

Abstract
The emergence of multidrug-resistant Gram-negative bacteria, including carbapenem-resistant Enterobacteriaceae, is a major health problem that necessitates the development of new antibiotics. Vancomycin inhibits cell-wall synthesis in Gram-positive bacteria, but is generally ineffective against Gram-negative bacteria and unable to penetrate the outer membrane barrier. In an effort to determine whether vancomycin and other antibiotics effective against Gram-positive bacteria could, through modification, be rendered effective against Gram-negative bacteria, we discovered that covalent attachment of a single arginine to vancomycin yielded conjugates with order-of-magnitude improvements in activity against Gram-negative bacteria, including pathogenic E. coli. The vancomycin-arginine conjugate (V-R) exhibited efficacy against actively-growing bacteria, induced loss of rod cellular morphology, and resulted in intracellular accumulation of peptidoglycan precursors, all consistent with cell-wall synthesis disruption as its mechanism of action. Membrane permeabilization studies demonstrated enhanced outer membrane permeability of V-R as compared to vancomycin. The conjugate exhibited no mammalian cell toxicity or hemolytic activity in MTT and hemolysis assays. Our study introduces a new vancomycin derivative effective against Gram-negative bacteria and underscores the broader potential of generating new antibiotics through combined mode-of-action and synthesis-informed design studies.
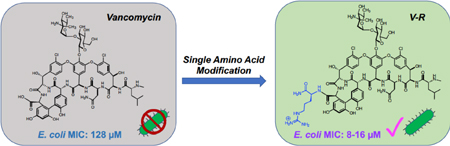
GRAPHICAL ABSTRACT
INTRODUCTION
Treatment of multidrug-resistant Gram-negative bacteria, including carbapenem-resistant Enterobacteriaceae (CRE), presents an enormous clinical challenge due to the dearth of antibiotics efficacious against these pathogens.1 It is estimated that CRE contributes to death in 30–50% of infected patients, emphasizing the urgent need for new strategies and agents to treat antibiotic-resistant Gram-negative bacteria.2,3 A key challenge in treating Gram-negative bacteria is their unique outer membrane and cell-surface modifications which serve as a barrier of entry to certain antibiotics. The glycopeptide antibiotics, including vancomycin as the first member of this class to be discovered, are effective clinically only against Gram-positive bacteria.4 Gram-positive bacteria surround themselves with a thick cell wall composed of peptidoglycan, as the major structural component, plus teichoic acids and cell wall associated proteins. Vancomycin binds to D-Ala-D-Ala termini of peptidoglycan precursors at the cell surface and cell division septa in Grampositive bacteria, sequestering Lipid II and preventing bacterial cell-wall assembly and thus inhibits cell growth and division.5, 6 Gram-negative bacteria produce a much thinner layer of peptidoglycan, which is surrounded by a unique outer membrane associated with lipopolysaccharide, proteins, and often other proteinaceous and carbohydrate components that provide protection from detergents and chemical assault.7 Thus, D-Ala-D-Ala binding sites for vancomycin are also present in E. coli and other Gram-negative pathogens, but are located in the periplasm and consequently inaccessible to vancomycin due to its inability to breach the outer membrane barrier.8
A promising approach to generate new treatment options for Gram-negative pathogens is to chemically modify antibiotics such as vancomycin to enable their translocation across the Gramnegative outer membrane to access intracellular targets. A classic example of this approach is the addition of an amine group to penicillin (generally effective only against Gram-positive organisms) to generate ampicillin, which can breach the Gram-negative outer membrane and inhibit peptidoglycan synthesis in the periplasm.9 This strategy has also been creatively implemented recently in notable efforts for arylomycin A-C16 and deoxynybomycin, where both compounds were chemically modified to improve antibiotic uptake.10, 11 In each of the modified compounds, one or more primary amine substitutions were incorporated, based on the hypothesis that non-sterically encumbered primary amines, along with molecular rigidity (≤5 rotatable bonds) and low globularity, are important factors in dictating compound entry in Gram-negative bacteria.12 These same principles do not appear to have been applied to chemically modify vancomycin for entry across the outer membrane. Efforts that have been reported involving other entry strategies include the incorporation of vancomycin in nanoparticles,13 nanotransformation of vancomycin into nanospheres using sonochemistry,14 and incorporation of a lipophilic alkyl chain terminated with a quaternary ammonium element,15 and improved efficacy against Gram-negative bacteria via outer and inner membrane perturbation.
We explored a new strategy to confer vancomycin with cell killing activity in Gram-negative bacteria through covalent conjugation of a molecular transporter to enable delivery across the cell envelope. We hypothesized that conjugation of vancomycin to a cell-penetrating, guanidinium-rich molecular transporter (GR-MoTr), D-octaarginine (r8), would render it effective against Gramnegative bacteria, enabling delivery to access periplasmic cell-wall precursors and arrest cell-wall synthesis. This hypothesis was based on the early observation that HIV-1 Tat, unlike most proteins, readily enters cells, a function we attributed to its arginine-rich domain (RKKRRQRRR).16–18 Through an extensive reverse engineering effort of this Tat-9-mer, we found that the transport process is driven by the number and spatial array of guanidinium groups and their initial electrostatic association with negatively charged cell surface carboxylic acids, sulfates and phosphates.19, 20 Our groups more recently discovered and reported the superior efficacy of a vancomycin D-octaarginine conjugate, V-r8, relative to vancomycin alone, in treating Grampositive bacteria, including clinically important MRSA biofilms and persister cells.21 However, the same improvement in efficacy was not observed for V-r8 in uropathogenic E. coli relative to r8 alone or a 1:1 mixture of r8+V, prompting our further evaluation of the use of GR-MoTrs for Gramnegative antibiotic delivery. Here we report on the design, synthesis, and evaluation of a panel of vancomycin-amino acid analogues, for which we discovered that addition of a single arginine residue significantly improved efficacy against Gram-negative bacteria.
RESULTS AND DISCUSSION
A vancomycin-octa-arginine conjugate, V-r8, was recently designed, synthesized and shown to exhibit extraordinary activity in killing Gram-positive bacteria, notably MRSA and vancomycin-resistant E. faecium.21 The conjugate was potent in killing S. aureus persister cells and biofilm-associated cells, thus exhibiting unique killing activity not exhibited by vancomycin itself. Here, we examined the efficacy of vancomycin conjugates against Gram-negative bacteria. V-r8 did not exhibit notable killing activity against a panel of Gram-negative bacteria, thus we designed, prepared and evaluated new vancomycin analogues and tested these first against a reference Gram-negative strain, E. coli 25922 (Figure 1). Vancomycin conjugates with 8- and 4-arginine sequences were ineffective (Table SI-1). Remarkably however, we discovered that a 2-arginine conjugate, V-RR, was more efficacious than vancomycin in E. coli 25922 (Figure 1), and a single arginine conjugate, V-R, was the most efficacious, exhibiting superior activity over vancomycin against E. coli 25922 and other Gram-negative organisms, including V. cholerae, A. baumannii, and P. aeruginosa and comparable activity against Gram-positive S. aureus (Table 1). MIC values for V-R treatment of E. coli strains were 8–16 μM, whereas the MIC for vancomycin is 128 μM. Notably, V-R was effective against the carbapenem-resistant E. coli BAA-2469 (Table 1). Covalent conjugation of vancomycin and arginine was essential for activity, as arginine did not exhibit antibacterial activity on its own (MIC > 64 μM) and a 1:1 mixture of vancomycin and arginine did not exhibit MIC synergism (MIC >64 μM, Table SI-1). Thus, in striking contrast to V-r8 and V-r4, we observed efficacy of V-R against Gram-negative bacteria. The efficacy of V-R and vancomycin were similar against Gram-positive bacteria.
Figure 1.

Preparation and antibacterial activity of vancomycin-amino acid conjugates. R corresponds to each amino acid amide connected to vancomycin (left). An MIC activity screen was performed in E. coli 25922 (right), where V-O, V-K, and V-D exhibited MICs of 32 μM or higher and were not further examined in subsequent experiments. Data for V-R, V-K, V-RR, and vancomycin represents median MICs obtained from 2–3 independent experiments, with ranges provided in parentheses.
Table 1.
Minimum Inhibitory Concentrations (MICs, μM)a of V and V-R
| Strain | V | V-R |
|---|---|---|
| E. coli 25922 | 128 | 8 (8–16) |
| E. coli UTI89 | 128 | 12 (8–16) |
| E. coli BAA-2469 (CR) | 128 (64–128) | 16 (8–16) |
| V. cholerae El Tor A1552R | 128 | 12 (8–16) |
| A. baumannii 19606 | 96 (64–128) | 32 |
| P. aeruginosa PA14 | 512 | 64 |
| S. aureus 29213 | 0.5 | 0.5 (0.25–0.5) |
Median MICs from 2–3 independent experiments, with ranges provided in parentheses.
The discovery of the antibacterial activity of V-R against Gram-negative organisms inspired our synthesis of a panel of amino acid analogues to investigate the role of the amino acid in contributing to antibiotic activity. The general scheme for V-R synthesis was implemented to generate the other analogues and enabled rapid, step economical and scalable synthesis of vancomycin conjugates (Figure 1). This approach enabled us to chemically tune and explore the possible contributions of charge and hydrogen bonding conferred by the amino acid addition, for example, as it could relate to enhanced cellular uptake. Indeed, added positive charge on an antibiotic could promote electrostatic association with negatively charged phosphate-containing lipopolysaccharides (LPS) at the Gram-negative outer membrane.19 Too few charges would allow only weak association and too many could deter membrane release. In general, we observed that conjugates with positively charged amino acids, specifically V-R and a vancomycin-lysine conjugate (V-K) were the most effective against a reference E. coli strain, 25922 and pathogenic isolates (Table SI-2). However, a V-threonine (V-T) conjugate yielded the same efficacy as the V-ornithine (V-O) conjugate, suggesting that properties other than amino acid charge, such as hydrogen bonding and steric effects, also appear to influence antibiotic activity. A vancomycin- aspartic acid (V-D) compound exhibited the least efficacy (Figure 1). These results demonstrate the sensitivity of presenting the amino acid at the C-terminus of vancomycin, where it appears to be positioned to contribute to hydrogen bonding and/or electrostatic interactions and impacts a conjugate’s efficacy against Gram-negative bacteria. Overall, V-R exhibited superior activity among the conjugates and was selected for further evaluation and mechanism-of-action studies.
We next designed a series of experiments to evaluate the mechanism of action of V-R and to test whether killing activity was consistent with enhanced vancomycin entry into cells and inhibition of cell-wall synthesis. The kinetics of cell killing activity were first considered through the determination of bacterial viability by enumeration of CFU/mL immediately after introduction of V-R to bacteria and again after 0.5, 2 and 4 hours. V-R killing activity was growth dependent and only exhibited efficacy once bacteria were given time to grow in the presence of compound (Figure 2A). Furthermore, vancomycin, at a matched 2X MIC threshold (256 μM vancomycin), yielded similar time-kill kinetics as observed for treatment with only 16 μM V-R in nutrient medium, indicating that the two compounds exhibit similar growth-dependent activity (Figure 2A).
Figure 2.

V-R targets actively growing E. coli and inhibits cell-wall synthesis. A) Time-kill kinetic analysis of V and V-R-treated UTI89 in Mueller-Hinton broth. Treatment concentrations were 16 μM for V-R and 256 μM for V. B) Both V and V-R treatment result in alteration of E. coli 25922 morphology, inducing rounding of bacterial cells (scale bar: 10 μm). Treatment concentrations were 16 μM for V-R and 256 μM for V. C & D) V-R treatment results in accumulation of UDP-MurNAc peptide in UTI89 as identified by HPLC and mass spectrometry. In C, treatment concentrations were 16 μM for V-R and 16 μM for V.
Interruption of peptidoglycan synthesis in E. coli impacts cell size and shape and vancomycin has been demonstrated to result in loss of rod morphology and blebbing in susceptible E. coli mutants and wild-type E. coli cells subjected to cold temperatures.22, 23 Thus, we next sought to compare the impact of vancomycin and V-R on cell morphology. Both vancomycin and V-R, at matched inhibitory concentrations (256 μM and 16 μM, respectively), induced rounding and blebbing of E. coli cells (Figure 2B), consistent with a mechanism of cell-wall synthesis inhibition for both compounds. To further investigate cell-wall synthesis inhibition on a molecular level, we performed HPLC-MS analysis of cells treated with vancomycin and V-R. HPLC-MS has previously been used to analyze accumulation of cell wall precursors upon treatment with vancomycin, wherein vancomycin treatment results in accumulation of UDP-MurNAc pentapeptide, a precursor that normally is incorporated into peptidoglycan during cell-wall synthesis.24–26 Upon treatment of E. coli with an equivalent concentration of vancomycin and V-R, we observed that both compounds resulted in accumulation of UDP-MurNAc pentapeptide relative to untreated control cells, and V- R resulted in increased accumulation of UDP-MurNAc pentapeptide relative to vancomycin (Figure 2C and 2D). Thus, V-R activity exhibits the anticipated hallmarks of cell-wall synthesis inhibition like vancomycin, yet V-R is significantly more effective, eradicating E. coli at 16-fold lower concentrations. We hypothesized that the improvement in activity was due to enhanced uptake of V-R relative to vancomycin, and we investigated one possible manifestation of this activity using membrane permeabilization assays.
Outer membrane permeabilization studies were performed with the probe 1-N-phenylnapthylamine (NPN), which exhibits fluorescence upon entry into permeabilized outer membranes27 Relative to vancomycin, V-R treatment resulted in modest 1-N-phenylnapthylamine fluorescence, indicative of modest perturbation to outer membrane integrity, but was not as potent as observed for a pore-forming peptide, polymyxin B (Figure 3). We also investigated inner membrane permeabilization activity of V-R using the probe propidium iodide, which exhibits fluorescence upon binding to cytoplasmic DNA.28 Neither vancomycin nor V-R exhibited inner membrane permeabilization activity relative to untreated cells. These results suggest that relative to vancomycin, V-R is able to more effectively breach the outer membrane and access its periplasmic target. Furthermore, the uptake of NPN upon V-R treatment is consistent with antibiotics that feature self-promoted cellular uptake, with molecular determinants to facilitate cellular association and eventual entry.29 Antibiotics that exhibit self-promoted uptake are typically inhibited by supplementation with Mg2+, where Mg2+ cations bridge neighboring LPS phosphate groups and can be competitively displaced by cationic antibiotics, for example, promoting enhanced cellular association.30 We found that supplementation of 5 mM Mg2+ in the growth medium reduced V-R efficacy and increased the MIC of V-R from 8 to 64 μM in E. coli. An increase in MIC for vancomycin is also observed, and vancomycin is positively charged near neutral pH. For comparison and as anticipated, Mg2+ supplementation does not impact the MIC of chloramphenicol, which enters E. coli via porins (Table SI-3).29 Finally, we sought to determine whether V-R would exhibit any obvious mammalian cell toxicity using MTT and hemolysis assays. Superficial bladder epithelial cells (HTB-9) were employed in the MTT assay towards the consideration of testing V-R in murine urinary tract infection models where agents can be delivered locally through transurethral introduction. Results with V-R were similar to those for vancomycin and exhibited no mammalian cell toxicity or hemolytic activity (Figure SI-1).
Figure 3.

Impacts of V-R on outer and inner membrane integrity in E. coli. A) Outer membrane permeabilization assay in UTI89 using the fluorescent probe, NPN. B) Outer and inner membrane permeabilization assay in UTI89, where propidium iodide was used as probe of membrane perturbation. In A and B, treatment concentrations were performed at 4X MIC (4 μM for polymyxin B, 512 μM for V and 64 μM for V-R).
In summary, vancomycin is one of the most widely studied and derivatized antibiotics in use today, directed to the treatment of Gram-positive infections. We previously reported that a conjugate of vancomycin and the cell penetrating peptide octaarginine exhibits superior activity relative to vancomycin against Gram-positive bacteria. Here we report on our design and discovery of a new vancomycin derivative with a single arginine modification that results in a compound with order of magnitude improved activity against Gram-negative E. coli. The arginine modification enables retainment of the mechanism of action of the parent antibiotic vancomycin in inhibiting peptidoglycan synthesis, but confers the antibiotic with enhanced activity. Our results suggest that V-R follows a self-promoted uptake mechanism, permitting entry in the E. coli periplasm and effective inhibition of cell-wall synthesis. Thus, V-R can now be used as a tool to conveniently inhibit cell-wall assembly and further study vancomycin-associated activity in Gram-negative bacteria. Conjugate synthesis is facile and readily amenable to other vancomycin derivatives. Our study highlights a simple chemical modification to vancomycin that generated an antibiotic active against critical carbapenem-resistant bacteria, furthering the development of new antibiotic strategies for resistant Gram-negative pathogens and inspiring the design of new antibiotic conjugates even for parent compounds that may seem to have been exhaustively studied.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the National Institutes of Health Grants R01GM117278 (L.C.) and NIH-CA031845 (P.A.W.). Support of this work through a fellowship from the Stanford Center for Molecular Analysis and Design (A.S.A.) is acknowledged. We acknowledge the Stanford University Mass Spectrometry Facility for instrumentation assistance.
Footnotes
SUPPORTING INFORMATION
The Supporting Information is available free of charge on the ACS Publications website.
• Synthetic procedures and characterization data and spectra
• Biological procedures
REFERENCES
- [1].Cerceo E, Deitelzweig SB, Sherman BM, and Amin AN (2016) Multidrug-Resistant Gram-Negative Bacterial Infections in the Hospital Setting: Overview, Implications for Clinical Practice, and Emerging Treatment Options, Microb Drug Resist 22, 412–431. [DOI] [PubMed] [Google Scholar]
- [2].Satlin MJ, Chen L, Patel G, Gomez-Simmonds A, Weston G, Kim AC, Seo SK, Rosenthal ME, Sperber SJ, Jenkins SG, Hamula CL, Uhlemann AC, Levi MH, Fries BC, Tang YW, Juretschko S, Rojtman AD, Hong T, Mathema B, Jacobs MR, Walsh TJ, Bonomo RA, and Kreiswirth BN (2017) Multicenter Clinical and Molecular Epidemiological Analysis of Bacteremia Due to Carbapenem-Resistant Enterobacteriaceae (CRE) in the CRE Epicenter of the United States, Antimicrob Agents Chemother 61, e02349–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].McConville TH, Sullivan SB, Gomez-Simmonds A, Whittier S, and Uhlemann AC (2017) Carbapenem-resistant Enterobacteriaceae colonization (CRE) and subsequent risk of infection and 90-day mortality in critically ill patients, an observational study, PLoS One 12, e0186195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Levine DP (2006) Vancomycin: a history, Clin Infect Dis 42 Suppl 1, S5–12. [DOI] [PubMed] [Google Scholar]
- [5].Reynolds PE (1989) Structure, biochemistry and mechanism of action of glycopeptide antibiotics, Eur J Clin Microbiol Infect Dis 8, 943–950. [DOI] [PubMed] [Google Scholar]
- [6].Walsh CT, Fisher SL, Park IS, Prahalad M, and Wu Z (1996) Bacterial resistance to vancomycin: five genes and one missing hydrogen bond tell the story, Chem Biol 3, 21–28. [DOI] [PubMed] [Google Scholar]
- [7].Silhavy TJ, Kahne D, and Walker S (2010) The bacterial cell envelope, Cold Spring Harb Perspect Biol 2, a000414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Nikaido H (1989) Outer membrane barrier as a mechanism of antimicrobial resistance, Antimicrob Agents Chemother 33, 1831–1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Delcour AH (2009) Outer membrane permeability and antibiotic resistance, Bba-Proteins Proteom 1794, 808–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Richter MF, Drown BS, Riley AP, Garcia A, Shirai T, Svec RL, and Hergenrother PJ (2017) Predictive compound accumulation rules yield a broad-spectrum antibiotic, Nature 545, 299–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Smith PA, Koehler MFT, Girgis HS, Yan D, Chen Y, Chen Y, Crawford JJ, Durk MR, Higuchi RI, Kang J, Murray J, Paraselli P, Park S, Phung W, Quinn JG, Roberts TC, Rouge L, Schwarz JB, Skippington E, Wai J, Xu M, Yu Z, Zhang H, Tan MW, and Heise CE (2018) Optimized arylomycins are a new class of Gram-negative antibiotics, Nature 561, 189–194. [DOI] [PubMed] [Google Scholar]
- [12].Richter MF, and Hergenrother PJ (2019) The challenge of converting Gram-positive-only compounds into broad-spectrum antibiotics, Ann N Y Acad Sci 1435, 18–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Gu HW, Ho PL, Tong E, Wang L, and Xu B (2003) Presenting vancomycin on nanoparticles to enhance antimicrobial activities, Nano Lett 3, 1261–1263. [Google Scholar]
- [14].Fernandes MM, Ivanova K, Hoyo J, Perez-Rafael S, Francesko A, and Tzanov T (2017) Nanotransformation of Vancomycin Overcomes the Intrinsic Resistance of Gram-Negative Bacteria, ACS Appl Mater Interfaces 9, 15022–15030. [DOI] [PubMed] [Google Scholar]
- [15].Yarlagadda V, Manjunath GB, Sarkar P, Akkapeddi P, Paramanandham K, Shome BR, Ravikumar R, and Haldar J (2016) Glycopeptide Antibiotic To Overcome the Intrinsic Resistance of Gram-Negative Bacteria, ACS Infect Dis 2, 132–139. [DOI] [PubMed] [Google Scholar]
- [16].Frankel AD, and Pabo CO (1988) Cellular Uptake of the Tat Protein from Human Immunodeficiency Virus, Cell 55, 1189–1193. [DOI] [PubMed] [Google Scholar]
- [17].Green M, and Loewenstein PM (1988) Autonomous Functional Domains of Chemically Synthesized Human Immunodeficiency Virus Tat Trans-Activator Protein, Cell 55, 1179–1188. [DOI] [PubMed] [Google Scholar]
- [18].Vives E, Brodin P, and Lebleu B (1997) A truncated HIV-1 Tat protein basic domain rapidly translocates through the plasma membrane and accumulates in the cell nucleus, J. Biol. Chem. 272, 16010–16017. [DOI] [PubMed] [Google Scholar]
- [19].Wender PA, Mitchell DJ, Pattabiraman K, Pelkey ET, Steinman L, and Rothbard JB (2000) The design, synthesis, and evaluation of molecules that enable or enhance cellular uptake: Peptoid molecular transporters, Proc. Natl. Acad. Sci. U.S.A. 97, 13003–13008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Stanzl EG, Trantow BM, Vargas JR, and Wender PA (2013) Fifteen Years of Cell-Penetrating Guanidinium-Rich Molecular Transporters: Basic Science, Research Tools, and Clinical Applications, Accounts Chem Res 46, 2944–2954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Antonoplis A, Zang X, Huttner MA, Chong KKL, Lee YB, Co JY, Amieva MR, Kline KA, Wender PA, and Cegelski L (2018) A Dual-Function Antibiotic-Transporter Conjugate Exhibits Superior Activity in Sterilizing MRSA Biofilms and Killing Persister Cells, J Am Chem Soc 140, 16140–16151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Huang KC, Mukhopadhyay R, Wen B, Gitai Z, and Wingreen NS (2008) Cell shape and cell-wall organization in Gram-negative bacteria, Proc Natl Acad Sci U S A 105, 19282–19287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Stokes JM, French S, Ovchinnikova OG, Bouwman C, Whitfield C, and Brown ED (2016) Cold Stress Makes Escherichia coli Susceptible to Glycopeptide Antibiotics by Altering Outer Membrane Integrity, Cell Chem Biol 23, 267–277. [DOI] [PubMed] [Google Scholar]
- [24].Ling LL, Schneider T, Peoples AJ, Spoering AL, Engels I, Conlon BP, Mueller A, Schaberle TF, Hughes DE, Epstein S, Jones M, Lazarides L, Steadman VA, Cohen DR, Felix CR, Fetterman KA, Millett WP, Nitti AG, Zullo AM, Chen C, and Lewis K (2015) A new antibiotic kills pathogens without detectable resistance, Nature 517, 455–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Okano A, Isley NA, and Boger DL (2017) Peripheral modifications of [Psi[CH2NH]Tpg(4)]vancomycin with added synergistic mechanisms of action provide durable and potent antibiotics, Proc Natl Acad Sci U S A 114, E5052-E5061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Lara B, Mengin-Lecreulx D, Ayala JA, and van Heijenoort J (2005) Peptidoglycan precursor pools associated with MraY and FtsW deficiencies or antibiotic treatments, Fems Microbiol Lett 250, 195–200. [DOI] [PubMed] [Google Scholar]
- [27].Loh B, Grant C, and Hancock REW (1984) Use of the Fluorescent-Probe 1-N-Phenylnaphthylamine to Study the Interactions of Aminoglycoside Antibiotics with the Outer-Membrane of Pseudomonas-Aeruginosa, Antimicrob Agents Ch 26, 546–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Huang E, and Yousef AE (2014) The Lipopeptide Antibiotic Paenibacterin Binds to the Bacterial Outer Membrane and Exerts Bactericidal Activity through Cytoplasmic Membrane Damage, Appl Environ Microb 80, 2700–2704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Hancock REW, and Bell A (1988) Antibiotic Uptake into Gram-Negative Bacteria, Eur J Clin Microbiol 7, 713–720. [DOI] [PubMed] [Google Scholar]
- [30].Farmer S, Li ZS, and Hancock REW (1992) Influence of Outer-Membrane Mutations on Susceptibility of Escherichia-Coli to the Dibasic Macrolide Azithromycin, J Antimicrob Chemoth 29, 27–33. [DOI] [PubMed] [Google Scholar]
- [31].Sundram UN, and Griffin JH (1995) General and Efficient Method for the Solution-Phase and Solid-Phase Synthesis of Vancomycin Carboxamide Derivatives, J Org Chem 60, 1102–1103. [Google Scholar]
- [32].Wiegand I, Hilpert K, and Hancock RE (2008) Agar and broth dilution methods to determine the minimal inhibitory concentration (MIC) of antimicrobial substances, Nat Protoc 3, 163–175. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.