

Abstract
Selective vulnerability of subpopulations of neurons is a striking feature of neurodegeneration. Mitochondrially transmitted diseases are no exception. In this study CNS tissues from a patient with myoclonus epilepsy and ragged red fibers (MERRF) syndrome, which results from an A to G transition of nucleotide (nt) 8344 in the mitochondrial tRNALys gene, were examined for the proportion of mutant mtDNA. Either individual neuronal somas or the adjacent neuropil and glia were microdissected from cryostat tissue sections of histologically severely affected brain regions, including dentate nuclei, Purkinje cells, and inferior olivary nuclei, and from a presumably less affected neuronal subpopulation, the anterior horn cells of the spinal cord. Mutant and normal mtDNA were quantified after PCR amplification with a mismatched primer and restriction enzyme digestion. Neurons and the surrounding neuropil and glia from all CNS regions that were analyzed exhibited high proportions of mutant mtDNA, ranging from 97.6 ± 0.7% in Purkinje cells to 80.6 ± 2.8% in the anterior horn cells. Within each neuronal group that was analyzed, neuronal soma values were similar to those in the surrounding neuropil and glia or in the regional tissue homogenate. Surprisingly, as compared with controls, neuronal loss ranged from 7% of the Purkinje cells to 46% of the neurons of the dentate nucleus in MERRF cerebellum. Thus, factors other than the high proportion of mutant mtDNA, in particular nuclear-controlled neuronal differences among various regions of the CNS, seem to contribute to the mitochondrial dysfunction and ultimate cell death.
Keywords: MERRF syndrome, CNS microdissection, PCR, neurodegeneration, tRNALys, mtDNA
Selective vulnerability of neuronal or glial subpopulations is a feature of many neurological diseases. The myoclonus epilepsy and ragged red fibers (MERRF) syndrome is characterized by myoclonic epilepsy, cerebellar ataxia, and progressive muscular weakness. In this maternally inherited disease (Wallace et al., 1988) the symptoms of myoclonic epilepsy, ataxia, and muscular weakness may reflect severe pathological changes in neurons of the dentato-rubral and the spinocerebellar pathways and of the inferior olivary nuclei and in skeletal muscle. The myopathic changes include the generation of ragged red fibers indicative of proliferation and subsarcolemmal accumulation of mitochondria.
The MERRF syndrome is most commonly the result of a single base pair substitution at position 8344 in the mitochondrial tRNALys gene (Shoffner et al., 1990, 1991; Yoneda et al., 1990; Chomyn et al., 1991; Hammans et al., 1993; Silvestri et al., 1993). The relationship between mtDNA genotype and phenotype of selective neuronal vulnerability has not been defined. It is not known whether the affected neurons contain more mutant mtDNA, either in their somas or in their processes within the surrounding neuropil, than neurons in the relatively spared regions, or, alternatively, whether differences in nuclear gene activity play a crucial role in determining the degree of damage to various neuronal populations. In particular, important variables differentially affecting cell survival may include demand for oxidative phosphorylation of different cell types and the terminally differentiated state of the neuron.
Previous quantifications of the mutant mtDNA proportion in the CNS have been made in tissue homogenates of anatomical regions (Tanno et al., 1993; Sanger and Jain, 1996). However, these samples contained not only neurons but the surrounding glia, including oligodendrocytes, astrocytes, and microglia as well as blood vessels. Furthermore, these analyses could not distinguish neuronal somas from dendrites, axons, and synaptic terminals that form the surrounding neuropil.
In the present study we have compared neuronal loss with the percentage of mutant mtDNA in the more severely and the less severely affected neuronal groups. Individual neuronal somas were microdissected from CNS tissue sections. Mutant and normal mtDNA were quantified after PCR amplification with a mismatched primer and restriction enzyme digestion (Zeviani et al., 1991; Yoneda et al., 1991). Four neuronal subpopulations were compared: Purkinje cells and neurons of the dentate nuclei of the cerebellum and neurons of the inferior olivary nuclei of the medulla, all heavily vulnerable, and motor neurons of the spinal cord anterior horns, a presumably less-affected neuronal subpopulation. Three types of samples from each neuronal group were analyzed: neuronal somas only, neuropil and glia only, and homogenates of regions, including entire neuronal groups (neuronal somas plus neuropil and glia).
MATERIALS AND METHODS
CNS tissue. CNS tissues were obtained 20 hr postmortem from a 14-year-old Hispanic female with clinical and neuropathologically confirmed MERRF syndrome (Sanger and Jain, 1996). Paraffin-embedded CNS tissue sections (7 μm) were stained with hematoxylin and eosin. Control tissues for histology were obtained postmortem from an age-matched patient who died from non-neurological causes.
Neuronal loss was compared in MERRF and control tissues in four sections from each paraffin block. Neurons were counted at 250× magnification in five fields of either dentate nuclei or inferior olivary nuclei, or in the entire bilateral anterior horn region of the lower cervical and upper thoracic spinal cord, or Purkinje cells in five foliar loops. Counts were confirmed in all sites by two independent observers. The mean values were calculated for each.
For PCR analysis, blocks of tissue (1 cm3) were dissected at autopsy, immediately snap-frozen in liquid nitrogen-chilled isopentane, and stored at −90°C. Four neuronal subpopulations were selectively analyzed: the Purkinje neurons and dentate nuclei neurons of the cerebellum, the inferior olivary nuclei neurons of the medulla, and motor neurons from the anterior horns of the spinal cord. Two types of samples were examined from each neuronal group: pooled neuronal somas and neuropil with glia. The dissected Purkinje cell somas also were analyzed individually. Maternal and patient’s leukocytes were each prepared for PCR, using the same procedure as for the CNS tissues. Snap-frozen postmortem CNS tissues from one neurologically normal patient (age 40) were used for PCR analysis.
Neuronal dissection. Cryostat tissue sections (35 μm) were stained with 1% toluidine blue in 1% Na-borate in water for 30 sec and then rinsed in water until the excess stain was removed. Stained slides were viewed with an inverted microscope. Individual neurons were dissected out and suctioned into a glass micropipette guided by a Narashige micromanipulator (Narashige Scientific Instrument Lab, Tokyo, Japan). The pipette system consisted of a siliconized, 1-mm-diameter thin glass micropipette (World Precision Instruments, Sarasota, FL). Fine tips for dissection were prepared, using a model P-77 Brown-Flaming micropipette puller (Sutter Instrument, San Francisco, CA) at a heater temperature setting of 30 for 7 sec. The tip was gently, manually broken to yield an opening of ∼50 μm diameter. The blunt end was inserted into Tygon SILASTIC tubing [0.02 inches inner diameter (i.d.)], which was connected to amber latex rubber tubing (1/8 inch i.d.). A disposable syringe (30 ml) tip was inserted into the latex tubing, and gentle suction was provided to the system. Before isolation of the neuronal somas, the field was moistened with one or two drops of TE buffer (10 mm Tris-HCl, pH 7.6, and 1 mm EDTA). Neuropil plus glia samples included the residual tissue immediately surrounding the site of the neuronal soma after its dissection. After isolation of the neuronal somas or neuropil, the samples were transferred into separate microfuge tubes and centrifuged at 12,800 × g for 2 min. The supernatant fluid was removed and replaced with DNA extraction solution (50 mmTris-HC1, pH 8.5, 1 mm EDTA, and 0.5% Tween 20, containing 200 μg/ml proteinase K). The samples were incubated at 55°C for 2 hr and then immediately heated at 95°C for 10 min to inactivate the proteinase K.
PCR amplification of mtDNA. For PCR analysis, 20–30 neuronal soma isolates from either a specific nucleus or neuronal subpopulation were pooled in 50 μl of digestion buffer. Thus, ∼2.5 soma equivalents were included in each reaction sample of 10 μl. Neuropil samples were obtained from sections after the removal of somas. For tissue samples, material from ∼100 μm2 areas that included neuronal somas, neuropil, and glia was isolated from a tissue section. For “single-cell” analyses two Purkinje cell soma sections (approximately one soma equivalent) were isolated in 25 μl of digestion buffer.
The PCR conditions were based on the method of Zeviani et al. (1991). For the PCR reaction the following pair of primers was used, corresponding to mtDNA positions (5′-3′) 8278–8297 and (3′-5′) 8385–8345 (according to the Cambridge sequence, Anderson et al., 1981). The backward primer introduced mismatches at positions 8352 (C-C) and 8353 (T-C) that create, in combination with the A→G transition at position 8344, a restriction site for the enzymeBglI in the amplified fragments from the mutant, but not the wild-type, mtDNA.
The extracted DNA was combined with 20 pmol of primer 1 and 40 pmol of primer 2, 1.5 mm MgCl2, and 0.4 μl (2 U) of Taq polymerase (Perkin-Elmer, Norwalk, CT). The mixture was brought to a final volume of 50 μl by the addition of distilled H20, and then it was submitted to an initial 30 cycle PCR amplification. The template DNA was added to the reaction mixture after it was heated to 90°C, before the thermal cycling was started. The PCR protocol involved an initial incubation at 95°C for 5 min before cycling. The cycling procedure then followed, with a 30 sec incubation at 94°C for denaturation, a 30 sec annealing step at 55°C, and a 30 sec elongation at 72°C. The addition of 1.5 U ofTaq polymerase was followed by 10 more PCR cycles. For some experiments the PCR product was purified after the initial 30 cycles by the Gene-Clean Kit II (BIO-101, Vista, CA). The DNA purification step, however, reduced significantly the final amount of the product. In such cases we then performed an additional 30 cycles as described above, after mixing 300 ng of purified DNA and 25 μl of a PCR reaction mixture and adding distilled H20 to a final volume of 50 μl.
The PCR product plus 300 ng of pBluescript II SK (pBS II) DNA, added to each tube as an internal marker to assess completeness of digestion, were digested with 25 U of BglI (Fisher, Pittsburgh, PA) from 2 hr to overnight at 37°C. Samples of the digestion products were electrophoresed on a 1% agarose gel for detection of the pBS II and on a nondenaturing 12% polyacrylamide gel (29:1 acrylamide/bis-acrylamide) to detect the mutant mtDNA. The molecular weight marker for the agarose gels was a 1 kb DNA ladder (Life Technologies, Gaithersburg, MD) and HinfI-digested φX174 (Promega, Madison, WI) for the polyacrylamide gel. The ethidium bromide-stained gels were photographed with Polaroid instant film (positive–negative), type 55.
To quantify the percentages of mutant and wild-type genomes, we used a BioImage laser densitometer. The data were corrected for the presence of heteroduplexes resistant to BglI digestion, formed in the last PCR cycle, by using a standard curve (Yoneda et al., 1994).
RESULTS
Typical histological features of MERRF syndrome, characterized by neuronal cytoplasmic eosinophilia, striking shrinkage of many of the remaining neurons, and astrocytic gliosis, were seen in all sites that were examined (Fig. 1). The most severe changes were in the dentate nucleus, and the Purkinje cells were the least involved. Neuronal counts (Fig. 2) indicated a 46% reduction in the number per unit area of dentate nucleus neurons as compared with an age-matched control. Furthermore, there was a 29% loss of inferior olivary nucleus neurons and a comparable reduction (28%) of spinal motor neurons of the lower cervical and upper thoracic segments. By contrast, there was a striking preservation of Purkinje cells, with no significant loss.
Fig. 1.
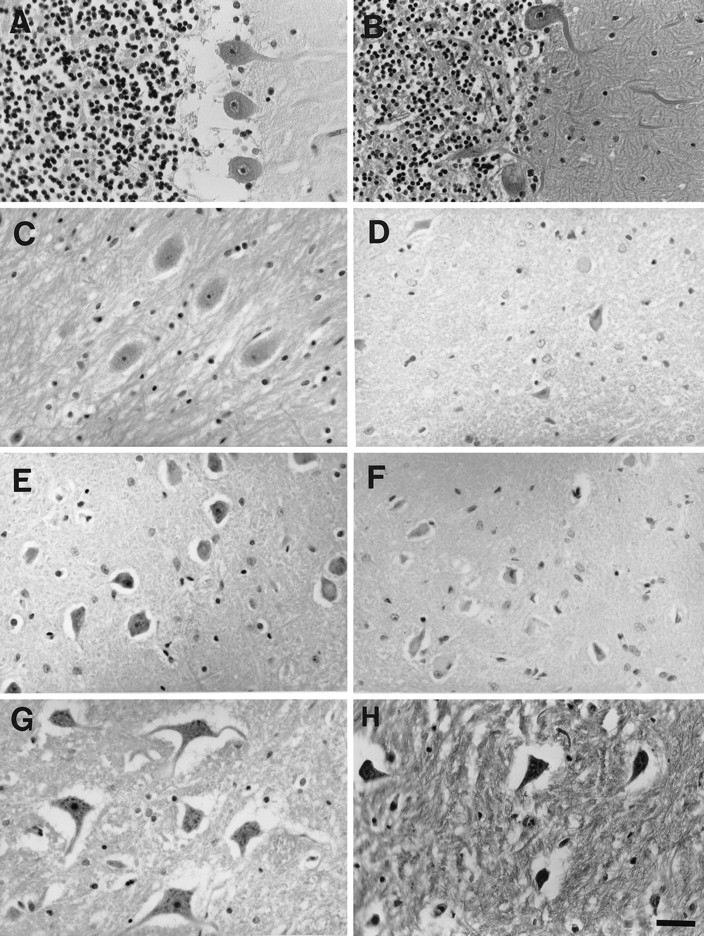
Tissue sections stained with hematoxylin and eosin of cerebellum (A, B), dentate nucleus (C, D), inferior olivary nucleus (E, F), and anterior horns of the thoracic spinal cord (G, H).A, C, E, and G show tissues from the neurologically normal individual; B, D, F, andH show tissues from the MERRF syndrome patient. In the diseased tissues neurons exhibit shrunken somas when compared with the normal control samples. In the control cerebellum there is an artifactual separation of the Purkinje cells from the underlying internal granule layer. Scale bar, 40 μm.
Fig. 2.
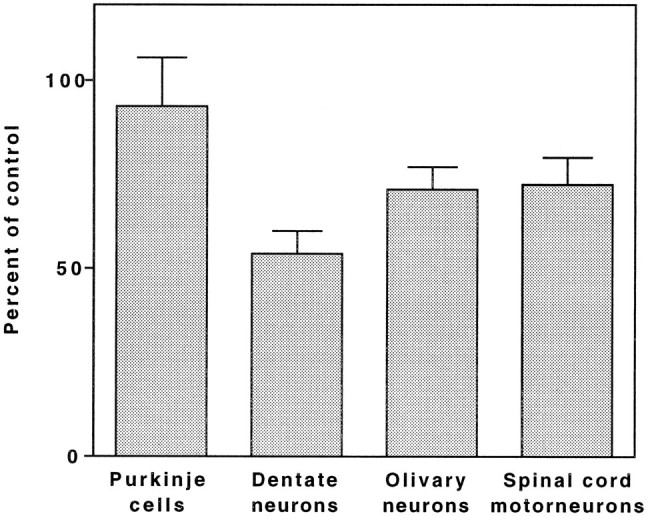
Bar graph representing the percentage of remaining neurons in different CNS regions. The number of neurons per microscopic field (at 250× magnification) was determined as described in Materials and Methods. The bars represent the ratios of the mean number in the MERRF sections to the mean number in the control sections. Error bars represent the SEM.
For PCR analysis Purkinje cells, neurons of the dentate and inferior olivary nuclei, and α motor neurons from the cervical or thoracic spinal cord were microdissected individually. As shown in Figure3, for Purkinje cells and neurons of the inferior olivary nucleus, individual somas were isolated intact with nearly total exclusion of the surrounding neuropil. The isolated Purkinje cells either were analyzed in pairs or were pooled in samples of 20–30 somas.
Fig. 3.

Microdissection procedure. The tissue sections of cerebellum are stained with 1% toluidine blue. A, The neuron to be removed (arrow) is targeted by the tip of a glass micropipette. B, The neuron is drawn into the glass micropipette. C, The same section is shown after suction of the neuronal soma. Scale bar, 70 μm. D, Neurons (arrows) in the inferior olivary nucleus.E, The same section after the neurons have been removed by syringe suction. Scale bar, 50 μm.
The results of BglI digestion of the PCR fragments amplified from the DNA of the four different regions of the CNS are shown in Figure 4. For each site the samples included pooled, isolated neuronal somas and homogenates of nondissected tissue sections (Fig. 4A–C). A sample of neuropil minus the neuronal somas is shown in Figure4B, and DNA from the equivalent of an individual Purkinje cell is shown in Figure 4C.
Fig. 4.

Results of BglI digestions of the PCR fragments amplified from the DNA of three different regions of the CNS. The top band is the 108 bp wild-type mtDNA fragment. The two lower bands are the 35 and 73 bp fragments derived from mutant mtDNA. A, Regional tissue compared with pooled neuronal isolates. Unmarked lane, Size marker, φX174 digested with HinfI. Lane 1, Uncut; lane 2, inferior olivary nucleus, tissue; lane 3, inferior olivary nucleus, neuronal somas; lane 4, dentate nucleus, tissue; lane 5, dentate nucleus, neuronal somas; lane 6, spinal cord, tissue; lane 7, spinal cord, motor neuron somas; lane 8, MERRF patient’s blood; lane 9, mother’s blood. B, Regional tissue compared with neuropil. Unmarked lane, Size marker, as inA. Lane 1, Dentate nucleus, tissue;lane 2, dentate nucleus, neuropil. C, Regional tissue compared with a single neuron. Lane 1, Cerebellar tissue; lane 2, single Purkinje cell equivalent; lane 3, primers only, no DNA (arrow indicates primer dimer). D, pBluescript II SK as DNA internal marker of digestion withBglI. Unmarked lane, Size marker, 1 kb DNA ladder.Lane 1, Complete digestion; lane 2, uncut. The bottom band is supercoiled DNA; thetop band is relaxed circular DNA.
The presence of mutant mtDNA with the A→G substitution at bp 8344 was observed in all MERRF tissue samples. Using ethidium bromide stain, we detected a pattern of three bands by polyacrylamide gel electrophoresis. Two bands corresponded to the expected 35 and 73 bp products of cleavage of the PCR fragments at the BglI site created from the mutant template. The third band corresponded to the 108 bp uncut PCR product derived from wild-type DNA (Fig.4A, lane 1). Depending on the tissue site and sample, the three bands were present in variable amounts (Fig.4A–C). The exclusive presence of pBluescript DNA cleavage products of 1694 and 1267 bp in the agarose gel indicates the virtually complete extent of BglI digestion (Fig.4D).
An additional band of ∼60 bp was seen occasionally, especially in samples with small quantities of DNA, such as those derived from individual Purkinje cells (Fig. 4D, lane 2) and in a control sample lacking template DNA but containing the primers (Fig.4C, lane 3). The 3′ ends of the two primers used for PCR were complementary in four of the five terminal bases. If the primers annealed to each other and were extended by the polymerase, a 56 bp product would have been expected. Therefore, the ≅60 bp band most likely resulted from primer dimerization rather than from degradation.
Quantification of the results revealed that the mean percentage of mutant mtDNA was high in all four anatomical areas, but it was the highest in the Purkinje neurons (96.7% ± 0.7% in the individual cells and 90.5 ± 0.5% in the pooled cells; Fig.5). The motor neurons of the anterior horn of the spinal cord had a statistically significant or nearly significant lower proportion of mutant mtDNA (84.3 ± 4.2%;p < 0.06, when compared with the pooled Purkinje cells; p < 0.01, when compared with the individual Purkinje cells). The neurons of the dentate nuclei of the cerebellum and those of the inferior olivary nuclei of the medulla contained intermediate proportions of mutant mtDNA (89.0 ± 1.5% and 86.2 ± 3.8%, respectively).
Fig. 5.

Bar graph representing the percentage of mutant mtDNA in different CNS regions. Each bar represents the mean of two to six determinations by PCR and gel analysis of the proportion of mutant DNA in the specified tissue or neuronal isolate. The gel analysis was done on at least two pools of each type of sample (some pools were sampled more than once for PCR and gel analysis), with the exception of blood. Seven determinations were made on one sample of the patient’s blood. neuron, Neuronal soma;Purkinje cells, Purkinje cell soma;neuropil, the material immediately surrounding the neuronal soma collected after the soma was removed;tissue, material including somas, neuropil, and glia from a tissue section. The bar labeled individual Purkinje cells represents the mean of six pairs of Purkinje soma halves. The proportion of mutant DNA in each pair is 97.3, 97.6, 96.5, 95.8, 97.0, and 95.6%. The error bars represent 2 SEM.
There were comparable values of the percentage of mutant mtDNA in tissue samples within each CNS region. Thus, samples of cerebellar cortical homogenates showed a mutant DNA proportion similar to that of the Purkinje cell component. Omitting the toluidine blue tissue stain caused no quantitative or qualitative changes in the PCR amplification or BglI cleavage of the PCR product (Fig. 5, lane 2). Internal granule cells exhibited slightly lower proportions of mutant DNA than Purkinje cells (88.8 ± 1.3%; Fig. 5). In the other CNS regions the mutant mtDNA proportion determined for the tissue homogenate or the neuropil was only slightly lower than that found in the isolated neurons.
DNA from individually microdissected Purkinje cells subjected in pairs to PCR amplification of the mtDNA component revealed remarkably similar proportions of mutant DNA from pair to pair, with mean values of 96.7% ± 0.7% (Fig. 5, lane 3). Such a small error suggests that the variability is minimal from cell to cell. Shrunken somas contained the same amount of the mutant form as histologically normal neighboring neurons (data not shown). There is no obvious explanation for the ∼5% difference between individual and pooled Purkinje cell somas. Overall, results pointed to a relatively homogeneous distribution of mutant mtDNA in a given region regardless of cell class or whether samples were restricted to neuronal somas or neuropil only.
DISCUSSION
In the present study regional and single neuronal PCR analyses have revealed high percentages of mutant mtDNA in all of the CNS neuronal populations tested. There were, however, differences in the proportion of mutant mtDNA among the neuronal populations analyzed, with the Purkinje cells exhibiting the highest proportion (90–97%), the motor neurons of the spinal cord anterior horns exhibiting a statistically significant lower proportion (84%), and the neurons of dentate nuclei and of the inferior olivary nuclei exhibiting intermediate percentages (89 and 86%, respectively). Tanno et al. (1993), using quantitative PCR analysis of whole tissue samples of specific brain regions, also found only small differences in the proportion of mutant mtDNA among different brain regions. Our results, obtained at a higher level of resolution, agree with their findings.
In the present work the separation of neuronal somas from the surrounding tissue also has allowed the analysis of the distribution of the mutant mtDNA among neuronal somas and neuropil. Thus, the slight differences observed in mutant mtDNA proportion between neuronal somas and the surrounding neuropil would tend to exclude a segregation of the mutant mtDNA exclusively or predominantly in neurons or in glia.
The microdissection of neuronal somas from specific regions of the brain from the MERRF patient also has permitted a comparison of the histopathology with the percentage of mutant mtDNA in these regions. Thus, the high proportion of mutant mtDNA did not correlate directly with the histopathology, including neuronal loss. This discrepancy was especially striking for Purkinje neurons. “Individual” somas of residual Purkinje cells consistently showed the highest percentage of mutant mtDNA among the brain regions analyzed (97%), yet there was remarkably little loss of these neurons (7%). There was no significant individual variability among these neurons in their proportion of mutant DNA. Only slightly lower proportions of mutant mtDNA were found in the neurons of the dentate nucleus, as compared with Purkinje cells. However, in striking contrast to the latter cells, >45% of neurons were lost in this nucleus. This loss may be relevant to the clinical manifestation of myoclonic epilepsy.
There are several possible explanations for the discrepancy between the proportion of mutant mtDNA and the neuronal loss discussed above. As evidenced by the selective vulnerability to hypoxia (Brierly et al., 1973), requirements for oxidative phosphorylation differ among neuronal subpopulations. In fact, differential use of the glycolytic pathway may exist among such populations. Other mechanisms, such as excitotoxicity, may be triggered by mitochondrial dysfunction in glutamatergic neurons (Schinder et al., 1996). It is also possible that a different vulnerability to apoptosis of neurons harboring mtDNA mutations from various CNS regions (Asoh et al., 1996) plays a role in the phenomenon observed here. In this connection it is worth noting that a nonuniform susceptibility to cell death also may account for the differences in proportion of mutant mtDNA found among the various neuronal subpopulations. Thus, it is possible that the neurons of the olivary nuclei and the motor neurons of the spinal cord would degenerate and die when a certain level of mutant mtDNA (85%) is reached. Therefore, the maximum proportion of mutant mtDNA would be lower in these neuronal populations than in other neurons, such as Purkinje cells, which could survive with a higher level of mutant mtDNA.
Recent studies by Enriquez et al. (1995) demonstrate that, in cells carrying the MERRF mutation in nearly homoplasmic form, the mutant mitochondrial tRNALys is present at moderately but significantly decreased levels. This tRNA is functional, although it exhibits a reduced capacity to be charged by the specific synthetase. However, the reduced level of charged tRNALys per cell, i.e., ≃50–60% of the normal level, is not sufficient to support normal respiratory chain function. An addition of a minimum of 10% wild-type mtDNA is required for full respiratory competence (Yoneda et al., 1994; Attardi et al., 1995). These observations were made on transmitochondrial cell lines carrying the MERRF mutation, which were constructed by transfer of the patient’s mitochondria into human mtDNA-less cells (Chomyn et al., 1991). A threshold effect also was reported by Larsson et al. (1992), who observed that >92% of mutant mtDNA was required for causing respiratory chain dysfunction in muscle. Boulet et al. (1992) obtained comparable findings in myoblast cultures derived from muscle biopsies of MERRF patients. Therefore, complementation of the mutation in individual heteroplasmic mitochondria and/or the residual functional capacity of the mutant mtDNA may support sufficient oxidative phosphorylation for maintaining cell viability in certain cell types.
The cellular model of the MERRF syndrome mentioned above has been used recently to study the rules that govern complementation of the mutation (Yoneda et al., 1994; Attardi et al., 1995). It is reasonable to think that the distribution of mutant mtDNA among mitochondria and the threshold of wild-type mtDNA required for complementation vary among different cell types in the CNS. Thus, the most plausible interpretation of the results obtained in the present work is that the nuclear gene activity plays an important role in the phenotypic manifestation of the MERRF mutation (Lertrit et al., 1992), as has been shown for other disease-causing mtDNA mutations (Harding et al., 1995;Guan et al., 1996). This possibility is supported by the fact that the nt8344 mutation can be associated with other phenotypes (Silvestri et al., 1993).
The sensitivity of PCR amplification extended to the single-cell level and the reliability of the dissection under direct visualization described in the present work provide a powerful method for analyzing, at the cellular level, the distribution of an mtDNA mutation, which is applicable to other mtDNA-linked diseases affecting the CNS. In contrast to in situ PCR, quantification of mutant mtDNA is possible in single neurons. Resolution of dissection at the light microscopic level followed by PCR is, in fact, higher than that achievable by applying PCR directly to sections (O’Leary et al., 1996). Application of single-cell dissection and analysis of mRNA expression have been made in cultures (Lambolez et al., 1992; Mackler et al., 1992). Recently, this method has been extended to formalin-fixed paraffin-embedded tissues obtained from CNS of patients with tuberous sclerosis, a disease that affects nuclear gene expression of tuberin (Crino et al., 1996).
Single muscle fiber PCR analysis has been applied previously to tissue sections from patients carrying either the mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes (MELAS) nt3243 mutation (Moraes et al., 1992; Petruzzella et al., 1994) or an mtDNA deletion (Sciacco et al., 1994). Either microcapillaries or a fine metal probe was used for cell retrieval. Analysis of serial sections of muscle from a MELAS patient histochemically stained for cytochrome c oxidase activity shows a direct correlation between the localized accumulation of the nt3243 MELAS mutation and the impairment of cytochrome c oxidase function of the single muscle fiber (Petruzzella et al., 1994). There is significant heterogeneity in the proportion of mutant mtDNA from fiber to fiber and along the length of the fiber. In apparent contrast to these results are those obtained by Shoubridge (1993), who applied the same method to the analysis of skeletal muscle from a MERRF patient carrying the 8344 mutation in the tRNALys genes. In this study the mtDNA was predominantly mutant in all the fibers, but the cytochromec oxidase activity varied from fiber to fiber, giving a fully positive or a deficient or virtually absent staining reaction. There seemed to be no correlation between genotype and phenotype. Similarly, Sparaco et al. (1995) found a mosaic pattern of staining of neurons, including Purkinje cells, from a MERRF patient who exhibited a moderate proportion of mutant DNA in brain (55% in brain total DNA and 59% in brain mitochondrial DNA; Lombes et al., 1992) when they used antisera directed against cytochrome c oxidase subunit II. The data that we have obtained in the present work on the Purkinje cell pairs would suggest that, among those cells, the level of mutant mtDNA varied very little from cell to cell. Thus, the mosaic phenotype observed by Sparaco et al. (1995) may have arisen from a failure of the residual wild-type DNA to complement the mutant DNA in some cells because of an unfavorable segregation of the two genotypes within the cells. In fact, previous evidence clearly indicates that, in addition to the proportion of mutant and wild-type mtDNAs in a cell, the way in which the two types of mtDNA are distributed among the organelles has an important role in determining the phenotypic manifestation of the mutation (Shoubridge, 1993; Yoneda et al., 1994; Attardi et al., 1995).
Further analysis by the method described here of CNS tissues from this and other patients carrying the MERRF or other mtDNA mutations, in conjunction with a determination of respiratory chain activity at the single-cell level, is needed to define the relationship of mitochondrial genotype to phenotype in mutant neurons. Sensitivity at the level of the single soma isolate is within reach with the present method and would allow for the identification of any heterogeneity of mtDNA distribution as well as an examination of the possible mechanisms leading to loss of cell function and death. These probably include a variety of nuclear gene-controlled factors that contribute to the partitioning of mutant mtDNA within the mitochondria in a cell, to neuroprotection, or to selective vulnerability of neurons to oxidative stress or apoptosis.
Footnotes
These studies were supported by Grants from the National Institute of Aging (P50-AG05142) and the National Institute of Mental Health (5R37-MH39145) to C.A.M. and from the National Institute of General Medical Sciences (GM-11726) to G.A. We are grateful to Ms. Jeanette Espinosa and Carol Church for their excellent secretarial assistance, to Dr. Roger Duncan of the School of Pharmacy, University of Southern California, for assistance with quantitative densitometry, and to Drs. Karen Jain and Terence Sanger for helpful discussions.
Correspondence should be addressed to Dr. Carol A. Miller, Department of Pathology, University of Southern California School of Medicine, 2011 Zonal Avenue, Los Angeles, CA 90033.
REFERENCES
- 1.Anderson S, Bankier AT, Barrell BG, deBruijn MHL, Coulson AR, Drovin J, Eperon IC, Nierlich DP, Roe BA, Sanger F, Schreier PH, Smith AJH, Staden R, Young IG. Sequence and organization of the human mitochondrial genome. Nature. 1981;290:457–465. doi: 10.1038/290457a0. [DOI] [PubMed] [Google Scholar]
- 2.Asoh S, Mori T, Hayashi J-I, Ohta S. Expression of the apoptosis mediator Fas is enhanced by dysfunctional mitochondria. J Biochem. 1996;120:600–607. doi: 10.1093/oxfordjournals.jbchem.a021455. [DOI] [PubMed] [Google Scholar]
- 3.Attardi G, Yoneda M, Chomyn A. Complementation and segregation behavior of disease-causing mitochondrial DNA mutations in cellular model systems. Biochim Biophys Acta. 1995;1271:241–248. doi: 10.1016/0925-4439(95)00034-2. [DOI] [PubMed] [Google Scholar]
- 4.Boulet L, Karpati G, Shoubridge EA. Distribution and threshold expression of the tRNALys mutation in skeletal muscle of patients with myoclonic epilepsy and ragged-red fibers (MERRF). Am J Hum Genet. 1992;51:1187–1200. [PMC free article] [PubMed] [Google Scholar]
- 5.Brierly JB, Meldrum BS, Brown AW. The threshold and neuropathology of cerebral “anoxic-ischemic” cell change. Arch Neurol. 1973;29:367–374. doi: 10.1001/archneur.1973.00490300029003. [DOI] [PubMed] [Google Scholar]
- 6.Chomyn A, Meola G, Bresolin N, Lai ST, Scarlato G, Attardi G. In vitro genetic transfer of protein synthesis and respiration defects to mitochondrial DNA-less cells with myopathy patient mitochondria. Mol Cell Biol. 1991;11:2236–2244. doi: 10.1128/mcb.11.4.2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Crino PB, Trojanowski JQ, Dichter MA, Eberwine J. Embryonic neuronal markers in tuberous sclerosis: single-cell molecular pathology. Proc Natl Acad Sci USA. 1996;93:14152–14157. doi: 10.1073/pnas.93.24.14152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Enriquez JA, Chomyn A, Attardi G. mtDNA mutation in MERRF syndrome causes defective aminoacylation of tRNALys and premature translation termination. Nat Genet. 1995;10:47–55. doi: 10.1038/ng0595-47. [DOI] [PubMed] [Google Scholar]
- 9.Guan M-X, Fischel-Ghodsian N, Attardi G. Biochemical evidence for nuclear gene involvement in phenotype of non-syndromic deafness associated with mitochondrial 12S rRNA mutation. Hum Mol Genet. 1996;5:963–971. doi: 10.1093/hmg/5.7.963. [DOI] [PubMed] [Google Scholar]
- 10.Hammans SR, Sweeney MG, Brockington M, Lennox GG, Lawton NF, Kennedy CR, Morgan-Hughes JA, Harding AE. The mitochondrial DNA transfer RNALys A→G(8344) mutation and syndrome of myoclonic epilepsy with ragged-red fibers (MERRF). Brain. 1993;116:617–632. doi: 10.1093/brain/116.3.617. [DOI] [PubMed] [Google Scholar]
- 11.Harding AL, Sweeney MG, Govan GG, Riordan-Eva P. Pedigree analysis in lobar hereditary optic neuropathy families with pathogenic mtDNA mutation. Am J Hum Genet. 1995;57:77–86. [PMC free article] [PubMed] [Google Scholar]
- 12.Lambolez B, Audinat E, Bochet P, Crepel E, Rossier J. AMPA receptor subunits expressed by single Purkinje cells. Neuron. 1992;9:247–258. doi: 10.1016/0896-6273(92)90164-9. [DOI] [PubMed] [Google Scholar]
- 13.Larsson NG, Tulinius MH, Holme F, Oldfois A, Anderson O, Wahlstrom J, Aasly J. Segregation and manifestations of the mtDNA tRNALys A→G (8344) mutation of myoclonus epilepsy and ragged-red fibers (MERRF) syndrome. Am J Hum Genet. 1992;51:1201–1212. [PMC free article] [PubMed] [Google Scholar]
- 14.Lertrit P, Noer AS, Byrne E, Marzuki S. Tissue segregation of a heteroplasmic mtDNA mutation in MERRF (myoclonic epilepsy with ragged-red fibers) encephalomyopathy. Hum Genet. 1992;90:251–254. doi: 10.1007/BF00220072. [DOI] [PubMed] [Google Scholar]
- 15.Lombes A, Diaz C, Romero NB, Ziegler F, Fardeau M. Analyses of the tissue distribution and inheritance of heteroplasmic mitochondrial DNA point mutation by denaturing gradient gel electrophoresis in MERRF syndrome. Neuromuscul Disord. 1992;2:323–330. doi: 10.1016/s0960-8966(06)80003-9. [DOI] [PubMed] [Google Scholar]
- 16.Mackler SA, Brooks BP, Eberwine JP. Stimulus-induced coordinate changes in mRNA abundance in single post-synaptic hippocampal CA-1 neurons. Neuron. 1992;9:539–548. doi: 10.1016/0896-6273(92)90191-f. [DOI] [PubMed] [Google Scholar]
- 17.Moraes C, Ricci E, Bonilla E, DiMauro S, Schon EA. The mitochondrial tRNAleu(UUR) mutation in mitochondrial encephalomyopathy, lactic acidosis, and strokelike episodes (MELAS): genetic, biochemical, and morphological correlations in skeletal muscle. Am J Hum Genet. 1992;50:934–949. [PMC free article] [PubMed] [Google Scholar]
- 18.O’Leary JJ, Chetty R, Graham AK, McGee JOD. In situ PCR: pathologist’s dream or nightmare? J Pathol. 1996;178:11–20. doi: 10.1002/(SICI)1096-9896(199601)178:1<11::AID-PATH459>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 19.Petruzzella V, Moraes CT, Sano MC, Bonilla E, DiMauro S, Schon EA. Extremely high levels of mutant mtDNAs co-localize with cytochrome c oxidase-negative ragged-red fibers in patients harboring a point mutation at nt3243. Hum Mol Genet. 1994;3:449–454. doi: 10.1093/hmg/3.3.449. [DOI] [PubMed] [Google Scholar]
- 20.Sanger TD, Jain K. MERRF syndrome with overwhelming lactic acidosis: case report and pedigree. Pediatr Neurol. 1996;14:57–60. doi: 10.1016/0887-8994(95)00226-x. [DOI] [PubMed] [Google Scholar]
- 21.Schinder A, Olson EC, Spitzer NC, Montal M. Mitochondrial dysfunction is a primary event in glutamate neurotoxicity. J Neurosci. 1996;16:6125–6133. doi: 10.1523/JNEUROSCI.16-19-06125.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sciacco M, Bonilla E, Schon EA, DiMauro S, Moraes CT. Distribution of wild-type and common deletion forms of mtDNA in normal and respiration-deficient muscle fibers from patients with mitochondrial myopathy. Hum Mol Genet. 1994;3:13–19. doi: 10.1093/hmg/3.1.13. [DOI] [PubMed] [Google Scholar]
- 23.Shoffner JM, Lott MT, Lezza AMS, Seibel P, Ballinger SW, Wallace DC. Myoclonic epilepsy and ragged-red fiber disease (MERRF) is associated with a mitochondrial DNA tRNALys mutation. Cell. 1990;61:931–937. doi: 10.1016/0092-8674(90)90059-n. [DOI] [PubMed] [Google Scholar]
- 24.Shoffner MJ, Lott MT, Wallace DC. MERRF: a model disease for understanding the principles of mitochondrial genetics. Rev Neurol (Paris) 1991;147:431–435. [PubMed] [Google Scholar]
- 25.Shoubridge EA. Molecular histology of mitochondrial diseases. In: DiMauro S, Wallace DC, editors. Mitochondrial DNA in human pathology, Chap 9. Raven; New York: 1993. pp. 109–123. [Google Scholar]
- 26.Silvestri G, Ciafaloni E, Santorelli FM, Shanski S, Servidei S, Graf WD, Sumi M, DiMauro S. Clinical features associated with the A→G transition at nucleotide 8344 of mtDNA (“MERRF mutation”). Neurology. 1993;43:1200–1206. doi: 10.1212/wnl.43.6.1200. [DOI] [PubMed] [Google Scholar]
- 27.Sparaco M, Schon EA, DiMauro S, Bonilla E. Myoclonic epilepsy with ragged-red fibers (MERRF): an immunohistochemical study of the brain. Brain Pathol. 1995;5:125–133. doi: 10.1111/j.1750-3639.1995.tb00586.x. [DOI] [PubMed] [Google Scholar]
- 28.Tanno Y, Yoneda M, Tanaka K, Kondo R, Hozomi I, Wakabayashi K, Yamada M, Fukuhara N, Ikuta F, Tsuji S. Uniform tissue distribution of tRNALys mutation in mitochondrial DNA in MERRF patients. Neurology. 1993;43:1198–1200. doi: 10.1212/wnl.43.6.1198. [DOI] [PubMed] [Google Scholar]
- 29.Wallace DC, Zheng X, Lott MT, Shoffner JM, Hodge JA, Kelley RI, Epstein CM, Hopkins LC. Familial mitochondrial encephalomyopathy (MERRF): genetic, pathophysiological, and biochemical characterization of a mitochondrial DNA disease. Cell. 1988;55:601–610. doi: 10.1016/0092-8674(88)90218-8. [DOI] [PubMed] [Google Scholar]
- 30.Yoneda M, Tanno Y, Horai S, Ozawa T, Miyatake T, Tsuji S. A common mitochondrial DNA mutation in the tRNALys of patients with myoclonus epilepsy associated with ragged-red fibers. Biochem Int. 1990;21:789–796. [PubMed] [Google Scholar]
- 31.Yoneda M, Tanno Y, Nonaka I, Miyatake T, Tsuji S. Simple detection of tRNALys mutation in myoclonus epilepsy associated with ragged-red fibers (MERRF) by polymerase chain reaction with a mismatched primer. Neurology. 1991;1:1838–1840. doi: 10.1212/wnl.41.11.1838. [DOI] [PubMed] [Google Scholar]
- 32.Yoneda M, Miyatake T, Attardi G. Complementation of mutant and wild-type human mitochondrial DNAs co-existing since the mutation event and lack of complementation of DNAs introduced separately into a cell within distinct organelles. Mol Cell Biol. 1994;14:2699–2712. doi: 10.1128/mcb.14.4.2699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zeviani M, Amati P, Bresolin N, Antozzi C, Piccolo G, Toscano A, DiDonato S. Rapid detection of the A→G(8344) mutation of mtDNA in Italian families with myoclonus epilepsy and ragged-red fibers (MERRF). Am J Hum Genet. 1991;48:203–211. [PMC free article] [PubMed] [Google Scholar]