

Abstract
Kainate binding proteins (KBPs) are highly homologous to ionotropic glutamate receptors; however, no ion channel function has been demonstrated for these proteins. To investigate possible reasons for the apparent lack of ion channel function we transplanted the ion channel domains of five KBPs into glutamate receptors GluR 6 and GluR1. In each case we obtained functional chimeric receptors in which glutamatergic agonists were able to open the KBP-derived ion channel with EC50 values identical to those of the subunit contributing the ligand binding domain. Maximal current amplitudes were significantly smaller than those of the parent clones, however. We also show that the KBP ion channels are highly permeable for calcium and have certain pharmacological properties that are distinct from all other glutamate receptor (GluR) subunits. Thus, all five known KBPs, in addition to their well characterized functional ligand binding sites, have functional ion permeation pathways. Our data suggest that the lack of ion channel function in wild-type KBPs results from a failure to translate ligand binding into channel opening. We interpret our findings to indicate the requirement for a modulatory protein or an additional subunit serving to alter the structure of the KBP subunit complex such that signal transduction is enabled from the ligand binding site to the intrinsically functional ion pore.
Keywords: kainate binding proteins, KBP, ion pore, domain transplantation, kainate receptors, GluR1, GluR6, calcium permeability, Xenopus oocytes
Ionotropic glutamate receptors (GluRs) are the prevalent excitatory neurotransmitter receptors in the CNS of vertebrates. Three pharmacologically distinct types have been identified through molecular cloning: AMPA receptors, kainate (KA) receptors, and NMDA receptors (for review, see Hollmann and Heinemann, 1994). In addition to GluR subunits, which form functional ion channels, several homologous subunits have been characterized that lack apparent intrinsic ion channel function and do not seem to form functional heteromeric complexes with other subunits. These include the kainate binding proteins (KBPs) (Gregor et al., 1989; Wada et al., 1989; Wo and Oswald, 1994, Ishimaru et al., 1996; for review, seeHenley, 1994) and several orphan receptors (Hollmann and Heinemann, 1994).
The KBPs are ∼50 kDa proteins that bind kainate receptor agonists such as Glu, KA, and domoate (Dom) (Wada et al., 1989); however, the physiological role of these proteins remains enigmatic (Henley, 1994). In the KBP subfamily, five different genes have been identified from five different species: one subunit each from the frog Rana pipiens [KBP(Rp)], the chicken Gallus domesticus[KBP(Gd)], the toad Xenopus laevis [KBP(Xl)], and the duck Anas domesticus [KBP(Ad)], and two different subunits from the goldfish Carassius auratus [KBP(Ca)α and KBP(Ca)β]. KBP(Gd) and KBP(Ad) are 92.8% identical at the amino acid level, indicating that they represent the same gene. The other subunits share between 49.8 and 67.9% sequence identity and thus are derived from different genes. Notably, no KBPs have been discovered in mammals.
There is significant sequence homology (35–40%) between KBPs and other GluRs, particularly the KA receptors. In addition, their transmembrane topology is believed to be identical to that of other GluRs (Hollmann et al., 1994; Wo and Oswald, 1994). The most compelling structural difference between KBPs and other GluRs is their short N-terminal domains (128–148 amino acids as opposed to ∼520 for other GluR subunits), which is characteristic for all KBPs (see Fig. 1).
Fig. 1.

Bar graph representation of the structural features of receptor subunits used for ion pore transplantation. KBPs of Rana pipiens (Rp), Gallus domesticus (Gd), Carassius auratus(Ca, two subunits, α and β), and Xenopus laevis(Xl) are compared with the AMPA receptor GluR1 and the KA receptor GluR6. The chimera GluR6N-KBP(Rp)C is a construct with an N-terminal transplantation between GluR6 (N-terminal part) and KBP(Rp) (C-terminal part); the black circle marks the junction between the two domains. Chimera GluR6-Rp-R6-Rp (bottom bar) is derived from GluR6N-KBP(Rp)C by exchanging the pore domain for that of GluR6(Q). The predicted signal peptides, the three transmembrane domains (TMD A, TMD B, TMD C) and the pore loop domains (the “TMD II” of previous topology models) are indicated by black rectangles.
To gain insight into the biological role of KBPs we set out to determine whether KBPs could potentially form functional ion channels. We chose a domain transplantation strategy that involved engineering of the ion channel domain of KBPs into functional GluR subunits. This approach was based on recent suggestions that GluRs may be modular proteins made up of building blocks derived from different precursor proteins (Seeburg et al., 1995; Wo and Oswald, 1995; Hollmann, 1996), a design that should allow domain exchange among subunits.
The domain transplantation experiments described in this study identified the putative ion channel domains of all five KBP genes [KBP(Rp), KBP(Gd), KBP(Ca)α, KBP(Ca)β, and KBP(Xl)] as sequences capable of forming functional cation conduction pathways with distinct pharmacological properties. Moreover, these domains are capable of coupling to glutamatergic ligand binding sites, and they permit the flow of calcium ions.
MATERIALS AND METHODS
Mutagenesis. To allow easy exchange of ion pores between subunits, unique restriction sites were introduced into both donor and acceptor subunits by PCR-based mutagenesis. Homologous positions within each sequence were chosen to engineer anEcoRI site downstream of the pore region at amino acids 211–213 (RII) in KBP(Rp) (the cDNA clone was kindly provided by Drs. K. Wada and R. Wenthold, National Institutes of Health) and at the corresponding amino acids in GluR6(Q) (603–605, RIV) and GluR1 (595–597, RIV). Numbering starts with the first codon of the mature protein. Upstream of the pore region an Nru I site was introduced at amino acids 163–165 (FLV) in KBP(Rp), at amino acids 548–560 (FVI) in GluR6(Q), and at amino acids 537–539 (FLV) in GluR1 (see Fig. 2). The sites were chosen such that the entire pore region including both adjacent intracellular loops (L1 and L2) (Hollmann et al., 1994) was transplanted. The resulting constructs were named “x-PCS,” where “x” stands for the clone modified and “PCS” means “pore cassette sites”. Mutagenetic oligonucleotide primers of 21–36 bp length were obtained from Eurogentec (Seraing, Belgium). A fragment containing both newly generated restriction sites was synthesized in a first round of PCR. The purified fragment was extended C-terminally in a second PCR using wild-type DNA as the template and a tailed primer binding downstream of the gene within the vector sequence. The vector used throughout this study was pSGEM, a modified version of pGEMHE in which we replaced the original multiple cloning site by the pBluescript (Stratagene, Heidelberg, Germany) polylinker; furthermore, two additional sites for template linearization (the eight-cutters PacI and SfiI) were inserted between the existing SphI and NheI sites. The original pGEMHE vector was kindly provided by Emily Liman and the late Peter Hess (Harvard Medical School). In a third round of PCR the second-round fragment was N-terminally extended using primers binding to the T7 promoter of pSGEM and the tail sequence generated in the second round of PCR, respectively, so that only the mutated strand will be amplified.
Fig. 2.

Amino acid sequence alignment of the transplanted ion pore regions (the sequence between the end of TMD Aand the start of TMD B) of the five KBPs (see legend to Fig. 1), GluR1, and GluR6(Q). Flanking the sequences to be exchanged are three amino acids shown in bold that were mutated to obtain the consensus sequences FAI and RIL shown at thebottom, thereby introducing unique restriction sites as indicated. The Q/R editing site of AMPA and KA receptors is marked by an arrow.
PCRs were set up as follows: 1 ng of template DNA, 200 mmTris-HCl, pH 8.8, 100 mm KCl, 100 mm(NH4)2SO4, 20 mm MgSO4, 1% Triton X-100, 1 mg/ml bovine serum albumin, 50 μm each dATP, dCTP, dTTP, and dGTP, 100 pmol of each primer, and 2 U Pfu polymerase (Stratagene). PCR conditions were 5 min/95°C for denaturation, 5 min/50°C for annealing, 5 min/72°C for elongation in the first cycle, followed by 28 cycles of 1 min/95°C, 2 min/50°C, and 2.5 min/72°C. The last cycle ended with a 10 min/72°C amplification step.
The final PCR fragments were cut with suitable restriction sites as close as possible to the region to be transplanted and were reinserted into the respective wild-type clones to minimize any PCR-generated sequence. The following cloning cassettes were used for shuttling:EcoRV-AccI [nucleotide (nt) 1685–1950] for GluR6(Q), BglII-BglII (nt 1810–2220) for GluR1, and AflIII-Xcm I (nt 455–720) for KBP(Rp). The resulting mutants were designated GluR6(Q)-PCS, GluR1-PCS, and KBP(Rp)-PCS, respectively, and were used as recipient clones of the pore regions to be transplanted. Next, PCR-amplified fragments of the pore domains of GluR6(Q), KBP(Rp), KBP(Ca)α, KBP(Ca)β, KBP(Gd), and KBP(Xl) were generated to include the required flankingEcoRI and Nru I sites, using the appropriate mutagenetic primers. These fragments were digested withEcoRI and Nru I and ligated into the appropriate recipient clones, GluR1-PCS, GluR6(Q)-PCS, or KBP(Rp)-PCS, which had been prepared for ligation by digestion with EcoRI andNru I. The original cDNA clones of the KBPs were used as starting material. Clones of these KBPs were generously provided by Dr. R. Oswald (Cornell University) (KBP(Ca)α and KBP(Ca)β) and Dr. V. Teichberg (Weizmann Institute) (KBP(Gd)). For the recently cloned KBP(Xl) (Ishimaru et al., 1996), a cDNA clone was not available to us. We therefore used an 865 bp PCR-generated fragment (nt 241–1105) amplified from Xenopus cDNA that had been reverse-transcribed from total brain mRNA isolated from 10 femaleXenopus laevis.
All mutant clones were sequenced across the PCR-generated fragment with the Sequenase 2.0 sequencing kit (United States Biochemicals, Braunschweig, Germany), which uses the dideoxynucleotide chain termination method (Sanger et al., 1977). Sequence data were analyzed with the University of Wisconsin software package (Devereux et al., 1984).
cRNA synthesis. Template DNA was linearized withNheI. cRNA was synthesized from 1 μg of linearized DNA using an in vitro transcription kit (Stratagene) with a modified protocol that uses 800 μm each nucleotide (except GTP, 200 μm), 800 μmm7GpppG (Pharmacia, Freiburg, Germany) for capping, and an extended reaction time of 3 hr with T7 polymerase. Trace labeling was performed with [32P]UTP to allow calculation of yields and transcript quality check by agarose gel electrophoresis.
Electrophysiological measurements in Xenopusoocytes. Oocytes of stages V–VI were surgically removed from the ovaries of Xenopus laevis anesthetized with 3-aminobenzoic acid ethylester (2.3 gm/l). The oocytes were incubated in calcium-free Barth’s solution (see below) containing 815 U/ml (=2.8 mg/ml) collagenase and 2200 U/ml (=0.15 mg/ml) trypsin for 2.75 hr while they were gently shaken to remove the follicular cell layer. Oocytes were washed five to six times in Barth’s solution (88 mm NaCl, 1.1 mm KCl, 2.4 mm NaHCO3, 0.3 mm Ca(NO)3, 0.3 mmCaCl2, 0.8 mm MgSO4, 15 mm HEPES-NaOH, pH 7.6). After selection the oocytes were kept in Barth’s solution containing 63 μg/ml penicillin, 40 μg/ml streptomycin, and 100 μg/ml gentamycin; 10 ng (=50 nl) of cRNA was injected into the oocytes using a 10 μl Drummond microdispenser. Two-electrode voltage-clamp recordings were performed with a TurboTec 10CD amplifier (npi, Tamm, Germany) 4–8 d after cRNA injection by superfusion of the oocyte with glutamatergic agonists (1–300 μm) prepared in normal frog Ringer’s solution (NFR) (115 mm NaCl, 2.5 mm KCl, 1.5 mmCaCl2, 10 mm HEPES-NaOH, pH 7.2). Voltage electrodes were filled with 3 m KCl and had resistances of ∼4 MΩ; current electrodes were filled with 3m CsCl and had resistances of ∼0.5–1.5 MΩ. Oocytes were held at −70mV. All measurements of clones based on the KA receptor GluR6 were performed after preincubation of the oocyte with 1 mg/ml concanavalin A (ConA) for 8 min. This treatment eliminates desensitization of KA receptors, particularly GluR6 (Egebjerg et al., 1991). Agonists were applied for 10 sec by superfusion of the oocyte at a flow rate of 10–14 ml/min. Current–voltage (I–V) relationships were determined with a 2 sec voltage ramp and analyzed using the PulseFit 7.62 program (HEKA Electronics, Lambrecht, Germany). To determine the EC50 values for KA and Glu, 8–10 different agonist concentrations were applied to the same oocyte, and steady-state values of the evoked currents were measured. Data from each oocyte were fitted separately, and EC50 values obtained this way from three to five oocytes were averaged. Calcium permeability tests were performed in low or high “Ca-Ringer” lacking any other permeable cation. Both 10 mm Ca-Ringer (10 mm HEPES, 10 mm CaCl2, 105.2 mmN-methyl-d-glucamine, pH 7.2, adjusted with concentrated HCl) and 80 mm Ca-Ringer (80 mm CaCl2, 10 mm HEPES, pH 7.2, adjusted with N-methyl-d-glucamine) were used.
Gel electrophoresis and immunoblotting. Batches of 12 oocytes were used for membrane preparations 6–8 d after cRNA injection, following a previously described protocol (Hollmann et al., 1994). For experiments in which only those proteins inserted into the plasma membrane were to be analyzed, membrane preparations were performed after biotinyl-ConA labeling of glycosylated surface proteins and streptavidin–Sepharose-mediated precipitation of labeled proteins. Briefly, the oocytes were incubated in 10 μmbiotinyl-ConA (Sigma, Munich, Germany) in NFR for 30 min at room temperature (RT). After five washes for 10 min each in NFR, oocytes were homogenized with a Teflon pestle in 240 μl H-buffer (100 mm NaCl, 20 mm Tris-HCl, pH 7.4, 1% Triton X-100, 1 mm phenylmethylsulfonyl fluoride plus a cocktail of additional proteinase inhibitors: 2.5 μg/ml leupeptin, 20 μg/ml aprotinin, 2.5 μg/ml pepstatin, and 20 μg/ml benzamidine hydrochloride) and kept on ice for 15 min. After centrifugation for 60 sec at 16,000 × g, the supernatants were supplemented with 20 μm washed streptavidin–Sepharose beads (Sigma) and incubated for 3 hr at 4°C on a rotator. The streptavidin–Sepharose beads were pelleted by a 60 sec spin and washed three times with H-buffer, and the final pellets were boiled in 40 μl/oocyte SDS-PAGE loading buffer (0.8 mβ-mercaptoethanol, 6% SDS, 20% glycerol, 25 mmTris-HCl, pH 6.8, 0.1% bromphenol blue).
Samples were run on 20 cm discontinuous SDS-PAGE gels (Laemmli, 1970) (5% stacking gel, 7.5% separating gel; running time 2.5 hr at 4°C). Prestained protein markers (Bio-Rad, Munich, Germany) were used to monitor separation on the gel as well as to identify the position of immunoreactive bands on blots. The gel was blotted (Towbin et al., 1979) onto Hybond ECL nitrocellulose membranes (Amersham, Braunschweig, Germany) at a constant current of 200 mA for 16 hr at 4°C. Filters were blocked for 2 hr at RT with blocking buffer [1× Roti-block (Roth, Karlsruhe, Germany) in 140 mm NaCl, 0.1% Triton X-100, 20 mm Tris-HCl, pH 7.6] and probed overnight at 4°C with affinity-purified rabbit antisera directed against the C termini of GluR6 (peptide sequence TFNDRRLPGKETMA) (Wenthold et al., 1994) or KBP(Rp) (peptide sequence KSPTSNSCDEVKA). Both antibodies were kindly provided by Dr. Robert Wenthold. Incubations were performed in 0.1× Roti-block, 0.1% Triton X-100, 140 mm NaCl, 20 mm Tris-HCl, pH 7.6. Peroxidase-labeled donkey anti-rabbit IgG (Dianova, Hamburg, Germany) was used as secondary antibody. Immunoreactive bands were visualized by the chemoluminescence method (ECL detection kit, Amersham).
Reagents. Restriction enzymes were purchased from Boehringer Mannheim (Mannheim, Germany), Promega (Mannheim, Germany), and New England Biolabs (Schwalbach, Germany). All nucleotides were from Pharmacia (Freiburg, Germany). Unless noted otherwise, all chemicals were from Sigma.
RESULTS
N-terminal elongation of KBPs
The most obvious structural difference between functional members of the GluR family and the nonfunctional KBPs is the short N-terminal domain of the latter (Fig. 1). To test whether an extended N-terminal domain would render KBPs functional, we engineered the N-terminal sequence of the KA receptor GluR6 (comprising amino acids 1–399) onto the KBP from Rana pipiens. Amino acid 22 (a lysine) of wild-type KBP(Rp) was used as the N-terminal point of connection to create the chimera GluR6N-KBP(Rp)C (Fig. 1). Unfortunately, this chimera was not functional on expression inXenopus oocytes (data not shown), although the protein was expressed and inserted into the oocyte plasma membrane (see Fig.4C). Coexpression of wild-type GluR6(Q) did not enhance expression levels (see Fig. 4C, right lane) and did not produce any alterations of the functional properties found for GluR6(Q) alone (data not shown).
Fig. 4.
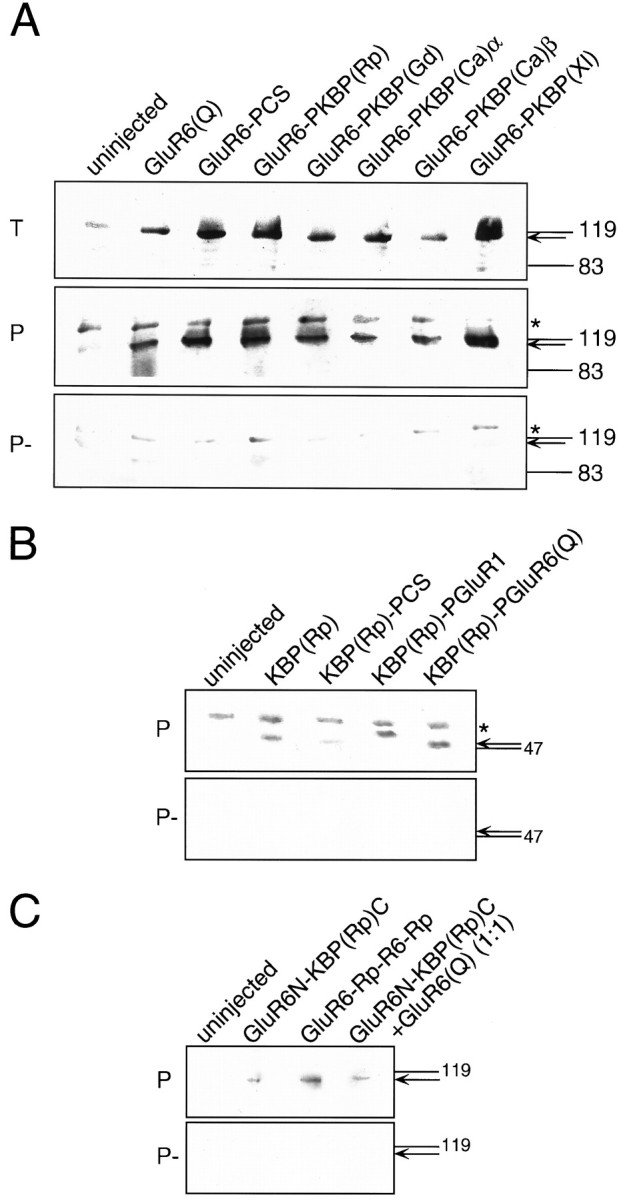
Western blots demonstrating protein expression of chimeric receptors. A, GluR6(Q) wild-type compared with pore transplantation chimeras of GluR6. B, KBP(Rp) wild-type compared with pore transplantation chimeras of KBP(Rp). C, N-terminus transplantation chimeras between GluR6 and KBP(Rp). Total oocyte proteins (T, 1 oocyte/lane), streptavidin-precipitated biotinyl-ConA-labeled plasma membrane proteins (P, 10 oocytes/lane), and nonbiotinylated controls (P−, 10 oocytes/lane) were separated electrophoretically, blotted, and probed with affinity-purified antibodies generated to the C-terminal peptide of GluR6 (A) or the C-terminal peptide of KBP(Rp) (B, C). Arrows point to the position of the ∼120 kDa GluR6 wild-type and pore transplantation mutant proteins in A, to the position of the ∼48 kDa KBP inB, and to the position of the ∼118 kDa N-terminal transplantation chimeras between GluR6 and KBP in C. Note that in A an unidentified band cross-reacting with the GluR6 antibody (marked by an asterisk) is present in all control precipitations and even in uninjected oocytes (panels P, P−). This band is absent from the total oocyte protein (panel T) because only a single oocyte was loaded in that case as opposed to 10 oocytes for panels P and P−. Note also that specific bands (arrows) are either totally absent (B, C) or only very weak (A) in precipitation control samples (P−) that were not biotinyl-ConA-labeled. This demonstrates that the immunoreactive protein identified in panel P is actually residing in the plasma membrane. KBP(Rp)-PGluR1 in B runs at a slightly larger molecular weight than expected. This erratic running behavior is not attributable to a sequence problem in the construct but likely reflects a conformational peculiarity of this particular construct.
Transplantation of KBP ion pores into functional channels
Another domain critically involved in channel function is obviously the ligand binding site. This domain, however, has already been well established as a functional site on all cloned KBPs through agonist binding experiments with 3H-KA (Gregor et al., 1989; Wada et al., 1989; Wo and Oswald, 1994, Ishimaru et al., 1996). An equally important domain that has not received nearly as much attention is the ion channel domain itself. If this domain was intrinsically defective or nonfunctional, obviously no ionic currents would be possible even after binding of the proper ligand. We therefore transplanted the putative ion channel domain of KBP(Rp) into functional GluR subunits to test the intrinsic functionality of the channel domain in the context of proven functional subunits. We chose the AMPA receptor GluR1 (45.9% sequence identity at the amino acid level) and the KA receptor subunit GluR6, which is the most closely related subunit (51.5% sequence identity), as donors for the ligand binding site. The excision points of the transplanted ion channel domain from KBP(Rp) and the corresponding insertion points in GluR1 and GluR6 were chosen such that the entire hairpin loop comprising the putative channel-lining segment could be transplanted together with the flanking intracellular loops L1 and L2 as defined in Hollmann et al. (1994). The transplanted sequence starts at the C-terminal end of transmembrane domain (TMD) A and runs up to the N-terminal end of TMD B. To facilitate ion pore exchange between various receptor subunits, we inserted unique restriction enzyme sites at the starting points and end point of the region to be transplanted. The resulting constructs made from GluR1, GluR6, and KBP(Rp) were named GluR1-PCS, GluR6-PCS, and KBP(Rp)-PCS, respectively (Fig. 2) (also see Materials and Methods). They served as the parent constructs for all ion channel transplantations performed for this study.
Both chimeric constructs, GluR1-PKBP(Rp) and GluR6-PKBP(Rp), gave functional ion channels that could be activated by KA, Glu, and Dom (Fig. 3B,D). Maximal current amplitudes of GluR1-PKBP(Rp) (6.3 ± 0.4 nA for KA currents;n = 7) and GluR6-PKBP(Rp) (9.03 ± 0.2 nA;n = 39) were rather small compared with those of wild-type GluR1 (3157 ± 711 nA for KA currents; n= 4) and wild-type GluR6 (14550 ± 1892 nA; n = 4), respectively (∼1%). They could be reproducibly measured in every oocyte tested, however, provided that current desensitization was minimized by ConA pretreatment of oocytes expressing GluR1-PKBP(Rp) and GluR6-PKBP(Rp) (see Materials and Methods). Additionally, cyclothiazide was coapplied for recordings of GluR1-PKBP(Rp), because this compound specifically blocks desensitization at AMPA receptors (Partin et al., 1993). Interestingly, despite the pronounced difference between wild-type GluR1 and GluR6 in maximal current amplitudes (the ratio found was 1:∼4.6), the two chimeras GluR1-PKBP(Rp) and GluR6-PKBP(Rp) had similar currents (a ratio of 1:∼1.4), indicating that the transplanted KBP pore rather than the sequence background of the ligand binding site donor subunit determined the maximal currents.
Fig. 3.

Sample current traces of the receptor constructs GluR1-PCS (A) and GluR6(Q)-PCS (C), which had been engineered for easy pore transplantation (see Materials and Methods), and of chimeric receptors harboring the ion pore domains of various KBPs (B, D–H). Agonists used were kainate (KA, 100 μm in C–H; 300 μm inA, B), glutamate (GLU, 300 μm), and domoate (DOM, 10 μm). To minimize desensitization, all clones were treated with 10 μm ConA before recording; in addition, for GluR1-derived clones (A, B), 100 μmcyclothiazide was coapplied with the agonist.
To test whether the ion channel domains of other KBPs were similarly capable of conducting currents, we engineered the respective domains of KBP(Xl) (Ishimaru et al., 1996), KBP(Ca)α and KBP(Ca)β (Wo and Oswald, 1994), and KBP(Gd) (Gregor et al., 1989) into GluR6 using the same strategy as described above for KBP(Rp) (for details, see Materials and Methods). All chimeras containing KBP ion channel transplants were expressed at the protein level (Fig.4A, panel T) and were inserted into the plasma membrane (Fig.4A, panel P), and all were functional (Fig. 3E–H). GluR6-PKBP(Ca)α gave the largest maximal amplitudes (Table 1), which were in the range of 7% of wild-type GluR6(Q) or 17% of GluR6(Q)-PCS (see Materials and Methods) for both Glu- and KA-evoked currents.
Table 1.
EC50 values of GluR6 and chimeras containing ion pores from KBPs of different species
| Clone | EC50KA | nH | Imax[nA] | n | EC50GLU | nH | Imax[nA] | n |
|---|---|---|---|---|---|---|---|---|
| GluR6(Q) wild type | 1.74 ± 0.72 | 1.09 ± 0.11 | 6224 ± 1487 | 3 | 34.4 ± 6.2 | 1.2 ± 0.12 | 9898 ± 2042 | 3 |
| GluR6(Q)-PCS | 2.2 ± 0.06 | 0.97 ± 0.06 | 667 ± 179 | 4 | 59.9 ± 4.8 | 1.2 ± 0.06 | 695 ± 132 | 3 |
| GluR6-PKBP(Rp) | 2.4 ± 0.15 | 1.4 ± 0.1 | 8.02 ± 1.13 | 4 | 35.7 ± 7.35 | 1.78 ± 0.2 | 6.47 ± 1.9 | 5 |
| GluR6-PKBP(Gd) | 6.0 ± 1.4 | 0.93 ± 0.08 | 6.0 ± 0.7 | 5 | 47.2 ± 1.6 | 1.35 ± 0.02 | 10.61 ± 1.2 | 3 |
| GluR6-PKBP(Ca)α | 2.16 ± 0.05 | 1.64 ± 0.26 | 55.9 ± 16.2 | 4 | 49.9 ± 2.8 | 1.47 ± 0.04 | 58.7 ± 10.8 | 4 |
| GluR6-PKBP(Ca)β | 2.9 ± 0.8 | 1.06 ± 0.1 | 11.03 ± 3.4 | 4 | 35.6 ± 2.3 | 1.26 ± 0.1 | 18.4 ± 5.5 | 4 |
| GluR6-PKBP(XI) | 2.25 ± 0.53 | 0.95 ± 0.04 | 7.9 ± 2.25 | 4 | 44.4 ± 7.5 | 1.1 ± 0.17 | 8.1 ± 1.12 | 3 |
Half-maximal efficient concentrations (EC50) for kainate (KA) and glutamate (Glu), Hill coefficients (nH), as well as maximal current amplitudes (Imax) are listed ± SEM; n, number of oocytes tested. PCS, Clone-engineered for easy pore cassette exchange (containing unique restriction sites flanking the pore; see Materials and Methods for details). GluR6-PKBP(Nn), GluR6 containing the pore domain of a KBP; “Nn” stands for the abbreviation of the Latin names of the species from which the KBP ion pore originated (Rp = Rana pipiens; Gd = Gallus domesticus; Ca = Carassius auratus, genes α and β; XI = Xenopus laevis).
We recorded dose–response curves for KA- and Glu-evoked currents of wild-type GluR6(Q), GluR6(Q)-PCS, and the five chimeras harboring KBP ion pores. The mutant GluR6(Q)-PCS, which carries the engineered sites for ion channel exchange but has the original ion channel domain of GluR6(Q), displays a small (1.7-fold) decrease in Glu affinity compared with wild-type GluR6(Q), whereas the affinity for KA is unchanged (Table 1). When we analyzed the chimeras we did not find significant differences in the EC50 values of KA-evoked currents compared with GluR6(Q)-PCS (Fig.5A,B), except for GluR6-PKBP(Gd), in which we measured a modest increase in the EC50 (approximately threefold) (Table 1). For Glu-evoked currents we observed no major changes in the EC50 values (Fig. 5C,D), with none of the EC50 deviating by more than a factor of 1.7 from the agonist affinity of GluR6(Q)-PCS (Table 1). Moreover, those chimeras deviating most from GluR6(Q)-PCS had EC50 values close to that of wild-type GluR6(Q). Therefore, construction of the chimeras had little effect on the affinities for any of the agonists and thus evidently had no major impact on the ligand binding site.
Fig. 5.

Comparison of dose–response curves of GluR6(Q)-PCS and chimeric GluR6 receptors harboring the ion pore domains of KBP(Ca)α (A, C), KBP(Ca)β (A, C), KBP(Rp) (B, D), KBP(Gd) (B, D), or KBP(Xl) (B, D). Note lack of significant changes in EC50 values with either KA (A, B) or Glu (C, D) as the agonist. Each datapoint represents the average of three to five oocytes ± SEM, as indicated. See Materials and Methods for details. For EC50 values and maximal currents, see Table1.
Channel properties of KBP-GluR6 chimeras
The wild-type GluR6(Q) is characterized by an inwardly rectifyingI–V relationship (Egebjerg and Heinemann, 1993), indicating that outward currents are blocked, presumably by endogenous compounds such as spermine (Bowie and Mayer, 1995). This block of outward current is determined mainly by the so-called Q/R-site, where either a glutamine (Q) or an arginine (R) residue is found to be located at the presumably most narrow position of the ion channel. The amino acid present at this site is determined by RNA editing (Seeburg, 1996). We recorded I–V curves of GluR6(Q)-PCS and the five chimeras (Fig. 6A,B) and determined that all of them had inwardly rectifying I–Vrelations. Thus, the ion channel of KBPs is susceptible to block of outward current just like the AMPA and KA receptor channels; however, the five chimeras, unlike GluR6(Q) and GluR6(Q)-PCS, did show a small but significant current at positive membrane potentials (Fig.6A,B). This small outward current is not attributable to the activation of calcium-dependent chloride channels, which are endogenous to the oocyte (and which have linear I–Vrelations), because they persist even in a buffer in which calcium has been replaced by magnesium (Fig. 6C, trace 2). We speculated that this outward current might be attributable to the fact that in all KBPs cloned so far a leucine is located at the site equivalent to the Q/R editing site of GluR6. We therefore mutated the glutamine at the Q/R-site of GluR6(Q) to a leucine residue and tested this mutant for outward current. As expected, GluR6(Q590L) had an inwardly rectifying I–V and indeed showed small but significant outward currents at positive holding potentials, just like the chimeras (Fig. 6C, trace 4). This outward current did not persist in calcium-free NFR (Fig.6C, trace 3), however, indicating that the leucine residue at the Q/R-site by itself cannot be the sole determinant of the outward current at positive membrane potentials. To investigate this further we constructed a mutant chimera, GluR6-PKBP(Rp)L583Q, which carries the KBP pore region transplanted into GluR6 but has a glutamine instead of a leucine residue at the Q/R site. This clone is functional, has a rectifying I–V curve, and shows distinct outward currents at positive membrane potentials (Fig. 6C, trace 5). Thus, determinants of the pore domain of KBPs other than the leucine at the Q/R site are responsible for the slight outward rectification observed on top of a basically inwardly rectifying I–V relation.
Fig. 6.
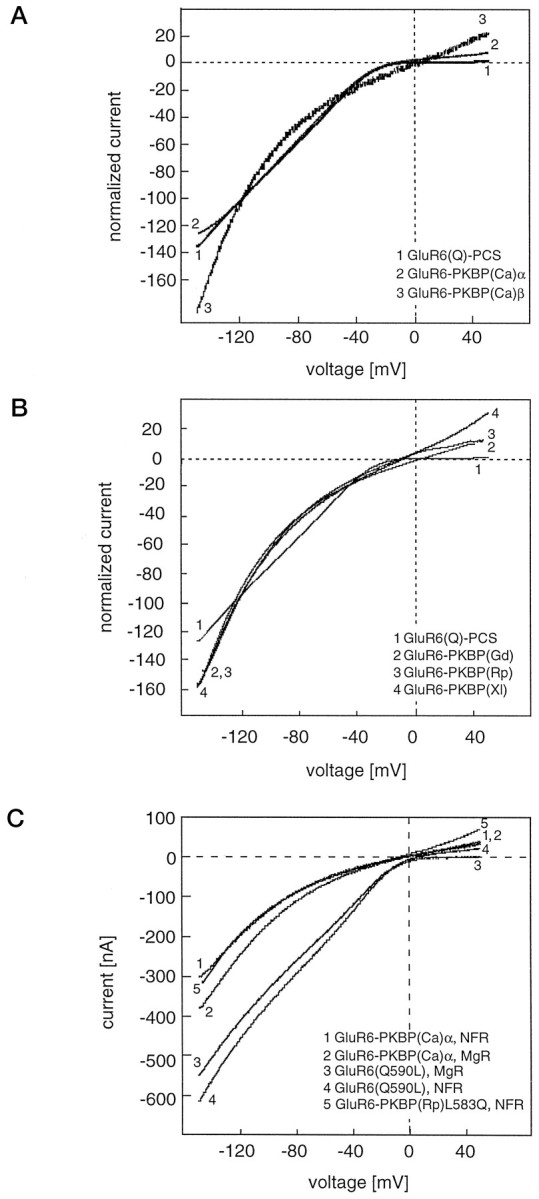
Comparison of I–V curves of GluR6(Q)-PCS and chimeric GluR6 receptors. Note that transplantation mutants harboring the ion pore domains of KBP(Ca)α (A), KBP(Ca)β (A), KBP(Rp) (B), KBP(Gd) (B), or KBP(Xl) (B) are all inwardly rectifying and not significantly different from GluR6(Q)-PCS. C, Comparison of I–V curves of GluR6-PKBP(Ca)α and GluR6(Q590L) in NFR and calcium-free, magnesium-substituted NFR (MgR), and I–V of the mutant chimera GluR6-PKBP(Rp)L583Q. Note that lack of outward rectification is observed only in GluR6(Q590L) in MgR. At least three oocytes were measured for each chimera and gave identical curves.
The Q/R-site not only determines the rectification properties of GluRs but also has a major impact on calcium permeability. AMPA receptors carrying an edited arginine residue at this position (such as GluR2) are virtually impermeable to calcium, whereas unedited subunits carrying a glutamine residue are calcium permeable (Hume et al., 1991). Similarly, for KA receptors, “Q” editing variants have a higher calcium permeability than “R” variants, although the latter are not entirely calcium impermeable (Egebjerg and Heinemann, 1993). Despite the large number of Q/R site mutants reported in the literature, no data are available on a GluR subunit carrying a leucine residue at this position. Consequently, it is difficult to predict whether the presence of a leucine residue at the Q/R site might allow calcium permeability of the ion channel. We therefore tested GluR6(Q)-PCS and GluR6-PKBP(Ca)α for calcium permeability using a sodium- and potassium-free modified NFR that contains Ca2+ as the sole cation capable of permeating the ion channel on agonist-mediated channel opening (Hollmann et al., 1991). For GluR6(Q)-PCS we found about the same permeability for Ca2+ as is seen in wild-type GluR6(Q). KA (300 μm) in 10 mm Ca-Ringer evoked ∼60% of the control current seen in NFR, and in 80 mm Ca-Ringer almost as much current can be recorded as in NFR (data not shown). GluR6-PKBP(Ca)α was selected for analysis of calcium permeability of KBP pores because this chimera yields the largest currents of the five chimeras. In 10 mm Ca-Ringer, GluR6-PKBP(Ca)α gave 5% of the current evoked by 300 μm KA in NFR, and in 80 mm Ca-Ringer it yielded 75% of the control current (Fig.7). Thus, the KBP(Ca)α ion channel domain is capable of fluxing Ca2+ ions to a considerable degree, although not quite to the extent of wild-type GluR6(Q) and GluR6(Q)-PCS. We also tested the pore of KBP(Xl) and found it to flux calcium to a similar degree (data not shown). Thus, the leucine residue at the Q/R site of the pore evidently is compatible with calcium permeating the ion channel. In keeping with these results, we determined that the mutant GluR6(Q590L), which contains a leucine residue at the Q/R site, is also permeable for calcium (data not shown).
Fig. 7.

The ion pore of KBP(Ca)α is permeable to calcium. KA-evoked currents were compared in normal frog Ringer’s solution (NFR, A) and Ca-Ringer containing either 80 mm (B) or 10 mm (C) calcium but no other cations. Note distinct currents in the absence of sodium and potassium (B, C). At least three oocytes were measured under each condition and gave identical results.
The KBP pore domains, just like the pores of GluR6, are not blocked by NMDA receptor channel blockers such as magnesium (Fig.6C, trace 2) or MK-801. We tested this for GluR6-PKBP(Rp), GluR6-PKBP(Ca)α, and GluR6-PKBP(Gd) (Fig.8A). The KBP pore domains, however, are blocked by zinc, which is a channel blocker at both NMDA and non-NMDA receptors (Westbrook and Mayer, 1987; Rassendren et al., 1990). We found that 100 μm zinc blocked 30% of KA-evoked (100 μm) currents at GluR6(Q) as well as GluR6-PKBP(Rp), GluR6-PKBP(Ca)α, and GluR6-PKBP(Ca)β. No block was seen with 1 μm zinc (data not shown). Interestingly, the KBP pore domains are not affected by philanthotoxin (PhTx), a wasp toxin (Fig. 8B), or by N-(4-hydroxyphenylpropanoyl)-spermine, a synthetic analog of wasp and spider toxins (data not shown). PhTx and spider toxins are known to efficiently block all NMDA receptors and all non-NMDA receptors with rectifying I–V relations (Blaschke et al., 1993;Herlitze et al., 1993; Washburn and Dingledine, 1996). We tested GluR6-PKBP(Rp), GluR6-PKBP(Ca)α, GluR6-PKBP(Ca)β, and GluR6-PKBP(Gd) and found no effect of the toxin or its synthetic analog. The toxin block had previously been shown to be determined by the amino acid located at the Q/R/N site, with “Q” and “N” versions being blocked and “R” versions not being blocked. Our data suggested that a leucine residue might also unexpectedly prevent channel block, althoug it does not carry the positive charge of an arginine residue. To verify this we tested the mutant GluR6(Q590L) for PhTx block and indeed found that the channel, which now has a leucine residue at the Q/R site, is no longer significantly affected by the toxin (Fig. 8B). To obtain further evidence we used our mutant GluR6-PKBP(Rp)L583Q, which tests the “reverse” situation in which the leucine has been converted into a glutamine. As expected, this mutant could now be blocked by PhTx (data not shown).
Fig. 8.
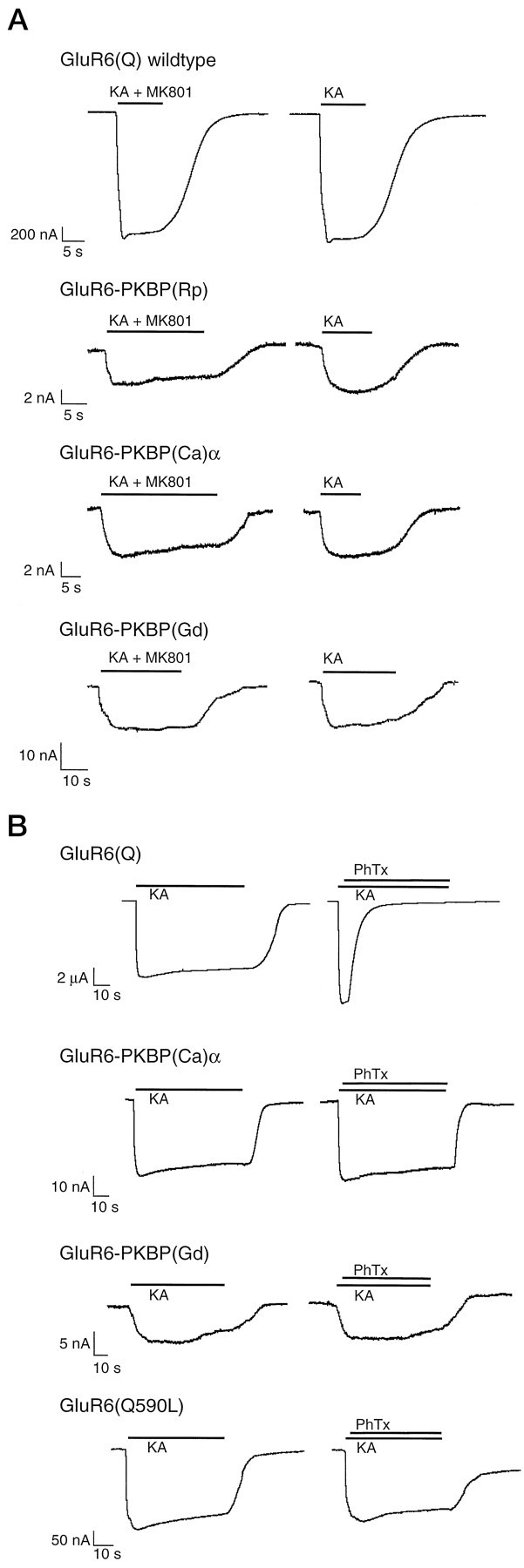
Effects of MK-801 and PhTx on KBP ion channel domains transplanted into GluR6. A, Absence of block by MK-801. MK-801 (1 μm) was coapplied with 100 μm KA for 20–30 sec. After a washout period, 100 μm KA was applied alone. Note lack of block of GluR6(Q) and three ion pore transplantation chimeras during coapplication of agonist and MK-801, and unaltered size of agonist-evoked response after washout. MK-801 (1 μm) did block NMDA receptors in control oocytes (data not shown). B, Absence of block by PhTx. KA (100 μm) was applied for 50–60 sec, followed by a brief wash and another application of 100 μm KA followed within 2 sec by additional application of the open channel blocker PhTx (0.5 μm). Note lack of block of two ion pore transplantation chimeras and mutant GluR6(Q590L), whereas GluR6(Q) is rapidly and efficiently blocked. Three to four oocytes were measured in each case and gave identical results.
Transplantation of functional ion pores into KBPs
We have demonstrated that the ion channel domains of KBPs are functional in the sequence background of both the KA receptor GluR6 and the AMPA receptor GluR1 but not in their own (KBP) sequence background. This suggested that most likely communication between the ligand binding site and the ion channel domain is disrupted in homomeric KBPs. If this is indeed the case, a functional channel domain such as that of GluR1 or GluR6 when inserted into a KBP should not be able to rescue function but rather should produce nonfunctional chimeric receptors. To test this we engineered the appropriate “reverse” chimeras, transplanting the ion channel domains of GluR6 and GluR1 into KBP(Rp). These chimeras when expressed in Xenopus oocytes did not produce any measurable currents on activation with the glutamatergic agonists Glu, KA, or Dom, although the chimeric proteins were clearly synthesized and inserted into the plasma membrane (Fig.4B). We also attempted to rescue function of the N-terminally elongated KBP construct GluR6N-KBP(Rp)C by transplanting the pore domain of GluR6 into this clone, creating the chimera GluR6-Rp-R6-Rp (Fig. 1, bottom bar graph). This construct was also nonfunctional, although it was expressed and transported to the plasma membrane (Fig. 4C).
Thus, even with a highly functional ion channel domain such as that of GluR6(Q), these receptor subunits failed to translate ligand binding into channel opening, supporting our conclusion that the gating mechanism rather than the ion pore is dysfunctional in homomeric KBPs.
DISCUSSION
N-terminal elongation does not rescue KBP function
N-terminal domains have been swapped between GluR6 and NMDAR1 GluR subunits without loss of function (Stern-Bach et al., 1994). Because even a remotely related subunit such as NMDAR1 (only 34.1% sequence identity with GluR6) evidently can serve as the donor of an N-terminal domain, it was not unreasonable to expect that the much more closely related KBP(Rp) (40.0% sequence identity) might be functionally rescued by a grafted N-terminal domain from GluR6; however, the N-terminal transplant did not render KBP(Rp) functional. The failure to produce a functional chimera indicates that the short N-terminal domain in KBPs is not likely the reason for lack of channel function.
Transplanted KBP ion pores are functional
The overall distribution of hydrophobic domains in KBPs is similar to that of functional GluRs (Fig. 1), and even the gene structure that has been elaborated for KBP(Gd) (Eshhar et al., 1992; Gregor et al., 1992) is quite similar. Furthermore, the topology that has been shown for KBP(Ca)α and KBP(Ca)β based on N-glycosylation studies (Wo and Oswald, 1994) is identical to that proposed for GluR1 (Hollmann et al., 1994), GluR3 (Bennett et al., 1995), and NMDAR1 (Wood et al., 1995). The obvious structural resemblance between KBPs and other GluRs suggested that domains from KBP subunits could potentially be exchanged for equivalent domains of functional GluRs, and vice versa. Indeed, when we inserted the hypothetical ion pore domains of KBPs in GluR1 or GluR6 we obtained functional chimeras, although maximal current amplitudes reached only 1–10% of the currents found in the parent clones. The domains transplanted consisted of the entire region between TMDs A and B (Fig. 2) rather than just the hydrophobic stretch that is thought to loop into the membrane to form the ion pore (Hollmann et al., 1994). This design was chosen because recent cysteine scanning mutagenesis data (Kuner et al., 1996) suggested that the sequences flanking the pore loop participate to some extent in establishing the structure of the pore.
Chimeras containing pores from KBPs had pharmacological characteristics and agonist affinities virtually indistinguishable from those of the ligand binding site donor subunit, demonstrating that the properties of the extracellular ligand binding site of GluRs are not dependent on the pore structure to which that site is connected. This conclusion is consistent with the work of Keinänen and colleagues, who reported construction of a functional soluble ligand binding site solely derived from the two extracellular halves of the ligand binding domain, without any pore structure in between (Kuusinen et al., 1995; Arvola and Keinänen, 1996).
KBP ion pores have distinct properties
The properties of the pore domains of the five known KBPs tested as domain transplants in GluR6 are very similar. This is not unexpected given the high amino acid sequence homology of these regions (56–76% among the five KBPs), and in particular the presence of a leucine residue in all five genes at a position that is homologous to the Q/R/N editing site in AMPA, KA, and NMDA receptors. All five pore domains have rectifying I–V values as might be predicted from the absence of an arginine residue at the Q/R/N editing site (Hume et al., 1991), and they all are presumably permeable to calcium, although we verified this only for GluR6-PKBP(Ca)α and GluR6-PKBP(Xl). The KBP pore domains are not blocked by NMDA receptor channel blockers, such as magnesium or MK-801, but are inhibited by zinc, which at high concentrations (>100 μm) does not discriminate between NMDA and non-NMDA receptors (Westbrook and Mayer, 1987; Rassendren et al., 1990). Interestingly, the KBP pore domains are not affected by the wasp toxin PhTx, other spider toxins, or their synthetic analogs, and we showed that this property is linked to the presence of a peculiar leucine residue at the Q/R/N site that is unique to KBPs. Thus, the properties of the KBP ion pore as reflected in the channel properties of the GluR6-KBP chimeras are distinct from those of AMPA, KA, and NMDA receptors, although they somewhat resemble properties of typical KA receptors.
KBPs are defective in gating
If all KBPs have functional agonist binding sites as has been shown previously (Henley, 1994), and if the pores of KBPs in principle are capable of conducting currents as our experiments showed, why then are these subunits nonfunctional as ligand-gated ion channels? Obviously, homomerically expressed KBP subunits fail to translate ligand binding into channel opening. Because ligand binding generally is assumed to cause a conformational change in the extracellular domain of the receptors (Mano et al., 1996; Laube et al., 1997), which presumably represents the gating step required to open the ion channel, it may be concluded that homomerically expressed KBPs either fail to generate the appropriate conformational change or fail to communicate it to the pore. This conclusion is backed by our observation that the functional pore domains of GluR6 or GluR1, when inserted into KBP(Rp), failed to generate functional ion channels.
KBPs likely lack an essential subunit
It seems unlikely that some secondary modification of the receptor protein such as phosphorylation, which has been shown for KBP(Rp) (Ortega and Teichberg, 1990; Ibarra and Ortega, 1995), or glycosylation is required to “switch on” the interrupted binding site-to-ion pore communication in KBPs. Such a mechanism most likely would have been detected in one of the various expression systems tried for the KBP clones, such as Xenopus oocytes, chinese hamster ovary cells, and human embryonic kidney cells (for review, see Henley, 1994). Rather, it seems likely that an additional subunit may be required that interacts with the KBPs to reestablish the connection between ligand binding site and ion channel. This modulatory subunit may be either an accessory protein or another subunit of the GluR family. Among GluRs, several cases of homomerically nonfunctional subunits have been observed that are rendered functional only on coassembly with another subunit. Examples are the NR2 subunits of the NMDA receptor subfamily (Monyer et al., 1992) or the KA2 subunit of the KA receptor subfamily (Herb et al., 1992). Recently it was reported that KBP(Xl) formed functional ion channels of a peculiar, novel pharmacology on coexpression with the NMDAR1 subunit of Xenopus laevis. None of the two subunits was functional by itself, but on coexpression formed channels that reportedly were activated by specific agonists for all pharmacological subclasses of GluRs, AMPA, KA, and NMDA, and KBP(Xl) was therefore dubbed “unitary” receptor (Soloviev et al., 1996). This report is in agreement with our finding that the pore of KBP(Xl) is functional. Other KBPs, however, have not been examined in coexpression experiments with NMDAR1 subunits from the same species, and our own attempts to coexpress KBPs with the rat NMDAR1 subunit in Xenopus oocytes did not yield functional receptors other than typical homomeric NMDA receptors (C. Villmann and M. Hollmann, unpublished data). Thus, although it is possible that KBPs from other species might also form functional receptors with NMDAR1 subunits, there is currently no evidence for this. KBP(Xl) differs from all other KBPs in binding both KA and AMPA with high affinity (Ishimaru et al., 1996). Therefore, it is possible that this subunit plays a quite different role and indeed has unique properties not shared by any of the other KBPs.
GluRs are modular proteins
Our data support the view that GluRs are modular proteins with structural features derived from a number of different precursor proteins (Seeburg, 1993; Wo and Oswald, 1995). In particular, the demonstration that pore regions can be transplanted between distantly related subunits without killing function strongly supports the hypothesis that GluRs indeed might have evolved from bacterial amino acid binding proteins by insertion of a pore domain in between the two ligand binding subdomains (O’Hara et al., 1993; Kuryatov et al., 1994;Stern-Bach et al., 1994).
In summary, we conclude that KBPs in addition to harboring perfectly functional ligand binding sites also have intrinsically functional ion channel domains that display inward rectification, allow significant calcium permeability, and have distinct pharmacological properties. Our findings suggest that the elusive physiological role of KBPs may indeed be that of Glu-activated ion channels. We speculate that one or more additional subunits or an accessory protein may be required to twist the subunits in a heteromeric receptor complex just enough to enable the occupied ligand binding site to successfully gate the ion channel. Furthermore, the chimera construction approach presented in this study should prove useful in analyzing the mechanism of channel gating in GluRs and defining the molecular pathway involved in signal transduction from the agonist binding site to the ion pore.
Footnotes
This work was supported by a Deutsche Forschungsgemeinschaft Heisenberg fellowship to M.H., the Starke-Werner Foundation (L.B.), and a PhD fellowship of the Max-Planck-Society to C.V. We thank Drs. Keiji Wada and Robert Wenthold (National Institute on Deafness and Other Communication Disorders-National Institutes of Health, Bethesda, MD) for the Rana pipiens KBP cDNA clone [KBP(Rp)], Dr. Robert Oswald (Cornell University, Ithaca, NY) for the GFKARα and GFKARβ clones [KBP(Ca)α and KBP(Ca)β], and Dr. Vivian Teichberg (Weizmann Institute, Rehovot, Israel) for the chicken KBP cDNA [KBP(Gd)]. Dr. Robert Wenthold generously donated the affinity-purified anti-GluR6 and anti-KBP(Rp) antisera used in this study, and Emily Liman and the late Peter Hess (Harvard Medical School, Boston, MA) provided the pGEMHE vector.
Correspondence should be addressed to Dr. Michael Hollmann, Glutamate Receptor Laboratory, Max-Planck-Institute for Experimental Medicine, Hermann-Rein-Strasse 3, D-37075 Göttingen, Germany.
REFERENCES
- 1.Arvola M, Keinänen K. Characterization of the ligand binding domains of glutamate receptor (GluR)-B and GluR-D subunits expressed in Escherichia coli as periplasmic proteins. J Biol Chem. 1996;271:15527–15532. doi: 10.1074/jbc.271.26.15527. [DOI] [PubMed] [Google Scholar]
- 2.Bennett JA, Escobar W, Dingledine R. Topological profile of GluR3: three transmembrane domains and a reentrant channel-lining domain. FASEB J. 1995;9:A390–390. doi: 10.1016/0896-6273(95)90293-7. [DOI] [PubMed] [Google Scholar]
- 3.Blaschke M, Keller BU, Rivosecchi R, Hollmann M, Heinemann S, Konnerth A. A single amino acid determines the subunit specific spider toxin block of alpha-amino-3-hydroxy-5-methylisoxazole-4-propionate/kainate receptor channels. Proc Natl Acad Sci USA. 1993;90:6528–6532. doi: 10.1073/pnas.90.14.6528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bowie D, Mayer ML. Inward rectification of both AMPA and kainate subtype glutamate receptors generated by polyamine-mediated ion channel block. Neuron. 1995;15:453–462. doi: 10.1016/0896-6273(95)90049-7. [DOI] [PubMed] [Google Scholar]
- 5.Devereux J, Haeberli P, Smithies O. A comprehensive set of sequence analysis programs for the VAX. Nucleic Acids Res. 1984;12:387–395. doi: 10.1093/nar/12.1part1.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Egebjerg J, Heinemann SF. Ca2+ permeability of unedited and edited versions of the kainate selective glutamate receptor GluR6. Proc Natl Acad Sci USA. 1993;90:755–759. doi: 10.1073/pnas.90.2.755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Egebjerg J, Bettler B, Hermans-Borgmeyer I, Heinemann S. Cloning of a cDNA for a glutamate receptor subunit activated by kainate but not AMPA. Nature. 1991;351:745–748. doi: 10.1038/351745a0. [DOI] [PubMed] [Google Scholar]
- 8.Eshhar N, Hunter C, Wenthold RJ, Wada K. Structural characterization and expression of a brain specific gene encoding chick kainate binding protein. FEBS Lett. 1992;297:257–262. doi: 10.1016/0014-5793(92)80551-q. [DOI] [PubMed] [Google Scholar]
- 9.Gregor P, Mano I, Maoz I, McKeown M, Teichberg VI. Molecular structure of the chick cerebellar kainate binding subunit of a putative glutamate receptor. Nature. 1989;342:689–692. doi: 10.1038/342689a0. [DOI] [PubMed] [Google Scholar]
- 10.Gregor P, Yang XD, Mano I, Takemura M, Teichberg VI, Uhl GR. Organization and expression of the gene encoding chick kainate binding protein, a member of the glutamate receptor family. Mol Brain Res. 1992;16:179–186. doi: 10.1016/0169-328x(92)90223-x. [DOI] [PubMed] [Google Scholar]
- 11.Henley JM. Kainate binding proteins: phylogeny, structures and possible functions. Trends Pharmacol Sci. 1994;15:182–190. doi: 10.1016/0165-6147(94)90146-5. [DOI] [PubMed] [Google Scholar]
- 12.Herb A, Burnashev N, Werner P, Sakmann B, Wisden W, Seeburg PH. The KA-2 subunit of excitatory amino acid receptors shows widespread expression in brain and forms ion channels with distantly related subunits. Neuron. 1992;8:775–785. doi: 10.1016/0896-6273(92)90098-x. [DOI] [PubMed] [Google Scholar]
- 13.Herlitze S, Raditsch M, Ruppersberg JP, Jahn W, Monyer H, Schoepfer R, Witzemann V. Argiotoxin detects molecular differences in AMPA receptor channels. Neuron. 1993;10:1131–1140. doi: 10.1016/0896-6273(93)90061-u. [DOI] [PubMed] [Google Scholar]
- 14.Hollmann M. The topology of glutamate receptors: sorting through the domains. In: Monaghan DT, Wenthold R, editors. The ionotropic glutamate receptors. Humana; Totowa, NJ: 1996. pp. 39–79. [Google Scholar]
- 15.Hollmann M, Heinemann S. Cloned glutamate receptors. Annu Rev Neurosci. 1994;17:31–108. doi: 10.1146/annurev.ne.17.030194.000335. [DOI] [PubMed] [Google Scholar]
- 16.Hollmann M, Hartley M, Heinemann S. Calcium permeability of KA-AMPA gated glutamate receptor channels depends on subunit composition. Science. 1991;252:851–853. doi: 10.1126/science.1709304. [DOI] [PubMed] [Google Scholar]
- 17.Hollmann M, Maron C, Heinemann S. N-glycosylation site tagging suggests a three transmembrane domain topology for the glutamate receptor GluR1. Neuron. 1994;13:1331–1343. doi: 10.1016/0896-6273(94)90419-7. [DOI] [PubMed] [Google Scholar]
- 18.Hume RI, Dingledine R, Heinemann SF. Identification of a site in glutamate receptor subunits that controls calcium permeability. Science. 1991;253:1028–1031. doi: 10.1126/science.1653450. [DOI] [PubMed] [Google Scholar]
- 19.Ibarra C, Ortega A. Interaction of guanine-nucleotides with the kainate binding protein from chick cerebellum. NeuroReport. 1995;6:1149–1152. doi: 10.1097/00001756-199505300-00019. [DOI] [PubMed] [Google Scholar]
- 20.Ishimaru H, Kamboj R, Ambrosini A, Henley JM, Soloviev MM, Sudan H, Rossier J, Abutidze K, Rampersad V, Usherwood PNR, Bateson AN, Barnard EA. A unitary non-NMDA receptor short subunit from Xenopus: DNA cloning and expression. Receptors Channels. 1996;4:31–49. [PubMed] [Google Scholar]
- 21.Kuner T, Wollmuth LP, Karlin A, Seeburg PH, Sakmann B. Structure of the NMDA receptor channel M2 segment inferred from the accessibility of substituted cysteines. Neuron. 1996;17:343–352. doi: 10.1016/s0896-6273(00)80165-8. [DOI] [PubMed] [Google Scholar]
- 22.Kuryatov A, Laube B, Betz H, Kuhse J. Mutational analysis of the glycine binding site of the NMDA receptor: structural similarity with bacterial amino acid binding proteins. Neuron. 1994;12:1291–1300. doi: 10.1016/0896-6273(94)90445-6. [DOI] [PubMed] [Google Scholar]
- 23.Kuusinen A, Arvola M, Keinänen K. Molecular dissection of the agonist binding site of an AMPA receptor. EMBO J. 1995;14:6327–6332. doi: 10.1002/j.1460-2075.1995.tb00323.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 25.Laube B, Hirai H, Sturgess M, Betz H, Kuhse J. Molecular determinants of agonist discrimination by NMDA receptor subunits: analysis of the glutamate binding site on the NR2B subunit. Neuron. 1997;18:493–503. doi: 10.1016/s0896-6273(00)81249-0. [DOI] [PubMed] [Google Scholar]
- 26.Mano I, Lamed Y, Teichberg VI. A venus flytrap mechanism for activation and desensitization of α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptors. J Biol Chem. 1996;271:15299–15302. doi: 10.1074/jbc.271.26.15299. [DOI] [PubMed] [Google Scholar]
- 27.Monyer H, Sprengel R, Schoepfer R, Herb A, Higuchi M, Lomeli H, Burnashev N, Sakmann B, Seeburg PH. Heteromeric NMDA receptors: molecular and functional distinction of subtypes. Science. 1992;256:1217–1221. doi: 10.1126/science.256.5060.1217. [DOI] [PubMed] [Google Scholar]
- 28.O’Hara PJ, Sheppard PO, Thogersen H, Venezia D, Haldeman BA, McGrane V, Houamed KM, Thomsen C, Gilbert TL, Mulvihill ER. The ligand binding domain in metabotropic glutamate receptors is related to bacterial periplasmic binding proteins. Neuron. 1993;11:41–52. doi: 10.1016/0896-6273(93)90269-w. [DOI] [PubMed] [Google Scholar]
- 29.Ortega A, Teichberg VI. Phosphorylation of the 49 kDa putative subunit of the chick cerebellar kainate receptor and its regulation by kainatergic ligands. J Biol Chem. 1990;265:21404–21406. [PubMed] [Google Scholar]
- 30.Partin KM, Patneau DK, Winters CA, Mayer ML, Buonanno A. Selective modulation of desensitization at AMPA versus kainate receptors by cyclothiazide and concanavalin A. Neuron. 1993;11:1069–1082. doi: 10.1016/0896-6273(93)90220-l. [DOI] [PubMed] [Google Scholar]
- 31.Rassendren F, Lory P, Pin J-P, Nargeot J. Zinc has opposite effects on NMDA and non-NMDA receptors expressed in Xenopus oocytes. Neuron. 1990;4:733–740. doi: 10.1016/0896-6273(90)90199-p. [DOI] [PubMed] [Google Scholar]
- 32.Sanger F, Nicklen S, Coulson AR. DNA sequencing with chain termination inhibitors. Proc Natl Acad Sci USA. 1977;74:5463–5467. doi: 10.1073/pnas.74.12.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Seeburg PH. The TIPS/Trends Neurosci lecture: the molecular biology of mammalian glutamate receptor channels. Trends Pharmacol Sci. 1993;14:297–303. doi: 10.1016/0165-6147(93)90047-N. [DOI] [PubMed] [Google Scholar]
- 34.Seeburg PH. The role of RNA editing in controlling glutamate receptor channel properties. J Neurochem. 1996;66:1–5. doi: 10.1046/j.1471-4159.1996.66010001.x. [DOI] [PubMed] [Google Scholar]
- 35.Seeburg PH, Burnashev N, Köhr G, Kuner T, Sprengel R, Monyer H. The NMDA receptor channel: molecular design of a coincidence detector. Recent Prog Horm Res. 1995;50:19–34. doi: 10.1016/b978-0-12-571150-0.50006-8. [DOI] [PubMed] [Google Scholar]
- 36.Soloviev MM, Brierley MJ, Shao ZY, Mellor IR, Volkova TM, Kamboj R, Ishimaru H, Sudan H, Harris J, Foldes RL, Grishin EV, Usherwood PNR, Barnard EA. Functional expression of a recombinant unitary glutamate receptor from Xenopus, which contains N-methyl-D-aspartate (NMDA) and non-NMDA receptor subunits. J Biol Chem. 1996;271:32572–32579. doi: 10.1074/jbc.271.51.32572. [DOI] [PubMed] [Google Scholar]
- 37.Stern-Bach Y, Bettler B, Hartley M, Sheppard PO, O’Hara PJ, Heinemann SF. Agonist selectivity of glutamate receptors is specified by 2 domains structurally related to bacterial amino acid binding proteins. Neuron. 1994;13:1345–1357. doi: 10.1016/0896-6273(94)90420-0. [DOI] [PubMed] [Google Scholar]
- 38.Towbin H, Stähelin T, Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci USA. 1979;76:4350–4354. doi: 10.1073/pnas.76.9.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wada K, Dechesne CJ, Shimasaki S, King RG, Kusano K, Buonanno A, Hampson DR, Banner C, Wenthold RJ, Nakatani Y. Sequence and expression of a frog brain complementary DNA encoding a kainate-binding protein. Nature. 1989;342:684–689. doi: 10.1038/342684a0. [DOI] [PubMed] [Google Scholar]
- 40.Washburn MS, Dingledine R. Block of alpha-amino-3-hydroxy-5-methyl-4-isoazole propionic acid (AMPA) receptors by polyamines and polyamine toxins. J Pharmacol Exp Ther. 1996;278:669–678. [PubMed] [Google Scholar]
- 41.Wenthold RJ, Trumpy VA, Zhu WS, Petralia RS. Biochemical and assembly properties of GluR6 and KA2, 2 members of the kainate receptor family, determined with subunit specific antibodies. J Biol Chem. 1994;269:1332–1339. [PubMed] [Google Scholar]
- 42.Westbrook GL, Mayer ML. Micromolar concentrations of Zn++ antagonize NMDA and GABA responses in hippocampal neurons. Nature. 1987;328:640–643. doi: 10.1038/328640a0. [DOI] [PubMed] [Google Scholar]
- 43.Wo ZG, Oswald RE. Transmembrane topology of two kainate receptor subunits revealed by N-glycosylation. Proc Natl Acad Sci USA. 1994;91:7154–7158. doi: 10.1073/pnas.91.15.7154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wo ZG, Oswald RE. Unraveling the modular design of glutamate-gated ion channels. Trends Neurosci. 1995;18:161–168. doi: 10.1016/0166-2236(95)93895-5. [DOI] [PubMed] [Google Scholar]
- 45.Wood MW, VanDongen HMA, VanDongen AMJ. Structural conservation of ion conduction pathways in K channels and glutamate receptors. Proc Natl Acad Sci USA. 1995;92:4882–4886. doi: 10.1073/pnas.92.11.4882. [DOI] [PMC free article] [PubMed] [Google Scholar]