

Abstract
To gain a molecular understanding of neuronal responses to amyloid-β peptide (Aβ), we have analyzed the effects of Aβ treatment on neuronal gene expression in vitro by quantitative reverse transcription-PCR and in situhybridization. Treatment of cultured rat cortical neurons with Aβ1–40 results in a widespread apoptotic neuronal death. Associated with death is an induction of several members of the immediate early gene family. Specifically, we (1) report the time-dependent and robust induction of c-jun,junB, c-fos, and fosB, as well as transin, which is induced by c-Jun/c-Fos heterodimers and encodes an extracellular matrix protease; these gene inductions appear to be selective because other Jun and Fos family members, i.e., junD and fra-1, are induced only marginally; (2) show that the c-juninduction is widespread, whereas c-fos expression is restricted to a subset of neurons, typically those with condensed chromatin, which is a hallmark of apoptosis; (3) correlate gene induction and neuronal death by showing that each has a similar dose–response to Aβ; and (4) demonstrate that both cell death and immediate early gene induction are dependent on Aβ aggregation state. This overall gene expression pattern during this “physiologically inappropriate” apoptotic stimulus is markedly similar to the pattern we previously identified after a “physiologically appropriate” stimulus, i.e., the NGF deprivation-induced death of sympathetic neurons. Hence, the parallels identified here further our understanding of the genetic alterations that may lead neurons to apoptosis in response to markedly different insults.
Keywords: amyloid, apoptosis, immediate early genes, Alzheimer’s disease, programmed cell death
During normal nervous system maturation, physiologically appropriate neuronal loss contributes to a sculpting process that removes approximately one-half of all neurons born during neurogenesis (Oppenheim, 1991). Neuronal loss subsequent to this developmental window is physiologically inappropriate for most systems and can contribute to neurological deficits, e.g., neurodegenerative diseases such as Alzheimer’s disease (AD) and conditions such as stroke. Elucidating the molecular mechanisms underlying neuronal death hence will contribute to our understanding of basic developmental biology and to human neuropathology.
Apoptosis is a type of cell death that represents the culmination of the naturally occurring, or programmed, cell death (PCD) pathway during normal development. Apoptosis is defined on the basis of accompanying cellular morphology, which includes cellular shrinkage as well as chromatin condensation and nucleosomal fragmentation (Wyllie et al., 1980; Clarke, 1990). Apoptosis also has been associated with the pathophysiology of AD (Su et al., 1994; Dragunow et al., 1995; Lassmann et al., 1995; Smale et al., 1995), Huntington’s disease (Dragunow et al., 1995; Portera-Calliau et al., 1995), and stroke (Goto et al., 1990; Shigeno, 1990; Linnik et al., 1993; MacManus et al., 1993,1994).
Elucidating patterns of gene expression during neuronal death may be critical to our understanding of underlying mechanisms. Indeed, studies with RNA and protein synthesis inhibitors have demonstrated that neuronal PCD in vitro (Martin et al., 1988) and in vivo (Oppenheim et al., 1990), as well as in certain other models (for review, see Freeman et al., 1993; Bredesen, 1995), is dependent on macromolecular synthesis. This has led to the hypothesis that PCD results from the activation of a genetic program. Recently, we identified a temporal cascade of gene expression in sympathetic neurons undergoing NGF deprivation-induced PCD (Estus et al., 1994; Freeman et al., 1994). Moreover, by microinjecting neutralizing antibodies, we implicated the protein product of one of the induced genes, c-Jun, as necessary for death (Estus et al., 1994), results that were confirmed by others (Ham et al., 1995). Hence, a genetic cascade appears to be necessary for NGF deprivation-induced neuronal PCD.
To evaluate whether neurons undergoing physiologically inappropriate neuronal cell death manifest a similar genetic cascade, we have analyzed rat cortical neurons undergoing amyloid-β (Aβ)-induced cell death. As will be discussed later, the relevance of this work includes the observations that Aβ aggregates accumulate in AD brain, induce neuronal apoptosis in vitro, and have been implicated as contributory to AD by a series of genetic and protein processing studies (for review, see Selkoe, 1997). Because neuronal apoptosis may be facilitated by altered gene expression and because genes induced by Aβ may be integral to neuropathological changes observed in AD brain, we have quantified Aβ-induced changes in neuronal gene expression.
Much of this work has been reported in abstract form (Estus et al., 1995, 1996).
MATERIALS AND METHODS
Primary rat cortical neurons
Primary rat cortical neuron cultures were established from embryonic day 18 rat fetuses. Cortical tissue was dissociated by incubation in trypsin/EDTA (0.05% trypsin plus 0.53 mmEDTA in HBSS; Life Technologies, Gaithersburg, MD) for 20 min at 37°C. Then trypsin was inactivated by resuspending the cells in serum-containing medium (DMEM/fetal bovine serum, Life Technologies): DMEM which contains 4.5 gm/l glucose, 1 mm sodium pyruvate, 1 mm glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin and supplemented with 10% heat-inactivated fetal bovine serum (JRH Biologicals, Lenexa, KA). Then cells were pelleted by centrifugation and resuspended in a chemically defined medium (DMEM/B27): DMEM containing B27 (Life Technologies) supplement in place of fetal bovine serum. Polyethyleneimine-coated 6.4 mm (96-well) dishes were rinsed with PBS, coated with DMEM/fetal bovine serum, and then seeded at 0.75–1.25 × 105 cells per well in 100 μl of DMEM/B27. Cultures were maintained in a humidified incubator with an atmosphere of 90% air/10% CO2 at 37°C. Serum replacement with B27 supplement yields nearly pure neuronal cultures, as judged by immunocytochemistry for glial fibrillary acidic protein (GFAP) and neuron-specific enolase (NSE) (Brewer et al., 1993).
Treatment with Aβ peptides
Aggregated Aβ1–40. Aggregated Aβ1–40 stock solutions were prepared as 1 mmstocks in sterile double-distilled water immediately before their addition to cultures. Aβ concentrations in all stock solutions were determined by amino acid analysis. Cultured rat cortical neurons were exposed to Aβ by removing the culture medium and replacing it with DMEM/N2 (Life Technologies; Bachem lots ZK840 and ZM482) or DMEM/B27 (Bachem lot ZM605) containing 5–40 μmAβ1–40. Qualitatively similar results were obtained with all three lots of Aβ1–40. Neuronal cultures were maintained for 2–4 d before neuronal survival was assessed visually by phase-contrast microscopy and quantified by measuring (1) the reduction of alamarBlue (Accumed/Alamar Biosciences, Sacramento, CA); (2) the reduction of the tetrazolium salt, 2,3-bis (2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide (XTT); and (3) the release of the cytoplasmic enzyme, lactate dehydrogenase (LDH).
Nonaggregated Aβ. Stock nonaggregated Aβ1–40 solutions were prepared by dissolving Aβ1–40 (Bachem lot ZM605) to 7.5 mm in dimethylsulfoxide (DMSO, Aldrich, Milwaukee, WI), sonicating for 30 min in a bath sonicator, and filtering through a 3 mm Teflon membrane filter (pore size 0.2 μm). Aβ concentration in the stock solution was determined by amino acid analysis. Aliquots were snap-frozen on dry ice and ethanol and stored at −80°C. Cultures were exposed to nonaggregated Aβ by removing the culture medium and replacing it with DMEM/B27 containing 40 μm Aβ1–40. For these experiments an equal volume of DMSO was added to control wells and to wells treated with aggregated Aβ1–40.
Measurement of Aβ aggregation in solution. The detection of Aβ aggregation in solution was based on the method of LeVine (LeVine, 1993). Thioflavin-T (ThT, Aldrich) stocks were made at 2.5 mm in DMSO, frozen on dry ice, and stored at −30°C. ThT stocks were diluted to 125 μm in DMEM/B27, and 11 μl of this solution was added to 100 μl/well of Aβ in DMEM/B27. ThT binding to aggregated Aβ1–40 was measured spectrofluorometrically, using a Millipore Cytofluor 2350 Scanner (excitation 440 nm, emission 485 nm) and CytoCalc software (Millipore, Bedford, MA), and normalized to ThT in DMEM/B27 without added Aβ1–40.
Neurotoxicity/apoptosis assays
AlamarBlue assay. The alamarBlue assay incorporates a proprietary fluorometric/colorimetric metabolic indicator. Viable cells convert alamarBlue from an oxidized (nonfluorescent, blue) form to a reduced (fluorescent, red) form. Assays were performed by replacing the culture media with a 10% alamarBlue solution in DMEM. Reduction of alamarBlue was determined spectrofluorometrically after 2 hr, using a Millipore Cytofluor 2350 Scanner (excitation, 560 nm; emission, 590 nm) and CytoCalc software. AlamarBlue reduction was directly proportional to neuronal cell number and was linear over 3 hr.
XTT colorimetric assay. The reduction of the XTT was determined by the Cell Proliferation Kit II (XTT) (Boehringer Mannheim, Indianapolis, IN). XTT is a substrate for intracellular and plasma membrane oxidoreductases, and its reduction is an indication of cellular metabolic activity. Assays were performed by replacing the culture medium with 150 μl of XTT reagent. The 96-well culture plate was read after 2 hr with a microplate reader (UVmax, Molecular Devices, Palo Alto, CA) and “SOFTmax” version 2.32 software. Absorbance at 450 nm minus absorbance at 650 nm was used to quantify the amount of soluble formazan dye formed from the XTT tetrazolium salt. The absorbance was directly proportional to neuronal cell number and was linear over this time period.
LDH kinetic assay. The release of the cytoplasmic enzyme LDH into the culture medium was used to quantify cell membrane integrity. Twenty microliters of culture supernatant were assayed with 200 μl of reconstituted LD-L 10 reagent (Sigma, St. Louis, MO). Samples were read every 30 sec over a 5 min time period with a kinetic microplate reader (UVmax, Molecular Devices) and “SOFTmax” version 2.32 software. Absorbance at 340 nm minus absorbance at 650 nm was used to determine the rate of formation of reduced nicotinamide adenine dinucleotide. The reaction rate was linear over this time period. The rate of reduced nicotinamide adenine dinucleotide formation is directly proportional to LDH activity in the sample. Fluorescent values were converted to units per milliliter by the inclusion of an LDH standard curve on each assay plate by using LDH controls (Sigma).
In situ labeling of nuclear DNA fragments. DNA fragmentation was assessed by using a fluorescent TUNEL technique (TdT-mediated dUTP-X nick end labeling; In Situ Cell Death Detection Kit, Boehringer Mannheim). Briefly, neurons were fixed with 4% paraformaldehyde, rinsed with PBS, and incubated with terminal deoxy-terminal transferase and digoxigenin-dUTP. Incorporated digoxigenin was detected with a fluorescein-labeled anti-digoxigenin antibody.
Nuclear staining. Chromatin integrity was examined by staining neurons with 2′-(4-hydroxyphenyl)-5-(4-methyl-1-piperazinyl)2,5′-bi-1H-benzimidazole trihydrochloride (bisbenzimide, Hoechst 33258; Sigma) at a concentration of 1 μg/ml in PBS for 10 min. After being rinsed with PBS, cultures prepared by the TUNEL method and costained with Hoechst 33258 were examined for DNA fragmentation and chromatin condensation by fluorescence microscopy.
Gene expression assays
cDNA preparation. Total RNA was prepared by using RNAeasy kits as directed by the manufacturer (Qiagen, Hilden, Germany), subjected to DNase treatment, and then repurified either by using the RNAeasy kit or by ethanol precipitation. RNA was reverse-transcribed by using random hexamers to prime Maloney murine leukemia virus reverse transcriptase (Superscript). In the initial time course experiments, 1 μg of total RNA was used in each reaction, whereas in later studies the RNA isolated from 300,000 initially plated cells was used; essentially identical results were obtained with each approach. For the reverse transcription, the RNA was mixed with 500 pmol of random hexamers (Boehringer Mannheim) in a volume of 20 μl, incubated at 95°C for 2 min, and then placed on ice. Then a stock solution was added such that the final reaction volume of 30 μl contained 200 U of Superscript, 500 μm dNTPs, 40 U of RNAsin, and 1× reaction buffer (Life Technologies). The solution was incubated at 20°C for 10 min and at 42°C for 50 min, and the Superscript was inactivated by heating to 95°C for 2 min. Sufficient cDNA was synthesized in each reaction for 30 separate PCR analyses.
PCR amplification. Stock PCR reaction mixtures (50 μl) were prepared on ice and contained 50 μm dCTP, 100 μm each of dGTP, dATP, and dTTP, 10 μCi of dCTP (3000 Ci/mmol), 1.5 mm MgCl2, 1× reaction buffer (Life Technologies), 1 μm each primer, 1 U ofTaq polymerase (Life Technologies), and 1/30th of the cDNA synthesized in the reverse transcription. Then the stock solutions were separated into three 14 μl aliquots that were covered with a drop of mineral oil and subjected to various numbers of cycles of PCR. The use of multiple cycles allowed us to determine the minimum number of cycles necessary to detect PCR product and thereby stay within the linear region of PCR amplification. Typical reaction conditions were 1 min at 94°C, 1 min at 55°C, and 2 min at 72°C. After amplification, the cDNAs were separated by polyacrylamide gel electrophoresis and visualized by autoradiography of the dried gels or by PhosphorImager technology (Molecular Dynamics, Sunnyvale, CA).
The sequences of the primers used in this study included the following:cyclophilin sense primer, 5′ ATG GTC AAC CCC ACC GTG TT 3′ and cyclophilin antisense primer, 5′ CGT GTG AAG TCA CCA CCC T 3′ (204 bp product); neurofilament M(NFM) sense primer, 5′ ACG CTG GAC TCG CTG GGC AA 3′ and NFM antisense primer, 5′ GCG AGC GCG CTG CGC TTG TA 3′ (156 bp product); NSE sense primer, 5′ ATC TTG GAC TCC CGT GGG AA 3′ and NSE antisense primer, 5′ TTT GGC AGT ATG GAG ATC CA 3′ (54 bp product); GFAP sense primer, 5′ GCG CTC AAT GCC GGC TTC AA 3′ and GFAP antisense primer, 5′ TTC TCG ATG TAG CTA GCA AA 3′ (88 bp product); c-jun sense primer, 5′ ACT CAG TTC TTG TGC CCC AA 3′ and c-jun antisense primer, 5′ CGC ACG AAG CCT TCG GCG AA 3′ (64 bp product); junB sense primer, 5′ GGG AAT TCA AAC CCA CCT TGG CGC TCA A 3′ and junBantisense primer, 5′ GCG GAT CCG GAC CCT TGA GAC CCC GAT A 3′ (69 bp product); junD sense primer, 5′ GGG AAT TCA GGC TGA TCA TCC AGT CCA A and junD antisense primer, 5′ GGG GAT CCG CCA CCT TCG GGT AGA GGA A 3′ (128 bp product); c-fos sense primer, 5′ AAT AAG ATG GCT GCA GCC AA and c-fos antisense primer, 5′ TTG GCA ATC TCG GTC TGC AA 3′ (116 bp product); fosB sense primer, 5′ GAG ATC GCC GAG CTG CAA AA 3′ and fosB antisense primer, 5′ TTG TGG GCC ACC AGG ACA AA 3′ (58 bp product);fra1 sense primer, 5′ GCC TTG AGC TGG TGC TGG AA 3′ andfra1 antisense primer, 5′ ATG CAG TGC TTC CGG TTC AA 3′ (175 bp product); transin sense primer, 5′ GGG AAT TCC TTT CCA GGT TCA CCC AA 3′ and transin antisense primer, 5′ GCG GAT CCT TCA GAG ATC CTG GAG AA 3′ (172 bp product); amyloid-β protein precursor (APP) sense primer, 5 CACCACAGAGTCTGTGGAAG and APP antisense primer, 5′ AGGTGTCTCGAGATACTTGT (these primers flank an alternative splice site such that APP695 produces an 87 bp product,APP751 produces a 255 bp fragment, andAPP770 produces a 312 bp product) (Golde et al., 1990). The identity of the amplified cDNAs was confirmed typically either by subcloning and then sequencing from the vector (some of the oligos contain restriction sites to facilitate subcloning) or, more recently, by direct sequencing, which was facilitated by purifying the cDNA by using a purification kit (Qiagen). We validated this RT-PCR assay in initial studies by showing that the PCR product yields were linear with respect to input RNA (Estus, 1997) and that the technique could be used to detect gene inductions in neuronal cultures, i.e., HSP70 in a heat–shock paradigm (Estus, 1997) and immediate early and delayed early gene inductions in NGF-treated PC12 cells (data not shown). Where shown, differences in gene expression were analyzed statistically by ANOVA comparison of treated versus control samples with a post hoc Fisher Protected Least Significant Difference (PLSD) Test for significance (StatView version 4.5, Abacus Concepts, Calabasas, CA).
In situ hybridization. Cells were maintained on polyethyleneimine-coated 16-well chamber slides, treated with Aβ for 24 or 48 hr, fixed with 4% paraformaldehyde in PBS for 30 min at room temperature, and processed for in situ hybridization (Wanaka et al., 1990), except that proteinase K treatment was omitted. Slides were hybridized at 55°C for 16–18 hr with 33P-labeled RNA probes (500,000 cpm/slide) corresponding to rat c-jun orc-fos (Estus et al., 1994). Antisense and sense riboprobes were synthesized by using T7 and T3 RNA polymerases (Stratagene, La Jolla, CA) and (α-33P)UTP. Sense probes served as specificity controls. After treatment with ribonuclease and high-stringency washes, slides were processed for emulsion autoradiography for 11–14 d. After development of the emulsion, the cells were stained for 10 min with Hoechst 33258 (1 μg/ml) (Molecular Probes, Eugene, OR) in water, followed by a 10 min water rinse. Slides were viewed with phase-contrast, dark-field, and fluorescence microscopy.
RESULTS
Time course of Aβ toxicity
To define the time course of Aβ toxicity, we quantified cell viability by three criteria. These assays were performed on neuronal cultures treated in parallel with Aβ or vehicle only, i.e., medium addition. Viability assays included measures of metabolic and membrane integrity. Metabolic integrity was assessed by both alamarBlue and XTT reduction, which quantify cellular reducing potential; by these measures, neuronal viability began to decline at 24 hr of treatment and approached a minimum by 48 hr. Membrane integrity was assessed by LDH release, which was lost after metabolic integrity, approaching only 30% of maximal by 48 hr (Fig. 1). We interpret these data as indicating that Aβ treatment induces a delayed neuronal cell death wherein a decline in metabolic activity occurs before disruption of plasma membrane integrity. This temporal pattern of loss of metabolic and membrane integrity is consistent with the majority of cell death occurring by an apoptotic mechanism.
Fig. 1.

Time course of Aβ neurotoxicity. Cultured rat cortical neurons were treated with Aβ1–40 (20 μm, Lot ZK840) for the indicated intervals, and cell viability was assayed by metabolic integrity (XTT reduction and alamarBlue reduction) and plasma membrane integrity (LDH release). Metabolic parameters decrease well before the loss of membrane integrity. Data are mean ± SD (error bars) values from triplicate wells and represent typical results obtained with these cells on a routine basis.
Aβ toxicity manifests the hallmarks of apoptosis
To assess whether the delayed Aβ-mediated neuronal death manifested other hallmarks of apoptosis, we compared Aβ-treated and control neurons for two characteristic changes in chromatin integrity, i.e., DNA fragmentation and chromatin condensation. These indices were colocalized by in situ DNA end labeling (Gavrieli et al., 1992) and with Hoechst 33258 staining, respectively. After Aβ treatment, large numbers of neurons manifested punctate and fragmented chromatin, changes that were not seen in control samples (Fig.2A–D). Hence, our results agree with those of others (Forloni et al., 1993; Loo et al., 1993; Gschwind and Huber, 1995), in that the Aβ-induced neuronal death as described here manifests apoptotic characteristics.
Fig. 2.

Aβ toxicity manifests the hallmarks of apoptosis. Cortical neuron preparations were treated with medium change alone (A, B) or Aβ1–40 (40 μm, lot ZM482) for 24 hr (C, D) and then assessed for chromatin integrity as discerned by Hoechst 33258 staining (A, C) and by DNA end labeling (B, D). The incidence of neurons manifesting punctate and fragmented chromatin is much higher in Aβ-treated neurons; note that neurons that manifest punctate chromatin also display an increased amount of fragmented DNA, as assessed by DNA end labeling.
Time-dependent changes in gene expression during Aβ treatment
To begin the analysis of Aβ-induced changes in gene expression over time, we established a baseline by quantifying changes in the expression of cellular markers: cyclophilin, expressed constitutively in all cell types; MAP-2, NSE, NFM, and PGP9.5, mRNAs unique to neurons; and GFAP, a marker for astrocytes, one of the non-neuronal cell types in the cultures. The resultant PCR data indicate that, although the neuronal mRNAs decreased with Aβ treatment, the levels of GFAP showed a modest increase (Fig.3A) consistent with the notion that, as the neurons died, a greater proportion of the RNA was derived from non-neuronal cells in the culture.
Fig. 3.
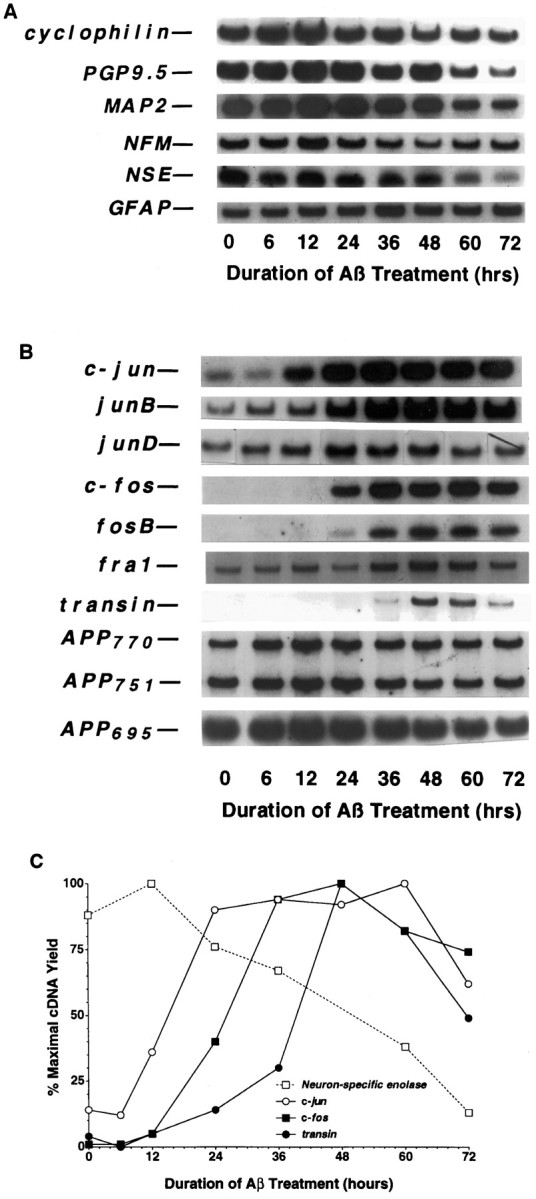
Time course of mRNA expression in rat cortical cultures undergoing Aβ-mediated neuronal apoptosis.A, Cellular marker genes. B, Jun and Fos family members and related genes. C, Quantification of changes in NSE, c-jun,c-fos, and transin expression. To assess changes in mRNA levels, we maintained primary rat cortical cultures (∼125,000 neurons/well) for 3–4 d and then treated them with Aβ1–40 (40 μm, lot ZM482), as described in Materials and Methods. After various times of Aβ treatment, total RNA was isolated, aliquots were converted to cDNA, and then 3% of the resultant cDNA was analyzed in each PCR sample. The data presented are from a single preparation of neuronal cultures, which were maintained and treated in parallel with those described in Figure 1. Each gene induction was confirmed in at least two independent neuronal culture preparations.
We have identified previously a cascade of gene induction in rat sympathetic neurons undergoing apoptosis after trophic factor deprivation (Estus et al., 1994). To evaluate the expression of these genes during Aβ treatment, we compared their patterns of expression with those of the baseline marker genes. In marked contrast to the baseline genes, the prototype immediate early genes c-junand c-fos were induced markedly after Aβ treatment, beginning at 12 and 24 hr, respectively (Fig. 3B,C). Among other Jun and Fos family members, junB and fosBalso were induced markedly, whereas junD andfra-1 were induced only modestly, each after thec-jun induction. The c-jun results per se confirm those of Anderson et al. (1995). The results overall are strikingly similar to the changes we observed in NGF-deprived sympathetic neurons in that the inductions of these “immediate early genes” occur many hours after Aβ treatment, and the induction of c-junprecedes that of the other genes.
To assess indirectly whether these induced genes produce functional protein, we quantified the expression of a prototype target gene of c-Jun/c-Fos heterodimers, i.e., transin (Angel and Karin, 1991; Cochran, 1993), which encodes an extracellular matrix protease (McDonnell et al., 1990). Indeed, transin was induced, beginning after c-fos (Fig. 3B,C) and peaking at 48 hr. We also quantified the expression of the three primary mRNAs encoding the APP (APP695, APP751, and APP770). Because the APP promotor has been reported to be activated by c-Jun (Trejo et al., 1994), this raises the possibility that Aβ exposure leads to increased APP mRNA and protein and thereby more Aβ, in a feed-forward cycle. However, this possibility was not supported by the data, because no increase was detected in the levels of any of the APP transcripts (Fig.3B). These results indicate that Aβ treatment leads to an increase in several transcription factors and a selective increase in a known target gene of these transcription factors. When these data are quantified, three waves of gene induction are observed, beginning withc-jun, followed by a group of genes that are nearly simultaneous with c-fos, and then transin (Fig.3C). Comparison of the temporal pattern of Aβ-mediated gene induction relative to the time course of neurotoxicity (Fig. 1) reveals that the first two waves precede dramatic changes in neuronal viability. The third wave, represented by transin, begins during the decline in metabolic parameters and peaks as cells begin to lose membrane integrity. These results are consistent with the possibility that these gene inductions are involved with the neuronal “decision” to die.
Dose–response relationship of gene induction and neuronal cell death
To begin to evaluate whether Aβ-induced apoptosis and immediate early gene induction may be separable events, we examined whether they displayed a similar dependence on Aβ concentration and aggregation; after neurons were treated with 5, 10, 20, or 40 μm Aβ for varying intervals, we quantified Aβ aggregation state, neuronal death, and mRNA levels. Aβ neurotoxicity was concentration-dependent; although little toxicity was observed in preparations treated with 5 μm Aβ for up to 72 hr, neuronal viability decreased earlier and more completely with increasing Aβ concentrations (Fig.4A). This difference in neurotoxicity correlated with Aβ aggregation state because the rate and extent of Aβ aggregation were also concentration-dependent (Fig.4B); aggregation continued through the time course of the experiment. Changes in gene expression were quantified in neurons treated in parallel. The magnitude of the inductions of the Jun and Fos family members showed a strong correlation with Aβ concentration (Fig. 4C–E). For c-fos,fosB, and junB, the timing of the induction was also concentration-dependent, beginning earlier with increasing Aβ concentration and correlating approximately with declining neuronal viability (Fig. 4A,C,E). In contrast, whereas the magnitude of c-juninduction was also concentration-dependent, the trend of thetiming of c-jun induction was less concentration-dependent, because c-jun levels began to increase significantly beginning 12 hr after treatment with Aβ at concentrations >5 μm. In summary, because the gene inductions and neuronal toxicity were overall similarly dependent on Aβ concentration, we interpret these data as consistent with the possibility that these gene inductions may be causally involved with neuronal death.
Fig. 4.

Gene induction and death are dependent on Aβ concentration. Cortical neuronal cultures were treated with Aβ1–40 (lot ZM605) for the indicated concentrations and times. Viability was determined by measuring alamarBlue reduction (A) and Aβ aggregation assessed by changes in ThT fluorescence (B). Changes in gene expression were assessed by RT-PCR (C), with quantification for c-jun (D) andc-fos (E). Values for the alamarBlue and ThT assays are expressed as mean ± SD (error bars) from triplicate wells, whereas those for c-jun andc-fos inductions are the mean ± SE (error bars) from triplicate determinations. The induction of c-jun,junB, c-fos, fosB,transin, and death shows a strong dependence on Aβ concentration and aggregation. The induction of c-junwas significant (p < 0.05) for 5 μm Aβ at the 48 and 72 hr time points and at every time point after 6 hr for the higher Aβ concentrations. The induction ofc-fos was significant at 72 hr for 5 μmAβ, at 48 and 72 hr for 10 and 20 μm Aβ, and at 24, 48, and 72 hr for 40 μm Aβ [ANOVA comparison of Aβ-treated vs control samples (n = 3), withpost hoc Fisher PLSD test].
Neuronal death and gene induction are dependent on Aβ aggregation
A second means to test the association between Aβ toxicity and altered gene expression was to examine directly whether they were both dependent on Aβ aggregation. Neurons were treated with Aβ that was dissolved initially in water (which promotes subsequent aggregation) or DMSO (which delays subsequent aggregation). After this initial solvation, the Aβ was diluted to 40 μm in medium as per our usual protocol; to control for possible effects of trace amounts of DMSO, we added DMSO back to the water-solvated samples such that the final concentration of DMSO was equal to that of samples treated with DMSO-solvated Aβ. We then quantified neuronal death, Aβ aggregation, and mRNA levels. This paradigm confirmed a previous observation that Aβ neurotoxicity was dependent on Aβ aggregation (Fig. 5A) (Pike et al., 1991;Simmons et al., 1994). Changes in gene expression were examined in RNA isolated from neurons treated in parallel. This analysis revealed thatc-jun, c-fos, junB, fosB, and transin were induced only in samples treated with aggregated Aβ and not in samples treated with nonaggregated Aβ (Fig. 5B,C). Although a transient rise in c-junand c-fos was observed 3 hr after replacement of the tissue culture medium, the sustained induction of the immediate early genes was observed only after treatment with aggregated Aβ (Fig.5C).
Fig. 5.
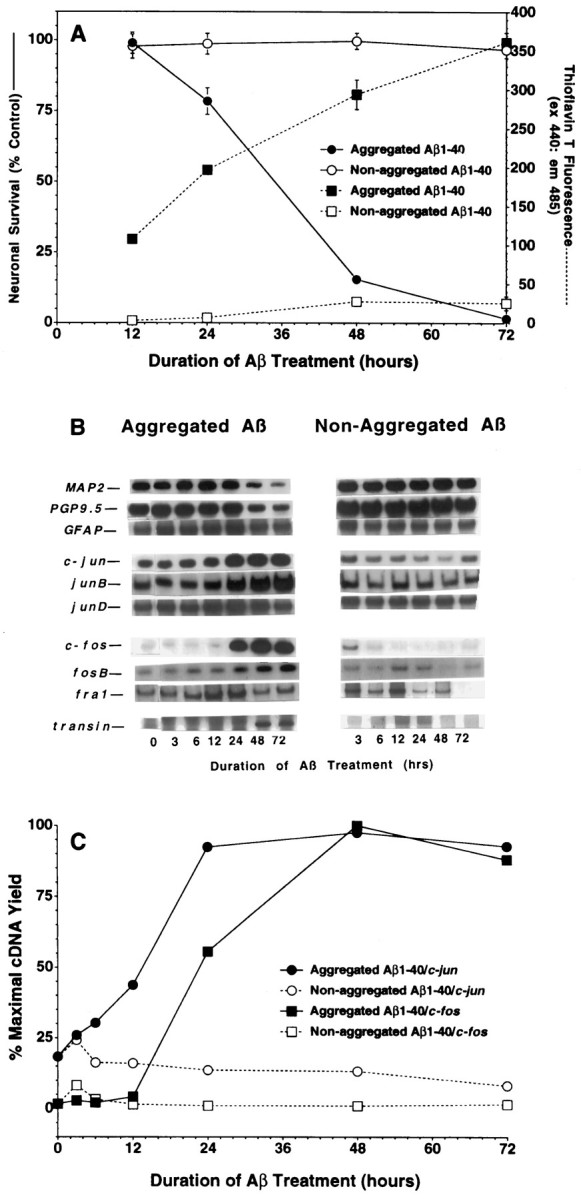
mRNA induction correlates with Aβ aggregation and Aβ neurotoxicity. Changes in neuronal viability and Aβ aggregation (A) and gene expression (B, C) were assessed in neurons treated with Aβ1–40(40 μm, lot ZM605) solvated initially in either water or DMSO. Values for the alamarBlue and ThT assays are expressed as mean ± SD (error bars) from triplicate wells. Similar results were observed in at least two independent neuronal culture preparations.
Immediate early gene induction is attributable to enhanced transcription
Because inhibition of protein synthesis, which might occur in dying neurons, increases immediate early gene mRNA levels by mRNA stabilization (Greenberg et al., 1986), we evaluated whether increases in the mRNA for c-fos, a prototype immediate early gene, were attributable to increases in transcription or stabilization. Because increased transcription, but not mRNA stabilization, leads to an increase in heteronuclear RNA (hnRNA), we compared the induction patterns of c-fos hnRNA and c-fos mRNA; RT-PCR was performed by using a pair of c-fos oligos designed such that the PCR product spanned an intron in hnRNA (Fig.6). We found that c-fos hnRNA and c-fos mRNA were enhanced in a parallel manner, strongly suggesting that the increase in immediate early gene mRNA levels is attributable, at least partially, to increased transcription.
Fig. 6.

Enhanced immediate early gene levels appear to be derived from increased transcription. To assess whether induction of a prototype immediate early gene, c-fos, resulted from increased transcription or mRNA stabilization, we designedc-fos oligos such that the PCR product spanned an intron in hnRNA (A). After Aβ1–40treatment (40 μm, lot ZM605), c-fosheteronuclear RNA (hnRNA) and c-fosmature mRNA were induced in parallel, as revealed by the autoradiograms (B) and PhosphorImager quantitation (C). That the hnRNA-associated PCR product was not a result of contaminating genomic DNA was demonstrated by showing that no product was detected in the absence of reverse transcription (data not shown). The parallel nature of the hnRNA and mRNA inductions indicates that enhanced transcription likely contributes toc-fos induction.
In situ analyses of gene expression and chromatin condensation
Although the cortical preparations are nearly pure neuronal cultures, we used in situ hybridization to verify that two representative genes, i.e., c-jun and c-fos, were expressed in neurons and not in the small number of astrocytes and, potentially, microglia, contaminating the cultures. This approach also allowed us to assess the heterogeneity of neuronal expression. To visualize chromatin, we stained neuronal cultures with Hoechst 33258. Aβ treatment led to a widespread induction of c-jun (Fig.7A,B). In contrast, the Aβ-induced increase in c-fos was restricted to a subset of neurons (Fig. 7C,D). Closer examination (Fig.7E–H) revealed that the chromatin ofc-fos-positive neurons was not distributed uniformly, i.e., chromatin condensation was observed in 79 ± 1% ofc-fos-positive cells (mean ± range, two separate experiments, 230 cells scored in total). However, many cells with abnormal chromatin were not positive for c-fos expression, suggesting that c-fos expression was not an obligate step in chromatin condensation (Fig. 7E–H). Thus,c-jun mRNA was expressed for extended periods in neurons undergoing Aβ treatment, whereas c-fos mRNA was induced transiently in a subset of neurons, frequently those with chromatin condensation.
Fig. 7.
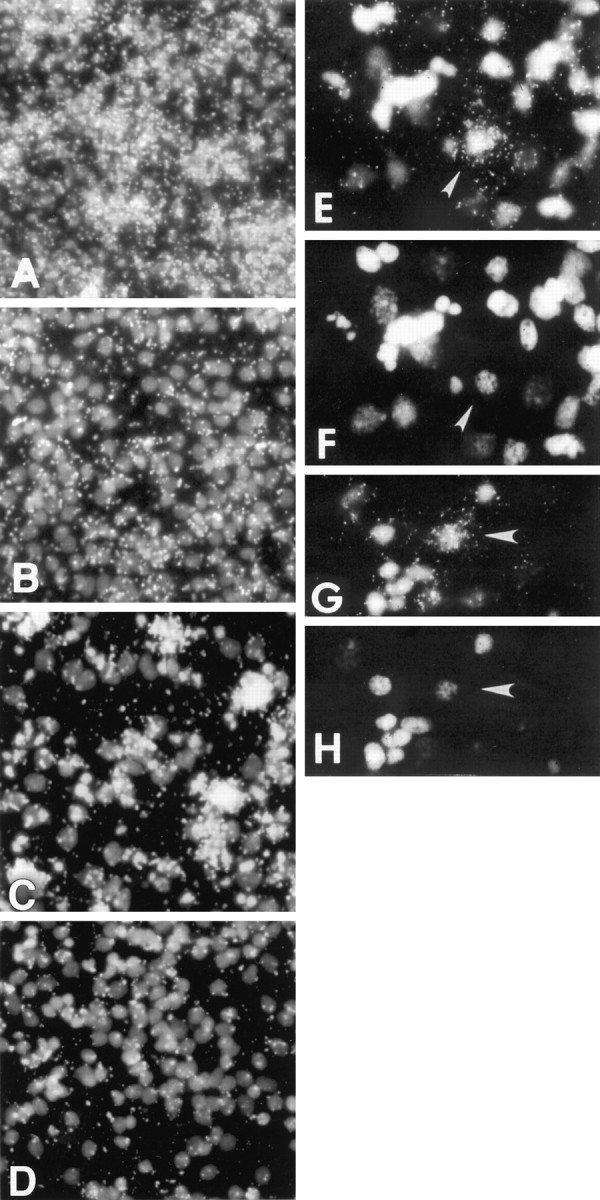
Patterns of c-jun andc-fos induction in situ. Cultures were analyzed for c-jun (A, B) orc-fos (C–H) expression by performing in situ hybridization and for chromatin integrity by staining with Hoechst 33258. Cultures were treated with Aβ1–40 (40 μm, lot ZM605) for 48 hr (A, C, E–H) or treated with medium change only (B, D). The depicted images represent dark-field-and-fluorescence (A–E, G) or fluorescence (F, H) microscopy. The neurons depicted inE and G are shown again inF and H, respectively; thearrows indicate c-fos-positive neurons and their associated chromatin. A–D originally were magnified 200×, whereas E and F were magnified 400×. Sense cRNA probes did not label above background. These results were replicated in at least two separate neuronal preparations. These genes also were induced at 24 hr after Aβ treatment (data not shown).
DISCUSSION
In the studies reported here, we have established a strong correlation among Aβ aggregation, altered gene expression, and Aβ-induced neuronal apoptosis. As such, these results constitute the second stage in ongoing efforts aimed at elucidating patterns of gene expression in neurons undergoing apoptosis that may be regarded as physiologically appropriate, e.g., induced by trophic factor deprivation (Estus et al., 1994), or physiologically inappropriate, e.g., induced by neurotoxic insult (this study). We anticipate that elucidating these genetic patterns will contribute to our understanding of basic developmental processes as well as potential mechanisms leading to neurodegenerative disease.
Indeed, the import of the work described here rests partially on the relevance of Aβ aggregates to AD, which is indicated by several observations, including (1) Aβ has been reported to induce apoptosis (Forloni et al., 1993; Loo et al., 1993; Gschwind and Huber, 1995) and necrosis (Behl et al., 1994a) in neurons in vitro, and this Aβ neurotoxicity depends on Aβ aggregation (Pike et al., 1991;Simmons et al., 1994); (2) Aβ aggregates accumulate in senile plaques found throughout the neocortex in AD; (3) that Aβ aggregation may actually be causal in AD is suggested by findings that the known mutations associated with familial Alzheimer’s disease all lead to an increased production of the more amyloidogenic Aβ1–42(Citron et al., 1992; Cai et al., 1993; Suzuki et al., 1994; Scheuner et al., 1996). Further support for a role of aggregated Aβ in AD-like pathology comes from the findings that transgenic mice that express high levels of mutant human Aβ precursor protein (APP with valine at residue 717 substituted by phenylalanine) manifest a phenotype that includes senile plaques and neuropathology markedly similar to that observed in AD (Games et al., 1995), and transgenic mice overexpressing the 695-amino acid isoform of human APP containing a Lys670→Asn, Met671→Leu familial AD mutation manifest a phenotype that includes senile plaques and memory deficits (Hsiao et al., 1996). Considered together, these data implicate aggregated Aβ as critical to AD in that aggregated Aβ accumulates in AD brain in vivo, aggregated Aβ induces neuronal cell death in vitro, and production of Aβ with a propensity to aggregate cosegregates with susceptibility to AD in humans or AD-like pathology in vivo. Hence, elucidating the patterns of altered gene expression induced during Aβ neurotoxicity may contribute to our understanding of AD.
Aβ-treated neuronal preparations manifest several trends in gene expression patterns that are markedly similar to those observed in NGF-deprived sympathetic neurons. The first general trend is the delayed but widespread induction of immediate early gene transcription factors. For NGF-deprived neurons, death begins at ∼24 hr, as quantified by crystal violet staining. The induction ofc-jun begins within 5 hr and is followed byc-fos, fosB, and junB and other genes at 15 hr (Deckwerth and Johnson, 1993; Estus et al., 1994). After Aβ treatment, death begins at ∼36–48 hr, as determined by LDH release, the induction of c-jun begins within 12 hr, and that is followed by c-fos, fosB, and junBbeginning at 24 hr (Figs. 1, 3). A further similarity between both models, and an indication that these mRNAs are translated into functional protein, was the induction after c-fos of a target gene of c-Jun/c-Fos heterodimers, i.e., transin, which encodes an extracellular matrix protease (McDonnell et al., 1990). Because we previously implicated c-Jun as well as the Fos family as necessary for neuronal apoptosis by using microinjected, neutralizing antibodies (Estus et al., 1994), and Ham and coworkers confirmed our results regarding c-Jun (Ham et al., 1995), these results are suggestive that these gene inductions may be relevant to neuronal death.
The second trend identified by two separate lines of evidence was that the c-jun induction correlated with initial neuronal responses to Aβ, whereas the c-fos induction, as well as genes coinduced with c-fos temporally, correlated with later stage(s) of apoptosis. First, whereas the c-jun induction began 12–24 hr after Aβ treatment, the timing, as well as the magnitude, of the induction of c-fos, fosB, andjunB showed a much greater dependence on Aβ concentration; the induction of these later genes coincided more closely with declining neuronal viability. Second, the in situhybridization studies showed that c-jun is induced in most neurons and that this induction is independent of overt signs of toxicity, i.e., chromatin changes. In contrast, the induction ofc-fos, and presumably genes coinduced temporally, was restricted mainly to neurons with condensed chromatin, a hallmark of apoptosis. Hence, c-jun generally was induced in a widespread manner and well before overt signs of neuronal toxicity. This suggests that c-jun expression may be more predictive of impending apoptosis than of apoptosis itself; alternatively, if c-Jun proves necessary for Aβ-induced death, c-Jun may act by accumulating over time, eventually reaching a threshold level that leads to the induction of later genes and concomitant loss of viability. This possibility is consistent with the observations of others regarding c-Jun reaching a critical level sufficient to act as a transcription factor (Trejo et al., 1992). Overall, c-jun is induced in most neurons before changes in viability, whereasc-fos is induced concomitant with an end stage of neuronal apoptosis.
The third trend discerned from these data is that Aβ aggregation per se is critical to changes in neuronal gene expression and resulting death; gene induction and neuronal cell death were not observed in cultures treated with nonaggregated Aβ. Hydrogen peroxide and free radical production have been implicated in mediating Aβ neurotoxicity in cultured neurons (Behl et al., 1994b; Hensley et al., 1994; Schubert et al., 1995), although quite recent studies indicate metabolic stress rather than oxidative stress as more likely to contribute to the cell death after exposure of neurons to Aβ (Zhang et al., 1996). Cellular stress is known to activate the Jun kinase (JNK) pathway (Kyriakis et al., 1994), and JNK activation has been reported to mediate neuronal apoptosis (Xia et al., 1995). We hypothesize that stress mediated by aggregated Aβ leads to JNK activation, which in turn phosphorylates cytoplasmic c-Jun, causing its activation and nuclear translocation, and subsequent activation of c-jun transcription. Current studies are addressing the role of JNK in Aβ-mediated neuronal apoptosis and identifying additional genes that may be involved more directly in neuronal cell death.
The temporal pattern of the immediate early gene response associated with Aβ-mediated neuronal apoptosis is remarkably similar to the immediate early gene response associated with neuronal apoptosis after NGF withdrawal. These findings demonstrate that similar genetic alterations are associated with markedly different apoptotic insults (toxic insult to cortical neurons and trophic factor deprivation of sympathetic neurons). Indeed, the results reported here lend further support that Aβ neurotoxicity in vitro is relevant to AD pathology in vivo; the induction of c-jun andc-fos by Aβ in vitro is consistent with the observation that c-Jun and c-Fos expression are induced in tangle-bearing neurons in AD (Anderson et al., 1994). Hence, the elucidation of molecular mechanisms underlying neuronal cell death in these model systems will likely contribute to our understanding of PCD and the development of therapeutic strategies for the treatment of neurodegenerative diseases such as AD.
Footnotes
This work was supported by grants (to S.E.) from the Alzheimer’s Association (Mr. Darrell Phillippi Pilot Research Grant), National Institutes of Health (NS35607), and the University of Kentucky. We thank F. Bard and G. Basi for helpful discussions and for assistance with the apoptotic characterization of neuronal cell death; we also thank F. Bard, E. M. Johnson, I. Lieberburg, P. A. K. Osborne, D. Schenk, and D. Selkoe for critically reviewing this manuscript.
Correspondence should be addressed to Dr. Steven Estus, Department of Physiology, Sanders-Brown Center on Aging, University of Kentucky, 800 South Limestone, Lexington, KY 40536-0230, or to Dr. Russell E. Rydel, Athena Neurosciences, 800 Gateway Boulevard, South San Francisco, CA 94080.
REFERENCES
- 1.Anderson AJ, Cummings BJ, Cotman CW. Increased immunoreactivity for Jun- and Fos-related proteins in Alzheimer’s disease: association with pathology. Exp Neurol. 1994;125:286–295. doi: 10.1006/exnr.1994.1031. [DOI] [PubMed] [Google Scholar]
- 2.Anderson AJ, Pike CJ, Cotman CW. Differential induction of immediate early gene proteins in cultured neurons by β-amyloid (Aβ): association of c-jun with Aβ-induced apoptosis. J Neurochem. 1995;65:1487–1498. doi: 10.1046/j.1471-4159.1995.65041487.x. [DOI] [PubMed] [Google Scholar]
- 3.Angel P, Karin M. The role of Jun, Fos, and the Ap-1 complex in cell proliferation and transformation. Biochim Biophys Acta. 1991;1072:129–157. doi: 10.1016/0304-419x(91)90011-9. [DOI] [PubMed] [Google Scholar]
- 4.Behl C, Davis JB, Klier FG, Schubert D. Amyloid beta peptide induces necrosis rather than apoptosis. Brain Res. 1994a;645:254–264. doi: 10.1016/0006-8993(94)91659-4. [DOI] [PubMed] [Google Scholar]
- 5.Behl C, Davis JB, Lesley R, Schubert D. Hydrogen peroxide mediates amyloid beta protein toxicity. Cell. 1994b;77:817–827. doi: 10.1016/0092-8674(94)90131-7. [DOI] [PubMed] [Google Scholar]
- 6.Bredesen DE. Neural apoptosis. Ann Neurol. 1995;38:839–851. doi: 10.1002/ana.410380604. [DOI] [PubMed] [Google Scholar]
- 7.Brewer GJ, Torricelli JR, Evege EK, Price PJ. Optimized survival of hippocampal neurons in B27-supplemented Neurobasal, a new serum-free medium combination. J Neurosci Res. 1993;35:567–576. doi: 10.1002/jnr.490350513. [DOI] [PubMed] [Google Scholar]
- 8.Cai XD, Golde TE, Younkin SG. Release of excess amyloid beta protein from a mutant amyloid beta protein precursor. Science. 1993;259:514–516. doi: 10.1126/science.8424174. [DOI] [PubMed] [Google Scholar]
- 9.Citron M, Oltersdorf T, Haass C, McConlogue L, Hung AY, Seubert P, Vigo-Pelfrey C, Lieberburg I, Selkoe DJ. Mutation of the beta-amyloid precursor protein in familial Alzheimer’s disease increases beta-protein production. Nature. 1992;360:672–674. doi: 10.1038/360672a0. [DOI] [PubMed] [Google Scholar]
- 10.Clarke PG. Developmental cell death: morphological diversity and multiple mechanisms. Anat Embryol (Berl) 1990;181:195–213. doi: 10.1007/BF00174615. [DOI] [PubMed] [Google Scholar]
- 11.Cochran BH. Regulation of immediate early gene expression. NIDA Res Monogr. 1993;125:3–24. [PubMed] [Google Scholar]
- 12.Deckwerth TL, Johnson EM., Jr Temporal analysis of events associated with programmed cell death (apoptosis) of sympathetic neurons deprived of nerve growth factor (NGF). J Cell Biol. 1993;123:1207–1222. doi: 10.1083/jcb.123.5.1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dragunow M, Faull RLM, Lawlor P, Beilharz EJ, Singleton K, Walker EB, Mee E. In situ evidence for DNA fragmentation in Huntington’s disease striatum and Alzheimer’s disease. NeuroReport. 1995;6:1053–1057. doi: 10.1097/00001756-199505090-00026. [DOI] [PubMed] [Google Scholar]
- 14.Estus S. Optimization and validation of RT-PCR as a tool to analyze apoptotic gene expression. In: Poirier J, editor. NeuroMethods 29: apoptosis techniques and protocols. Humana; Totowa, NJ: 1997. pp. 67–84. [Google Scholar]
- 15.Estus S, Zaks W, Freeman R, Gruda M, Bravo R, Johnson E. Altered gene expression in neurons during programmed cell death: identification of c-jun as necessary for neuronal apoptosis. J Cell Biol. 1994;127:1717–1727. doi: 10.1083/jcb.127.6.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Estus S, van Rooyen C, Mattson M, Brigham EF, Rydel RE. Comparison of altered gene expression during neuronal death induced by NGF withdrawal, amyloid β-protein treatment, or glucose deprivation. Soc Neurosci Abstr. 1995;21:1721. [Google Scholar]
- 17.Estus S, van Rooyen C, Tucker HM, Wright S, Brigham EF, Rydel RE. Altered gene expression during neuronal death induced by amyloid β-protein treatment. Soc Neurosci Abstr. 1996;22:2114. [Google Scholar]
- 18.Forloni G, Chiesa R, Smiroldo S, Verga L, Salmona M, Tagliavini F, Angeretti N. Apoptosis-mediated neurotoxicity induced by chronic application of β amyloid fragment 25–35. NeuroReport. 1993;4:523–526. doi: 10.1097/00001756-199305000-00015. [DOI] [PubMed] [Google Scholar]
- 19.Freeman RS, Estus S, Horigome K, Johnson EM., Jr Cell death genes in invertebrates and (maybe) vertebrates. Curr Opin Neurobiol. 1993;3:25–31. doi: 10.1016/0959-4388(93)90031-s. [DOI] [PubMed] [Google Scholar]
- 20.Freeman RS, Estus S, Johnson EM., Jr Analysis of cell cycle-related gene expression in postmitotic neurons: selective induction of cyclin D1 during programmed cell death. Neuron. 1994;12:343–355. doi: 10.1016/0896-6273(94)90276-3. [DOI] [PubMed] [Google Scholar]
- 21.Games D, Adams D, Alessandrini R, Barbour R, Berthelette P, Blackwell C, Carr T, Clemens J, Donaldson T, Gillespie F. Alzheimer-type neuropathology in transgenic mice overexpressing V717F beta-amyloid precursor protein. Nature. 1995;373:523–527. doi: 10.1038/373523a0. [DOI] [PubMed] [Google Scholar]
- 22.Gavrieli Y, Sherman Y, Ben-Sasson SA. Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J Cell Biol. 1992;119:493–501. doi: 10.1083/jcb.119.3.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Golde TE, Estus S, Usiak M, Younkin LH, Younkin SG. Expression of beta amyloid protein precursor mRNAs: recognition of a novel alternatively spliced form and quantitation in Alzheimer’s disease using PCR. Neuron. 1990;4:253–267. doi: 10.1016/0896-6273(90)90100-t. [DOI] [PubMed] [Google Scholar]
- 24.Goto K, Ishige A, Sekiguchi K, Iizuka S, Sugimoto A, Yuzurihara M. Effects of cycloheximide on delayed neuronal death in rat hippocampus. Brain Res. 1990;534:299–302. doi: 10.1016/0006-8993(90)90144-z. [DOI] [PubMed] [Google Scholar]
- 25.Greenberg ME, Hermanowski AL, Ziff EB. Effect of protein synthesis inhibitors on growth factor activation of c-fos, c-myc, and actin gene transcription. Mol Cell Biol. 1986;6:1050–1057. doi: 10.1128/mcb.6.4.1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gschwind M, Huber G. Apoptotic cell death induced by β-amyloid 1–42 peptide is cell type-dependent. J Neurochem. 1995;65:292–300. doi: 10.1046/j.1471-4159.1995.65010292.x. [DOI] [PubMed] [Google Scholar]
- 27.Ham J, Babij C, Whitfield J, Pfarr CM, Lallemand D, Yaniv M, Rubin LL. A c-Jun dominant negative mutant protects sympathetic neurons against programmed cell death. Neuron. 1995;14:927–939. doi: 10.1016/0896-6273(95)90331-3. [DOI] [PubMed] [Google Scholar]
- 28.Hensley K, Carney JM, Mattson MP, Aksenova M, Harris M, Wu JF, Floyd RA, Butterfield DA. A model for beta-amyloid aggregation and neurotoxicity based on free radical generation by the peptide: relevance to Alzheimer disease. Proc Natl Acad Sci USA. 1994;91:3270–3274. doi: 10.1073/pnas.91.8.3270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Coel G. Correlative memory deficits, Aβ elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- 30.Kyriakis JM, Banerjee P, Nikolakaki E, Dai T, Rubie EA, Ahmad MF, Avruch J, Woodgett JR. The stress-activated protein kinase subfamily of c-Jun kinases. Nature. 1994;369:156–160. doi: 10.1038/369156a0. [DOI] [PubMed] [Google Scholar]
- 31.Lassmann H, Bancher C, Breitschopf H, Wegiel J, Bobinski M, Jellinger K, Wisniewski HM. Cell death in Alzheimer’s disease evaluated by DNA fragmentation in situ. Acta Neuropathol (Berl) 1995;89:35–41. doi: 10.1007/BF00294257. [DOI] [PubMed] [Google Scholar]
- 32.LeVine H. Thioflavine T interaction with synthetic Alzheimer’s disease beta-amyloid peptides: detection of amyloid aggregation in solution. Protein Sci. 1993;2:404–410. doi: 10.1002/pro.5560020312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Linnik MD, Zobrist RH, Hatfield MD. Evidence supporting a role for programmed cell death in focal cerebral ischemia in rats. Stroke. 1993;24:2002–2009. doi: 10.1161/01.str.24.12.2002. [DOI] [PubMed] [Google Scholar]
- 34.Loo DT, Copani A, Pike CJ, Whittemore ER, Walencewicz AJ, Cotman CW. Apoptosis is induced by β-amyloid in cultured central nervous system neurons. Proc Natl Acad Sci USA. 1993;90:7951–7955. doi: 10.1073/pnas.90.17.7951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.MacManus JP, Buchan AM, Hill IE, Rasquinha I, Preston E. Global ischemia can cause DNA fragmentation indicative of apoptosis in rat brain. Neurosci Lett. 1993;164:89–92. doi: 10.1016/0304-3940(93)90864-h. [DOI] [PubMed] [Google Scholar]
- 36.MacManus JP, Hill IE, Huang ZG, Rasquinha I, Xue D, Buchan AM. DNA damage consistent with apoptosis in transient ischaemic neocortex. NeuroReport. 1994;5:493–496. doi: 10.1097/00001756-199401120-00031. [DOI] [PubMed] [Google Scholar]
- 37.Martin DP, Schmidt RE, DiStefano PS, Lowry OH, Carter JG, Johnson EM., Jr Inhibitors of protein synthesis and RNA synthesis prevent neuronal death caused by nerve growth factor deprivation. J Cell Biol. 1988;106:829–844. doi: 10.1083/jcb.106.3.829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McDonnell SE, Kerr LD, Matrisian LM. Epidermal growth factor stimulation of stromelysin mRNA in rat fibroblasts requires induction of proto-oncogenes c-fos and c-jun and activation of protein kinase C. Mol Cell Biol. 1990;10:4284–4293. doi: 10.1128/mcb.10.8.4284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Oppenheim RW. Cell death during development of the nervous system. Annu Rev Neurosci. 1991;14:453–501. doi: 10.1146/annurev.ne.14.030191.002321. [DOI] [PubMed] [Google Scholar]
- 40.Oppenheim RW, Prevette D, Tytell M, Homma S. Naturally occurring and induced neuronal death in the chick embryo in vivo requires protein and RNA synthesis: evidence for the role of cell death genes. Dev Biol. 1990;138:104–113. doi: 10.1016/0012-1606(90)90180-q. [DOI] [PubMed] [Google Scholar]
- 41.Pike C, Walencewicz A, Glabe C, Cotman C. In vitro aging of β-amyloid protein causes peptide aggregation and neurotoxicity. Brain Res. 1991;563:311–314. doi: 10.1016/0006-8993(91)91553-d. [DOI] [PubMed] [Google Scholar]
- 42.Portera-Calliau C, Hedreen JC, Price DL, Koliatsos VE. Evidence for apoptotic cell death in Huntington disease and excitotoxic animal models. J Neurosci. 1995;15:3775–3787. doi: 10.1523/JNEUROSCI.15-05-03775.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Scheuner D, Eckman C, Jensen M, Song X, Citron M, Suzuki N, Bird TD, Hardy J, Hutton M, Kukull W, Larson E, Levy-Lahad E, Viitanen M, Peskind E, Poorkaj P, Schellenberg G, Tanzi R, Wasco W, Lannfelt L, Selkoe D, Younkin SG. Secreted amyloid β-protein similar to that in the senile plaques of Alzheimer’s disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer’s disease. Nat Med. 1996;2:864–870. doi: 10.1038/nm0896-864. [DOI] [PubMed] [Google Scholar]
- 44.Schubert D, Behl C, Lesley R, Brack A, Dargusch R, Sagara Y, Kimura H. Amyloid peptides are toxic via a common oxidative mechanism. Proc Natl Acad Sci USA. 1995;92:1989–1993. doi: 10.1073/pnas.92.6.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Selkoe DJ. Alzheimer’s disease: genotypes, phenotype, and treatments. Science. 1997;275:630–631. doi: 10.1126/science.275.5300.630. [DOI] [PubMed] [Google Scholar]
- 46.Shigeno T. Reduction of delayed neuronal death by inhibition of protein synthesis. Neurosci Lett. 1990;120:117–119. doi: 10.1016/0304-3940(90)90182-9. [DOI] [PubMed] [Google Scholar]
- 47.Simmons LK, May PC, Tomaselli KJ, Rydel RE, Fuson KS, Brigham EF, Wright S, Lieberburg I, Becker GW, Brems DN. Secondary structure of amyloid beta peptide correlates with neurotoxic activity in vitro. Mol Pharmacol. 1994;45:373–379. [PubMed] [Google Scholar]
- 48.Smale G, Nichols NR, Brady DR, Finch CE, Horton WE. Evidence for apoptotic cell death in Alzheimer’s disease. Exp Neurol. 1995;133:225–230. doi: 10.1006/exnr.1995.1025. [DOI] [PubMed] [Google Scholar]
- 49.Su JH, Anderson AJ, Cummings BJ, Cotman CW. Immunohistochemical evidence for apoptosis in Alzheimer’s disease. NeuroReport. 1994;5:2529–2533. doi: 10.1097/00001756-199412000-00031. [DOI] [PubMed] [Google Scholar]
- 50.Suzuki N, Cheung TT, Cai XD, Odaka A, Otvos LJ, Eckman C, Golde TE, Younkin SG. An increased percentage of long amyloid beta protein secreted by familial amyloid beta protein precursor (beta APP717) mutants. Science. 1994;264:1336–1340. doi: 10.1126/science.8191290. [DOI] [PubMed] [Google Scholar]
- 51.Trejo J, Chambard J-C, Karin M, Brown JH. Biphasic increase in c-jun mRNA is required for induction of AP-1-mediated gene transcription: differential effects of muscarinic and thrombin receptor activation. Mol Cell Biol. 1992;12:4742–4750. doi: 10.1128/mcb.12.10.4742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Trejo J, Massamiri T, Deng T, Dewji NN, Bayney RM, Brown JH. A direct role for protein kinase C and the transcription factor Jun/AP-1 in the regulation of the Alzheimer’s beta-amyloid precursor protein gene. J Biol Chem. 1994;269:21682–21690. [PubMed] [Google Scholar]
- 53.Wanaka A, Johnson EM, Jr, Milbrandt J. Localization of FGF receptor mRNA in the adult rat central nervous system by in situ hybridization. Neuron. 1990;5:267–281. doi: 10.1016/0896-6273(90)90164-b. [DOI] [PubMed] [Google Scholar]
- 54.Wyllie AH, Kerr JFR, Currie AR. Cell death: the significance of apoptosis. Int Rev Cytol. 1980;68:251–306. doi: 10.1016/s0074-7696(08)62312-8. [DOI] [PubMed] [Google Scholar]
- 55.Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science. 1995;270:1326–1331. doi: 10.1126/science.270.5240.1326. [DOI] [PubMed] [Google Scholar]
- 56.Zhang Z, Rydel RE, Drzewiecki GJ, Fuson K, Wright S, Wogulis M, Audia JE, May PC, Hyslop PA. Amyloid β-mediated oxidative and metabolic stress in rat cortical neurons: no direct evidence for a role for H2O2 generation. J Neurochem. 1996;67:1595–1606. doi: 10.1046/j.1471-4159.1996.67041595.x. [DOI] [PubMed] [Google Scholar]