

Abstract
Cell death was examined by studying the spinal cords of rats subjected to traumatic insults of mild to moderate severity. Within minutes after mild weight drop impact (a 10 gm weight falling 6.25 mm), neurons in the immediate impact area showed a loss of cytoplasmic Nissl substances. Over the next 7 d, this lesion area expanded and cavitated. Terminal deoxynucleotidyl transferase (TdT)-mediated deoxyuridine triphosphate–biotin nick end labeling (TUNEL)-positive neurons were noted primarily restricted to the gross lesion area 4–24 hr after injury, with a maximum presence at 8 hr after injury. TUNEL-positive glia were present at all stages studied between 4 hr and 14 d, with a maximum presence within the lesion area 24 hr after injury. However 7 d after injury, a second wave of TUNEL-positive glial cells was noted in the white matter peripheral to the lesion and extending at least several millimeters away from the lesion center. The suggestion of apoptosis was supported by electron microscopy, as well as by nuclear staining with Hoechst 33342 dye, and by examination of DNA prepared from the lesion site. Furthermore, repeated intraperitoneal injections of cycloheximide, beginning immediately after a 12.5 mm weight drop insult, produced a substantial reduction in histological evidence of cord damage and in motor dysfunction assessed 4 weeks later. Present data support the hypothesis that apoptosis dependent on active protein synthesis contributes to the neuronal and glial cell death, as well as to the neurological dysfunction, induced by mild-to-moderate severity traumatic insults to the rat spinal cord.
Keywords: acute SCI, apoptosis, cell death, contusion injury, spinal cord, rat, cycloheximide, motor function
Traumatic insults to the spinal cord induce both immediate mechanical damage and subsequent tissue degeneration, the latter progressing in a setting of ischemia, hemorrhage, and edema (Allen, 1914; Ducker et al., 1971; Fairholm and Turnbull, 1971; Green and Wagner, 1973; Osterholm, 1974; Senter and Venes, 1978; Young, 1993). There has been a long-standing, primarily implicit assumption that such trauma-induced spinal cord cell death, like the death of brain cells induced by several acute insults, represents the form of death called “necrosis” (Kerr et al., 1972; Balentine, 1978a,b;Selina et al., 1989). This assumption is consistent with the more recent implication of excitotoxicity in the pathogenesis of traumatic spinal cord damage (Faden and Simon, 1988; Panter et al., 1990;Wrathall et al., 1992), because excitotoxicity is typically marked by early neuronal cell swelling (Coyle et al., 1981) and likely leads preferentially to necrosis (Csernansky et al., 1994; Gwag et al., 1996).
Over the past few years, however, growing evidence has suggested that both global (Goto et al., 1990; Shigeno et al., 1990; Papas et al., 1992; Heron et al., 1993; Okamoto et al., 1993; Roberts-Lewis et al., 1993; Kihara et al., 1994; Nitatori et al., 1995) and focal (Linnik et al., 1993; Tominaga et al., 1993; MacManus et al., 1994;Charriaut-Marlangue et al., 1995; Li et al., 1995; Du et al., 1996a,b) ischemic brain cell loss may in part reflect programmed cell death, resulting in apoptosis (Kerr et al., 1972; Wyllie et al., 1980; Arends and Wyllie, 1991; Johnson et al., 1995). In cortical cell cultures deprived of oxygen and glucose, neurons die predominantly by excitotoxic necrosis (Goldberg and Choi, 1993), but if excitotoxicity is blocked by the combined application of NMDA receptor and AMPA/kainate receptor antagonists, then neurons undergo apoptosis (Gwag et al., 1995) as do cultured sympathetic neurons exposed to hypoxia (Rosenbaum et al., 1994). Apoptosis of brain cells likely also occurs after traumatic insults (Rink et al., 1995) and may occur in the setting of neurodegenerative diseases (Portera-Cailliau et al., 1995;Thompson, 1995).
It seemed therefore plausible that apoptosis might also contribute to the spinal cord cell loss occurring after traumatic insults. We initiated the present study to look for apoptosis in the spinal cords of rats subjected to moderate severity traumatic insults using a well characterized injury model (Gruner, 1992). In the meantime, independent evidence supporting this idea has been reported from other laboratories (see Discussion). An abstract on our current research has been published previously (Liu et al., 1996).
MATERIALS AND METHODS
Spinal cord injury (SCI). Impact injury was induced using the weight drop device developed at New York University (Gruner, 1992). Adult Long–Evans female rats (Simonsen Lab, Gilroy, CA) were anesthetized with pentobarbital (50 mg/kg, i.p.). During surgery, rectal temperature was maintained at 37.0 ± 0.2°C by a thermostatically regulated heating pad (Versa-Therm 2156; Cole-Parmer, Chicago, IL). A laminectomy was performed at the T9–T10 level, exposing the cord underneath without disrupting the dura. After the spinous processes of T8 and T11 were clamped to stabilize the spine, the exposed dorsal surface of the cord was subjected to weight drop impact using a 10 gm rod (2.5 mm in diameter) dropped at a height of either 6.25 or 12.5 mm and following the procedure guidelines established by a multicenter consortium (Multicenter Animal Spinal Cord Injury Study; Basso et al., 1995, 1996a,b). Animals subjected to the 6.25 mm insult developed near complete recovery of hindlimb motor function 4 weeks after injury. Therefore, the 12.5 mm insult was used for testing of therapeutic intervention.
After the injury, the muscles and skin were closed in layers, and rats were placed in a temperature- and humidity-controlled chamber (Thermocare, Incline Village, NV) overnight. Manual bladder expression was performed three times per day until reflex bladder emptying was established. All of the surgical interventions and presurgical and postsurgical animal care were provided in accordance with the Laboratory Animal Welfare Act, the Guide for the Care and Use of Laboratory Animals (National Research Council, 1996), and the Guidelines and Policies for Rodent Survival Surgery provided by the Animal Studies Committee of Washington University School of Medicine.
Cycloheximide administration. Cycloheximide (Sigma, St. Louis, MO) was dissolved in sterile saline at a concentration of 1 mg/ml. Rats receiving the 12.5 mm insult were assigned randomly to receive either cycloheximide (1 mg/kg) or vehicle that was injected intraperitoneally immediately after insult and then every third day for 4 weeks.
Behavioral tests. Behavioral tests were performed by investigators blinded to the treatment groups and using the Basso–Beattie–Bresnahan (BBB) scales. Testing began 1 d after the 12.5 mm weight drop injury and then continued twice per week for 4 weeks.
Histopathology. After receiving behavioral testing for 4 weeks, 24 animals subjected to the 12.5 mm injury were given a lethal overdose of pentobarbital and perfused intracardially with normal saline followed by 4% paraformaldehyde in PBS, pH 7.4. For histological evaluation, a 15 mm cord segment centered at the injury site was removed from the vertebral canal, was placed in the same fixative overnight, and was embedded in paraffin. Serial 10 μm cross-sections were cut and stained with hematoxylin and eosin. The spared areas of the spinal cord at the epicenter (0), at 750 and 1500 μm rostral (−750 and −1500 μm), and at 750 and 1500 μm caudal (+750 and +1500 μm) to the epicenter were measured using a Metamorph image analysis system (Universal Imaging orporation, West Chester, PA). Measurements were performed and analyzed by an investigator blind to treatment group assignment.
Another 64 rats were subjected to the 6.25 mm injury. Four animals were euthanized at 5 min, 4, 8, and 24 hr, and 3, 7, 14, and 30 d after insult using the perfusion and embedding procedure described above. Serial 7 μm longitudinal (coronal) sections were cut. These sections through the central canal were Nissl-stained and examined under an Olympus BX 60 microscope. Longitudinal sections through the anterior horn were used for terminal deoxynucleotidyl transferase (TdT)-mediated deoxyuridine triphosphate (dUTP)–biotin nick end labeling (TUNEL), for Hoechst 33342 staining, or for staining for neuronal-specific enolase. Camera lucida drawings of lesion size and TUNEL-positive cells were performed using a drawing tube attached to the microscope. The rest of the rats were perfused as described above, and the spinal cords were post-fixed for 4 hr and were then transferred in 30% sucrose overnight. Fourteen micrometer longitudinal sections through the anterior horn were cut for rip immunohistochemistry. All the histological analyses were performed blinded to experimental condition.
TUNEL staining. Paraffin-embedded cord sections were deparaffinized in two changes of xylene for 5 min each. The sections were washed sequentially in 100, 95, and 75% ethanol before being incubated with 20 μg/ml proteinase K (Sigma, St. Louis, MO) for 5 min to strip off nuclear proteins. TUNEL was accomplished using the Apoptagin situ kit obtained from Oncor (Gaithersburg, MD). After immersion in equilibration buffer for 10 min, sections were incubated with TdT and dUTP-digoxigenin in a humidified chamber at 37°C for 1 hr and then incubated in the stop/wash buffer at 37°C for 30 min to stop the reaction. The sections were washed with PBS once before incubation in antidigoxigenin–peroxidase solution for 30 min. They were colorized with diaminobenzidine–H2O2solution (0.2 mg/ml tetrachloride and 0.005% H2O2 in 50 mm Tris-HCl buffer) and then counterstained with methyl green. Control sections were treated similarly but incubated in the absence of TdT enzyme, dUTP-digoxigenin, or anti-digoxigenin antibody.
Double staining. Sections from different time points were double-stained by TUNEL and by immunohistochemical staining with a polyclonal antibody directed against neuron-specific enolase (Dako, Carpinteria, CA) or with the oligodendrocyte-specific monoclonal antibody rip (Developmental Studies Hybridoma Bank, Iowa City, IA) (Friedman et al., 1989). To identify neurons, we first subjected fixed sections to TUNEL and then blocked the sections with 5% horse serum, washed them in PBS, and incubated them with mouse neuron-specific enolase antibody. The sections were again washed in PBS and incubated in biotinylated horse anti-mouse IgG (Vector Laboratories, Burlingame, CA) and in avidin–biotin complex (Vector Laboratories). Peroxidase was demonstrated with a Vector SG kit (Vector Laboratories). Negative controls were stainings performed without primary antibody. To identify oligodendrocytes, we incubated frozen sections overnight in rip diluted 1:200. Sections were washed with PBS for 10 min, incubated with secondary anti-mouse IgG antibody conjugated to fluorescein, and then subjected to TUNEL using a secondary antibody conjugated to rhodamine. Sections were examined under epifluorescence illumination on a Olympus BX 60 microscope.
Hoechst 33342 staining. Paraffin sections were deparaffinized with xylene two times for 5 min each and then rinsed with PBS. The sections were first stained with 10 mg/ml Hoechst 33342 from Molecular Probes (Eugene, OR) for 5 min, washed with PBS, and then stained with propidium iodide (1:1000) from Molecular Probes for 5 min.
Transmission electron microscopy (EM). Cross-sections of the spinal cord were cut serially every 500 μm through the epicenter of the injury in rats that received the 6.25 mm injury and were perfused at 4, 8, and 24 hr after injury. Samples were fixed in 2.5% glutaraldehyde in 0.1 m cacodylate buffer, pH 7.4, for 60 min at 4°C and then washed in the same buffer for 80 min. After post-fixation in 1% osmium tetroxide in 0.1 m cacodylate buffer, pH 7.4, for 60 min at room temperature, the tissues were dehydrated in graded ethanol and were embedded in Spurr’s epoxy resin (Spurr, 1969). One micrometer semithin plastic sections were stained with 1% toluidine blue for light microscopic observations. Ultrathin sections of the same specimen were cut and stained with uranyl acetate and lead citrate and examined with a Zeiss 108 electron microscope.
Quantitation of histone-associated DNA fragmentation. The extent of histone-associated DNA fragmentation was assessed using an ELISA kit (Cell Death Detection ELISA) obtained from Boehringer Mannheim (Indianapolis, IN). The assay is based on the quantitative sandwich enzyme immunoassay principle with mouse monoclonal antibodies directed against DNA and histones, respectively and detects mononucleosomes and oligonucleosomes.
Rats subjected to the 6.25 mm injury were euthanized under deep anesthesia at 4 and 24 hr and 3 and 7 d after injury (n = 3 animals at each time point). A 5 mm segment of the injured cord at the epicenter or of the normal cord (n = 3) was dissected, homogenized, and centrifuged (14,890 × g for 10 min). The supernatant was diluted 1:200 and used as an antigen source in sandwich ELISA with a primary anti-histone antibody coated to the microliter plate and a secondary anti-DNA antibody coupled to peroxidase.
DNA gel electrophoresis. Spinal cord DNA was isolated according to the method of Sambrook et al. (1989) with a few modifications. Briefly, rats receiving the 6.25 mm injury were euthanized at 4, 8, and 24 hr, and 50 mg of fresh cord tissue was removed and homogenized individually in an Eppendorf tube containing 300 μl of cell lysis buffer (10 mm Tris-HCl, 100 mm EDTA, and 0.5% SDS). After mixing with an additional 300 μl of cell lysis buffer, the sample was incubated for 1 hr at 65°C and then incubated with proteinase K (final concentration, 100 μg/ml) overnight at 55°C. Extraction was performed with an equal volume of phenol, equilibrated with 0.5 m Tris-HCl, and phenol/chloroform/amyl alcohol (25:24:1). Total DNA contained in the aqueous phase was precipitated with ethanol. The DNA pellet was washed twice with 70% ethanol and dissolved in 25 μl Tris–EDTA buffer (10 mm Tris-HCl, pH 8.0, and 1 mm EDTA, pH 8.0). The DNA was then treated with DNase-free RNase (10 mg/ml) for 1 hr at 37°C and assayed by optical absorption at 260 nm. Equal amounts of DNA samples were subjected to 1.5% agarose gel electrophoresis.
Statistical analysis. Data are expressed as mean ± SEM. To analyze differences in open field locomotor scores or in volumes of spared tissue between groups, we used a repeated measures ANOVA with Tukey’s studentized range test to correct for multiple comparisons. A two-way ANOVA was also used for comparison of the extent of histone-associated DNA fragmentation between groups. For the difference between groups at each time point, we used a pairwisepost hoc Tukey’s studentized range test.p < 0.05 was considered significant.
RESULTS
Morphological features
Five minutes after a weight drop insult (a 10 gm rod, 2.5 mm in diameter, falling 6.25 mm), a well defined area of gray matter injury, defined by loss of neuronal Nissl staining and petechiae, was apparent (Fig. 1) with a clear boundary between damaged and morphologically normal neurons. The injured region had a longitudinal extent of 2.31 ± 0.08 mm (mean ± SEM, n = 4 animals) and a transverse extent of 1.17 ± 0.13 mm. Little gross damage to white matter was initially apparent.
Fig. 1.
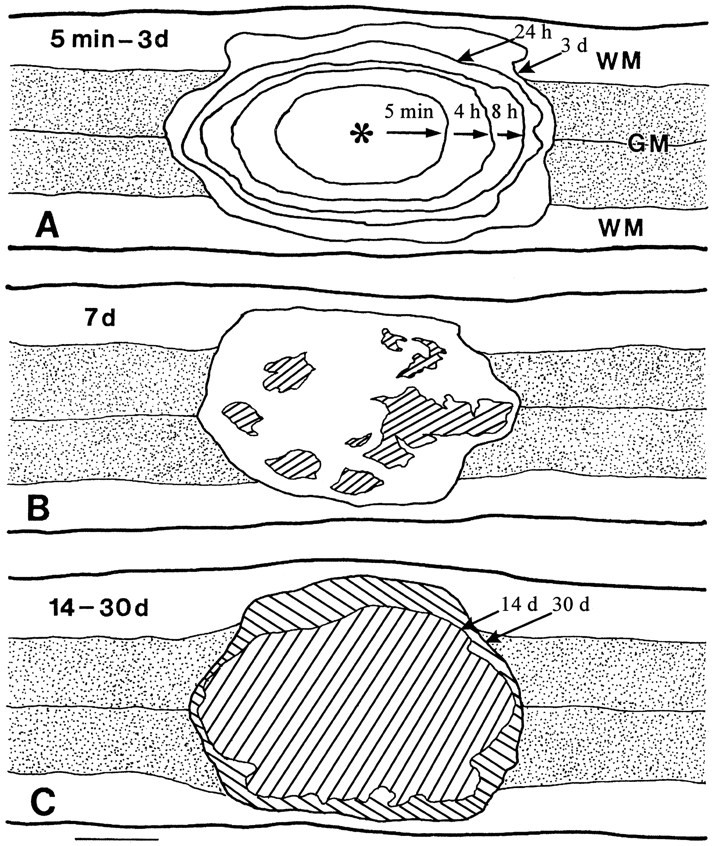
Schematic drawings showing longitudinal (coronal) sections through the central canal in animals receiving the 6.25 mm impact injury and being perfused at 5 min, 4, 8, and 24 hr, and 3 d (A); 7 d (B); and 14 and 30 d (C). Each contour is the average from four animals. Progressive expansion of the lesion was seen, initially defined by the disappearance of Nissl substance from neurons (5 min–4 hr), later defined by the breaking down of axonal segments and myelin as well as the invasion of blood cells into white matter, and still later defined by gross cavitation (7–30 d,cross-hatched areas). By 14 d, the lesion consisted entirely of a cavity and was somewhat smaller than the lesion defined at 3 d; this cavity was somewhat increased by 30 d.WM, White matter; GM, gray matter; *Site of impact. Scale bar, 1 mm.
The gross lesion area expanded over time. By 3 d, the lesion area extended to 4.62 ± 0.08 mm along the cord axis and 2.60 ± 0.05 mm transversely (Fig. 1). White matter injury became grossly apparent 8 hr after injury, delineated by the breaking down of axonal segments and myelin as well as the invasion of blood cells into white matter, although changes in white matter could be seen 4 hr after injury by EM. EM changes, including axonal swelling, degenerating axonal segments, and thinning or loss of myelin sheaths, were observed, in agreement with earlier studies (Balentine, 1978b; Bresnahan, 1978;Blight, 1983; Kao et al., 1983; Blight and Decrescito, 1986; Beattie et al., 1988).
By 7 d, multiple cavitations were noted within the lesion area. By 14 d, a large central cavity formed that expanded modestly further by 30 d.
TUNEL
Five min after injury, no TUNEL-positive cells were observed. After 4 hr, many darkly TUNEL-positive cells were present in the gray matter, confined to the lesion area as defined by loss of Nissl staining and petechiae. Morphology as well as double staining for neuronal-specific enolase indicated that many of these TUNEL-positive cells were neurons (Figs.2, 3, 4). These TUNEL-positive neurons typically exhibited shrinkage of both cytoplasm and nucleus, creating pericellular space, and nuclear fragmentation. The mean nuclear diameter of TUNEL-labeled neurons was 6.74 ± 0.34 μm (mean ± SD, n = 36), approximately half the size of TUNEL-negative neurons in the same sections (13.01 ± 0.59 μm, mean ± SD; n = 42). Neuronal TUNEL positivity was maximal at 8 hr after injury and decreased subsequently (Fig. 4). By 24 hr after injury, few TUNEL-positive neurons were seen (Fig. 4).
Fig. 2.

TUNEL-labeled (A–F) and Hoechst 33342-stained (G–I) cells in the spinal cord after a 6.25 mm injury. A, Longitudinal section of the cord showing numerous TUNEL-positive cells (arrowheads) present within the injury area 8 hr after injury. B, Higher magnification of demarcated region in A showing two TUNEL-labeled neurons with chromatin condensation. C, Section showing TUNEL-labeled neuron) (arrowhead) that can be distinguished from labeled glial cells (thin arrows) by its relatively larger nucleus, its prominent cytoplasm, and the pericellular space created by its shrinkage (thick arrows). The labeled nucleus of this neuron is smaller than the nuclei of neighboring morphologically normal neurons (D). E, Section showing breakdown of the nucleus of a glial cell in the white matter near the injury epicenter into several fragments. F, Section showing TUNEL-labeled glial cell, presumably an oligodendrocyte (thick arrow), in the peripheral white matter away from the injury epicenter. Note the close association of the cell with the space (asterisks) likely created by a degenerated axon. A morphologically normal oligodendrocyte nucleus (thin arrow) and a degenerating axonal segment (arrowhead) are also marked for comparison. G–I, Hoechst 33342-stained sections. These show nuclear fragmentation of a probable neuron (H, arrow) and a glial cell (I, arrow) in the spinal cord 24 hr after injury. The nuclear labeling of a morphologically normal neuron (G, arrow) is presented for comparison.WM, White matter; GM, gray matter. Scale bars: A, 50 μm; B–F, 10 μm; G–I, 10 μm.
Fig. 3.

Double staining of cells in the spinal cord after a 6.25 mm injury. TUNEL-positive cells were also positive for neuronal-specific enolase or rip. A, Section from rat euthanized 8 hr after injury showing two shrunken neurons within the lesion area that were labeled simultaneously by TUNEL (brown) and neuronal-specific enolase immunohistochemistry (blue gray). B, Two other neurons in the same section used in A but outside the lesion area that were labeled for neuronal-specific enolase but were TUNEL negative. C, Section from rat euthanized 7 d after injury showing representative immunofluorescence micrograph showing a spinal cord oligodendrocyte cell body that was located in white matter labeled for TUNEL (yellow) as well as rip (green). Scale bars: A,B, 10 μm; C, 10 μm.
Fig. 4.

Schematic drawing of TUNEL-stained longitudinal (coronal) sections of the spinal cord through the ventral horns, after a 6.25 mm weight drop insult and euthanization of the rats at the indicated times after injury. Five minutes after insult, no TUNEL-labeled cells were observed. By 8 hr, many TUNEL-positive neurons (large dots) and glial cells (small dots) were observed mostly within the lesion area (defined by loss of Nissl substance from neurons). By 24 hr, TUNEL-positive neurons were no longer found, but many TUNEL-positive glial cells were seen within the injury area. By 7 d, a second wave of TUNEL-positive glial cells was observed mostly outside of the lesion area in the lateral funiculus extending the entire length of the section (1.5 cm). By 14 d, fewer TUNEL-labeled glia were found. WM, White matter;GM, gray matter; *Site of impact. Scale bar, 1 mm.
Apoptotic changes in presumptive glial cells were also observed beginning 4 hr after injury and with a maximum at 24 hr after injury (Fig. 4, data for 4 hr not shown). Most of these TUNEL-positive glial cells were found within the lesion area, although some were present in the neighboring white matter (Fig. 4). The cells typically exhibited small, fragmented nuclei with little visible cytoplasm surrounding them (Fig. 2). The mean nuclear diameter of TUNEL-positive glial cells was 4.53 ± 0.25 μm (mean ± SD, n = 42), compared with 7.76 ± 0.25 μm (mean ± SD, n = 36) in TUNEL-negative glia. By 3 (data not shown) and 7 d after injury, the number of TUNEL-positive glial cells within the lesion area had fallen sharply (Fig. 4; data not shown).
However, by 7 d, a second wave of TUNEL-positive glial cells was observed in white matter extending the entire 1.5 cm length of the coronal section (taken through the ventral horn, Fig. 4). Few TUNEL-positive cells were seen in the white matter at the 3 d time point (data not shown). These TUNEL-positive white matter cells typically showed a close association with degenerating axons (Fig.2F) and stained for the oligodendrocyte-specific marker rip (Fig. 3C) (Friedman et al., 1989).
EM
Transmission EM revealed coexistent apoptotic and necrotic changes in cells within the lesion area 4–24 hr after injury. Apoptotic changes were characterized by cytoplasmic shrinkage, plasma membrane infolding, coarse chromatin condensation, and breakdown of the nucleus into discrete, membrane-bounded bodies (Fig.5A). Occasionally, well preserved apoptotic bodies containing fragmented nuclear chromatin were found within the cytoplasm of macrophages (data not shown). Necrotic changes were characterized by cell, nuclear, and mitochondrial swelling with loosely textured chromatin aggregation and dilation of rough endoplasmic reticulum (Fig. 5B).
Fig. 5.

Electron micrograph showing a representative apoptotic cell and a representative necrotic cell in the gray matter of the cord 8 hr after injury. A, Apoptosis. The nucleus has fragmented into several membrane-bounded, highly condensed bodies (thick arrow), and the cell body has shrunk with an intact, infolded cell membrane (arrowheads).B, Necrosis. The nucleus (Nu) is swollen (arrowhead) with scattered granular aggregations of chromatin (thick arrows), and the cell is swollen with dilation of rough endoplasmic reticulum (thin arrows) and mitochondria (asterisks). RBC, Red blood cell. Scale bars, 1 μm.
DNA laddering
DNA prepared from lesion site tissue 4–24 hr after injury and run on an agarose gel revealed a progressive increase in internucleosomal laddering over this time (Fig. 6).
Fig. 6.

Ethidium bromide-stained agarose gel showing a DNA ladder from a rat spinal cord after a weight drop insult. Data are from rats euthanized: in Lane 1, after no injury (normal control); in lane 2, 4 hr after injury; inlane 3, 8 hr after injury; and in lane 4, 24 hr after injury. Molecular weight markers are shown to theleft of lane 1. DNA laddering is apparent in lane 4 (arrows).
Quantitation of DNA breakdown
Tissue DNA breakdown was quantitated using a commercial ELISA that measures histone-associated mononucleosomes or oligonucleosomes. DNA fragmentation in lesion site tissue from rats receiving 6.25 mm insults was detectable as early as 4 hr after injury, reached a large peak by 24 hr, and then declined. DNA fragmentation was reduced by cycloheximide treatment (1 mg/kg, i.p.) that began immediately after injury and continued once every third day until the animals were euthanized (Fig. 7).
Fig. 7.

Enrichment of mononucleosomes and oligonucleosomes in the cytoplasmic fraction after a 6.25 mm injury (enrichment factor, absorbance of the injured tissue/absorbance of the normal control tissue). An ELISA kit (see Materials and Methods) was used to detect mononucleosomes and oligonucleosomes in the cytoplasmic fraction of spinal cord tissue at the indicated times after injury. Evidence of internucleosomal DNA fragmentation peaked 24 hr after injury and was reduced by cycloheximide treatment. Error bars indicate SEM; *p < 0.05; n = 3 animals per group.
Protective effect of cycloheximide administration
We turned to the protein synthesis inhibitor cycloheximide to try to reduce apoptotic cell loss in this injury model (Martin et al., 1988, 1992; Ciutat et al., 1996; Yaginuma et al., 1996). Having detected evidence of oligodendrocyte apoptosis 7 d after insult, we decided to use a recurrent injection paradigm. Pavlik and Teisinger (1980) showed that subcutaneous injection of a single 0.6 mg/kg dose of cycloheximide resulted in transient (∼12 hr) suppression of brain protein synthesis in rats. We administered 1 mg/kg intraperitoneally immediately after a 12.5 mm weight drop injury and followed with 1 mg/kg intraperitoneally every third day thereafter, a regimen that was well tolerated by the rats presumably because protein synthesis was intermittently released from inhibition. This treatment regimen resulted in the gross sparing of spinal cord tissue compared with vehicle-treated controls measured 4 weeks after injury (Figs. 8,9). The tissue sparing consisted of a wider rim of tissue appearing to approximate normal architecture surrounding a hypercellular, vascularized lesion area with many macrophages (Fig. 10). In addition, open field motor testing using the BBB Locomotor Rating Scale (Basso et al., 1995,1996a,b) showed that this cycloheximide treatment substantially improved hindlimb function compared with vehicle-treated controls (Fig.11). By 4 weeks after injury, the cycloheximide-treated rats had improved to a BBB score of 19 ± 1.71, compared with 14.75 ± 3.17 (mean ± SEM, n = 12) in vehicle-treated controls (a BBB score of 21 is normal). This difference in BBB score reflects a grossly apparent improvement in gait, although there is coordinated plantar stepping at both scores of 15 and 19. At a score of 15, there is little toe clearance during forward limb advancement, and paws are parallel to the body only at initial contact. At a score of 19, there is consistent toe clearance during forward limb advancement, and paws are parallel to the body both at initial contact and at liftoff.
Fig. 8.

Hematoxylin- and eosin-stained horizontal cross-sections taken 4 weeks after injury in rats treated with either vehicle (left) or cycloheximide (right).B, E, Sections through the lesion epicenter (0). A, D, Sections 750 μm rostral to the epicenter (−750 μm). C,F, Sections 750 μm caudal to the epicenter (+750 μm). Scale bar, 100 μm.
Fig. 9.

Quantitation of histological sparing produced by cycloheximide in animals 4 weeks after a 12.5 mm weight drop insult. The rim of nearly normal tissue (some vacuoles can be seen) was measured in both vehicle- and cycloheximide-treated animals. The hypercellular core in cycloheximide-treated animals was considered part of the lesion area and thus not counted as spared tissue. A beneficial effect of cycloheximide on tissue sparing was seen at all levels examined. Error bars indicate SEM; *p < 0.05;n = 12 animals per group.
Fig. 10.

Hematoxylin- and eosin-stained transverse right hemisection at the lesion epicenter in a rat treated with cycloheximide. A, Section showing a wide rim of spared cord tissue (arrow). In the central region, hypercelluar, vascularized tissue with many macrophage-like cells is seen. B, Higher magnification of demarcated region inA showing a border between the peripheral rim of spared cord tissue and this central hypercellular tissue (dotted line). BV, Blood vessel.Arrowheads indicate probable macrophages. Scale bars, 100 μm.
Fig. 11.

Long-term beneficial effect of cycloheximide on the hindlimb neurological function of rats after a 12.5 mm weight drop injury, assessed by the BBB Locomotor Rating Scale. Error bars indicate SEM; *p < 0.05; n = 12 animals per group.
DISCUSSION
Data presented here suggest that apoptosis, involving both neurons and glia, contributes to spinal cord tissue damage after traumatic insults of mild to moderate severity. Our determination of apoptosis relies on multiple criteria: morphology under both light and electron microscopic examination, nuclear chromatin staining with Hoechst 33342 dye and with TUNEL, DNA laddering on gel electrophoresis, an increase in histone-associated mononucleosomes and oligonucleosomes by ELISA, and the sensitivity of gross tissue damage to inhibition of protein synthesis (Wyllie et al., 1980; Kerr and Harmon, 1991; Johnson et al., 1995). Impact-induced spinal cord cell apoptosis was not confined in space to the immediate impact site or in time to the immediate postinjury period. Although there was a burst of neuronal and glial apoptosis in gray and white matter at the lesion site within approximately the first 24 hr, a delayed plethora of oligodendrocyte apoptosis occurred in distant white matter several days later. Because apoptosis is typically a rapid process, over in hours (Bursch et al., 1990), this late apoptosis likely reflects a true wave of delayed oligodendrocyte death.
The idea of delayed oligodendrocyte apoptosis in the spinal cord after traumatic insults was recently proposed by Li et al. (1996), who observed TUNEL-positive, glial fibrillary acidic protein-negative cells in the white matter of rat spinal cords subjected to compression injury, and by Bresnahan et al. (1996), who observed apoptotic oligodendrocytes closely associated with dying axons in monkey spinal cords subjected to contusion injury. Perhaps reflecting specific model differences between their injury model and the model used here, Li et al. (1996) found little TUNEL positivity in gray matter, although Katoh et al. (1996) recently reported the occurrence of TUNEL positivity in cells from both gray and white matter after extradural weight compression injury to the rat spinal cord. Furthermore, morphological evidence for spinal cord cell apoptosis in rats injured with the same New York University impactor used here has been reported in abstracts by Crowe et al. (1995), Shuman et al. (1996), Swoboda et al. (1996), and Yong et al. (1996).
The evolution of the gross lesion observed here is consistent with older studies of impact trauma to the spinal cord, which have described the progressive enlargement of an initial lesion in central gray matter over a period of hours to days to involve contiguous white matter, eventually leading to central cavitation at higher levels of impact severity (Allen, 1914; Goodkin and Campbell, 1969; Ducker et al., 1971;White, 1975). Balentine (1978a,b) performed a detailed study of lesion evolution using both light and electron microscopic methods to examine the spinal cords of Sprague Dawley rats subjected to weight drop injury. He emphasized the occurrence of immediate (3–5 min) multifocal petechial hemorrhages in central gray matter that were followed over the next hours by tissue edema and by the necrosis deaths of both neurons and glia. These necrosis deaths were marked by the loss of cytoplasmic detail and by the swelling of organelles. White matter exhibited progressive “extracellular swelling” thought to represent edema fluid. By 8–72 hr after insult, axons were swollen and granular. The only changes described between 1 and 4 weeks after insult were reactive gliosis, phagocytosis of necrotic debris by macrophages, deposits of calcium and hemosiderin, and the formation of multiloculated cysts.
Did apoptosis occur in the study of Balentine (1978a,b)? Possibly not. He did not comment on the possibility, and although his model was not identical to ours, he used a more severe insult that likely shifted cell death away from apoptosis toward necrosis. On the other hand, coarse condensation of nuclear chromatin, reminiscent of apoptosis, can be seen in his cell electron micrographs (Balentine, 1978a, his Figs. 12, 15).
The idea that a delayed wave of oligodendrocyte apoptosis occurs in spinal cord white matter after traumatic insults is especially intriguing in light of other evidence suggesting that poor myelination of axons can persist long after experimental (Blight, 1985) or human (Bunge et al., 1993) spinal cord injury. Further studies will be needed to identify the mechanisms responsible for this delayed oligodendrocyte death. Most likely, it was triggered by evolving axonal degeneration and subsequent loss of axonally derived survival signals (Barres et al., 1993). Alternatively, delayed oligodendrocyte apoptosis may occur as a result of slowly evolving adverse changes in the cellular milieu distant to the impact site, for example, as a result of inflammatory events (Hsu and Dimitrijevic, 1990). In this latter formulation, all cells might be exposed to low levels of some injury, but oligodendrocytes would be especially vulnerable and would succumb selectively.
Apoptosis in the CNS has been classically considered to occur only during development, in which it plays a vital role in the size matching of cell populations and in the formation of proper synaptic connections. Growing evidence that apoptosis contributes importantly to pathological CNS loss raises the exciting possibility that measures aimed at blocking apoptosis may find therapeutic use in various disease states. To our knowledge, the data reported here are the first to provide direct support for the idea that an antiapoptotic treatment can improve outcome after spinal cord injury. Specifically, intraperitoneal injections of cycloheximide at 3 d intervals produced substantial preservation of tissue, reduced central cavitation, and improved recovery of function. However, the possibility cannot be presently excluded that the beneficial effects of cycloheximide observed here were mediated by mechanisms not related to direct inhibition of programmed cell death. Some alternative mechanisms might be enhancement of cellular glutathione levels because of reduced cysteine use (Ratan et al., 1994) or suppression of neutrophil chemotaxis (Tanabe et al., 1994). Yet another possible mechanism is raised by the interesting finding that low concentrations of cycloheximide that produce only limited inhibition of protein synthesis can induce the production ofBcl2 and antioxidant enzymes (Furukawa et al., 1997). However, we think that the 1 mg/kg dose of cycloheximide used here should have achieved considerable suppression of protein synthesis (Pavlik and Teisinger, 1980).
Whether indeed the beneficial effect of cycloheximide treatment results from reduction of spinal cord cell apoptosis, it will be important to determine the extent to which specific populations of spinal cord cells can be preserved by this treatment, as well as the contribution of each cycloheximide-preserved population to functional benefit. It is possible, for example, that most or even all of the functional benefit is unrelated to lesion size reduction and reflects improved axonal myelination caused by preservation of the oligodendrocyte population. It will also be important to determine whether inhibition of apoptosis can be therapeutically effective against insults more severe than those used here (in controls, sparing <20% of spinal cord tissue at the lesion core but reducing the BBB score to only 15 after recovery). On the optimistic side, however, the current regimen of cycloheximide administration, being intermittent, may not have achieved complete inhibition of protein synthesis-dependent apoptosis. Testing of more specific inhibitors of apoptosis, such as the interleukin-converting enzyme family inhibitors (Kondo et al., 1996; Pronk et al., 1996), as well as combined antiapoptotic and antiexcitotoxic measures (Du et al., 1996b), offers additional goals for future study that may lead to the development of practical clinical therapies.
Footnotes
This work was supported by National Institute of Neurological Disorders and Stroke Grant 32636 to D.W.C., as well as by grants from the American Paralysis Association to C.Y.H. and D.W.C. and from the Daniel Heumann Fund for Spinal Cord Research to X.M.X. We thank Dr. Q. C. Yu from the University of Chicago for helpful discussion of EM data and Dr. Michael Province from Washington University for assistance with statistical analyses. The rip monoclonal antibody developed by B. Friedman was obtained from the Developmental Studies Hybridoma Bank maintained by the Department of Pharmacology and Molecular Sciences, Johns Hopkins University School of Medicine (Baltimore, MD) and by the Department of Biological Sciences, University of Iowa (Iowa City, IA) under Contract N01-HD-2-3144 from the National Institute of Child Health and Human Development.
Correspondence should be addressed to Dr. Dennis W. Choi, Center for the Study of Nervous System Injury and Department of Neurology, Washington University School of Medicine, 660 South Euclid Avenue, Box 8111, Saint Louis, MO 63110-1093.
REFERENCES
- 1.Allen AR. Remarks on the histopathological changes in the spinal cord due to impact. An experimental study. J Nerv Ment Dis. 1914;41:141–147. [Google Scholar]
- 2.Arends MJ, Wyllie AH. Apoptosis: mechanisms and roles in pathology. Int Rev Exp Pathol. 1991;32:223–254. doi: 10.1016/b978-0-12-364932-4.50010-1. [DOI] [PubMed] [Google Scholar]
- 3.Balentine JD. Pathology of experimental spinal cord trauma. I. The necrotic lesion as a function of vascular injury. Lab Invest. 1978a;39:236–253. [PubMed] [Google Scholar]
- 4.Balentine JD. Pathology of experimental spinal cord trauma. II. Ultrastructure of axons and myelin. Lab Invest. 1978b;39:254–266. [PubMed] [Google Scholar]
- 5.Barres BA, Jacobson MD, Schmid R, Sendtner M, Raff MC. Does oligodendrocyte survival depend on axons? Curr Biol. 1993;3:489–497. doi: 10.1016/0960-9822(93)90039-q. [DOI] [PubMed] [Google Scholar]
- 6.Basso DM, Beattie MS, Bresnahan JC. A sensitive and reliable locomotor rating scale for open field testing in rats. J Neurotrauma. 1995;12:1–21. doi: 10.1089/neu.1995.12.1. [DOI] [PubMed] [Google Scholar]
- 7.Basso DM, Beattie MS, Bresnahan JC. Graded histological and locomotor outcomes after spinal cord contusion using the NYU weight drop device versus transection. Exp Neurol. 1996a;139:244–256. doi: 10.1006/exnr.1996.0098. [DOI] [PubMed] [Google Scholar]
- 8.Basso DM, Beattie MS, Bresnahan JC, Anderson DK, Faden AI, Gruner JA, Holford TR, Hsu CY, Noble LJ, Nockels R, Perot PL, Salzman SK, Young W. MASCIS evaluation of open field locomotor scores: effects of experience and teamwork on reliability. J Neurotrauma. 1996b;13:343–359. doi: 10.1089/neu.1996.13.343. [DOI] [PubMed] [Google Scholar]
- 9.Beattie MS, Stokes BT, Bresnahan JC. Experimental spinal cord injury: strategies for acute and chronic intervention based on anatomic, physiological, and behavioral studies. In: Stein DG, Sabel BA, editors. Pharmacological approaches to the treatment of brain and spinal cord injury. Plenum; New York: 1988. pp. 43–74. [Google Scholar]
- 10.Blight AR. Axonal physiology of chronic spinal cord injury in the cat: intracellular recording in vitro. Neuroscience. 1983;10:1471–1486. doi: 10.1016/0306-4522(83)90128-8. [DOI] [PubMed] [Google Scholar]
- 11.Blight AR. Delayed demyelination and macrophage invasion: a candidate for secondary cell damage in spinal cord injury. Cent Nerv Syst Trauma. 1985;2:299–315. doi: 10.1089/cns.1985.2.299. [DOI] [PubMed] [Google Scholar]
- 12.Blight AR, Decrescito V. Morphometric analysis of experimental spinal cord injury in the cat: the relation of injury intensity to survival of myelinated axons. Neuroscience. 1986;19:321–341. doi: 10.1016/0306-4522(86)90025-4. [DOI] [PubMed] [Google Scholar]
- 13.Bresnahan JC. An electron-microscopic analysis of axonal alterations following blunt contusion of the spinal cord of the rhesus monkey (Macaca Mulatta). J Neurol Sci. 1978;37:59–82. doi: 10.1016/0022-510x(78)90228-9. [DOI] [PubMed] [Google Scholar]
- 14.Bresnahan JC, Shuman SL, Beattie MS. Evidence for apoptosis of oligodendroglia in long tracts undergoing wallerian degeneration after spinal cord injury (SCI) in monkeys. Soc Neurosci Abstr. 1996;22:1185. [Google Scholar]
- 15.Bunge RP, Puckett WR, Becerra JL, Marcillo A, Quencer RM. Observations on the pathology of human spinal cord injury: a review and classification of 22 new cases with details from a case of chronic cord compression with extensive focal demyelination. Adv Neurol. 1993;59:75–89. [PubMed] [Google Scholar]
- 16.Bursch W, Kleine L, Tenniswood M. The biochemistry of cell death by apoptosis. Biochem Cell Biol. 1990;68:1071–1074. doi: 10.1139/o90-160. [DOI] [PubMed] [Google Scholar]
- 17.Charriaut-Marlangue C, Margaill I, Plotkine M, Ben-Ari Y. Early endonuclease activation following reversible focal ischemia in the rat brain. J Cereb Blood Flow Metab. 1995;15:385–388. doi: 10.1038/jcbfm.1995.48. [DOI] [PubMed] [Google Scholar]
- 18.Ciutat D, Caldero J, Oppenheim RW, Esquerda JE. Schwann cell apoptosis during normal development and after axonal degeneration induced by neurotoxins in the chick embryo. J Neurosci. 1996;16:3979–3990. doi: 10.1523/JNEUROSCI.16-12-03979.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Coyle JT, Bird SJ, Evans RH, Gulley RL, Nadler JV, Nicklas WJ, Olney JW. Excitatory amino acid neurotoxins: selectivity, specificity, and mechanisms of action. Neurosci Res Program Bull. 1981;19:1–427. [PubMed] [Google Scholar]
- 20.Crowe MJ, Shuman SL, Masters JN, Bresnahan JC, Beattie MS. Morphological evidence suggesting apoptotic nuclei in spinal cord injury. Soc Neurosci Abstr. 1995;21:232. [Google Scholar]
- 21.Csernansky CA, Canzoniero LMT, Sensi SL, Yu SP, Choi DW. Delayed application of aurintricarboxylic acid reduces glutamate-induced cortical neuronal injury. J Neurosci Res. 1994;38:101–108. doi: 10.1002/jnr.490380113. [DOI] [PubMed] [Google Scholar]
- 22.Du C, Hu R, Csernansky CA, Hsu CY, Choi DW. Very delayed infarction after mild focal cerebral ischemia: a role for apoptosis? J Cereb Blood Flow Metab. 1996a;16:195–201. doi: 10.1097/00004647-199603000-00003. [DOI] [PubMed] [Google Scholar]
- 23.Du C, Hu R, Csernansky CA, Liu XZ, Hsu CY, Choi DW. Additive neuroprotective effects of dextrorphan and cycloheximide in rats subjected to transient focal cerebral ischemia. Brain Res. 1996b;718:233–236. doi: 10.1016/0006-8993(96)00162-x. [DOI] [PubMed] [Google Scholar]
- 24.Ducker TB, Kindt GW, Kempe LG. Pathological findings in acute experimental spinal cord trauma. J Neurosurg. 1971;35:700–708. doi: 10.3171/jns.1971.35.6.0700. [DOI] [PubMed] [Google Scholar]
- 25.Faden AI, Simon RP. A potential role for excitotoxins in the pathophysiology of spinal cord injury. Ann Neurol. 1988;23:623–626. doi: 10.1002/ana.410230618. [DOI] [PubMed] [Google Scholar]
- 26.Fairholm DJ, Turnbull IM. Microangiographic study of experimental spinal cord injuries. J Neurosurg. 1971;35:277–286. doi: 10.3171/jns.1971.35.3.0277. [DOI] [PubMed] [Google Scholar]
- 27.Friedman B, Hockfield S, Black JA, Woodruff KA, Waxman SG. In situ demonstration of mature oligodendrocytes and their processes: an immunocytochemical study with a new monoclonal antibody, rip. Glia. 1989;2:380–390. doi: 10.1002/glia.440020510. [DOI] [PubMed] [Google Scholar]
- 28.Furukawa K, Estus S, Fu WM, Mark RJ, Mattson MP. Neuroprotective action of cycloheximide involves induction of BCL-2 and antioxidant pathways. J Cell Biol. 1997;136:1137–1149. doi: 10.1083/jcb.136.5.1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Goldberg MP, Choi DW. Combined oxygen and glucose deprivation in cortical cell culture: calcium-dependent and calcium-independent mechanisms of neuronal injury. J Neurosci. 1993;13:3510–3524. doi: 10.1523/JNEUROSCI.13-08-03510.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Goodkin R, Campbell JB. Sequential pathologic changes in spinal cord injury: a preliminary report. Surg Forum. 1969;20:430–432. [PubMed] [Google Scholar]
- 31.Goto K, Ishige A, Sekiguchi K, Iizuka S, Sugimoto A, Yuzurihara M, Aburada M, Hosoya E, Kogure K. Effects of cycloheximide on delayed neuronal death in rat hippocampus. Brain Res. 1990;534:299–302. doi: 10.1016/0006-8993(90)90144-z. [DOI] [PubMed] [Google Scholar]
- 32.Green BA, Wagner FC., Jr Evolution of edema in the acutely injured spinal cord: a fluorescence microscopic study. Surg Neurol. 1973;1:98–101. [PubMed] [Google Scholar]
- 33.Gruner JA. A monitored contusion model of spinal cord injury in the rat. J Neurotrauma. 1992;9:123–128. doi: 10.1089/neu.1992.9.123. [DOI] [PubMed] [Google Scholar]
- 34.Gwag BJ, Lobner D, Koh JY, Wie MB, Choi DW. Blockade of glutamate receptors unmasks neuronal apoptosis after oxygen-glucose deprivation in vitro. Neuroscience. 1995;68:615–619. doi: 10.1016/0306-4522(95)00232-8. [DOI] [PubMed] [Google Scholar]
- 35.Gwag BJ, Koh JY, DeMaro JA, Ying HS, Jacquin M, Choi DW. Slowly-triggered excitotoxicity occurs by necrosis in cortical cultures. Neuroscience. 1997;77:393–401. doi: 10.1016/s0306-4522(96)00473-3. [DOI] [PubMed] [Google Scholar]
- 36.Heron A, Pollard H, Dessi F, Moreau J, Lasbennes F, Ben-Ari Y, Charriaut-Marlangue C. Regional variability in DNA fragmentation after global ischemia evidenced by combined histological and gel electrophoresis observations in the rat brain. J Neurochem. 1993;61:1973–1976. doi: 10.1111/j.1471-4159.1993.tb09843.x. [DOI] [PubMed] [Google Scholar]
- 37.Hsu CY, Dimitrijevic MR. Methylprednisolone in spinal cord injury: the possible mechanism of action. J Neurotrauma. 1990;7:115–119. doi: 10.1089/neu.1990.7.115. [DOI] [PubMed] [Google Scholar]
- 38.Johnson EM, Jr, Greenlund LJ, Akins PT, Hsu CY. Neuronal apoptosis: current understanding of molecular mechanisms and potential role in ischemic brain injury. J Neurotrauma. 1995;12:843–852. doi: 10.1089/neu.1995.12.843. [DOI] [PubMed] [Google Scholar]
- 39.Kao CC, Wrathall JR, Kyoshima K. Axonal reaction to transection In: Spinal cord reconstruction (Kao CC, ed), pp 41–57. Raven; New York: 1983. [Google Scholar]
- 40.Katoh K, Ikata T, Katoh S, Hamada Y, Nakauchi K, Sano T, Niwa M. Induction and its spread of apoptosis in rat spinal cord after mechanical trauma. Neurosci Lett. 1996;216:9–12. doi: 10.1016/0304-3940(96)12999-2. [DOI] [PubMed] [Google Scholar]
- 41.Kerr JFR, Harmon BV. Definition and incidence of apoptosis: an historical perspective. In: Tomei LD, Cope FO, editors. Apoptosis: the molecular basis of cell death. Cold Spring Harbor Laboratory; New York: 1991. pp. 5–29. [Google Scholar]
- 42.Kerr JFR, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. 1972;26:239–257. doi: 10.1038/bjc.1972.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kihara S, Shiraishi T, Nakagawa S, Toda K, Tabuchi K. Visualization of DNA double strand breaks in the gerbil hippocampal CA1 following transient ischemia. Neurosci Lett. 1994;175:133–136. doi: 10.1016/0304-3940(94)91097-9. [DOI] [PubMed] [Google Scholar]
- 44.Kondo S, Kondo Y, Yin D, Barnett GH, Kaakaji R, Peterson JW, Morimura T, Kubo H, Takeuchi J, Barna BP. Involvement of interleukin-1 beta-converting enzyme in apoptosis of bFGF-deprived murine aortic endothelial cells. FASEB J. 1996;10:1192–1197. doi: 10.1096/fasebj.10.10.8751721. [DOI] [PubMed] [Google Scholar]
- 45.Li GL, Brodin G, Farooque M, Funa K, Holtz A, Wang WL, Olsson Y. Apoptosis and expression of BCL-2 after compression trauma to rat spinal cord. J Neuropathol Exp Neurol. 1996;55:280–289. doi: 10.1097/00005072-199603000-00003. [DOI] [PubMed] [Google Scholar]
- 46.Li Y, Sharov VG, Jiang N, Zaloga C, Sabbah HN, Chopp M. Ultrastructural and light microscopic evidence of apoptosis after middle cerebral artery occlusion in the rat. Am J Pathol. 1995;146:1045–1051. [PMC free article] [PubMed] [Google Scholar]
- 47.Linnik MD, Zobrist RH, Hatfield MD. Evidence supporting a role for programmed cell death in focal cerebral ischemia in rats. Stroke. 1993;24:2002–2009. doi: 10.1161/01.str.24.12.2002. [DOI] [PubMed] [Google Scholar]
- 48.Liu XZ, Xu XM, Hu R, Du C, Fan GS, Hsu CY, Choi DW. Effect of cycloheximide on DNA breakdown, tissue loss, and behavioral outcome after spinal cord impact injury. Soc Neurosci Abstr. 1996;22:1185. [Google Scholar]
- 49.MacManus JP, Hill IE, Huang ZG, Rasquinha I, Xue D, Buchan AM. DNA damage consistent with apoptosis in transient focal ischemic neocortex. NeuroReport. 1994;5:493–496. doi: 10.1097/00001756-199401120-00031. [DOI] [PubMed] [Google Scholar]
- 50.Martin DP, Schmidt RE, DiStefano PS, Lowry OH, Carter JG, Johnson EM., Jr Inhibitors of protein synthesis and RNA synthesis prevent neuronal death caused by nerve growth factor deprivation. J Cell Biol. 1988;106:829–844. doi: 10.1083/jcb.106.3.829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Martin DP, Ito A, Horigome K, Lampe PA, Johnson EM., Jr Biochemical characterization of programmed cell death in NGF-deprived sympathetic neurons. J Neurobiol. 1992;23:1205–1220. doi: 10.1002/neu.480230911. [DOI] [PubMed] [Google Scholar]
- 52.Nitatori T, Sato N, Waguri S, Karasawa Y, Araki H, Shibanai K, Kominami E, Uchiyama Y. Delayed neuronal death in the CA1 pyramidal cell layer of the gerbil hippocampus following transient ischemia is apoptosis. J Neurosci. 1995;15:1001–1011. doi: 10.1523/JNEUROSCI.15-02-01001.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Okamoto M, Matsumoto M, Ohtsuki T, Taguchi A, Mikoshiba K, Yanagihara T, Kamada T. Internucleosomal DNA cleavage involved in ischemia-induced neuronal death. Biochem Biophys Res Commun. 1993;196:1356–1362. doi: 10.1006/bbrc.1993.2402. [DOI] [PubMed] [Google Scholar]
- 54.Osterholm JL. The pathophysiological response to spinal cord injury. The current status of related research. J Neurosurg. 1974;40:5–33. [PubMed] [Google Scholar]
- 55.Panter SS, Yum SW, Faden AI. Alteration in extracellular amino acids after traumatic spinal cord injury. Ann Neurol. 1990;27:96–99. doi: 10.1002/ana.410270115. [DOI] [PubMed] [Google Scholar]
- 56.Papas S, Crepel V, Hasboun D, Jorquera I, Chinestra P, Ben-Ari Y. Cycloheximide reduces the effects of anoxic insult in vivo and in vitro. Eur J Neurosci. 1992;4:758–765. doi: 10.1111/j.1460-9568.1992.tb00185.x. [DOI] [PubMed] [Google Scholar]
- 57.Pavlik A, Teisinger J. Effect of cycloheximide administered to rats in early postnatal life: prolonged inhibition of DNA synthesis in the developing brain. Brain Res. 1980;192:531–541. doi: 10.1016/0006-8993(80)90903-8. [DOI] [PubMed] [Google Scholar]
- 58.Portera-Cailliau C, Hedreen JC, Price DL, Koliatsos VE. Evidence for apoptotic cell death in Huntington disease and excitotoxic animal models. J Neurosci. 1995;15:3775–3787. doi: 10.1523/JNEUROSCI.15-05-03775.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pronk GJ, Ramer K, Amiri P, Williams LT. Requirement of an ICE-like protease for induction of apoptosis and ceramide generation by REAPER. Science. 1996;271:808–810. doi: 10.1126/science.271.5250.808. [DOI] [PubMed] [Google Scholar]
- 60.Ratan RR, Murphy TH, Baraban JM. Macromolecular synthesis inhibitors prevent oxidative stress-induced apoptosis in embryonic cortical neurons by shunting cysteine from protein synthesis to glutathione. J Neurosci. 1994;14:4385–4392. doi: 10.1523/JNEUROSCI.14-07-04385.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rink A, Fung KM, Trojanowski JQ, Lee VM, Neugebauer E, McIntosh TK. Evidence of apoptotic cell death after experimental traumatic brain injury in the rat. Am J Pathol. 1995;147:1575–1583. [PMC free article] [PubMed] [Google Scholar]
- 62.Roberts-Lewis JM, Marcy VR, Zhao Y, Vaught JL, Siman R, Lewis ME. Aurintricarboxylic acid protects hippocampal neurons from NMDA- and ischemia-induced toxicity in vivo. J Neurochem. 1993;61:378–381. doi: 10.1111/j.1471-4159.1993.tb03583.x. [DOI] [PubMed] [Google Scholar]
- 63.Rosenbaum DM, Michaelson M, Batter DK, Doshi P, Kessler JA. Evidence for hypoxia-induced, programmed cell death of cultured neurons. Ann Neurol. 1994;36:864–870. doi: 10.1002/ana.410360610. [DOI] [PubMed] [Google Scholar]
- 64.Sambrook J, Fritsch EF, Maniatis T. Isolation of DNA from mammalian cells: protocol I. In: Sambrook J, editor. Molecular cloning: a laboratory manual, Ed 2, Chap 9. Cold Spring Harbor Laboratory; New York: 1989. pp. 16–19. [Google Scholar]
- 65.Selina CC, McIntosh TK, Noble LJ. Experimental fluid percussion brain injury: vascular disruption and neuronal and glial alterations. Brain Res. 1989;482:271–282. doi: 10.1016/0006-8993(89)91190-6. [DOI] [PubMed] [Google Scholar]
- 66.Senter HJ, Venes JL. Altered blood flow and secondary injury in experimental spinal cord trauma. J Neurosurg. 1978;49:569–578. doi: 10.3171/jns.1978.49.4.0569. [DOI] [PubMed] [Google Scholar]
- 67.Shigeno T, Yamasaki Y, Kato G, Kusaka K, Mima T, Takakura K, Graham DI, Furukawa S. Reduction of delayed neuronal death by inhibition of protein synthesis. Neurosci Lett. 1990;120:117–119. doi: 10.1016/0304-3940(90)90182-9. [DOI] [PubMed] [Google Scholar]
- 68.Shuman SL, Bresnahan JC, Beattie MS. Morphological evidence of glial involvement in apoptotic cell death following spinal cord contusion. Soc Neurosci Abstr. 1996;22:1185. [Google Scholar]
- 69.Spurr AR. A low-viscosity epoxy resin embedding medium for electron microscopy. J Ultrastruct Res. 1969;26:31–43. doi: 10.1016/s0022-5320(69)90033-1. [DOI] [PubMed] [Google Scholar]
- 70.Swoboda TK, Li Y, Nockels RP, Chopp M. Evidence of apoptosis in an acute spinal cord injury model. Soc Neurosci Abstr. 1996;22:20. [Google Scholar]
- 71.Tanabe J, Watanabe M, Kondoh S, Mue S, Ohuchi K. Possible roles of protein kinases in neutrophil chemotactic factor production by leucocytes in allergic inflammation in rats. Br J Pharmacol. 1994;113:1480–1486. doi: 10.1111/j.1476-5381.1994.tb17163.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Thompson CB. Apoptosis in the pathogenesis and treatment of disease. Science. 1995;267:1456–1462. doi: 10.1126/science.7878464. [DOI] [PubMed] [Google Scholar]
- 73.Tominaga T, Kure S, Narisawa K, Yoshimoto T. Endonuclease activation following focal ischemic injury in the rat brain. Brain Res. 1993;608:21–26. doi: 10.1016/0006-8993(93)90768-i. [DOI] [PubMed] [Google Scholar]
- 74.White RJ. Pathology of spinal cord injury in experimental lesions. Clin Orthop. 1975;112:16–26. [PubMed] [Google Scholar]
- 75.Wrathall JR, Teng YD, Choiniere D, Mundt DJ. Evidence that local non-NMDA receptors contribute to functional deficits in contusive spinal cord injury. Brain Res. 1992;586:140–143. doi: 10.1016/0006-8993(92)91384-q. [DOI] [PubMed] [Google Scholar]
- 76.Wyllie AH, Kerr JFR, Currie AR. Cell death: the significance of apoptosis. Int Rev Cytol. 1980;68:251–306. doi: 10.1016/s0074-7696(08)62312-8. [DOI] [PubMed] [Google Scholar]
- 77.Yaginuma H, Tomita M, Takashita N, McKay SE, Cardwell C, Yin QW, Oppenheim RW. A novel type of programmed neuronal death in the cervical spinal cord of the chick embryo. J Neurosci. 1996;16:3685–3703. doi: 10.1523/JNEUROSCI.16-11-03685.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yong C, Arnold PM, Zoubine MN, Citron BA, Festoff BW. Apoptosis in injured spinal cord of rat. Soc Neurosci Abstr. 1996;22:20. [Google Scholar]
- 79.Young W. Secondary injury mechanisms in acute spinal cord injury. J Emerg Med. 1993;11:13–22. [PubMed] [Google Scholar]