Pennartz et al., 1992; O’Donnell and Grace, 1993).
The ability of dopamine to depress EPSCs is stimulus-dependent
To test the possibility that the depressant effect of D1 receptor activation on the EPSC was attributable to an indirect mechanism, we first examined whether the depression of the EPSC by dopamine required glutamatergic synaptic transmission for its expression. Stimulation was ceased during the first 8 min of dopamine application, which was sufficient time to observe a maximal depression by dopamine under normal conditions of stimulation (Harvey and Lacey, 1996c) (see also Fig. 1B). When stimulation was resumed with dopamine still present, the EPSC amplitude was initially the same as before the addition of dopamine but declined only on subsequent stimulation, recovering to control levels on washout (n = 3; Fig. 1A). Thus these data indicate that ongoing glutamatergic synaptic transmission is required for dopamine to depress EPSCs.
Fig. 1.
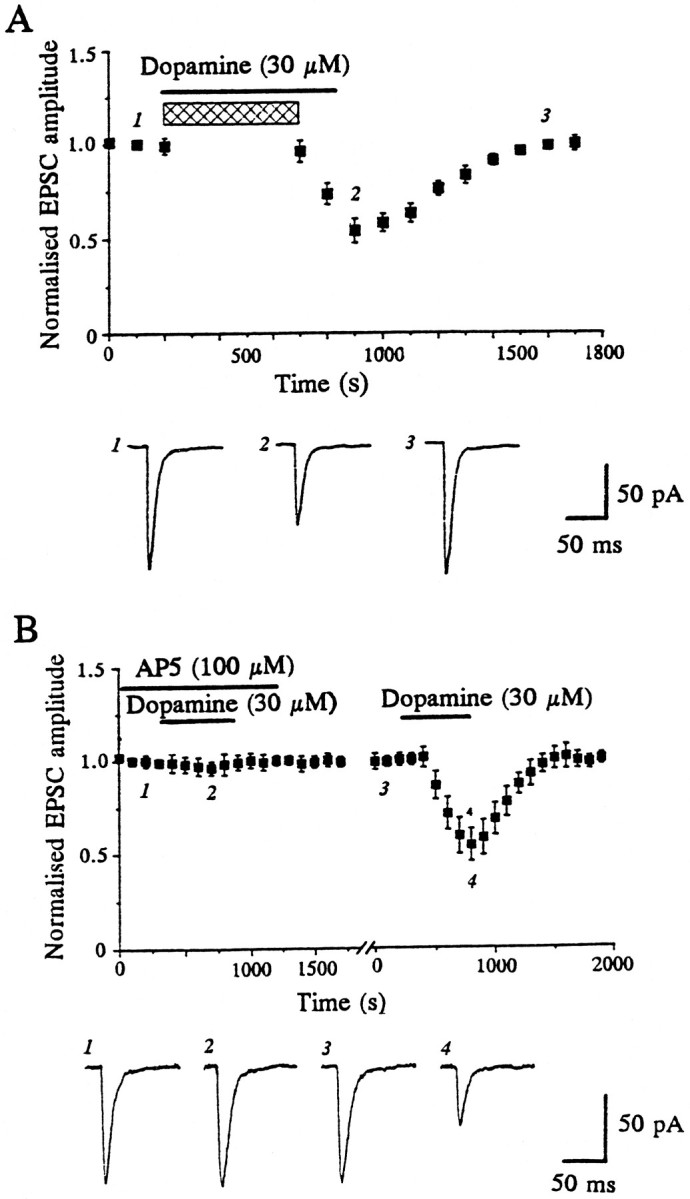
The dopamine-induced depression of the EPSC is dependent on synaptic activation of NMDA receptors. A, Single-shock electrical stimulation (to evoke EPSCs) was stopped during the period shown by the hatched bar, during which time dopamine (30 μm, filled bar) was applied. Only once stimulation was resumed did depression of EPSCs commence. Data were pooled from three separate experiments. Each point on the graph is the average of five consecutive records, such as those shownbelow the graph (taken from a single experiment), and is normalized with respect to the 5 min period immediately before the addition of dopamine. B, Depression of EPSCs by dopamine was reversibly blocked by the NMDA receptor antagonist AP5 (100 μm). The plot (top panel) illustrates data pooled from four individual neurons, and thex-axis break, variable between experiments, represents 10–20 min. Bottom panel, Sample records from one experiment, taken at the times indicated.
Depression of the EPSC by dopamine is dependent on the synaptic activation of NMDA receptors
Previous reports of a facilitatory influence of D1receptors on NMDA responses in the dorsal striatum (Cepeda et al., 1993; Colwell and Levine, 1995; Levine et al., 1996) prompted us to examine whether glutamate acting on NMDA receptors was involved in the depressant action of dopamine. In nine cells the competitive NMDA receptor antagonist AP5 (100 μm) reduced the depression induced by dopamine (30 μm) from 47 ± 3.0 to 4.1 ± 1.4% in a reversible manner (Fig. 1B). Thus the EPSC depression induced by dopamine seems to be dependent on the synaptic activation of NMDA receptors.
Adenosine is released after activation of NMDA receptors
In support of this indication that synaptic NMDA receptors might be involved in the depression of the EPSC, application of NMDA (20 μm) itself also reversibly depressed EPSC amplitude by 49 ± 1.9% (n = 8; Fig. 2A,B). One possible consequence of NMDA receptor activation is the formation of nitric oxide (NO; Schuman and Madison, 1994; Garthwaite and Boulton, 1995), which itself can promote release of a variety of neuroactive substances in the striatum (Guevara-Guzman et al., 1994) and modulate synaptic transmission in other parts of the brain by a trans-synaptic action (see Schuman and Madison, 1994; Garthwaite and Boulton, 1995). A role for NO was explored by examining the effect of a specific inhibitor of NO synthase, l-NARG, on the EPSC depressions caused by NMDA application. Application of NMDA caused a depression of 49 ± 2.7% (n = 3), which was readily reversible on washout (Fig. 2A). However, 15–20 min after the addition of l-NARG (100 μm), the ability of NMDA (20 μm) to depress EPSCs was unaffected (mean depression of 48 ± 3.2%; p > 0.05; Fig.2A), which suggests that NO is not involved in this process.
Fig. 2.

Adenosine A1 receptor activation is required for EPSC depression by both NMDA and dopamine, indicating that adenosine release results from NMDA receptor activation.A, Application of NMDA caused a reversible depression of EPSCs, but this was unaffected by the nitric oxide synthase (NOS) inhibitor l-nitroarginine. The plot shows that NMDA (20 μm) depressed EPSCs in a reversible manner. After perfusion of the NOS inhibitor l-nitroarginine (100 μm) for 15–20 min, reapplication of NMDA caused a similar depression. Data were pooled from three separate experiments, and sample records of EPSCs from one experiment are displayedbelow the plot. B, The reversible depression of EPSCs induced by NMDA (20 μm) was reduced considerably by the A1 receptor antagonist DPCPX (200 nm), which itself increased EPSC amplitude. The plot shows data pooled from five neurons, and the x-axis break corresponds to 3–5 min before the addition of DPCPX. C, DPCPX also blocks depression of EPSCs by dopamine. Dopamine (30 μm) reduced the amplitude of EPSCs, and this effect was blocked in the presence of DPCPX (200 nm). DPCPX itself caused a clear facilitation of EPSC amplitude. Data were pooled from five individual neurons; the x-axis break corresponds to 3–8 min. D, The EPSC depression caused by the A1 receptor agonist CPA (200 nm) is unaffected by the NMDA receptor antagonist AP5 (100 μm). Data were obtained from five experiments; the x-axis break corresponds to 10–20 min.
Another possible consequence of NMDA receptor activation that might depress the EPSC is the release of adenosine. Adenosine depresses glutamatergic synaptic transmission in many regions of the CNS (for review, see Fredholm, 1995), including the NAcc (Uchimura and North, 1991), and it is released after NMDA receptor activation in cortical tissue (Craig and White, 1993) and also in the hippocampus (Manzoni et al., 1994). The effects of the selective adenosine A1receptor antagonist DPCPX (Fredholm, 1995) on NMDA-induced depressions, therefore, were examined. Application of NMDA (20 μm) reduced the amplitude of EPSCs by 34 ± 5.7% (n = 5), which was reduced to a depression of only 4.4 ± 2.5% (n = 5) in the presence of the DPCPX (200 nm; Fig. 2B). This suggests that NMDA receptor activation promotes release of adenosine, which inhibits glutamate release at this synapse via an action on A1adenosine receptors.
Depression of the EPSC by dopamine requires activation of adenosine A1 receptors
Because NMDA receptor activation leads to the release of adenosine and NMDA receptor activation is critical for dopamine-induced depressions of the EPSC, we explored the possible involvement of adenosine in this effect of dopamine, using the selective adenosine A1 receptor antagonist DPCPX. The ability of dopamine (30 μm) to depress EPSCs was reduced by DPCPX (200 nm) from a depression of 51 ± 3.5 to 2.0 ± 0.8% (n = 7; Fig. 2C). In a further three cells, depressions induced by the selective D1 receptor agonist SKF 38393 (10 μm) also were reduced significantly from 47 ± 3.9 to 4.5 ± 1.3% by DPCPX (200 nm). However, in contrast to the actions of dopamine, the ability of the A1 receptor-selective agonist CPA (200 nm; Fredholm, 1995) to depress EPSCs was unaffected by AP5 (100 μm) in all five cells tested (Fig.2D). Therefore depression of the EPSC by A1 receptors is itself independent from NMDA receptor activation. Taken together, these findings indicate that the ability of dopamine to depress glutamate receptor-mediated synaptic currents in the nucleus accumbens requires the activation of adenosine A1 as well as D1 receptors, and that activation of NMDA receptors is a critical intermediate step for the production of adenosine.
In all cells examined, DPCPX (200 nm) itself caused a rapid and pronounced increase in EPSC amplitude (by 74 ± 14%,n = 28; Figs. 2B,C, 3A,B), which was not accompanied by any discernible change in the holding current or membrane conductance. This effect did not reverse readily on washout of the drug (for up to 30 min). This suggests that glutamate receptor-mediated synaptic transmission is also subject to tonic inhibition by endogenous adenosine, which is relieved by DPCPX.
Fig. 3.

Both endogenous and exogenous adenosine depressed EPSCs via activation of presynaptic adenosine A1 receptors.A, The ability of adenosine to depress EPSCs is blocked by DPCPX (200 nm). The upper graphs are plots of EPSC amplitude (squares, top graph), input conductance (circles,middle graph), and holding current (diamonds, bottom graph) during an experiment on a single neuron, voltage-clamped at −90 mV. Thelower panel shows synaptic currents evoked at specific points (1–5) during the same experiment. Neither the reduction in synaptic transmission induced by adenosine nor the enhancement by DPCPX was accompanied by any change in the holding current or input conductance of the neuron. B, Similarly, the selective A1 receptor agonist CPA depressed EPSCs in a DPCPX-sensitive manner. CPA (200 nm) depressed EPSCs (squares, top graph) in the absence of any effect on the input conductance (circle,middle graph) or the holding current (diamond, bottom graph). Data were obtained from a single neuron voltage-clamped at −90 mV.C, Paired-pulse facilitation is increased when EPSCs are depressed by the A1 receptor agonist CPA. CPA (200 nm) produced a reversible enhancement in the paired-pulse ratio evoked with a 50 msec interstimulus interval. The pairs of EPSCs in the lower panel were obtained in control conditions (1) and in the presence of 200 nm CPA (2). In the right trace the first EPSC in2 has been scaled to match the size of the first EPSC in1. D, Paired-pulse facilitation is decreased when EPSCs are facilitated by the A1 receptor antagonist DPCPX (200 nm). Thus the depressant actions of both endogenous and applied adenosine are attributable to a presynaptic mechanism.
Adenosine depresses EPSCs by activation of presynaptic adenosine A1 receptors
The next series of experiments was aimed at establishing the site of action of adenosine at this synapse, because adenosine seems to be critical for dopamine- and NMDA-induced depression of the EPSC. In all 13 cells tested, application of adenosine (30–100 μm) concentration dependently and reversibly depressed the peak amplitude of EPSCs. At a concentration of 60 μm, adenosine depressed the EPSCs by 46 ± 4.4% (n = 7; Fig.3A). Depressions were evident 1–2 min after adenosine additions, were sustained for the duration of its application, and reversed within 8–12 min of drug washout. In all 13 cells examined, adenosine caused no significant change in either the holding current or input conductance (measured during the +10 mV voltage step; Fig. 3A). Thus, in agreement with Uchimura and North (1991), adenosine (60 μm) depressed glutamatergic synaptic transmission in the nucleus accumbens in the absence of any detectable change in the postsynaptic membrane properties of the cell under study. Similarly, the selective adenosine A1 receptor agonists (Fredholm, 1995) CPA (200 nm; Figs.2D, 3B) and R-PIA (200 nm; data not shown) both reversibly depressed the peak amplitude of EPSCs by 55 ± 9.4% (n = 9) and 47 ± 6.5% (n = 8), respectively, also without any detectable change in postsynaptic membrane conductance. This effect was evident 1–2 min after agonist application and completely reversed 10–12 min after washout. Depressions of EPSCs induced by adenosine (60 μm; n = 3), CPA (200 nm;n = 5), and R-PIA (200 nm;n = 3) all were blocked completely by the selective adenosine A1 receptor antagonist DPCPX (200 nm; Fig. 3A,B). Taken together, these data indicate that adenosine depresses EPSCs via an action at A1receptors.
To investigate further the locus of action of both applied and endogenous adenosine, we used a paired-pulse stimulation protocol (50 msec interpulse interval). While reducing the amplitude of both EPSCs, adenosine (60 μm) reversibly enhanced the ratio of the second EPSC to the first by 32 ± 3.2% in five cells. In a further five cells CPA (200 nm) also increased the paired-pulse ratio by 98 ± 3.2% (Fig. 3C). These findings are consistent with a presynaptic locus for the adenosine A1 receptors involved in depressing synaptic transmission. Furthermore, the EPSC facilitation induced by DPCPX (200 nm) was accompanied by a decrease of 25 ± 5.1% (n = 6) in the corresponding paired-pulse ratio (Fig.3D), indicating that the EPSC depression attributed to endogenous adenosine also is mediated by presynaptic A1receptors.
Postsynaptic potentiation of NMDA currents by D1receptor activation
Having established that synaptically activated NMDA receptors are a critical link between the activation of D1 receptors and the release of adenosine that results in presynaptic inhibition, we then sought to determine more directly whether dopamine could modulate postsynaptic NMDA receptors in the NAcc. This was explored initially by applying dopamine in conjunction with a submaximal dose of NMDA. In three cells voltage-clamped between −80 and −90 mV, inward currents evoked in response to application of NMDA (20 μm) were enhanced by 60 ± 4.7% by coapplication with dopamine (30 μm). In the presence of TTX (200 nm), which completely blocked synaptic transmission (data not shown), the selective D1 receptor agonist SKF 38393 (10 μm) also caused an enhancement (by 71 ± 7.9%,n = 5; Fig. 4) of NMDA receptor-mediated inward currents that was readily reversible on washout in the three cells examined. Thus these data indicate that dopamine acting on D1 receptors facilitates postsynaptic NMDA receptor-mediated currents, as shown previously in the dorsal striatum (Cepeda et al., 1993; Levine et al., 1996).
Fig. 4.

Dopamine D1 receptor activation enhances postsynaptic NMDA receptor-mediated inward currents independently of synaptic transmission. A, Data pooled from five cells showing that bath application of NMDA (20 μm) induced an inward current. When NMDA was reapplied in the same five cells 5–10 min after application of the D1receptor agonist SKF 38393 (10 μm), NMDA-induced currents were enhanced. Cells were voltage-clamped at −80 to −90 mV with tetrodotoxin (200 nm) present throughout. B,Continuous record of membrane current from an individual neuron (1 of the 5 in A; voltage-clamped at −90 mV) showing the reversible enhancement of the NMDA-mediated current by SKF 38393 (10 μm).
Dopamine enhances the NMDA receptor-mediated component of the EPSC
To see whether dopamine could modulate NMDA-induced currents evoked by synaptic activation, as well as those caused by NMDA application, we examined the effect of dopamine on the pharmacologically isolated NMDA receptor-mediated component of synaptic transmission (EPSCN). When cells were voltage-clamped at −50 mV in the presence of CNQX (10 μm), the antagonist of AMPA-type glutamate receptors, single-shock electrical stimulation resulted in a slower synaptic current that was blocked completely by AP5 (100 μm) in all four cells tested (Fig. 5A). In a manner similar to its action on fast AMPA receptor-mediated synaptic currents, dopamine (30 μm) reversibly depressed the peak amplitude of the EPSCN by 51 ± 3.1% (n = 8; Fig. 5B). However, in the presence of DPCPX (200 nm), dopamine (30 μm) caused a clear and readily reversible enhancement of EPSCN amplitude (by 28 ± 4.5%) in all nine cells examined (Fig. 5B). The adenosine A1 receptor antagonist DPCPX itself caused a rapid facilitation of the EPSCN amplitude (by 74 ± 12%, n = 9; Fig. 5B), similar to that observed with the EPSC (e.g., Fig. 2B). These data suggest that, in the absence of the inhibitory action of endogenous adenosine, dopamine facilitates, rather than depresses, the isolated NMDA receptor-mediated component of synaptic transmission. This is likely to be attributable to a postsynaptic interaction between D1 and NMDA receptors.
Fig. 5.

Adenosine A1 receptor blockade prevents dopamine-mediated depression of NMDA receptor-mediated EPSCs (EPSCN) and reveals that dopamine enhances the EPSCN. A, The EPSCN was isolated by application of CNQX (10 μm) to block AMPA receptors and by voltage-clamping at −50 mV. Under these conditions the residual component of the EPSC was blocked completely by the NMDA receptor antagonist AP5 (100 μm). B, Top panel, Dopamine (30 μm) reversibly reduced the EPSCN amplitude. In the presence of the A1receptor antagonist DPCPX (200 nm), which itself increased EPSCN amplitude, dopamine caused a clear and reversible facilitation of the EPSCN. The plot shows data pooled from nine cells, and the x-axis break corresponds to 3–8 min before the addition of DPCPX. Bottom panel, Sample records from one experiment, taken at the times indicated on the plot.
Biochemical mechanism of the depression of the EPSC by dopamine
It is well established that striatal D1 receptors couple to adenylyl cyclase, and their activation results in the stimulation of cAMP formation and subsequent activation of protein kinase A (PKA; Stoof and Kebabian, 1981). However, stimulation of adenylyl cyclase with forskolin and elevation of intracellular cAMP levels with either dibutyryl cAMP or inhibitors of cAMP-specific phosphodiesterases such as IBMX or rolipram caused facilitation, rather than depression, of the EPSC (Harvey and Lacey, 1996c). Therefore we have proposed that D1 receptor-mediated inhibition of glutamatergic synaptic transmission in the nucleus accumbens does not involve a cAMP-dependent process (Harvey and Lacey, 1996c). Furthermore, in the presence of the PKA inhibitor H-89 (1 μm), dopamine (30 μm) depressed the EPSC by 49 ± 3.3%, which was not significantly different from the depression (48 ± 3.9%) evoked in control conditions (n = 4; p > 0.05; Fig.6A). H-89 (1 μm) was clearly active in this system, however, because it prevented the increase in EPSC amplitude by forskolin (10 μm;n = 3; Fig. 6A). This particular effect of forskolin has been described previously (Colwell and Levine, 1995; Harvey and Lacey, 1996c) and attributed to PKA-dependent phosphorylation of postsynaptic neostriatal AMPA receptors (Colwell and Levine, 1995). Similarly, dopamine (30 μm) depressed synaptic currents by 42 ± 3.0 and 43 ± 2.6% (n = 3, p > 0.05) in the presence and absence of the nonselective protein kinase inhibitor H-7 (10 μm), respectively. Thus these data provide further evidence that a PKA-independent process underlies the EPSC depression induced by dopamine.
Fig. 6.
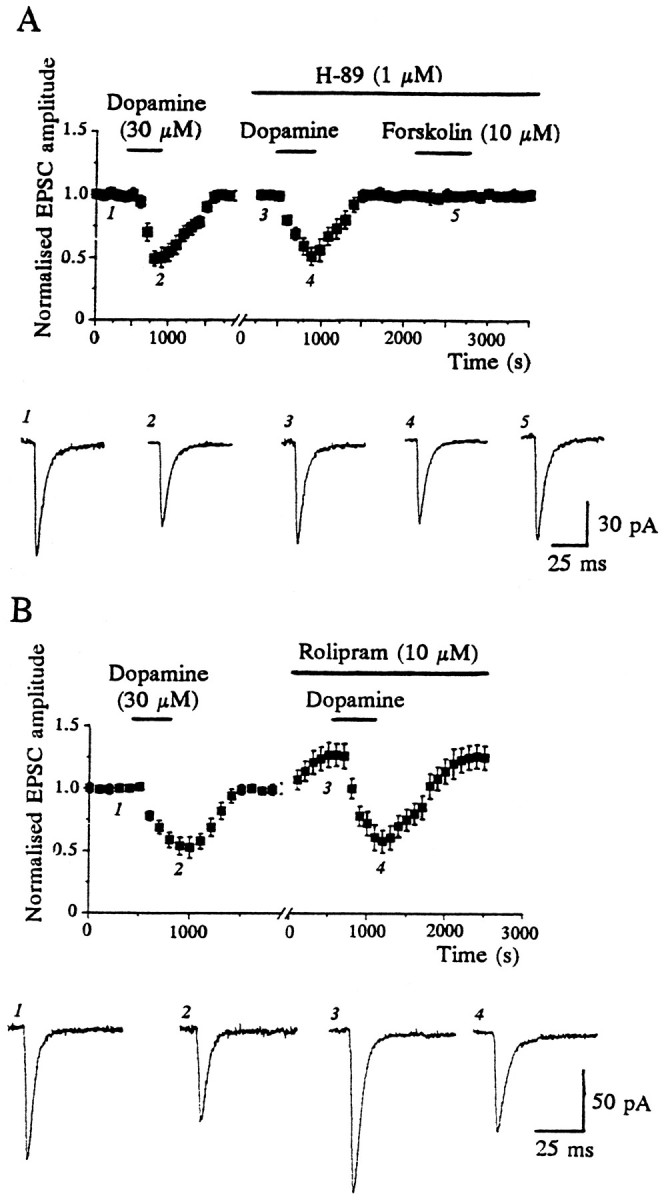
Dopamine depresses EPSCs independently of protein kinase A or metabolism of cAMP. A, Dopamine-induced depressions were unaffected by the protein kinase A inhibitorH-89 (1 μm). However, the ability of forskolin (10 μm) to enhance EPSCs was blocked by H-89. The plot consists of data pooled from three cells, and thex-axis break corresponds to 3–5 min. B, Rolipram (10 μm), a cAMP-dependent phosphodiesterase inhibitor, failed to affect dopamine-induced depressions. The data in the plot are pooled from three cells, and thex-axis break corresponds to 3–6 min.
In other brain regions presynaptic inhibition of synaptic transmission after metabotropic receptor activation has been attributed to adenosine produced by the metabolism of cAMP by cAMP-specific phosphodiesterases (Gereau and Conn, 1994; Bonci and Williams, 1996). If the formation of cAMP and its subsequent metabolism was the principal biochemical process underlying dopamine-induced depressions of the EPSC, then rolipram, a specific inhibitor of cAMP-specific phosphodiesterase (Beavo and Reifsnyder, 1990), might be expected to alter the ability of dopamine to reduce synaptic currents. However, although rolipram (10 μm) caused a rapid enhancement in EPSC amplitude (by 26 ± 4.3%), as reported previously (Harvey and Lacey, 1996c), it did not alter the ability of dopamine to depress synaptic transmission (n = 3; Fig. 6B). Moreover, when the facilitatory action of forskolin on the EPSC was abolished completely by H-89 (1 μm; n = 3), no other effects of forskolin were observed (Fig. 6A), although production of cAMP would be unimpaired under these conditions. Together these findings do not support a role for cAMP, PKA, or a metabolite of cAMP in the dopamine-induced depression of the EPSC.
DISCUSSION
We have shown that the depression of glutamatergic synaptic transmission in the NAcc by dopamine involves a novel indirect process, central to which is an interaction between D1 and NMDA receptors and subsequent adenosine release. Adenosine in turn acts as a retrograde messenger and inhibits glutamate release via activation of presynaptic adenosine A1 receptors (Fig. 7). This mechanism argues against the presynaptic localization of D1 receptors proposed in previous electrophysiological studies (Pennartz et al., 1992; Harvey and Lacey, 1996c; Nicola et al., 1996), but not the anatomical evidence for postsynaptic D1receptors in the striatum (Hersch et al., 1995).
Fig. 7.

Diagram of a glutamatergic synapse onto a dendritic spine on a medium spiny NAcc output neuron, illustrating the processes operating to permit presynaptic modulation of glutamate release by postsynaptic dopamine D1 receptors. Glutamate released from the cortical afferent (left) activates postsynaptic glutamatergic AMPA and NMDA receptors, resulting in the EPSC. Concurrent activation of D1 receptors amplifies the current caused by the synaptic activation of NMDA receptors, thereby promoting release of adenosine (or a precursor) into the extracellular space. Adenosine in turn acts on presynaptic inhibitory A1receptors to reduce glutamate release. This sequence of events occurs independently of adenylyl cyclase stimulation and production of cyclic AMP.
A pivotal role for adenosine A1 receptors in the presynaptic inhibition of glutamatergic synaptic transmission
EPSC depressions induced by dopamine and the selective D1 receptor agonist SKF 38393 and also by NMDA were abolished by the A1 receptor antagonist DPCPX. In agreement with a previous report (Uchimura and North, 1991), adenosine reversibly depressed excitatory synaptic transmission in the NAcc in all cells examined. This was attributable to activation of A1receptors, because the selective adenosine A1 receptor agonists CPA and R-PIA both mimicked the actions of adenosine, and DPCPX, a specific adenosine A1 receptor antagonist, completely blocked depressions induced by all the A1 receptor agonists.
In agreement with Uchimura and North (1991), A1 receptor agonists at concentrations capable of depressing EPSCs had no discernible effect on the postsynaptic membrane properties (holding current or input conductance) of NAcc neurons. In addition, depressions induced by adenosine and CPA were accompanied by an increase in the paired-pulse ratio, whereas a reduction in the degree of paired-pulse facilitation accompanied the EPSC augmentation induced by DPCPX. These changes in paired-pulse ratio indicate a presynaptic locus for the A1 receptors regulating glutamatergic synaptic transmission in the NAcc. Although A1 receptor mRNA has been located in a minority of striatal neurons (Ferré et al., 1996), postsynaptic A1 receptors play no obvious role in the effects of adenosine, NMDA, and dopamine described here. Thus glutamate release is likely to be inhibited presynaptically not by D1 receptors, but by A1 receptors, which are activated indirectly by dopamine.
NMDA receptor activation is critical for the effect of dopamine
Synaptic activation of NMDA receptors is critical for dopamine-induced EPSC depressions, because the ability of dopamine to depress glutamatergic EPSCs is both stimulus-dependent and blocked by the competitive NMDA receptor antagonist AP5. Thus it follows that there must be significant activation of NMDA receptors in the presence of dopamine, although NMDA receptors apparently contribute little to the EPSC (Harvey and Lacey, 1996c) (see also Figs.1B, 2D). However, glutamatergic synaptic potentials (EPSPs) in NAcc neurons do exhibit an NMDA component (Martin et al., 1997), particularly at membrane potentials less negative to −80 mV. Therefore a significant activation of synaptic NMDA receptors in neurons other than the (voltage-clamped) cell under study probably occurs under our experimental conditions. The postsynaptic facilitatory interaction between D1 and NMDA receptors was demonstrated by the potentiation by dopamine and the selective D1 receptor agonist SKF 38393 of inward currents evoked by applied NMDA and also was demonstrable on the EPSCN. Similar findings have been reported in dorsal striatal neurons (Cepeda et al., 1993; Colwell and Levine, 1995; Levine et al., 1996), and they also resemble in some respects the augmentation of the EPSPN in NAcc by μ-opioid receptor agonists (Martin et al., 1997). This postsynaptic D1/NMDA receptor interaction in medium spiny projection neurons constitutes an attractive candidate mechanism for promoting the EPSC depression.
NMDA itself, as well as D1 receptor agonists, depressed EPSCs in a DPCPX-sensitive manner, also supporting a role for NMDA receptors “downstream” of the D1 receptor. In the absence of dopamine, NMDA receptor-dependent release of adenosine during single-shock stimulation seems not to occur, because AP5 does not alter the EPSC amplitude (Harvey and Lacey, 1996c). However, higher frequencies of stimulation, which probably lead to greater NMDA receptor activation, do induce a short-term A1receptor-dependent depression of glutamatergic synaptic transmission in the dorsal striatum (Lovinger and Choi, 1995). A similar phenomenon, attributable to NMDA receptor-dependent adenosine release, occurs in the hippocampus (Manzoni et al., 1994). In the NAcc, dopamine seems to enhance sufficiently the level of NMDA receptor activation during single-shock stimulation to promote the release of adenosine.
Release of adenosine after NMDA receptor activation
The precise mechanism whereby extracellular adenosine levels become raised by NMDA receptor activation is unclear. In slices of cortical tissue NMDA causes Ca2+-dependent release of a substrate for ecto-5′-nucleotidase, which then is converted to adenosine (Craig and White, 1993). The inhibitory tone revealed by the EPSC enhancement by DPCPX is unlikely to be NMDA receptor-dependent, however, because AP5 was without effect on EPSC amplitude. Because adenosine may be extruded actively from neurons (Brundege and Dunwiddie, 1996) and formed from a variety of precursors, including ATP, which may itself be released by electrical stimulation (Hamann and Attwell, 1996), there are several possible sources for this endogenous adenosine tone.
Biochemical mechanism coupling D1 receptors to depression of the EPSC
We have found no evidence to suggest that the D1receptor-mediated attenuation of EPSCs involves a cAMP- or PKA-dependent process or requires metabolism of cAMP to adenosine (Harvey and Lacey, 1996c; present study). This perhaps is unexpected, given the considerable evidence for D1 receptor-stimulated adenylyl cyclase in striatum (Stoof and Kebabian, 1981). Indeed, the observation that forskolin potentiates both NMDA and AMPA receptor-mediated depolarizations and excitatory synaptic potentials in the neostriatum (Colwell and Levine, 1995) certainly suggests a possible role for D1-stimulated adenylyl cyclase in the NAcc. Nonetheless, although we also observed a PKA-dependent augmentation of the EPSC with forskolin, this was not observed with dopamine, even when feedback inhibition was blocked by DPCPX. Therefore, although regulation of postsynaptic striatal glutamate receptors via PKA may indeed be possible, any stimulation of adenylyl cyclase resulting from D1 receptor activation was without effect in our experimental paradigm. The effects of dopamine we did observe, which most likely stem from the enhancement of NMDA receptor currents, must arise from a PKA-independent mechanism.
Several recent reports point to the possibility of NMDA current enhancement by neurotransmitter receptors coupled to phospholipase C (PLC; Ben-Ari et al., 1992; Markram and Segal, 1992; Harvey and Collingridge, 1993; Rahman and Neuman, 1996; Pisani et al., 1997). PLC activation by D1-like receptors has been reported in the striatum (Mahan et al., 1990; Wang et al., 1995), renal tissue (Felder et al., 1993; Yu et al., 1995), and retinal cells (Rodrigues and Dowling, 1990), and this may underlie the dopamine-induced enhancement of NMDA currents in dorsal and ventral striatum (Cepeda et al., 1993;Levine et al., 1996; present study). Alternatively, production of arachidonic acid, also associated with D1 receptor activation (Piomelli et al., 1991), may increase NMDA-induced currents (Miller et al., 1992).
Physiological and behavioral significance
D1 receptor activation can induce expression of immediate-early genes in the striatum and nucleus accumbens via NMDA receptor-dependent means (Keefe and Gerfen, 1996; Konradi et al., 1996;Wang and McGinty, 1996), and this may contribute to the long-term plastic changes underlying the behavioral sensitization and dependence associated with addictive drugs (Hyman, 1996). However, because these processes involve D1 receptor-stimulated adenylyl cyclase and cAMP-dependent phosphorylation, the effects observed in this study may not be related to these behavioral changes.
Postsynaptic NMDA receptors contribute significantly to the regulation of synaptic strength within the NAcc and also play a role in the direct excitation of projection neurons. Although coincident activation of D1 and NMDA receptors serves initially to enable this process, it is limited subsequently by the reduction in glutamate release resulting from the retrograde action of adenosine. However, because inhibitory GABAergic synaptic transmission in NAcc, which is driven at least partly by glutamatergic synaptic input onto intrinsic GABAergic NAcc neurons, also is depressed by D1 receptor activation (Pennartz et al., 1992), the net effect of dopamine on the synaptic drive onto NAcc neurons is harder to evaluate. Perhaps because of this, the present findings are not integrated easily with behavioral studies of dopamine/glutamate interactions in NAcc.
Intra-accumbal NMDA receptor antagonists block the orofacial stereotypy (Kelley and Delfs, 1994) and locomotor activity (Pulvirenti et al., 1991; Burns et al., 1994) produced by dopa-minergic drugs, mirroring the D1/NMDA receptor interaction observed in this study. In contrast, NMDA receptor antagonists in the NAcc mimic dopaminergic function in reward (Carlezon and Wise, 1996). Moreover, although dopamine receptor antagonists in the NAcc may be beneficial in schizophrenia (Wan et al., 1995), NMDA receptor antagonists tend to promote psychosis (see Iversen, 1995). Indeed, glutamate receptor agonists, rather than antagonists, have been suggested as an alternative therapy to dopamine receptor antagonists in the treatment of schizophrenia (Carlsson and Carlsson, 1990). Clearly, our understanding of how regulation of NAcc neurons might influence behavior is incomplete (see Pennartz et al., 1994). However, the present cellular and molecular model of an interaction between dopaminergic and glutamatergic neurotransmission in the NAcc, which contains both synergistic and antagonistic components, begins to account for such interactions observed in behavioral studies.
Footnotes
We are grateful to the Medical Research Council (Grant G9208513) and to Glaxo-Wellcome Research and Development, Greenford, UK, for financial support.
Correspondence should be addressed to Dr. Michael G. Lacey, Department of Pharmacology, The Medical School, University of Birmingham, Vincent Drive, Edgbaston, Birmingham B15 2TT, UK.
Dr. Harvey’s present address: Department of Biomedical Sciences, Institute of Medical Sciences, University of Aberdeen, Foresterhill, Aberdeen AB25 2ZD, Scotland, UK.
REFERENCES
- 1.Beavo JA, Reifsnyder DH. Primary sequences of cyclic nucleotide phosphodiesterase isoenzymes and the design of selective inhibitors. Trends Pharmacol Sci. 1990;11:150–155. doi: 10.1016/0165-6147(90)90066-H. [DOI] [PubMed] [Google Scholar]
- 2.Ben-Ari Y, Aniksztejn L, Bregestovski P. Protein kinase C modulation of NMDA currents: an important link for LTP induction. Trends Neurosci. 1992;15:333–339. doi: 10.1016/0166-2236(92)90049-e. [DOI] [PubMed] [Google Scholar]
- 3.Blanton MG, LoTurco JJ, Kreigstein AR. Whole-cell recording from neurons in slices of reptilian and mammalian cerebral cortex. J Neurosci Methods. 1989;30:203–210. doi: 10.1016/0165-0270(89)90131-3. [DOI] [PubMed] [Google Scholar]
- 4.Bonci A, Williams JT. A common mechanism mediates long-term changes in synaptic transmission after chronic cocaine and morphine. Neuron. 1996;16:631–639. doi: 10.1016/s0896-6273(00)80082-3. [DOI] [PubMed] [Google Scholar]
- 5.Brundege JM, Dunwiddie TV. Modulation of excitatory synaptic transmission by adenosine released from single hippocampal pyramidal neurons. J Neurosci. 1996;16:5603–5612. doi: 10.1523/JNEUROSCI.16-18-05603.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Burns LH, Everitt BJ, Kelley AE, Robbins TW. Glutamate–dopamine interactions in the ventral striatum: role in locomotor activity and responding with conditioned reinforcement. Psychopharmacology. 1994;115:516–528. doi: 10.1007/BF02245576. [DOI] [PubMed] [Google Scholar]
- 7.Carlezon WA, Jr, Wise RA. Rewarding actions of phencyclidine and related drugs in nucleus accumbens shell and frontal cortex. J Neurosci. 1996;16:3112–3122. doi: 10.1523/JNEUROSCI.16-09-03112.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carlsson M, Carlsson A. Interactions between glutamatergic and monoaminergic systems within the basal ganglia–implications for schizophrenia and Parkinson’s disease. Trends Neurosci. 1990;13:272–276. doi: 10.1016/0166-2236(90)90108-m. [DOI] [PubMed] [Google Scholar]
- 9.Cepeda C, Buchwald NA, Levine MS. Neuromodulatory actions of dopamine in the neostriatum are dependent upon the excitatory amino acid receptor subtypes activated. Proc Natl Acad Sci USA. 1993;90:9576–9580. doi: 10.1073/pnas.90.20.9576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Colwell CS, Levine MS. Excitatory synaptic transmission in neo-striatal neurons: regulation by cyclic AMP-dependent mechanisms. J Neurosci. 1995;15:1704–1713. doi: 10.1523/JNEUROSCI.15-03-01704.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Craig CG, White TD. N-methyl-d-aspartate- and non-N-methyl-d-aspartate-evoked adenosine release from rat cortical slices: distinct purinergic sources and mechanisms of release. J Neurochem. 1993;60:1073–1080. doi: 10.1111/j.1471-4159.1993.tb03256.x. [DOI] [PubMed] [Google Scholar]
- 12.Deutch AY, Cameron DS. Pharmacological characterization of dopamine systems in the nucleus accumbens core and shell. Neuroscience. 1992;46:49–56. doi: 10.1016/0306-4522(92)90007-o. [DOI] [PubMed] [Google Scholar]
- 13.Felder CC, Albrecht FE, Campbell T, Eisner GM, Jose PA. cAMP-independent, G-protein-linked inhibition of Na+/H+ exchange in renal brush border by D1 dopamine agonists. Am J Physiol. 1993;264:F1032–F1037. doi: 10.1152/ajprenal.1993.264.6.F1032. [DOI] [PubMed] [Google Scholar]
- 14.Ferré S, O’Connor WT, Svenningsson P, Bjorklund L, Lindberg J, Tinner B, Stromberg I, Goldstein M, Ogren SO, Ungerstedt U, Fredholm BB, Fuxe K. Dopamine D1 receptor-mediated facilitation of GABAergic neurotransmission in the rat strioentopeduncular pathway and its modulation by adenosine A1 receptor-mediated mechanisms. Eur J Neurosci. 1996;8:1545–1553. doi: 10.1111/j.1460-9568.1996.tb01617.x. [DOI] [PubMed] [Google Scholar]
- 15.Fibiger HC, Phillips AG. Reward, motivation, cognition: psychobiology of mesentelencephalic dopamine systems. In: Mountcastle VB, Bloom FE, editors. Handbook of physiology, Vol IV, Intrinsic regulatory systems of the brain, Sec 1, The nervous system. American Physiological Society; Bethesda, MD: 1986. pp. 647–675. [Google Scholar]
- 16.Fredholm BB. Purinoceptors in the nervous system. Pharmacol Toxicol. 1995;76:228–239. doi: 10.1111/j.1600-0773.1995.tb00135.x. [DOI] [PubMed] [Google Scholar]
- 17.Garthwaite J, Boulton CL. Nitric oxide signaling in the central nervous system. Annu Rev Physiol. 1995;57:683–706. doi: 10.1146/annurev.ph.57.030195.003343. [DOI] [PubMed] [Google Scholar]
- 18.Gereau RW, Conn PJ. Potentiation of cAMP responses by metabotropic glutamate receptors depresses excitatory synaptic transmission by a kinase-independent mechanism. Neuron. 1994;12:1121–1129. doi: 10.1016/0896-6273(94)90319-0. [DOI] [PubMed] [Google Scholar]
- 19.Gerfen CR, Engber TM, Mahan LC, Susel Z, Chase TN, Monsma FJJ, Sibley DR. D1 and D2 dopamine receptor-regulated gene expression of striatonigral and striatopallidal neurons. Science. 1990;250:1429–1432. doi: 10.1126/science.2147780. [DOI] [PubMed] [Google Scholar]
- 20.Guevara-Guzman R, Emson PC, Kendrick KM. Modulation of in vivo striatal transmitter release by nitric oxide and cyclic GMP. J Neurochem. 1994;62:807–810. doi: 10.1046/j.1471-4159.1994.62020807.x. [DOI] [PubMed] [Google Scholar]
- 21.Hamann M, Attwell D. Nonsynaptic release of ATP by electrical stimulation in slices of rat hippocampus, cerebellum, and habenula. Eur J Neurosci. 1996;8:1510–1515. doi: 10.1111/j.1460-9568.1996.tb01613.x. [DOI] [PubMed] [Google Scholar]
- 22.Harvey J, Collingridge GL. Signal transduction pathways involved in the acute potentiation of NMDA responses by 1S,3R-ACPD in rat hippocampal slices. Br J Pharmacol. 1993;109:1085–1090. doi: 10.1111/j.1476-5381.1993.tb13733.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Harvey J, Lacey MG. Tonic attenuation of excitatory synaptic transmission in the nucleus accumbens via presynaptic adenosine A1 receptors in rat brain slices. J Physiol (Lond) 1996a;495:P54–P55. [Google Scholar]
- 24.Harvey J, Lacey MG. Dopamine depresses EPSCs in the nucleus accumbens via NMDA receptor-dependent release of adenosine. Soc Neurosci Abstr. 1996b;22:1739. [Google Scholar]
- 25.Harvey J, Lacey MG. Endogenous and exogenous dopamine depress EPSCs in rat nucleus accumbens in vitro via D1 receptor activation. J Physiol (Lond) 1996c;492:143–154. doi: 10.1113/jphysiol.1996.sp021296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hersch SM, Ciliax BJ, Gutekunst C-A, Rees HD, Heilman CJ, Yung KKL, Bolam JP, Ince E, Yi H, Levey AI. Electron microscopic analysis of D1 and D2 dopamine receptor proteins in the dorsal striatum and their synaptic relationships with motor corticostriatal afferents. J Neurosci. 1995;15:5222–5237. doi: 10.1523/JNEUROSCI.15-07-05222.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Higashi H, Inanaga K, Nishi S, Uchimura N. Enhancement of dopamine actions on rat nucleus accumbens neurones in vitro after methamphetamine pre-treatment. J Physiol (Lond) 1989;408:587–603. doi: 10.1113/jphysiol.1989.sp017478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hyman SE. Addiction to cocaine and amphetamine. Neuron. 1996;16:901–904. doi: 10.1016/s0896-6273(00)80111-7. [DOI] [PubMed] [Google Scholar]
- 29.Iversen SD. Interactions between excitatory amino acids and dopamine systems in the forebrain: implications for schizophrenia and Parkinson’s disease. Behav Pharmacol. 1995;6:478–491. [PubMed] [Google Scholar]
- 30.Kalivas PW, Sorg BA, Hooks MS. The pharmacology and neural circuitry of sensitization to psychostimulants. Behav Pharmacol. 1993;4:315–334. [PubMed] [Google Scholar]
- 31.Keefe KA, Gerfen CR. D1 dopamine receptor-mediated induction of zif268 and c-fos in the dopamine-depleted striatum: differential regulation and independence from NMDA receptors. J Comp Neurol. 1996;367:165–176. doi: 10.1002/(SICI)1096-9861(19960401)367:2<165::AID-CNE1>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 32.Kelley AE, Delfs JM. Excitatory amino acid receptors mediate the orofacial stereotypy elicited by dopaminergic stimulation of the ventrolateral striatum. Neuroscience. 1994;60:85–95. doi: 10.1016/0306-4522(94)90205-4. [DOI] [PubMed] [Google Scholar]
- 33.Konradi C, Leveque JC, Hyman SE. Amphetamine and dopamine-induced immediate early gene expression in striatal neurons depends on postsynaptic NMDA receptors and calcium. J Neurosci. 1996;16:4231–4239. doi: 10.1523/JNEUROSCI.16-13-04231.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Koob GF. Drugs of abuse: anatomy, pharmacology, and function of reward pathways. Trends Pharmacol Sci. 1992;13:177–184. doi: 10.1016/0165-6147(92)90060-j. [DOI] [PubMed] [Google Scholar]
- 35.Le Moine C, Bloch B. D1 and D2 dopamine receptor gene expression in the rat striatum: sensitive cRNA probes demonstrate prominent segregation of D1 and D2 mRNAs in distinct neuronal populations of the dorsal and ventral striatum. J Comp Neurol. 1995;355:418–426. doi: 10.1002/cne.903550308. [DOI] [PubMed] [Google Scholar]
- 36.Levine MS, Altemus KL, Cepeda C, Cromwell HC, Crawford C, Ariano MA, Drago J, Sibley DR, Westphal H. Modulatory actions of dopamine on NMDA receptor-mediated responses are reduced in D1A-deficient mutant mice. J Neurosci. 1996;16:5870–5882. doi: 10.1523/JNEUROSCI.16-18-05870.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lovinger DM, Choi S. Activation of adenosine A1 receptors initiates short-term synaptic depression in rat striatum. Neurosci Lett. 1995;199:9–12. doi: 10.1016/0304-3940(95)12024-x. [DOI] [PubMed] [Google Scholar]
- 38.Mahan LC, Burch RM, Monsma FJJ, Sibley DR. Expression of striatal D1 dopamine receptors coupled to inositol phosphate production and Ca2+ mobilization in Xenopus oocytes. Proc Natl Acad Sci USA. 1990;87:2196–2200. doi: 10.1073/pnas.87.6.2196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Manzoni OJ, Manabe T, Nicoll RA. Release of adenosine by activation of NMDA receptors in the hippocampus. Science. 1994;265:2098–2101. doi: 10.1126/science.7916485. [DOI] [PubMed] [Google Scholar]
- 40.Markram H, Segal M. The inositol 1,4,5-trisphosphate pathway mediates cholinergic potentiation of rat hippocampal neuronal responses to NMDA. J Physiol (Lond) 1992;447:513–533. doi: 10.1113/jphysiol.1992.sp019015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Martin G, Nie Z, Siggins GR. μ-Opioid receptors modulate NMDA receptor-mediated responses in nucleus accumbens neurons. J Neurosci. 1997;17:11–22. doi: 10.1523/JNEUROSCI.17-01-00011.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Miller B, Sarantis M, Traynelis SF, Attwell D. Potentiation of NMDA receptor currents by arachidonic acid. Nature. 1992;355:722–725. doi: 10.1038/355722a0. [DOI] [PubMed] [Google Scholar]
- 43.Nicola SH, Kombian SB, Malenka RC. Psychostimulants depress excitatory synaptic transmission in the nucleus accumbens via presynaptic D1-like dopamine receptors. J Neurosci. 1996;16:1591–1604. doi: 10.1523/JNEUROSCI.16-05-01591.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.O’Donnell P, Grace AA. Physiological and morphological properties of accumbens core and shell neurons recorded in vitro. Synapse. 1993;13:135–160. doi: 10.1002/syn.890130206. [DOI] [PubMed] [Google Scholar]
- 45.Pennartz CMA, Dolleman-Van der Weel MJ, Kitai ST, Lopes da Silva FH. Presynaptic dopamine D1 receptors attenuate excitatory and inhibitory limbic inputs to the shell region of the rat nucleus accumbens studied in vitro. J Neurophysiol. 1992;67:1325–1334. doi: 10.1152/jn.1992.67.5.1325. [DOI] [PubMed] [Google Scholar]
- 46.Pennartz CMA, Groenewegen HJ, Lopes da Silva FH. The nucleus accumbens as a complex of functionally distinct neuronal ensembles: an integration of behavioural, electrophysiological, and anatomical data. Prog Neurobiol. 1994;42:719–761. doi: 10.1016/0301-0082(94)90025-6. [DOI] [PubMed] [Google Scholar]
- 47.Pijnenberg AJJ, Van Rossum JM. Stimulation of locomotor activity following injections of dopamine into the nucleus accumbens. J Pharm Pharmacol. 1973;25:1003–1005. doi: 10.1111/j.2042-7158.1973.tb09995.x. [DOI] [PubMed] [Google Scholar]
- 48.Piomelli D, Pilon C, Giros B, Sokoloff P, Martres MP, Schwartz JC. Dopamine activation of the arachidonic acid cascade as a basis for D1/D2 receptor synergism. Nature. 1991;353:164–167. doi: 10.1038/353164a0. [DOI] [PubMed] [Google Scholar]
- 49.Pisani A, Calabresi P, Centonze D, Bernardi G. Enhancement of NMDA responses by group I metabotropic glutamate receptor activation in striatal neurones. Br J Pharmacol. 1997;120:1007–1014. doi: 10.1038/sj.bjp.0700999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pulvirenti L, Swerdlow NR, Koob GF. Nucleus accumbens NMDA antagonist decreases locomotor activity produced by cocaine, heroin, or accumbens dopamine, but not caffeine. Pharmacol Biochem Behav. 1991;40:841–845. doi: 10.1016/0091-3057(91)90095-j. [DOI] [PubMed] [Google Scholar]
- 51.Rahman S, Neuman RS. Characterization of metabotropic glutamate receptor-mediated facilitation of N-methyl-d-aspartate depolarization of neocortical neurones. Br J Pharmacol. 1996;117:675–683. doi: 10.1111/j.1476-5381.1996.tb15243.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Robbins TW, Everitt BJ. Neurobehavioural mechanisms of reward and motivation. Curr Opin Neurobiol. 1996;6:228–236. doi: 10.1016/s0959-4388(96)80077-8. [DOI] [PubMed] [Google Scholar]
- 53.Rodrigues P, Dowling JE. Dopamine induces neurite retraction in retinal horizontal cells via diacylglycerol and protein kinase C. Proc Natl Acad Sci USA. 1990;87:9693–9697. doi: 10.1073/pnas.87.24.9693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schuman EM, Madison DV. Nitric oxide and synaptic function. Annu Rev Neurosci. 1994;17:153–183. doi: 10.1146/annurev.ne.17.030194.001101. [DOI] [PubMed] [Google Scholar]
- 55.Stoof JC, Kebabian JW. Opposing roles for D1 and D2 dopamine receptors in efflux of cyclic AMP from rat neostriatum. Nature. 1981;294:366–368. doi: 10.1038/294366a0. [DOI] [PubMed] [Google Scholar]
- 56.Uchimura N, North RA. Baclofen and adenosine inhibit synaptic potentials mediated by gamma-aminobutyric acid and glutamate release in rat nucleus accumbens. J Pharmacol Exp Ther. 1991;258:663–668. [PubMed] [Google Scholar]
- 57.Wan FJ, Geyer MA, Swerdlow NR. Presynaptic dopamine–glutamate interactions in the nucleus accumbens regulate sensorimotor gating. Psychopharmacology (Berl) 1995;120:433–441. doi: 10.1007/BF02245815. [DOI] [PubMed] [Google Scholar]
- 58.Wang HY, Undie AS, Friedman E. Evidence for the coupling of Gq protein to D1-like dopamine sites in rat striatum: possible role in dopamine-mediated inositol phosphate formation. Mol Pharmacol. 1995;48:988–994. [PubMed] [Google Scholar]
- 59.Wang JQ, McGinty JF. Acute methamphetamine-induced zif/268, preprodynorphin, and preproenkephalin mRNA expression in rat striatum depends on activation of NMDA and kainate/AMPA receptors. Brain Res Bull. 1996;39:349–357. doi: 10.1016/0361-9230(96)00002-0. [DOI] [PubMed] [Google Scholar]
- 60.Wilson CJ, Kawaguchi Y. The origins of two-state spontaneous membrane potential fluctuations of neostriatal spiny neurons. J Neurosci. 1996;16:2397–2410. doi: 10.1523/JNEUROSCI.16-07-02397.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wise RA. Neurobiology of addiction. Curr Opin Neurobiol. 1996;6:243–251. doi: 10.1016/s0959-4388(96)80079-1. [DOI] [PubMed] [Google Scholar]
- 62.Yu PY, Asico LD, Eisner GM, Jose PA. Differential regulation of renal phospholipase C isoforms by catecholamines. J Clin Invest. 1995;95:304–308. doi: 10.1172/JCI117656. [DOI] [PMC free article] [PubMed] [Google Scholar]