

Abstract
The expression of voltage-gated calcium (Ca2+) channel activity in brain cells is known to be important for several aspects of neuronal development. In addition, excessive Ca2+ influx has been linked clearly to neurotoxicity both in vivo and in vitro; however, the temporal relationship between the development of Ca2+ channel activity and neuronal survival is not understood. Over a period spanning 28 d in vitro, progressive increases in high voltage-activated whole-cell Ca2+ current and L-type Ca2+channel activity were observed in cultured hippocampal neurons. On the basis of single-channel analyses, these increases seem to arise in part from a greater density of functionally available L-type Ca2+ channels. An increase in mRNA for the α1 subunit of L-type Ca2+ channels occurred over a similar time course, which suggests that a change in gene expression may underlie the increased channel density. Parallel studies showed that hippocampal neuronal survival over 28 d was inversely related to increasing Ca2+ current density. Chronic treatment of hippocampal neurons with the L-type Ca2+ channel antagonist nimodipine significantly enhanced survival. Together, these results suggest that age-dependent increases in the density of Ca2+ channels might contribute significantly to declining viability of hippocampal neurons. The results also are analogous to patterns seen in neurons of aged animals and therefore raise the possibility that long-term primary neuronal culture could serve as a model for some aspects of aging changes in hippocampal Ca2+ channel function.
Keywords: hippocampal neurons, calcium currents, cell death, cell culture, L-type calcium channels, aging
The early survival and development of neurons is dependent on low-to-moderate levels of Ca2+ influx through voltage-gated Ca2+ channels (VGCCs) (Kater et al., 1988; Koike et al., 1989; Lipton and Kater, 1989; Larmet et al., 1992). Critical developmental processes such as the appearance of neurotransmitters and ion channels, neurite outgrowth, synaptogenesis, and intrinsic firing patterns all depend in part on developmental changes in voltage-dependent Ca2+ conductances (Mattson and Kater, 1987; Gruol et al., 1992; Basarsky et al., 1994;Spitzer, 1994; Rusanescu et al., 1995; Turrigiano et al., 1995).
Although low-to-moderate levels of Ca2+ influx are important for numerous developmental processes, it is also well established that excessive Ca2+ influx can be toxic to neurons both in vivo and in vitro (Rothman and Olney, 1987; Choi, 1988, 1995). Although neurons die during early development (Oppenheim, 1991) and also become more vulnerable with aging (Coleman and Flood, 1987), the role of time-dependent changes in voltage-gated Ca2+ currents in relation to neuronal death is not well understood.
Hippocampal neurons seem to be particularly appropriate cells in which to examine the relationship of Ca2+ currents to neuronal vulnerability. The hippocampus is important for memory and cognitive processing (Morris et al., 1982; Zola-Morgan and Squire, 1990) and is highly vulnerable to aging and neurodegenerative influences (Wisniewski and Terry, 1973; Coleman and Flood, 1987;Barnes, 1991; deToledo-Morrell and Morrell, 1991; Landfield et al., 1992; Disterhoft et al., 1994; West et al., 1994; Geinisman et al., 1995). In addition, hippocampal neurons are characterized by a high density of voltage-activated Ca2+ currents (Wong and Prince, 1981; Fisher et al., 1990; Brown and Jaffe, 1994) and contain the multiple types of high voltage-activated (HVA) Ca2+ channels that are present in many other neurons (Tsien et al., 1988; 1995; Fisher et al., 1990; Mogul and Fox, 1991;Llinás et al., 1992; Mintz et al., 1992; Eliot and Johnston, 1994; Dunlap et al., 1995; Elliot et al., 1995).
Most studies on the role of excessive Ca2+ influx in cell death have focused on Ca2+ entry via ligand-gated channels (Rothman and Olney, 1987; Choi, 1988); however, it is becoming increasingly recognized that VGCCs, particularly of the L-type, can be a route for toxic levels of Ca2+influx after a number of insults (Scriabine et al., 1989; Uematsu et al., 1989; Lobner and Lipton, 1993; Lipton, 1994; Stuiver et al., 1996). Furthermore, VGCCs represent a major Ca2+entry pathway even during the physiological activation of ligand-gated Ca2+ channels (Miyakawa et al., 1992; Jaffe et al., 1994; Regehr and Tank, 1994; Stuart and Sakmann, 1994; Magee and Johnston, 1995; Yuste and Denk, 1995). It also is becoming clear that Ca2+ influx via different routes (voltage vs ligand-gated channel influx) may not be functionally interchangeable (Gallin and Greenberg, 1995; Tsien et al., 1995). Therefore, it seems important to determine the nature of any changes that may occur in VGCCs over time in hippocampal neurons and to test the possibility that some aspects of these changes are related to increasing vulnerability and greater probability of death.
MATERIALS AND METHODS
Cell culture. Fetal rat hippocampal cell cultures were established from pregnant (embryonic day 18) Fischer 344 rats using slight modifications of established methods (Banker and Cowan, 1977; Ransom et al., 1977). After the pregnant rats were killed with CO2 and cervical dislocation, fetuses were removed, and the hippocampi were dissected and placed in ice-cold Ca2+- and Mg2+-free HBSS. The tissue was then transferred to 10 ml HBSS containing 0.25% trypsin and 1 mm EDTA and treated for 10 min at room temperature. The hippocampi were subsequently washed three times with 15 ml vol of Minimum Essential Medium (MEM; supplemented with additional 30 mm glucose) and then dispersed by repeated trituration. The cell suspension was diluted with MEM to a final concentration of 3–5 × 105 cells/ml, and 1 ml was added to poly-l-lysine-coated (100 μg/ml) plastic culture dishes (35 mm) (Corning, Corning, NY), which contained 1 ml MEM with 10% fetal bovine and 10% horse sera (previously incubated overnight at 36°C in a humidified atmosphere of 5% CO2 and 95% air). Cells were placed in the incubator, and on the next day half of the medium was exchanged for medium containing only MEM and 10% horse serum (MEM/H). To inhibit proliferation of non-neuronal cells, at 3 d in vitro (DIV) half the medium was replaced with MEM/H containing 5-fluoro-2′-deoxy-uridine (15 μg/ml). Uridine (35 μg/ml) was also included to prevent the inhibition of RNA synthesis. Feedings, in which half the medium was exchanged for fresh MEM/H, were carried out subsequently on 10, 15, and 22 DIV. All solutions, media, and sera were obtained from Life Technologies (Grand Island, NY) or HyClone (Logan, UT). In cases in which there had been medium exchanges, cells were allowed to equilibrate for at least 3 hr before recording.
Recording solutions. For whole-cell patch-clamp recordings of voltage-gated Ca2+ currents, the external solution had the following composition (in mm): 75 choline chloride, 50 TEA-Cl, 20 BaCl2, 5 CsCl, 2 MgCl2, 10 glucose, 10 HEPES, pH 7.3 with TEA-OH, and 0.001 TTX. The whole-cell recording pipette contained (in mm): 145 methanesulfonic acid, 11 EGTA, 10 TEA-Cl, 5 MgCl2, 5 Mg-ATP, 1 CaCl2, 0.1 leupeptin, and 10 HEPES, pH 7.3 with CsOH.
For cell-attached single-channel patch recording, the extracellular bath solution contained (in mm): 140 potassium gluconate, 15 NaCl, 3 MgCl2, 10 EGTA, 10 glucose, 10 HEPES, and 0.001 TTX, pH 7.4 with KOH; this solution zeroes the membrane potential (Nowycky et al., 1985). The recording pipette solution contained (in mm): 90 choline chloride, 20 BaCl2, 10 TEA-Cl, and 10 HEPES, pH 7.3 with TEA-OH. Bay K 8644 (500 nm), a potent L-type Ca2+ channel agonist, was added to the cell-attached pipette to enhance recording of L-type channels (Nowycky et al., 1985; Tsien et al., 1988; Bean, 1989). To facilitate comparisons between the whole-cell and single-channel recordings, equivalent concentrations of the charge carrier BaCl2 (20 mm) were used for both the whole-cell and cell-attached patch recording modes. This relatively low concentration of Ba2+ (for single-channel studies) substantially increases the frequency of repolarization openings (ROs) of L-type channels at resting potential (Thibault et al., 1993). The osmolarity of all recording solutions was adjusted to 320–325 mOsm as necessary, by the addition of sucrose. All chemicals were obtained from Sigma (St. Louis, MO).
Electrophysiology. All recording studies were performed on cultured rat embryonic hippocampal neurons from ages 1–28 DIV. The growth medium of a culture was exchanged for 2 ml of the extracellular bath solution immediately before recording. Cell-attached recording pipettes were pulled from glass capillary tubes (Drummond Scientific, Broomall, PA) using a micropipette puller (model P-87; Sutter Instruments, Novato, CA). Pipettes were coated with Sylgard (Dow Corning, Midland, MI) and fire-polished immediately before use (Corey and Stevens, 1983). The resistance of cell-attached patch-recording pipettes was 2–3 MΩ. Whole-cell recording electrodes were pulled from capillary tubes (Fisher Scientific, Pittsburgh, PA) and coated with polystyrene Q-dope, and had resistances of 1–2 MΩ. Recordings were obtained according to standard patch-clamp methods (Hamill et al., 1981) using an Axopatch 200 amplifier (Axon Instruments, Foster City, CA). Voltage commands were generated, and current responses were recorded and analyzed using a computerized acquisition and storage system (pCLAMP; Axon Instruments). For cell-attached patch recordings, current responses were low-pass filtered at 2 kHz and digitized at 5–8 kHz. Whole-cell recordings were filtered at 1 kHz and digitized at 2–3 kHz. Electrophysiological recordings were performed at room temperature (22–24°C).
Whole-cell studies. HVA Ca2+ currents were evoked by depolarizing pulse commands 150 msec in duration, and except for voltage-dependence studies were evoked from a holding potential (Vh) of −70 mV to a command potential (Vc) of +10 mV. A fractional (P/N) method, using five fractionally scaled hyperpolarizing subpulses, was used to leak-subtract whole-cell current traces on-line. In the recording solutions used, these leak current responses appeared to be linear functions of the hyperpolarizing voltage steps for all age groups. Junction potentials were nulled in the bath, and pipette capacitance was compensated. Whole-cell pipettes generally had access resistances of ∼5 MΩ; however, series resistance and whole-cell compensation at the amplifier were not applied routinely throughout these studies because we have found consistently that compensation does not alter the shape or amplitudes of activated currents under our conditions (also see Randall and Tsien, 1995). Whole-cell membrane capacitance (pF) was calculated by integrating the area of the capacitive transient evoked by a 150 msec duration, 5 mV hyperpolarizing pulse from a Vh of −70 mV. Current density (pA/pF) was determined by dividing the peak current evoked during a depolarizing command by the cell membrane capacitance. Half-maximal activation voltages (V1/2) were derived by fitting data obtained from current–voltage (I–V) relationships to the Boltzmann equation of the form y = peak I/{1 + exp[(V1/2 −V)/k]}, where V is the maximal voltage from the I–V relationship and krepresents the steepness of the sigmoid curve. At the later ages, cells became fragile and recordings were sometimes unstable. Electrophysiological results presented for age 28 DIV are only from neurons that were defined as healthy by our criteria (high seal resistance, stable holding current). Data were recorded from 5–12 cells per age point. Values are represented as the mean ± SEM.
Single-channel studies and analysis. Ensemble average (pseudomacroscopic) current responses were obtained for each multichannel patch from a series of 15 depolarizing pulses (150 msec duration) evoked from Vh = −70 mV toVc = +10mV. Leak and capacitive currents were subtracted using averaged currents obtained from hyperpolarizing steps. Most patches were run through an I–V series, with one evoked response at each command voltage (30 sec interpulse intervals). Average total patch current (I) was determined by integrating the leak-subtracted ensemble average current trace from the zero baseline to the inward current envelope during the pulse and dividing the integral by the duration of the pulse (150 msec). Total patch current, I, is given by I = N Po i, where N is the number of available channels, Po is the probability of opening of a channel, and i is the amplitude of a single channel (Nowycky et al., 1985).
To estimate i directly as a function of age in culture, the amplitudes of clearly resolvable single-channel openings reflecting L-type channel activity (openings >5 msec) were measured in patches at 2, 10, and 28 DIV from the I–V series. Single, unobscured openings could be measured readily at more negative potentials (−40 mV to −20 mV) but were more difficult to resolve at positive steps at which numerous channel openings overlapped; however, measurable openings (n = 5–20) were obtained for each voltage at each age point.
N was estimated directly in all patches by the method of maximal simultaneous openings (Horn, 1991; Sigworth and Zhou, 1992). In this method, the maximal multichannel current amplitude (Imax) obtained in a series of repeated depolarizations (largest peak current) is divided by current amplitude of a single channel (i) at the same voltage (N = Imax/i). This method is highly accurate when Po is relatively great; in the present studies, Po for L-type channels was quite high because of the use of the agonist Bay K 8644 and step depolarizations to maximally activating voltages (Thibault and Landfield, 1996). For each patch,Imax was taken as the largest instantaneous peak current in a series of 15 pulses to +10 mV. The average i at +10 mV for each age group was used with each patch to calculateN from Imax (i did not vary with age at any voltage). The density of available L-type Ca2+ channels per unit membrane area (N/μm2) was calculated from the estimate of N and pipette resistance (R), according to a regression equation of Sakmann and Neher (1983):a = 12.6 (1/R + 0.018), where ais the membrane area (μm2). Data were recorded from 4–12 cells per time point. Values are expressed as the mean ± SEM.
Probe synthesis. Antisense riboprobes (cRNA) were synthesized from cDNA clones for use in ribonuclease protection assays (RPAs). The cDNA clones for the α1C and α1DL-type Ca2+ channel subunits were generated from rat hippocampal RNA by reverse transcriptase-PCR (RT-PCR) using primers specific for each respective mRNA. The resulting PCR cDNA fragments were then cloned into the pGEMEX-1 vector (Promega, Madison, WI), and the cDNA clones were confirmed by sequencing. The α1CcDNA was a 333 bp fragment (nt 3306–3638) (Snutch et al., 1991), and the α1D cDNA was a 284 bp fragment (nt 2902–3185) (Hui et al.,1991) spanning a region at the II–III linker.
Labeled cRNA probes were made after an in vitrotranscription protocol (Promega). The labeling reaction consisted of 50 μCi [32P] CTP (specific activity 800 Ci/mmol), 1× transcription buffer, 15 mm DTT, 200 μmGTP, ATP, and CTP or UTP, 20 U placental RNase inhibitor (40 U/μl), 1 μg linearized plasmid DNA, and 20 U of appropriate RNA polymerase (SP6). Reactions were incubated at 37°C for 45 min. Subsequently, RNase-free DNase I was added at 1 U/μg DNA and incubated for 15 min at 37°C to digest the DNA template. The reaction mix was diluted to 50 μl with diethylpyrocarbamate-treated water and extracted with acid phenol, and probes were purified with G-50 spin columns (5 Prime → 3 Prime). Enzymes and nucleotides were obtained from Boehringer Mannheim (Indianapolis, IN).
RPA. Total RNA preparations from hippocampal neurons in culture were hybridized with cRNA probes followed by digestion with RNases using the protocol and reagents supplied in an RNase protection kit (Boehringer Mannheim). Briefly, 5 μg of total RNA was co-precipitated with 1–3 × 105 cpm32P-labeled cRNA, redissolved in 20 μl hybridization buffer, and heated at 95°C for 5 min followed by incubation overnight at 45°C. A mixture of RNaseA/T1 was then added to digest the single-stranded RNA, while the cRNA probes hybridized to the sense mRNA were protected from RNase digestion. The RNase digestion was terminated by the addition of proteinase K and 4 m guanidine thiocyanate solution (Bordonaro et al., 1994). The protected cRNA fragments were precipitated with isopropanol, and the pellet was redissolved in 6 μl of gel-loading buffer, denatured at 95°C, and loaded onto a 5% acrylamide/8 m urea TBE gel. The gel was fixed, dried, and analyzed on a phosphoimager (Molecular Dynamics, Sunnyvale, CA). Both cRNA probes for α1C and α1D were added in each hybridization reaction, and the protected fragments for each probe were quantitated using the Image QN program (Molecular Dynamics). Results were normalized to the total RNA concentration, determined from the optical density of each aliquot before gel loading.
Cell counts.To evaluate the survival of hippocampal neurons in culture over time, parallel studies were performed in 11 dishes with hippocampal neurons plated on marked and gridded glass coverslips (Eppendorf CELLocate, Madison, WI). The grids allowed the same group of neurons to be followed over age in culture.
Central grid fields (120× magnification; one field per dish) in each dish were selected and followed throughout the study. These grid fields were photographed on 1, 2, 3, 6, 10, 15, and 28 DIV corresponding to times at which recordings of VGCCs and currents had been performed in parallel cultures. Viable cells were defined as those that had phase-bright cell bodies. Counts of viable cells were performed on coded photomicrographs in a blind fashion by three independent scorers, and the values were averaged. In a separate series of experiments, cell survival was assessed after cultures were chronically exposed to nimodipine (5 μm).
To confirm the accuracy of the counting procedure, control experiments were performed in which we compared results obtained by counting phase-bright cell bodies with those obtained using the fluorescent dye fluorescein diacetate, which is incorporated into the cell bodies of viable cells (Novelli et al., 1988). In these experiments the difference between the counts obtained by the two different methods was <5%.
Pharmacology. Stock solutions of Bay K 8644 (RBI, Natick, MA) and nimodipine (RBI) were prepared in 100% ethanol, aliquoted, and frozen at −20°C until subsequent use. The final concentration of ethanol in experimental drug solutions was 0.1%.
RESULTS
Morphological patterns
As described previously, hippocampal neurons in primary culture undergo several phases of morphological development (Banker and Cowan, 1977; Goslin and Banker, 1991). In the present studies, these were highly consistent and apparent on visual inspection. Pyramidal neurons from different hippocampal subfields (CA1–3) were present but could not be distinguished consistently, whereas dentate granule cells have not developed yet (Banker and Cowan, 1977; Bayer, 1980). Figure1 shows photomicrographs of rat primary hippocampal neurons at different ages in culture. At 1 DIV, neurons were attached to the dish and began to send out processes. By 3 DIV, growth of the soma was evident, as was elongation of the processes; processes often made contact with neighboring neurons. At 6 DIV, cell bodies were even larger, and the processes began to form extensive networks. As cells aged further in culture (10, 15, and 28 DIV), the dendritic processes became thicker, and the process networks became denser. The number of surviving cells also declined as a function of age in culture (see below). At 28 DIV, swollen and unhealthy cell bodies could be seen consistently among the living cells.
Fig. 1.
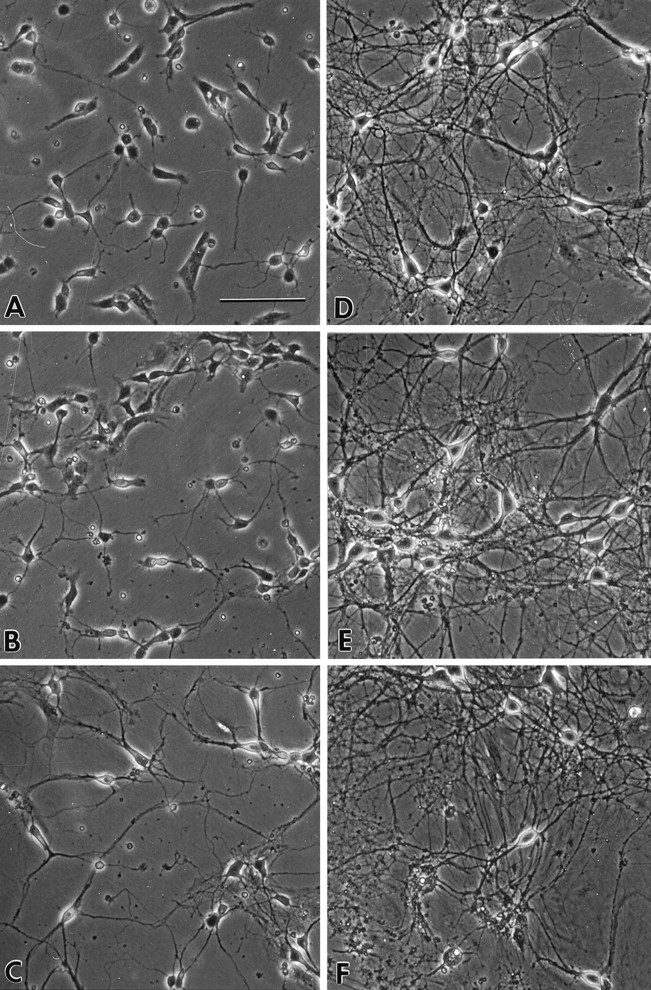
Photomicrographs of rat primary hippocampal neurons in culture. A, B, andC show neurons of ages 1, 3, and 6 DIV, respectively. An increase in the size of the soma and the length of neurites during the first week in culture is accompanied by a rapid decline in cell number. Examples of hippocampal neurons 10, 15, and 28 DIV in culture are represented in D, E, andF, respectively. Scale bar (shown in A): 100 μm.
Whole-cell recordings
Whole-cell HVA Ca2+ currents were recorded from hippocampal neurons on 1, 2, 3, 6, 10, 15, and 28 DIV. The amplitude of whole-cell Ca2+ currents changed substantially as a function of age in culture. Representative whole-cell Ca2+ current traces recorded from hippocampal neurons 1–28 DIV are shown in Figure 2. The average amplitudes of peak current were calculated for cells at each age point (Fig. 3A). Peak current increased substantially as cells aged in culture (p < 0.0001; ANOVA), and the rate of increase in current was greatest between 1–3 DIV and 10–28 DIV. Initial peak current evoked in cells that were 1 DIV was 48 ± 12 pA (mean ± SEM). By 6 DIV, peak current increased ∼10-fold (441 ± 64 pA) over that observed at 1 DIV.
Fig. 2.

HVA whole-cell Ca2+ currents increase with age in culture. Representative whole-cell Ca2+ current traces from hippocampal neurons are shown at different ages in culture. Currents were evoked during a 150 msec depolarization step from Vh = −70 mV to Vc = +10 mV. RCs, recorded at −70 mV after the depolarization step, typically do not appear until cells are 3 DIV. The voltage step and calibration bars are shown at thebottom.
Fig. 3.

Rate of increase of HVA whole-cell Ca2+ current exceeds that of the capacitance of the cell with age in culture. A, Peak whole-cell Ca2+ current (•) and corresponding cell capacitance (♦) measured over time in culture. Currents were evoked during a 150 msec depolarization from Vh = −70 mV to Vc = +10 mV. Cell capacitance was estimated from a capacitive transient evoked by a 5 mV hyperpolarizing step from Vh = −70 mV. B, Ca2+ current density for cells shown inA was obtained by dividing the peak whole-cell current by the cell capacitance. Current density increased rapidly during the first 1–3 DIV, remained stable until 10 DIV, and then continued to rise until 28 DIV. Values are mean ± SEM.
In these neurons, the HVA current is consistently followed by a very long tail current, or repolarization current (RC), that appears approximately at age 3 DIV (Fig. 2). These long repolarization Ca2+ currents are also seen in adult hippocampal slice neurons (Gähwiler and Brown, 1987; Pitler and Landfield, 1987; Kerr et al., 1992). The long tails resemble space-clamp artifact currents but do not originate solely from the large apical dendrites (Thibault et al., 1995). Single L-type Ca2+ channel openings on the soma have been found after repolarization (ROs) in hippocampal and cerebellar neurons (Fisher et al., 1990; Slesinger and Lansman, 1991; Forti and Pietrobon, 1993; Thibault et al., 1993;Kavalali and Plummer, 1995), particularly when the divalent charge carrier (Ca2+ or Ba2+) is low and approaches physiological concentrations (e.g., 5–20 mm) (Thibault et al., 1993). It is not clear, however, that ROs can account for all of the whole-cell RCs, and therefore it is possible that some of this current is generated by axial currents from unclamped small dendrites. Because the origin of these RCs is not yet fully resolved, they were not analyzed in detail in the present study.
To determine whether the increase in HVA Ca2+current in culture was related simply to the growth of cells, cell size with age was estimated from the capacitance of each cell. As cells aged in culture, cell size also increased significantly (p < 0.0001; ANOVA) (Fig. 3A). Average cell capacitance of cells at age 1 DIV was 13.9 ± 1.1 pF. By age 6 DIV, cells approximately doubled in size, and the cell capacitance was 23.8 ± 1.7 pF. At 28 DIV, cell capacitance was 105 ± 4.8 pF, representing an overall increase in membrane capacitance from age 1 DIV of approximately eightfold.
Ca2+ current density was estimated by dividing the Ca2+ current amplitude for each neuron by the capacitance value for that cell (pA/pF). This analysis showed a clear but nonlinear increase in Ca2+ current density with age in culture (Fig. 3B). The most rapid increase in current density occurred between ages 1 and 3 DIV, followed by a relatively stable phase and then a slower, gradual rise in the older cells (10–28 DIV). Overall, from 1 DIV to 28 DIV current density increased approximately sixfold.
I–V relationships also were studied to evaluate the voltage dependence of Ca2+ current as a function of age in culture (Fig. 4). In 20 mmBa2+, the voltage threshold for HVA Ca2+ current typically was between −50 mV and −40 mV. Half-maximal activation voltages (V1/2) calculated from theI–V curves shown in Figure 4 were −11.6 mV, −11.2 mV, −16.7 mV, −19.9 mV, −17.5 mV, and −17.4 mV for cells that were ages 1, 2, 3, 6, 10, and 15 DIV, respectively. TheV1/2 for neurons 28 DIV was −22.0 mV. Overall, with increasing age in culture, there appeared to be a modest shift in voltage dependence (∼10 mV) to more negative potentials for cells that were 28 DIV compared with those that were only 1–2 DIV.
Fig. 4.

I–V curves for hippocampal neurons of different ages in culture. Average I–V curves from cells recorded at different times in culture are superimposed to show the relative change in HVA whole-cell Ca2+ current amplitude with age. Voltage dependence is shifted to slightly more negative potentials as cells age in culture.
Possible voltage-clamp errors with age in culture
The substantial growth and process elaboration and the increased current of cultured neurons with age (Figs. 1, 2, 3) raise the possibility that age differences in voltage-clamp errors could arise from at least three sources: inadequate space clamp, series resistance, and junction potentials. It is well established that the adequacy of space clamp diminishes as the dendritic trees of neurons increase (Brown and Johnston, 1983; Johnston and Brown, 1983). Series and access resistance errors also increase with larger currents (Armstrong and Gilly, 1992). Furthermore, the diffusion and time to equilibrium between pipette solutions and cellular contents that result in junction potentials may be delayed in larger cells (Neher, 1992). These errors could conceivably affect results differently in different age groups; however, several considerations suggest that these factors did not influence the main findings. In terms of space-clamp difficulties related to the large dendritic tree, we have found that severing or separately voltage-clamping the large apical dendrite has very little effect on current waveforms at the soma and does not eradicate the long RC (which also is present in 3-d-old cells) (Thibault et al., 1995). In addition, the age-in-culture differences in current amplitude were observed with command steps well above the maximum inward current–voltage point on the I–V curve (Fig. 4). Thus, the larger amplitude current with age in culture does not seem to result from space-clamp error. As noted, series resistance compensation at the amplifier had almost no effect on amplitude or time course of either small or large currents, which may simply reflect the inadequacy of compensation methods for correcting fast currents; however, the degree of possible series resistance error even in the oldest cells would not seem able to account for the observed magnitude of amplitude differences. Liquid junction potentials could differ in rate of onset in larger cells, but I–V series were run only after maximal current values had stabilized, usually ∼15 min after the beginning of recording. Thus, although it is possible that some of the results are affected differentially by voltage-clamp error, particularly the small shift in voltage dependence in the larger cells (Fig. 4), the main results of increased current amplitude and increased current density with age do not seem to be substantially affected by these factors. In addition, perhaps the strongest validation of these whole-cell results is their similarity to data obtained with parallel single-channel methods (see below), which are not subject to comparable voltage-clamp errors.
Single-channel cell-attached patch
Single-channel studies were conducted in sister cultures using the cell-attached patch-clamp method. Representative current traces of L-type Ca2+ channels from multichannel patches of hippocampal neurons at different ages in culture are shown in Figure5. As in the whole-cell experiments, the charge carrier was 20 mm Ba2+. There was a clear trend apparent for both peak (Imax) and average total patch current (I) to increase with age in culture (Fig. 5). By analogy with whole-cell RCs, L-type Ca2+ channel ROs were observed infrequently in patches from cells that were 1–2 DIV (Fig. 5), and only 10–15% of these patches had RO activity. For cells that were 3 DIV or older, however, ROs were observed in >50% of the patches. Thus, ROs on the soma appeared at approximately the same day at which whole-cell repolarization tail currents appeared (Fig. 2).
Fig. 5.

Single L-type Ca2+ channel activity in hippocampal neurons increases with age in culture. Representative current traces of L-type Ca2+channels in cell-attached, multichannel patches recorded at different ages in culture. Currents were evoked during a 150 msec depolarization from Vh = −70 mV toVc = +10 mV. ROs, recorded at −70 mV after the depolarization step, rarely appear before 3 DIV.
Average total patch current (I) in multichannel patches (evoked from Vh = −70 mV to Vc = +10 mV) was 0.3 ± 0.1 pA at 1 DIV and 3.2 ± 0.6 pA for cells 28 DIV, representing an approximate 10-fold increase (p < 0.0001; ANOVA). The peak multichannel current (Imax) in cell-attached patches also increased with age in culture (p < 0.0001; ANOVA) (Fig.6A) and in a manner approximately similar to that observed for the peak whole-cell currents (Fig.3A). Pipette resistance also was measured for each patch and did not vary with age in culture (Fig. 6A).
Fig. 6.

Maximal patch current (Imax) and Ca2+channel density (N/μm2) increase with age in culture. A, Imax(•) evoked in cell-attached patches from hippocampal cells of different age in culture. Recordings were evoked during a 150 msec depolarization from Vh = −70 mV toVc = +10 mV. Patch-pipette resistance (♦) did not vary over the course of the study. B, Slope conductance of L-type Ca2+ channels was not altered as a function of age in culture. Amplitudes (i) of clearly resolvable single L-type Ca2+ channels were measured during depolarization to multiple test voltages [depolarization openings (DOs)] or on repolarization (ROs) to −70 mV in cell-attached patches from hippocampal cells 2 (•), 10 (▪), and 28 (▴) DIV. The slope conductance was determined from the regression line obtained by dividing the average amplitude by the indicated voltages for four to six patches per time point. C, L-type Ca2+ channel density increases as hippocampal neurons age in culture. As described in Materials and Methods,N (the number of channels per patch) was calculated by dividing Imax by i; the area of the patch membrane, μm2, was calculated from the pipette resistance.
As noted, total patch current (I) is given byI = N Po i. To determine which of these factors might be responsible for the overall increase in I, we measured i and Ndirectly. Single-channel amplitude (i) was measured at multiple test voltages in patches with clearly resolvable openings and did not differ with DIV at any test pulse, for either depolarization pulse openings (DOs) or ROs (Fig. 6B). In addition, the slope conductance for each patch was determined from values ofi obtained at multiple voltages as a function of age in culture. The average slope conductance ranged between 20.1 ± 1.5 and 21.2 ± 2.0 pS and did not differ for cells 2, 10, and 28 DIV (Fig. 6B).
The density of L-type Ca2+ channels per membrane area (N/μm2) was determined by the method of maximal simultaneous openings and pipette resistance (see Materials and Methods). The density of L-type Ca2+channels increased significantly by fivefold (p< 0.0001; ANOVA) over 28 DIV (Fig. 6C) and generally followed a pattern similar to that of whole-cell current density (Fig.3B). As with whole-cell Ca2+ current density, there was an initial rapid increase in channel density during the first 1–3 DIV. This was followed by a period of relatively constant channel density levels in cell-attached patches during the second week in culture, leading into a second phase of more gradual increase up until 28 DIV.
Ca2+ channel subunit mRNA levels
mRNA levels for the pore-forming subunits of two L-type Ca2+ channels, α1C and α1D, were measured in mRNA isolated from hippocampal neurons on 3, 10, 15, and 28 DIV using RPA analysis. The mRNA levels for the α1C subunit were severalfold greater than for the α1D subunit at all time points measured (Table 1). This is consistent with immunocytochemical studies in which the relative abundance of the neuronal α1C subunit protein was shown to be greater than that of the α1D (Hell et al., 1993). mRNA for the brain-specific α1D subunit of the L-type Ca2+ channel increased significantly by 2.6-fold over the time course of the study (3 DIV vs 28 DIV; p < 0.05; ANOVA) (Table 1). Although the α1C subunit mRNA showed the same trend toward an increase over the 28 DIV time period, this trend was not quite significant (p = 0.08). Thus, levels of mRNA for L-type channel subunits were increased concomitantly with the increased functional expression of L-type channels.
Table 1.
Ca2+ channel mRNA levels in hippocampal neurons
| Age in culture | α1D mRNA | α1C mRNA |
|---|---|---|
| 3 DIV | 258 ± 25 | 1790 ± 103 |
| 10 DIV | 544 ± 112 | 3391 ± 713 |
| 15 DIV | 632 ± 144* | 2918 ± 659 |
| 28 DIV | 680 ± 104* | 3606 ± 450 |
A significant increase in mRNA levels for the α1Dsubunit of the L-type Ca2+ channel was detected as a function of age in culture (*p < 0.05 vs 3 DIV; ANOVA and post hoc Student–Newman–Keuls comparisons test). Values are arbitrary density units for the specific message divided by the total RNA per preparation and are expressed as the mean ± SEM. n = 3–4 samples per time point.
Hippocampal cell death
Accompanying the observed changes in Ca2+channel patterns, hippocampal neuronal death increased significantly as cells aged in culture (Fig. 7A) (p < 0.0001; ANOVA). The decrease in cell survival seemed to occur in two general phases. An initial rapid phase of cell death was observed that occurred by 6 DIV. This rapid phase was followed by a slower more gradual phase of cell death that continued until 28 DIV. The onset of the initial rapid phase of death in the first week and the later slower phase of cell death (Fig.7A) were generally correlated temporally with the initial rapid phase (1–3 DIV) and the more gradual later (10–28 DIV) phase, respectively, of increasing Ca2+ current density (Fig. 7B).
Fig. 7.

Phases of neuronal death correlate closely with increases in Ca2+ current density. A, Two phases of neuronal death in culture are observed: an initial rapid phase that occurs by 6 DIV and then a slower more gradual phase that continues until 28 DIV. In the cell survival studies, the same groups of cells were followed in long-term culture (see Materials and Methods). Whole-cell current density (B) is plotted again to show the relationship of current density to cell survival (A) of sister cultures. Ca2+ current density rises rapidly between 1 and 3 DIV, stabilizes, and then rises again from 10 to 28 DIV.
These temporal correlations raise the clear possibility that increased Ca2+ current influx may contribute to neuronal death in hippocampal cell culture. To test this more directly, neuronal survival with age in culture also was assessed in hippocampal neurons after chronic treatment with the L-type Ca2+ channel antagonist nimodipine (5 μm). Cell cultures were treated chronically with nimodipine (in ethanol or DMSO vehicle), and cell survival was assessed at 17 DIV, 48 hr after the last treatment with nimodipine. Photomicrographs were taken and viable cells were counted using two methods: phase contrast or fluorescence microscopy. Highly similar results were obtained from both methods. In numerous comparisons we have found that ethanol at these concentrations does not reduce cell survival compared with untreated controls (unpublished observations), although the DMSO vehicle increased cell death relative to the ethanol control (Table 2). Chronic treatment of hippocampal neurons with nimodipine significantly increased cell survival regardless of whether the nimodipine was in ethanol or DMSO vehicle (Table 2). Although culture dishes were plated at the same initial density, DMSO-treated cultures were approximately threefold lower in density than EtOH-treated cultures at 17 DIV; however, nimodipine was neuroprotective under both conditions and restored neuronal survival in DMSO to levels seen with the nimodipine/EtOH-treated cells. These results are consistent with the interpretation that Ca2+ influx through L-type channels can chronically modulate survival in long-term hippocampal cultures.
Table 2.
Effects of nimodipine on survival of hippocampal neurons in culture
| Number of viable neurons | ||
|---|---|---|
| Phase contrast | Fluorescence | |
| EtOH vehicle (0.1%) | 18.3 ± 3.6 | 19.0 ± 3.1 |
| Nimodipine/EtOH | 33.2 ± 2.0* | 31.8 ± 1.9* |
| DMSO vehicle (0.1%) | 5.2 ± 2.0* | 6.0 ± 0.8* |
| Nimodipine/DMSO | 32.1 ± 4.02-160 | 30.8 ± 3.72-160 |
Cells were treated on 3, 6, 8, 10, 13, and 15 DIV with either the indicated vehicle or nimodipine (5 μm). Cell survival was assessed on 17 DIV. Viable cells were quantified by counting phase-bright or fluorescent cells (see Materials and Methods) on a gridded coverslip in a 120× field. Values represent the mean ± SEM (n = 4 sister cultures per group).
p < 0.05 compared with EtOH vehicle by ANOVA andpost hoc Bonferroni tests.
F2-160: p < 0.001 compared with DMSO vehicle.
DISCUSSION
Development of Ca2+ channels over time
The present studies show that hippocampal neurons in culture exhibit progressive and gradual increases in HVA Ca2+ current density that continue well after the early periods of cell growth and development. Absolute Ca2+ current amplitude increased in three phases: early rapid, middle slow, and late rapid (Fig. 3A). After correction for cell size, however, it was apparent thatcurrent density increased primarily in two phases: early rapid (1–3 DIV) and late slow (10–28 DIV) (Fig. 3B). These results confirm and extend to later ages other findings (see introductory remarks) that the electrophysiological properties of cultured neurons may change dramatically across only a few days in culture.
The results from single-channel analyses of the cell-attached patch recordings (Fig. 6C) indicate that an increase in the density of functionally available L-type Ca2+channels may account for much of the increase in total Ca2+ current density observed at the whole-cell level; however, the results do not preclude contributions from increases in other channel types. Basarsky et al. (1994) have shown that concomitantly with the development of mature synaptic function, Ca2+ influx through N-type Ca2+channels also increases in hippocampal neurons between 4 and 12 DIV. Using Ca2+ imaging techniques, they found that influx through L-type channels remains relatively stable during this period (consistent with the present study) (e.g., Figs. 3B,6A) and that at both 4 and 12 DIV, most of the Ca2+ influx is through L-type channels (Basarsky et al., 1994). In addition, in other types of primary neurons a decrease in survival with age in culture also has been observed and is accompanied by an increase in NMDA-evoked whole-cell currents (Dawson et al., 1993) and NMDA receptor-associated proteins (Xia et al., 1995). Thus, other Ca2+ channel types also seem to contribute to temporal changes in overall Ca2+influx. Moreover, it seems that both ligand-gated channels and VGCCs may contribute to declining neuronal viability.
Relationship to neuronal death
Accompanying the alterations in Ca2+ channel density are ongoing morphological changes in neurons, including the extension of processes and the growth of the soma, which are quite rapid in the first week (Banker and Cowan, 1979; Basarsky et al., 1994;Fletcher et al., 1994) (Figs. 1, 3A). Significant numbers of the neurons died during this initial growth phase, but cells also continued to die at a slower rate for the remainder of the 28-d cycle (Fig. 7A). Under our conditions, most neurons have died by age 28 d in vitro.
Previous electrophysiological studies of cultured hippocampal or cerebellar neurons have noted an increase in voltage-gated Ca2+ current with age in culture (Meyers and Barker, 1989; Gruol et al., 1992; Basarsky et al., 1994; Randall and Tsien, 1995), but the time-dependent relationship of Ca2+current to cell death has not been studied directly or for the extended periods examined in the present study. The finding of a significant positive temporal correlation between Ca2+ current density and neuronal death suggests that increased Ca2+ current and cell death may be functionally related (Fig. 7B). Consistent with this, chronic exposure to the L-type Ca2+ channel antagonist nimodipine blocked a significant amount of hippocampal cell loss in culture (Table2). These studies, therefore, point to changes in VGCCs, particularly of the L-type, as potential modulators of neuronal death over extended periods.
Possible differential survival
One caveat to the above conclusions is the possibility that neurons with more rather than less (L-type) Ca2+channels preferentially survive the early phase of death. This alternative explanation also could account for why cells with greater Ca2+ channel density are seen at later ages. This seems unlikely, however, because the initial rapid increase in Ca2+ current density precedes the initial phase of cell death, rather than vice versa (Fig. 7). Furthermore, the second phase of current density increase is gradual and does not seem to be attributable to differential survival of a subpopulation of cells with high Ca2+ current density; few if any cells with high current density were seen at the beginning of this later phase (e.g., at 10 DIV). In addition, the L-type Ca2+ channel antagonist nimodipine exerted a neuroprotective action (Table 2), suggesting that greater L-channel density reduces rather than favors neuronal survival. It is clear, however, that further studies will be needed to confirm that increased Ca2+ channels enhance the probability of death and to clarify the contributions of time-dependent changes in other channel types to neuronal survival.
Different phases of cell death in long-term culture
There is evidence of apoptotic and necrotic death of neuronsin vitro and in vivo, and both types of cell death are associated with a number of acute and chronic neurodegenerative conditions (Ankarcrona et al., 1995; Choi, 1995;Thompson, 1995). Conceivably, the rapid phase of cell death occurring during the first 3–6 DIV is more analogous to apoptotic cell death, which occurs during early development (Oppenheim, 1991). During this early phase, hippocampal neurons in culture are relatively immature and show little evoked synaptic activity (Basarsky et al., 1994; Fletcher et al., 1994); however, we also consistently observed evidence of necrosis, particularly for neurons 15–28 DIV, as indicated by the remnants of swollen cell bodies. It seems feasible that the initial increase in Ca2+ current density may contribute to both the early growth phase and developmentally programmed apoptosis, whereas the later gradual phase of Ca2+ current density increase may eventually result in heightened vulnerability of neurons to degenerative or necrotic death.
Mechanism of Ca2+ current increase
The single-channel analyses suggest that an increase in functionally available L-type VGCCs underlies much of the increase in whole-cell Ca2+ current density over age in culture. This could be accounted for by the formation of new channels, the formation of new subunits that alter stoichiometry of channel configuration (and activity), or the recruitment of previously silent channels. In these studies, RPA analyses of mRNA levels for the pore-forming subunits of the L-type Ca2+ channel indicated that increased expression of the α1D (and possibly the α1C) L-type subunit may have played an important role in the increased Ca2+ channel density observed as a function of age in culture (Table 1). Immunocytochemical studies have shown that the α1D and α1C subunits are roughly colocalized on the soma and proximal dendrites of neurons (Hell et al., 1993), suggesting that our cell-attached patch recordings included activity from channels with α1D and α1C subunits.
Relationship to aged neurons in vivo
Aspects of time-dependent changes in Ca2+channel patterns seen here in cultured hippocampal neurons are strikingly similar to those seen previously in hippocampal neurons of aged animals. There is considerable evidence that altered Ca2+ homeostasis plays a role in normal brain aging and Alzheimer’s disease (Gibson and Peterson, 1987; Khachaturian, 1989; Landfield et al., 1992; Disterhoft et al., 1994; Michaelis, 1994), and one aspect of this altered Ca2+homeostasis involves an increase in voltage-gated Ca2+ potentials and currents, particularly of the L-type (Landfield and Pitler, 1984; Moyer et al., 1992; Disterhoft et al., 1994; 1996; Landfield, 1994; Campbell et al., 1996). Single-channel studies indicate that this L-current increase in hippocampal aging is associated specifically with an increase in the membrane density of functional L-type VGCCs in hippocampal neurons (Thibault and Landfield, 1996), as is seen here in culture at 4 weeksin vitro. Furthermore, as in cultured neurons (Table 1), the expression of the α1D L-type subunit mRNA is significantly increased in CA1 hippocampal neurons of 25-month-old Fischer 344 rats (Chen et al., 1995). Thus, an increase in gene expression for L-type Ca2+ channel subunits could underlie the greater Ca2+ current density with age, both in vivo and in vitro.
These analogies seem to raise the remarkable possibility that changes in Ca2+ channel expression over 28 DIV might be similar to patterns in hippocampal neurons in vivo that develop and age through more than a 700 d life cycle. It seems conceivable that some of the same Ca2+ channel changes that are seen in hippocampal neurons in vivo only in the later stages of the lifespan (Thibault and Landfield, 1996)might be accelerated and occur earlier in cultured neurons. The in vitro environment presumably lacks many of the factors, dense connections, and contact inhibition present in vivo (Goslin and Banker, 1991; Oppenheim, 1991). It also is clearly important to emphasize that there are many differences other than time course between the in vitro system and in vivo aging as well, including the extensive cell death of hippocampal neurons in culture. Although it is widely thought that aging confers vulnerability on brain neurons (e.g., Gallagher et al., 1996; Landfield et al., 1996) and clearly increases the probability of neurodegenerative (Alzheimer’s) disease (Katzman and Saitoh, 1991), the extensive cell death seen in culture differs in degree from normal aging (Coleman and Flood, 1987; West et al., 1994; Gallagher et al., 1996); the cell death in culture presumably reflects developmental factors and unfavorable environmental conditions, in addition to possible age-dependent increases in neuronal vulnerability. The occurrence, however, of two primary phases (early and late) of both Ca2+ current density increase and cell death seems to suggest the possible analogy of the late phase with gradual in vivo aging changes. This hypothesis is consistent with comparative studies of antagonistic pleiotropy showing that many normal physiological processes and patterns of gene expression that provide a critical selective advantage to an organism in early developmental and adult stages can continue as residual processes without selective value and become disadvantageous as the organism ages (Finch and Rose, 1995;Campisi, 1996). Regardless of the degree of analogy with in vivo aging, the strong similarities in hippocampal Ca2+ channel density changes in vitro andin vivo suggest that this culture system may be a highly useful model system for defining the mechanistic role of time-dependent changes in specific Ca2+ conductances in the processes of neuronal death.
Footnotes
This work was supported in part by grants from the National Institute on Aging (AG04542 and AG10836) and the Kentucky Spinal Cord and Head Injury Trust. We thank Elsie Barr and Jeanise Staton for their excellent technical assistance.
Correspondence should be addressed to Dr. Nada M. Porter, University of Kentucky, Department of Pharmacology, MS-315 UKMC, Lexington, KY 40536.
REFERENCES
- 1.Ankarcrona M, Dypbukt JM, Bonfoco E, Zhivotovsky B, Orrenius S, Lipton SA, Nicotera P. Glutamate-induced neuronal death: a succession of necrosis or apoptosis depending on mitochondrial function. Neuron. 1995;15:961–973. doi: 10.1016/0896-6273(95)90186-8. [DOI] [PubMed] [Google Scholar]
- 2.Armstrong CM, Gilly WF. Access resistance and space clamp problems associated with whole-cell patch clamping. In: Rudy B, Iverson LE, editors. Methods in enzymology Vol 207. Academic; San Diego, CA: 1992. pp. 100–122. [DOI] [PubMed] [Google Scholar]
- 3.Banker GA, Cowan WM. Rat hippocampal neurons in dispersed cell culture. Brain Res. 1977;126:397–425. doi: 10.1016/0006-8993(77)90594-7. [DOI] [PubMed] [Google Scholar]
- 4.Banker GA, Cowan WM. Further observations on hippocampal neurons in dispersed cell culture. J Comp Neurol. 1979;187:469–494. doi: 10.1002/cne.901870302. [DOI] [PubMed] [Google Scholar]
- 5.Barnes CA. Memory changes with age: neurobiological correlates. In: Martinez JR Jr, Kesner RP, editors. Learning and memory: a biological view. Academic; New York: 1991. pp. 259–296. [Google Scholar]
- 6.Basarsky TA, Parpura V, Haydon PG. Hippocampal synaptogenesis in cell culture: developmental time course of synapse formation, calcium influx, and synaptic protein distribution. J Neurosci. 1994;14:6402–6411. doi: 10.1523/JNEUROSCI.14-11-06402.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bayer SA. Development of the hippocampal region of the rat. I. Neurogenesis examined with 3H-thymidine autoradiography. J Comp Neurol. 1980;190:87–114. doi: 10.1002/cne.901900107. [DOI] [PubMed] [Google Scholar]
- 8.Bean BP. Classes of calcium channels in vertebrate cells. Annu Rev Physiol. 1989;51:367–384. doi: 10.1146/annurev.ph.51.030189.002055. [DOI] [PubMed] [Google Scholar]
- 9.Bordonaro M, Saccomanno CF, Nordstrom JL. An improved T1/A ribonuclease protection assay. BioTechniques. 1994;16:428–430. [PubMed] [Google Scholar]
- 10.Brown TH, Jaffe DB. Calcium imaging in hippocampal neurons using confocal microscopy. In: Disterhoft JF, Gispen WH, Traber J, Khachaturian ZS, editors. Calcium hypothesis of aging and dementia, Vol 747. New York Academy of Sciences; New York: 1994. pp. 313–324. [DOI] [PubMed] [Google Scholar]
- 11.Brown TH, Johnston D. Voltage-clamp analysis of mossy fiber synaptic input to hippocampal neurons. J Neurophysiol. 1983;50:487–507. doi: 10.1152/jn.1983.50.2.487. [DOI] [PubMed] [Google Scholar]
- 12.Campbell LW, Hao S-Y, Thibault O, Blalock EM, Landfield PW. Aging changes in voltage-gated calcium currents in hippocampal CA1 neurons. J Neurosci. 1996;16:6286–6295. doi: 10.1523/JNEUROSCI.16-19-06286.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Campisi J. Replicative senescence: an old live’s tale? Cell. 1996;84:497–500. doi: 10.1016/s0092-8674(00)81023-5. [DOI] [PubMed] [Google Scholar]
- 14.Chen KC, Herman JP, Rucker D, Kaminker P, Morrison DG, Booze RM, Landfield PW. Altered expression of hippocampal adrenocorticoid receptor and voltage dependent Ca2+ channel subunit mRNAs in aged rats. Soc Neurosci Abstr. 1995;21:573. [Google Scholar]
- 15.Choi D. Calcium-mediated neurotoxicity: relationship to specific channel types and role in ischemic damage. Trends Neurosci. 1988;11:465–469. doi: 10.1016/0166-2236(88)90200-7. [DOI] [PubMed] [Google Scholar]
- 16.Choi D. Calcium: still center-stage in hypoxic-ischemic neuronal death. Trends Neurosci. 1995;18:58–60. [PubMed] [Google Scholar]
- 17.Coleman PD, Flood DG. Neuron numbers and dendritic extent in normal aging and Alzheimer’s disease. Neurobiol Aging. 1987;8:521–545. doi: 10.1016/0197-4580(87)90127-8. [DOI] [PubMed] [Google Scholar]
- 18.Corey DP, Stevens CF. Science and technology of patch-recording electrodes. In: Sakmann B, Neher E, editors. Single-channel recording. Plenum; New York: 1983. pp. 53–58. [Google Scholar]
- 19.Dawson VL, Dawson TM, Bartley DA, Uhl GR, Snyder SH. Mechanisms of nitric oxide-mediated neurotoxicity in primary brain cultures. J Neurosci. 1993;13:2651–2661. doi: 10.1523/JNEUROSCI.13-06-02651.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.deToledo-Morrell L, Morrell F. Age-related alterations in long-term potentiation and susceptibility to kindling. In: Morrell F, editor. Kindling and synaptic plasticity: the legacy of Graham Goddard. Birkhäuser; Boston: 1991. pp. 160–175. [Google Scholar]
- 21.Disterhoft JF, Moyer JR, Jr, Thompson LT. The calcium rationale in aging and Alzheimer’s disease: evidence from an animal model of normal aging. In: Disterhoft JF, Gispen WH, Traber J, Khachaturian ZS, editors. Calcium hypothesis of aging and dementia, Vol 747. New York Academy of Sciences; New York: 1994. pp. 382–406. [DOI] [PubMed] [Google Scholar]
- 22.Disterhoft JF, Thompson LT, Moyer JR, Jr, Mogul DJ. Calcium-dependent afterhyperpolarization and learning in young and aging hippocampus. Life Sci. 1996;59:413–420. doi: 10.1016/0024-3205(96)00320-7. [DOI] [PubMed] [Google Scholar]
- 23.Dunlap K, Luebke JI, Turner TJ. Exocytotic Ca2+ channels in mammalian central neurons. Trends Neurosci. 1995;18:89–98. [PubMed] [Google Scholar]
- 24.Eliot LE, Johnston D. Multiple components of calcium current in acutely dissociated dentate gyrus granule neurons. J Neurophysiol. 1994;27:762–777. doi: 10.1152/jn.1994.72.2.762. [DOI] [PubMed] [Google Scholar]
- 25.Elliot EM, Malouf AT, Catterall WA. Role of calcium channel subtypes in calcium transients in hippocampal CA3 neurons. J Neurosci. 1995;15:6433–6444. doi: 10.1523/JNEUROSCI.15-10-06433.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Finch CE, Rose MR. Hormones and the physiological architecture of life history evolution. Q Rev Biol. 1995;70:1–52. doi: 10.1086/418864. [DOI] [PubMed] [Google Scholar]
- 27.Fisher RE, Gray R, Johnston D. Properties and distribution of single voltage-gated calcium channels in adult hippocampal neurons. J Neurophysiol. 1990;64:91–104. doi: 10.1152/jn.1990.64.1.91. [DOI] [PubMed] [Google Scholar]
- 28.Fletcher TL, De Camilli P, Banker G. Synaptogenesis in hippocampal cultures: evidence indicating that axons and dendrites become competent to form synapses at different stages of neuronal development. J Neurosci. 1994;14:6695–6706. doi: 10.1523/JNEUROSCI.14-11-06695.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Forti L, Pietrobon D. Functional diversity of L-type calcium channels in rat cerebellar neurons. Neuron. 1993;10:437–450. doi: 10.1016/0896-6273(93)90332-l. [DOI] [PubMed] [Google Scholar]
- 30.Gähwiler BH, Brown DA. Effects of dihydropyridines on calcium currents in CA3 pyramidal cells in slice cultures of rat hippocampus. Neuroscience. 1987;20:731–738. doi: 10.1016/0306-4522(87)90236-3. [DOI] [PubMed] [Google Scholar]
- 31.Gallagher M, Landfield PW, McEwen B, Meaney MJ, Rapp PR, Sapolsky R, West MJ. Hippocampal neurodegeneration in aging. Science. 1996;274:481. doi: 10.1126/science.274.5287.484. [DOI] [PubMed] [Google Scholar]
- 32.Gallin WJ, Greenberg ME. Calcium regulation of gene expression in neurons: the mode of entry matters. Curr Opin Neurobiol. 1995;5:367–374. doi: 10.1016/0959-4388(95)80050-6. [DOI] [PubMed] [Google Scholar]
- 33.Geinisman Y, deToledo-Morrell L, Morrell F, Heller RE. Hippocampal markers of age-related memory dysfunction: behavioral, electrophysiological and morphological perspectives. Prog Neurobiol. 1995;45:223–252. doi: 10.1016/0301-0082(94)00047-l. [DOI] [PubMed] [Google Scholar]
- 34.Gibson GE, Peterson C. Calcium and the aging nervous system. Neurobiol Aging. 1987;8:329–343. doi: 10.1016/0197-4580(87)90072-8. [DOI] [PubMed] [Google Scholar]
- 35.Goslin K, Banker G. Rat hippocampal neurons in low-density culture. In: Banker G, Goslin K, editors. Culturing nerve cells. MIT; Cambridge, MA: 1991. pp. 251–281. [Google Scholar]
- 36.Gruol DL, Deal CR, Yool AJ. Developmental changes in calcium conductances contribute to the physiological maturation of cerebellar Purkinje neurons in culture. J Neurosci. 1992;12:2838–2848. doi: 10.1523/JNEUROSCI.12-07-02838.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hamill OP, Marty E, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflügers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- 38.Hell JW, Westenbroek RE, Warner C, Ahlijanian MK, Prystary W, Gilbert MM, Snutch TP, Catterall WA. Identification and differential subcellular localization of the neuronal class C and class D L-type calcium channel α1 subunits. J Cell Biol. 1993;123:949–962. doi: 10.1083/jcb.123.4.949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Horn R. Estimating the number of channels in patch recordings. Biophys J. 1991;60:433–439. doi: 10.1016/S0006-3495(91)82069-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hui AS, Ellinor PT, Wang J, Schwartz A. Molecular cloning of multiple subtypes of a novel rat brain isoform of the alpha-1 subunit of the voltage dependent calcium channel. Neuron. 1991;7:35–44. doi: 10.1016/0896-6273(91)90072-8. [DOI] [PubMed] [Google Scholar]
- 41.Jaffe DB, Fisher SA, Brown TH. Confocal laser scanning microscopy reveals voltage-gated calcium signals within hippocampal dendritic spines. J Neurobiol. 1994;25:220–233. doi: 10.1002/neu.480250303. [DOI] [PubMed] [Google Scholar]
- 42.Johnston D, Brown TH. Interpretation of voltage-clamp measurements in hippocampal neurons. J Neurophysiol. 1983;43:409–419. doi: 10.1152/jn.1983.50.2.464. [DOI] [PubMed] [Google Scholar]
- 43.Katzman R, Saitoh T. Advances in Alzheimer’s disease. FASEB J. 1991;5:278–286. [PubMed] [Google Scholar]
- 44.Kavalali ET, Plummer MR. Multiple voltage-dependent mechanisms potentiate calcium channel activity in hippocampal neurons. J Neurosci. 1995;16:1072–1082. doi: 10.1523/JNEUROSCI.16-03-01072.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kater SB, Mattson MP, Cohan C, Connor J. Calcium regulation of the neuronal growth cone. Trends Neurosci. 1988;11:315–321. doi: 10.1016/0166-2236(88)90094-x. [DOI] [PubMed] [Google Scholar]
- 46.Kerr DS, Campbell LW, Thibault O, Landfield PW. Hippocampal glucocorticoid receptor activation enhances voltage-dependent Ca2+ conductances: relevance to brain aging. Proc Natl Acad Sci USA. 1992;89:8527–8531. doi: 10.1073/pnas.89.18.8527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Khachaturian ZS. The role of calcium regulation in brain aging: reexamination of a hypothesis. Aging. 1989;1:17–34. doi: 10.1007/BF03323872. [DOI] [PubMed] [Google Scholar]
- 48.Koike T, Martin DP, Johnson EM., Jr Role of Ca2+ channels in the ability of membrane depolarization to prevent neuronal death induced by trophic-factor deprivation: evidence that levels of internal Ca2+ determine nerve growth factor dependence of sympathetic ganglion cells. Proc Natl Acad Sci USA. 1989;86:6421–6425. doi: 10.1073/pnas.86.16.6421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Landfield PW. Increased hippocampal Ca2+ channel activity in brain aging and dementia: hormonal and pharmacologic modulation. In: Disterhoft JF, Gispen WH, Traber J, Khachaturian ZS, editors. Calcium hypothesis of aging and dementia, Vol 747. New York Academy of Sciences; New York: 1994. pp. 351–364. [DOI] [PubMed] [Google Scholar]
- 50.Landfield PW, Pitler TA. Prolonged Ca2+-dependent afterhyperpolarizations in hippocampal neurons of aged rats. Science. 1984;226:1089–1092. doi: 10.1126/science.6494926. [DOI] [PubMed] [Google Scholar]
- 51.Landfield PW, Thibault O, Mazzanti ML, Porter NM, Kerr DS. Mechanisms of neuronal death in brain aging and Alzheimer’s Disease: role of endocrine-mediated calcium dyshomeostasis. J Neurobiol. 1992;23:1247–1260. doi: 10.1002/neu.480230914. [DOI] [PubMed] [Google Scholar]
- 52.Landfield PW, McEwen BS, Sapolsky RM, Meaney MJ. Hippocampal cell death. Science. 1996;272:1247. doi: 10.1126/science.272.5266.1249b. [DOI] [PubMed] [Google Scholar]
- 53.Larmet Y, Dolphin AC, Davies AM. Intracellular calcium regulates the survival of early sensory neurons before they become dependent on neurotrophic factors. Neuron. 1992;9:563–574. doi: 10.1016/0896-6273(92)90193-h. [DOI] [PubMed] [Google Scholar]
- 54.Lipton SA. AIDS-related dementia and calcium homeostasis. In: Disterhoft JF, Gispen WH, Traber J, Khachaturian ZS, editors. Calcium hypothesis of aging and dementia, Vol 747. New York Academy of Sciences; New York: 1994. pp. 205–224. [DOI] [PubMed] [Google Scholar]
- 55.Lipton SA, Kater SB. Neurotransmitter regulation of neuronal outgrowth, plasticity and survival. Trends Neurosci. 1989;12:265–270. doi: 10.1016/0166-2236(89)90026-x. [DOI] [PubMed] [Google Scholar]
- 56.Llinás R, Sugimori M, Hillman DE, Cherksey B. Distribution and functional significance of the P-type, voltage-dependent Ca2+ channels in the mammalian central nervous system. Trends Neurosci. 1992;15:351–355. doi: 10.1016/0166-2236(92)90053-b. [DOI] [PubMed] [Google Scholar]
- 57.Lobner D, Lipton P. Intracellular calcium levels and calcium fluxes in the CA1 region of the rat hippocampal slice during in vitro ischemia: relationship to electrophysiological cell damage. J Neurosci. 1993;13:4861–4871. doi: 10.1523/JNEUROSCI.13-11-04861.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Magee JC, Johnston D. Synaptic activation of voltage-gated channels in the dendrites of hippocampal pyramidal neurons. Science. 1995;268:301–304. doi: 10.1126/science.7716525. [DOI] [PubMed] [Google Scholar]
- 59.Mattson MP, Kater SB. Calcium regulation of neurite elongation and growth cone motility. J Neurosci. 1987;7:4034–4043. doi: 10.1523/JNEUROSCI.07-12-04034.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Meyers DER, Barker JL. Whole-cell patch-clamp analysis of voltage-dependent calcium conductances in cultured embryonic neurons. J Neurophysiol. 1989;61:467–477. doi: 10.1152/jn.1989.61.3.467. [DOI] [PubMed] [Google Scholar]
- 61.Michaelis M. Ion transport systems and Ca2+ regulation in aging neurons. In: Disterhoft JF, Gispen WH, Traber J, Khachaturian ZS, editors. Calcium hypothesis of aging and dementia, Vol 747. New York Academy of Sciences; New York: 1994. pp. 407–418. [DOI] [PubMed] [Google Scholar]
- 62.Mintz IM, Adams ME, Bean BP. P-type calcium channels in rat central and peripheral neurons. Neuron. 1992;9:85–95. doi: 10.1016/0896-6273(92)90223-z. [DOI] [PubMed] [Google Scholar]
- 63.Miyakawa H, Ross WN, Jaffe D, Callaway JC, Lasser-Ross N, Lisman JE, Johnston D. Synaptically activated increases in Ca2+ concentration in hippocampal CA1 pyramidal cells are primarily due to voltage-gated Ca2+ channels. Neuron. 1992;9:1163–1173. doi: 10.1016/0896-6273(92)90074-n. [DOI] [PubMed] [Google Scholar]
- 64.Mogul DJ, Fox AP. Evidence for multiple types of Ca2+ channels in acutely isolated hippocampal CA3 neurones of the guinea-pig. J Physiol (Lond) 1991;433:259–281. doi: 10.1113/jphysiol.1991.sp018425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Morris RGM, Garrud P, Rawlins JNP, O’Keefe J. Place navigation impaired in rats with hippocampal lesions. Nature. 1982;297:681–683. doi: 10.1038/297681a0. [DOI] [PubMed] [Google Scholar]
- 66.Moyer JR, Thompson LT, Black JP, Disterhoft JF. Nimodipine increases excitability of rabbit CA1 pyramidal neurons in an age- and concentration-dependent manner. J Neurophysiol. 1992;68:2100–2109. doi: 10.1152/jn.1992.68.6.2100. [DOI] [PubMed] [Google Scholar]
- 67.Neher E. Correction for liquid junction potentials in patch clamp experiments. In: Rudy B, Iverson LE, editors. Methods in enzymology, Vol 207. Academic; San Diego: 1992. pp. 123–131. [DOI] [PubMed] [Google Scholar]
- 68.Novelli A, Reilly JA, Lysko PG, Henneberry RC. Glutamate becomes neurotoxic via the N-methyl-d-aspartate receptor when intracellular energy levels are reduced. Brain Res. 1988;451:205–212. doi: 10.1016/0006-8993(88)90765-2. [DOI] [PubMed] [Google Scholar]
- 69.Nowycky MC, Fox AP, Tsien RW. Long-opening mode of gating of neuronal calcium channels and its promotion by the dihydropyridine calcium agonist Bay K 8644. Proc Natl Acad Sci USA. 1985;82:2178–2182. doi: 10.1073/pnas.82.7.2178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Oppenheim RW. Cell death during development of the nervous system. Annu Rev Neurosci. 1991;14:453–501. doi: 10.1146/annurev.ne.14.030191.002321. [DOI] [PubMed] [Google Scholar]
- 71.Pitler TA, Landfield PW. Probable Ca2+-mediated inactivation of Ca2+ currents in mammalian brain neurons. Brain Res. 1987;410:147–153. doi: 10.1016/s0006-8993(87)80037-9. [DOI] [PubMed] [Google Scholar]
- 72.Randall A, Tsien RW. Pharmacological dissection of multiple types of Ca2+ channel currents in rat cerebellar granule neurons. J Neurosci. 1995;15:2995–3012. doi: 10.1523/JNEUROSCI.15-04-02995.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ransom BR, Neale E, Henkat M, Bullock PN, Nelson PG. Mouse spinal cord in cell culture. I. Morphology and intrinsic neuronal electrophysiologic properties. J Neurophysiol. 1977;40:1132–1150. doi: 10.1152/jn.1977.40.5.1132. [DOI] [PubMed] [Google Scholar]
- 74.Regehr WG, Tank DW. Dendritic calcium dynamics. Curr Opin Neurobiol. 1994;4:373–382. doi: 10.1016/0959-4388(94)90099-x. [DOI] [PubMed] [Google Scholar]
- 75.Rothman SM, Olney JW. Excitotoxicity and the NMDA receptor. Trends Neurosci. 1987;10:299–302. doi: 10.1016/0166-2236(95)93869-y. [DOI] [PubMed] [Google Scholar]
- 76.Rusanescu G, Qi H, Thomas SM, Brugge JS, Halegoua S. Calcium influx induces neurite growth through a Src-Ras signaling cassette. Neuron. 1995;15:1415–1425. doi: 10.1016/0896-6273(95)90019-5. [DOI] [PubMed] [Google Scholar]
- 77.Sakmann B, Neher E. Geometric parameters of pipettes and membrane patches. In: Sakmann B, Neher E, editors. Single channel recording. Plenum; New York: 1983. pp. 37–51. [Google Scholar]
- 78.Scriabine A, Schuurman T, Traber J. Pharmacological basis for the use of nimodipine in central nervous system disorders. FASEB J. 1989;3:1799–1806. [PubMed] [Google Scholar]
- 79.Segal M. Rat hippocampal neurons in culture: responses to electrical and chemical stimuli. J Neurophysiol. 1983;50:1249–1264. doi: 10.1152/jn.1983.50.6.1249. [DOI] [PubMed] [Google Scholar]
- 80.Sigworth F, Zhou J. Analysis of nonstationary single-channel currents. In: Rudy B, Iverson LE, editors. Methods in enzymology, Vol 207. Academic; San Diego: 1992. pp. 746–762. [DOI] [PubMed] [Google Scholar]
- 81.Slesinger PA, Lansman JB. Reopening of Ca2+ channels in mouse cerebellar neurons at resting membrane potentials during recovery from inactivation. Neuron. 1991;7:755–762. doi: 10.1016/0896-6273(91)90278-8. [DOI] [PubMed] [Google Scholar]
- 82.Snutch TP, Tomlinson WJ, Leonard JP, Gilbert MM. Distinct calcium channels are generated by alternative splicing and are differentially expressed in the mammalian CNS. Neuron. 1991;7:45–57. doi: 10.1016/0896-6273(91)90073-9. [DOI] [PubMed] [Google Scholar]
- 83.Spitzer NC. Spontaneous Ca2+ spikes and waves in embryonic neurons: signalling systems for differentiation. Trends Neurosci. 1994;17:115–118. doi: 10.1016/0166-2236(94)90120-1. [DOI] [PubMed] [Google Scholar]
- 84.Stuart GJ, Sakmann B. Active propagation of somatic action potentials into neocortical pyramidal cell dendrites. Nature. 1994;367:69–72. doi: 10.1038/367069a0. [DOI] [PubMed] [Google Scholar]
- 85.Stuiver BT, Douma BRK, Bakker R, Nyakas C, Luiten PGM. In vivo protection against NMDA-induced neurodegeneration by MK-801 and nimodipine: combined therapy and temporal course of protection. Neurodegeneration. 1996;5:153–159. doi: 10.1006/neur.1996.0022. [DOI] [PubMed] [Google Scholar]
- 86.Thibault O, Landfield PW. Increase in single L-type calcium channels in hippocampal neurons during aging. Science. 1996;272:1007–1020. doi: 10.1126/science.272.5264.1017. [DOI] [PubMed] [Google Scholar]
- 87.Thibault O, Porter NM, Landfield PW. Low Ba2+ and Ca2+ induce a sustained high frequency of repolarization openings of L-type Ca2+ channels in hippocampal neurons: physiological implications. Proc Natl Acad Sci USA. 1993;90:11792–11796. doi: 10.1073/pnas.90.24.11792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Thibault O, Porter NM, Mazzanti-Rose ML, Campbell LW, Blalock EM, Landfield PW. Dual patch pipette recordings in hippocampal neurons: evidence that long Ca2+ tail currents reflect Ca2+ channel activity at resting potential. Soc Neurosci Abstr. 1995;21:1577. [Google Scholar]
- 89.Thompson CB. Apoptosis in the pathogenesis and treatment of disease. Science. 1995;267:1456–1462. doi: 10.1126/science.7878464. [DOI] [PubMed] [Google Scholar]
- 90.Tsien RW, Lipscombe D, Madison DV, Bley KR, Fox AP. Multiple types of neuronal calcium channels and their selective modulation. Trends Neurosci. 1988;11:431–437. doi: 10.1016/0166-2236(88)90194-4. [DOI] [PubMed] [Google Scholar]
- 91.Tsien RW, Lipscombe D, Madison D, Bley K, Fox A. Reflections on Ca2+-channel diversity, 1988–1994. Trends Neurosci. 1995;18:52–54. [PubMed] [Google Scholar]
- 92.Turrigiano G, LeMasson G, Marder E. Selective regulation of current densities underlies spontaneous changes in the activity of cultured neurons. J Neurosci. 1995;15:3640–3652. doi: 10.1523/JNEUROSCI.15-05-03640.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Uematsu D, Greenberg JH, Hickey WF, Reivich M. Nimodipine attenuates both increase in cytosolic free calcium and histologic damage following focal cerebral ischemia and reperfusion in cats. Stroke. 1989;20:1531–1537. doi: 10.1161/01.str.20.11.1531. [DOI] [PubMed] [Google Scholar]
- 94.West MJ, Coleman PD, Flood DG, Troncoso JC. Differences in the pattern of hippocampal neuronal loss in normal ageing and Alzheimer’s disease. Lancet. 1994;344:769–772. doi: 10.1016/s0140-6736(94)92338-8. [DOI] [PubMed] [Google Scholar]
- 95.Wisniewski HM, Terry RD. Morphology of the aging brain, human and animal. In: Ford DM, editor. Progress in brain research, Vol 40. Elsevier; Amsterdam: 1973. pp. 167–186. [DOI] [PubMed] [Google Scholar]
- 96.Wong RK, Prince DA. Afterpotential generation in hippocampal pyramidal cells. J Neurophysiol. 1981;45:86–97. doi: 10.1152/jn.1981.45.1.86. [DOI] [PubMed] [Google Scholar]
- 97.Xia Y, Ragan RE, Seah EEC, Michaelis ML, Michaelis EK. Developmental expression of N-methyl-d-aspartate (NMDA)-induced neurotoxicity, NMDA receptor function, and the NMDAR1 and glutamate-binding protein subunits in cerebellar granule cells in primary cultures. Neurochem Res. 1995;20:617–629. doi: 10.1007/BF01694545. [DOI] [PubMed] [Google Scholar]
- 98.Yuste R, Denk W. Dendritic spines as basic functional units of neuronal integration. Nature. 1995;375:682–684. doi: 10.1038/375682a0. [DOI] [PubMed] [Google Scholar]
- 99.Zola-Morgan SM, Squire LR. The primate hippocampal formation: evidence for a time-limited role in memory storage. Science. 1990;250:288–290. doi: 10.1126/science.2218534. [DOI] [PubMed] [Google Scholar]