

Abstract
In the present study we investigated whether the neuropeptide nociceptin/orphanin FQ (N/OFQ), previously implicated in the pathogenesis of Parkinson's disease, also affects l-DOPA-induced dyskinesia. In striatal slices of naive rodents, N/OFQ (0.1–1 μm) prevented the increase of ERK phosphorylation and the loss of depotentiation of synaptic plasticity induced by the D1 receptor agonist SKF38393 in spiny neurons. In vivo, exogenous N/OFQ (0.03–1 nmol, i.c.v.) or a synthetic N/OFQ receptor agonist given systemically (0.01–1 mg/Kg) attenuated dyskinesias expression in 6-hydroxydopamine hemilesioned rats primed with l-DOPA, without causing primary hypolocomotive effects. Conversely, N/OFQ receptor antagonists worsened dyskinesia expression. In vivo microdialysis revealed that N/OFQ prevented dyskinesias simultaneously with its neurochemical correlates such as the surge of nigral GABA and glutamate, and the reduction of thalamic GABA. Regional microinjections revealed that N/OFQ attenuated dyskinesias more potently and effectively when microinjected in striatum than substantia nigra (SN) reticulata, whereas N/OFQ receptor antagonists were ineffective in striatum but worsened dyskinesias when given in SN. Quantitative autoradiography showed an increase in N/OFQ receptor binding in striatum and a reduction in SN of both unprimed and dyskinetic 6-hydroxydopamine rats, consistent with opposite adaptive changes of N/OFQ transmission. Finally, the N/OFQ receptor synthetic agonist also reduced dyskinesia expression in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-treated dyskinetic macaques without affecting the global parkinsonian score. We conclude that N/OFQ receptor agonists may represent a novel strategy to counteract l-DOPA-induced dyskinesias. Their action is possibly mediated by upregulated striatal N/OFQ receptors opposing the D1 receptor-mediated overactivation of the striatonigral direct pathway.
Introduction
Nociceptin/orphanin FQ (N/OFQ) (Meunier et al., 1995; Reinscheid et al., 1995) and its receptor (NOP) represent a neuropeptide system bearing structural and functional analogies with classical opioid systems but unique pharmacological profile (Calò et al., 2000). NOP receptor expression and binding are widespread throughout the rodent and primate brain, supporting the role of the N/OFQ-NOP receptor system in the modulation of central functions such as sensory nociceptive processing, learning and memory, reward, mood, feeding, stress, and movement (Mogil and Pasternak, 2001; Lambert, 2008).
Preclinical and clinical studies revealed a link between N/OFQ and Parkinson's disease (PD) (Marti et al., 2005, 2010). Indeed, an increase of N/OFQ expression (Marti et al., 2005, 2010; Gouty et al., 2010) and release (Marti et al., 2005) in the substantia nigra (SN) of parkinsonian animals was found, together with an elevation of N/OFQ levels in the CSF of parkinsonian patients (Marti et al., 2010). Consistent with a pathogenic role of endogenous N/OFQ, NOP receptor antagonists reversed parkinsonian-like motor deficits (Marti et al., 2005, 2008; Viaro et al., 2008; Volta et al., 2010), also synergizing with l-3,4-dihydroxyphenylalanine (l-DOPA) (Marti et al., 2007; Visanji et al., 2008; Viaro et al., 2010). Contrary to substantia nigra reticulata (SNr), a reduction in N/OFQ expression was found in the dopamine (DA)-depleted striatum (Marti et al., 2010), suggesting a different adaptive response of striatal N/OFQ transmission in PD. The striatal N/OFQ-NOP receptor system has so far received little attention, possibly due to the low expression of N/OFQ and NOP receptor in the rodent striatum (Neal et al., 1999a,b). Nonetheless, N/OFQ impairs the firing activity of ascending DA (Marti et al., 2004) and serotonin (Tao et al., 2007; Nazzaro et al., 2009) neurons, and inhibits DA (Flau et al., 2002; Olianas et al., 2008) and serotonin (Sbrenna et al., 2000) release presynaptically. N/OFQ also postsynaptically counteracts the D1 receptor-stimulated cAMP accumulation in striatal neurons (Olianas et al., 2008), overall suggesting that endogenous N/OFQ might control striatal function. This control might be more relevant in the primate caudate/putamen due to the much greater expression of NOP receptors (Berthele et al., 2003; Bridge et al., 2003). We therefore investigated whether NOP receptor agonists and antagonists affect the expression of l-DOPA-induced dyskinesias (LID) in rats (Cenci et al., 1998) and nonhuman primates (Bézard et al., 2003). Indeed, LID are a major motor complication of l-DOPA pharmacotherapy thought to originate from aberrant striatal plasticity (Calabresi et al., 2010). LID are involuntary choreodystonic movements (Nutt and Gancher, 1994) that develop as a consequence of DA denervation and nonphysiological DA release from both residual DA and serotonin striatal terminals (Carta et al., 2007; Navailles et al., 2010). This leads to “pulsatile” DA receptor stimulation and upregulation of D1 signaling (Andersson et al., 1999; Aubert et al., 2005), increased activity along the Ras/MEK/ERK kinase pathway (Valjent et al., 2005; Feyder et al., 2011) and loss of neuronal depotentiation after long-term potentiation (LTP) induction in striatonigral spiny neurons (Picconi et al., 2003).
Materials and Methods
In vitro experiments
Immunohistochemistry
Anesthetized 2-month-old C57BL/6 mice were decapitated and the brains rapidly removed. Slices (200 μm thick) were cut using a vibratome keeping the brain submerged in ice-cold carboxygenated sucrose-based dissecting solution containing the following (in mm): 87 NaCl, 2.5 KCl, 7 MgCl2, 1 NaH2PO4, 75 sucrose, 25 NaHCO3, 10 d-glucose, 0.5 CaCl2, and 2 kynurenic acid. The slices were transferred into BSC1 chambers (Scientific System Design) and constantly perfused with carboxygenated artificial CSF solution containing the following (in mm): 124 NaCl, 5 KCl, 1.3 MgSO4, 1.2 NaH2PO4, 25 NaHCO3, 10 d-glucose, and 2.4 CaCl2 at a constant rate of 2 ml/min at 32°C for 1 h. Slices were then stimulated for 10 min with 100 μm SKF38393, 1 μm N/OFQ, or their combination, and fixed in 4% paraformaldehyde in 0.1 m sodium phosphate buffer, pH 7.4, at room temperature for 15 min. Slices were then rinsed three times for 20 min in 0.1 m sodium phosphate buffer, pH 7.4, at room temperature and cryoprotected in 30% sucrose overnight at 4°C.
Eighteen micrometer cryosections were cut and washed three times for 10 min with Dulbecco's PBS (D-PBS). After blocking in D-PBS containing 5% normal goat serum and 0.1% Triton X-100 for 1 h at room temperature, sections were incubated overnight at 4°C with the primary antibodies: anti-phospho-S6 ribosomal protein (Thr235/236) (1:200; Cell signaling Technology) and anti-NeuN (1:1000; Chemicon). Sections were then washed three times for 10 min with D-PBS and incubated for 1 h at room temperature with the secondary antibodies: Alexa Fluor 546 goat anti-mouse (1:200) and Alexa Fluor 488 goat anti-rabbit (1:500) (Invitrogen).
Following the incubation with the secondary antibodies, the sections were washed three times for 10 min with D-PBS and the coverslips were mounted using the fluorescent mounting medium (Dako).
Single- and double-labeled images were obtained using a laser scanning confocal microscopy (Leica SP2), equipped with the corresponding lasers and the appropriate filter sets to avoid cross talk between the fluorophores. Neuronal quantification was performed with ImageJ software on images taken at 40× magnification by counting phospho-S6-immunoreactive neurons among NeuN positive neurons in each slice.
Electrophysiology
Corticostriatal coronal slices (270 μm thick) were cut from 1- to 2-month-old male Wistar rats (n = 12; Harlan) using a vibratome, as previously described (Calabresi et al., 1998; Picconi et al., 2003). A single slice was then transferred to a recording chamber and submerged in a continuously flowing (2.5–3 ml/min) carboxygenated (95% O2-5% CO2) Krebs solution (in mm): 126 NaCl, 2.5 KCl, 1.2 MgCl2, 1.2 NaH2PO4, 2.4 CaCl2, 10 glucose, and 25 NaHCO3 kept at 34°C. Drugs were bath applied by switching the solution to one containing known concentrations of drugs. Total replacement of the medium in the chamber occurred within 1 min. Intracellular recordings of striatal medium spiny neurons (MSNs) were obtained with sharp microelectrodes pulled from borosilicate glass pipettes backfilled with 2 M KCl (30–60 MΩ). An Axoclamp 2B amplifier (Molecular Devices) was connected in parallel to an oscilloscope to monitor the signal in “bridge” mode and to a PC for acquisition of the traces using a pClamp9 software (Molecular Devices). Only neurons electrophysiologically identified as MSNs were considered for experiment (Calabresi et al., 1992).
A glutamatergic EPSP was evoked every 10 s by means of a bipolar electrode connected to a stimulator unit (Grass Telefactor). The stimulating electrode was located in the cortical areas close to the recording electrode or in the white matter between the cortex and the striatum to activate corticostriatal fibers. The recording electrodes were invariably placed within the striatum. The experiments were conducted in a Mg2+-free Krebs' solution to unmask the NMDA glutamate (Glu) receptor component of the EPSP. After 10–15 min the acquisition of EPSPs of stable amplitude, a high-frequency stimulation protocol (HFS), consisting of three trains of stimuli at 100 Hz (20 s intertrain interval), was delivered to the slice to induce LTP of the EPSP (Calabresi et al., 1992). Twenty minutes after the HFS protocol a low-frequency stimulation protocol (LFS), consisting of repeated stimuli applied at 2 Hz for 10 min, was applied to induce depotentiation of the EPSP amplitude to pre-HFS conditions (Picconi et al., 2003). Quantitative data are expressed as a percentage of the EPSP amplitudes in respect to the relative control amplitude values, the latter representing the mean of responses recorded during a stable period. EPSP amplitudes were measured 20 min after HFS or LFS and compared with pre-HFS baseline. Off-line analysis was performed using Clampfit (Molecular Devices) and GraphPad Prism 5 software.
Autoradiography
Twenty micrometer coronal sections were obtained from the frozen brains of sham-operated, untreated 6-hydroxydopamine (OHDA) lesioned and dyskinetic 6-OHDA lesioned (killed 48 h after last l-DOPA injection) male Sprague Dawley rats (see below). Sections were cut at two different brain levels from bregma: anteroposterior (AP) −0.30 to −0.92 and −3.60 to −5.80 (Paxinos and Watson, 1986) using a cryostat (13−15°C). Sets of five slides (three slides for total binding and two slides for nonspecific binding) were used for each group. Iodination of [Arg14Lys15Tyr18(di)iodo]N/OFQ-NH2 was performed according to the Chloramine T method (Hunter and Greenwood, 1962). Briefly, 5 μg of peptide was incubated in 0.05 m phosphate buffer, pH 7.4, for 30 s in the presence of 0.5 mCi (18.5 MBq) of Na125I and 220 nmol of Chloramine T in a total volume of 85 μl. The mono-iodinated peptide was then immediately purified by HPLC on a C18 Jupiter 300 column (250 × 4,6 mm; Phenomenex) with 0.1% trifluoroacetic acid and acetonitrile as mobile phases. The specific activity of the iodinated peptides corresponds to 2000 cpm/fmol or 1212 Ci/mmol.
The binding method used in this study was adapted from (Bridge et al., 2003). Tissue sections were thawed at room temperature and preincubated for 30 min in a solution containing: 50 mm Tris-HCl, pH 7.4; 5 mm MgCl2; and 0.2% bovine serum albumin (BSA). They were then incubated at room temperature for 90 min in the same buffer solution, to which 5 mg/ml Bacitracin and 150 pm 125I[Tyr14]N/OFQ were added. Nonspecific binding was determined by adding 1 μm cold ligand to the mixture. Slides were then rinsed in the preincubation solution without BSA (four rinses, 1 min each), then in distilled water at 4°C (15 s, to remove the excess of salts) and air dried. Kodak Scientific Biomax MS films were juxtaposed onto the slides for 3 d at room temperature. Films were developed in D-19 (Kodak developer) and fixed in Kodak fixer. Image analysis was performed using a computerized image analysis system (MCID; InterFocus GmbH). Specific binding was determined by subtracting nonspecific binding from total binding taken from adjacent sections. Calibration was performed on a standard curve generated using polymer [C14]-microscale standards (GE Healthcare), thereby enabling conversion of relative optical intensity units, directly measured from autoradiograms, to binding site density expressed in fmol/mg.
In vivo experiments in rats
Male Sprague Dawley rats (150 g; Harlan) were kept under regular lighting conditions (12 h light/dark cycle) and given food and water ad libitum. The experimental protocols performed in rodents were in accordance with the European Communities Council Directive of November 24, 1986 (86/609/EEC), and were approved by the Italian Ministry of Health (license #194/2008-B) and the Ethical Committee of the University of Ferrara. Adequate measures were taken to minimize the number of animals used and animal pain and discomfort.
Experimental design
Experiments in rats.
A total of 126 6-OHDA hemilesioned dyskinetic, 10 6-OHDA hemilesioned naive and 14 sham-operated rats were used. Pharmacological treatments were randomized and usually administered every 3 d. A maintenance dose of l-DOPA was administered the day before treatment, and animals were scored. In experiment using NOP agonists (see Fig. 3), 20 dyskinetic animals implanted with an intracerebroventricular cannula received either saline or three doses of N/OFQ, ON and OFF l-DOPA (n = 10 each condition). Another group of 20 animals was treated with saline or three different doses of Ro 65–6570 (i.p.), ON and OFF l-DOPA (n = 10 each condition). In microdialysis experiments (see Fig. 5), six dyskinetic animals were implanted with a cannula (i.c.v.) and two microdialysis probes in SNr and the ventromedial thalamus (VMTh). Microdialysis was performed for 4 d and treatments (saline, l-DOPA, N/OFQ, and l-DOPA + N/OFQ) were given in a randomized fashion. In experiments using NOP antagonists (see Fig. 6), 10 dyskinetic animals implanted with an intracerebroventricular cannula received l-DOPA in combination with saline or UFP-101 (i.c.v.), whereas another group of 10 animals received l-DOPA in combination with saline or J-113397 (i.p.). In separate experiments, saline, UFP-101 and J-113397 were also given OFF l-DOPA to measure the impact of NOP receptor antagonists alone on rotarod performance. Control group was injected with saline in both areas. In experiments using regional microinjections (see Fig. 7) 20 dyskinetic rats had a cannula implanted either in the SNr or dorsolateral striatum (DLS) (n = 10 each group) and received saline or three doses of N/OFQ in combination with l-DOPA. Another group of 10 animals was implanted both in the SNr and DLS, and received a single dose of N/OFQ in DLS, SNr, or both. In combination experiments (see Fig. 8), 20 dyskinetic animals implanted with a cannula in DLS or SNr (n = 10 each) received saline, N/OFQ (0.1 nmol), UFP-101 (10 nmol), or N/OFQ + UFP-101, in combination with l-DOPA. In autoradiography experiment (see Fig. 9) brain slices were obtained from 8 sham-operated, 10 6-OHDA hemilesioned, and 10 dyskinetic rats.
Figure 3.

NOP receptor agonists attenuated LID. Effect of N/OFQ (0.03–1 nmol, i.c.v.) and Ro 65–6570 (0.01–1 mg/kg, i.p.) on ALO AIMs induced by l-DOPA (6 mg/kg plus benserazide 15 mg/kg, i.p.). N/OFQ (A) or Ro 65–6570 (D), given 5 and 30 min before l-DOPA, respectively, attenuated the severity of dyskinesias in a dose-related fashion. N/OFQ and Ro 65–6570 improved rotarod performance ON l-DOPA (B, E) but worsened it OFF l-DOPA (C, F). Data are expressed as percentage of the l-DOPA effect measured in the same animal in the last training session, and represent the mean ± SEM of 10 determinations. Statistical analysis was performed by conventional (A, D) or RM (B, C, E, F) one-way ANOVA followed by the Newman–Keuls test for multiple comparisons. A, Significant effect of treatment (F(11,153) = 24.78, p < 0.0001). B, Significant effect of treatment (F(3,114) = 77.29, p < 0.0001), time (F(2,114) = 35.08, p < 0.0001), and time × treatment interaction (F(6,114) = 5.42, p < 0.0001). C, Significant effect of treatment (F(3,114) = 400.64, p < 0.0001), but not time (F(2,114) = 1.21, p = 0.30) or time × treatment interaction (F(6,114) = 0.64, p = 0.70). D, Significant effect of treatment (F(11,147) = 11.27, p < 0.0001). E, Significant effect of treatment (F(3,114) = 68.86, p < 0.0001) and time (F(2,114) = 18.24, p < 0.0001) but not time × treatment interaction (F(6,114) = 1.10, p = 0.36). F, Significant effect of treatment (F(3,114) = 387.52, p < 0.0001), time (F(2,114) = 20.66, p < 0.0001), and time × treatment interaction (F(6,114) = 5.39, p < 0.0001). *p < 0.05, **p < 0.01, significantly different from saline.
Figure 5.
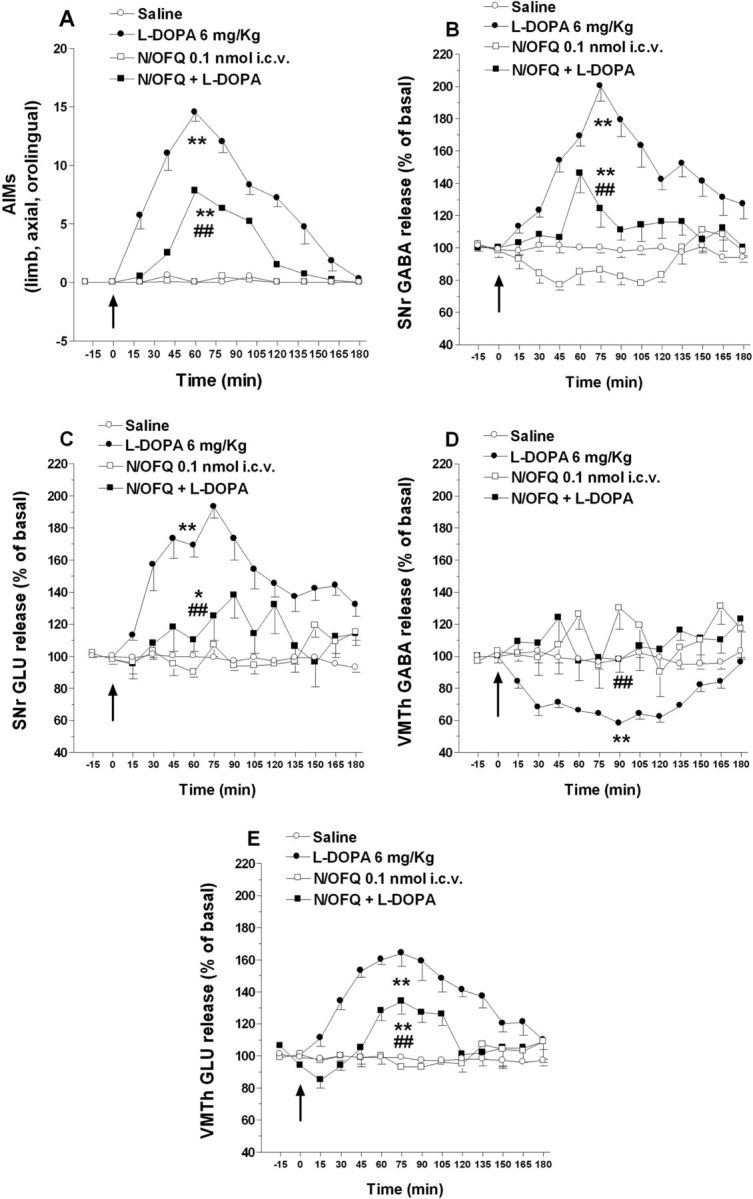
N/OFQ attenuated the elevation of nigral amino acids and the reduction of thalamic GABA associated with LID. Dyskinetic rats were implanted with one microdialysis probe in the lesioned SNr and another in ipsilateral VMTh. Twenty-four hours later, rats were acutely challenged with saline, l-DOPA (6 mg/kg plus 15 mg/kg benserazide, i.p.), N/OFQ (0.1 nmol, i.c.v.), or their combination (N/OFQ given 5 min before). In control rats, saline was injected (i.c.v.) and, 5 min later, administered (i.p.). ALO AIMs in arbitrary units (A) were monitored (for 1 min every 15 min) simultaneously with GABA and Glu levels in SNr (B, C) and VMTh (D, E) up to 180 min from l-DOPA administration. Microdialysis data were expressed as percentage of basal pretreatment levels (calculated as the mean of the two samples preceding the treatment). Data are means ± SEM of six animals. Basal dialysate GABA and Glu levels were 13.4 ± 1.0 and 190.6 ± 9.7 nm, respectively, in SNr, and 16.7 ± 0.9 and 204.8 ± 8.7 nm, respectively, in VMTh. Statistical analysis was performed on area-under-the-curve values by two-way ANOVA followed by Newman–Keuls test for multiple comparisons. A, Main effect of N/OFQ (F(1,1) = 80.75, p < 0.0001), l-DOPA (F(1,1) = 472.44, p < 0.0001), and their interaction (F(1,20) = 78.63, p < 0.0001). B, Main effect of N/OFQ (F(1,1) = 56.64, p < 0.0001), l-DOPA (F((1,1) = 158.69, p < 0.0001), and their interaction (F(1,20) = 21.64, p < 0.0001). C, Main effect of N/OFQ (F(1,1) = 24.30, p < 0.0001), l-DOPA (F(1,1) = 88.86, p < 0.0001), and their interaction (F(1,20) = 32.30, p < 0.0001). D, Main effect of N/OFQ (F(1,1) = 40.09, p < 0.0001), l-DOPA (F(1,1) = 17.42, p < 0.0001), and their interaction (F(1,20) = 12.01, p < 0.0001). E, Main effect of N/OFQ (F(1,1) = 33.25, p < 0.0001), l-DOPA (F(1,1) = 169.20, p < 0.0001), and their interaction (F(1,20) = 46.30, p < 0.0001). *p < 0.05, **p < 0.01 significantly different from saline. ##p < 0.01, significantly different from l-DOPA.
Figure 6.

NOP receptor antagonists worsened LID. Effect of UFP-101 (10 nmol, i.c.v.) and J-113397 (3 mg/kg, i.p.) on ALO AIMs induced by l-DOPA (6 mg/kg plus benserazide 15 mg/kg, i.p.). UFP-101 (A) and J-113397 (D), given 5 and 15 min before l-DOPA, respectively, worsened limb AIMs without affecting axial and orolingual AIMs. UFP-101 also slightly worsened rotarod performance ON l-DOPA (B) while J-113397 left it unaffected (E). Both compounds improved rotarod performance OFF l-DOPA (C, F). Data are expressed as percentage of the l-DOPA effect measured in the same animal in the last training session, and represent the mean ± SEM 10 determinations. Statistical analysis was performed by conventional (A, D) or RM (B, C, E, F) one-way ANOVA followed by the Newman–Keuls test for multiple comparisons. A, Significant effect of treatment (F(5,60) = 5.99, p < 0.0001). B, Significant effect of treatment (F(1,54) = 7.02, p = 0.0106) and time (F(2,54) = 12.95, p < 0.0001) but not time × treatment interaction (F(2,54) = 2.83, p = 0.0678). C, Significant effect of treatment (F(1,60) = 115.65, p < 0.0001), time (F(2,60) = 17.11, p < 0.0001), and time × treatment interaction (F(2,60) = 3.29, p = 0.0441). D, Significant effect of treatment (F(5,84) = 3.056, p < 0.0001). E. Significant effect of time (F(2,54) = 5.41, p = 0.0072) but not treatment (F(1,54) = 0.79, p = 0.37) or time × treatment interaction (F(2,54) = 0.37, p = 0.69). F, Significant effect of treatment (F(1,54) = 175.12, p < 0.0001) but not time (F(2,54) = 1.09, p = 0.3448) or time × treatment interaction (F(2,54) = 1.95, p = 0.15). *p < 0.05, **p < 0.01, significantly different from saline.
Figure 7.
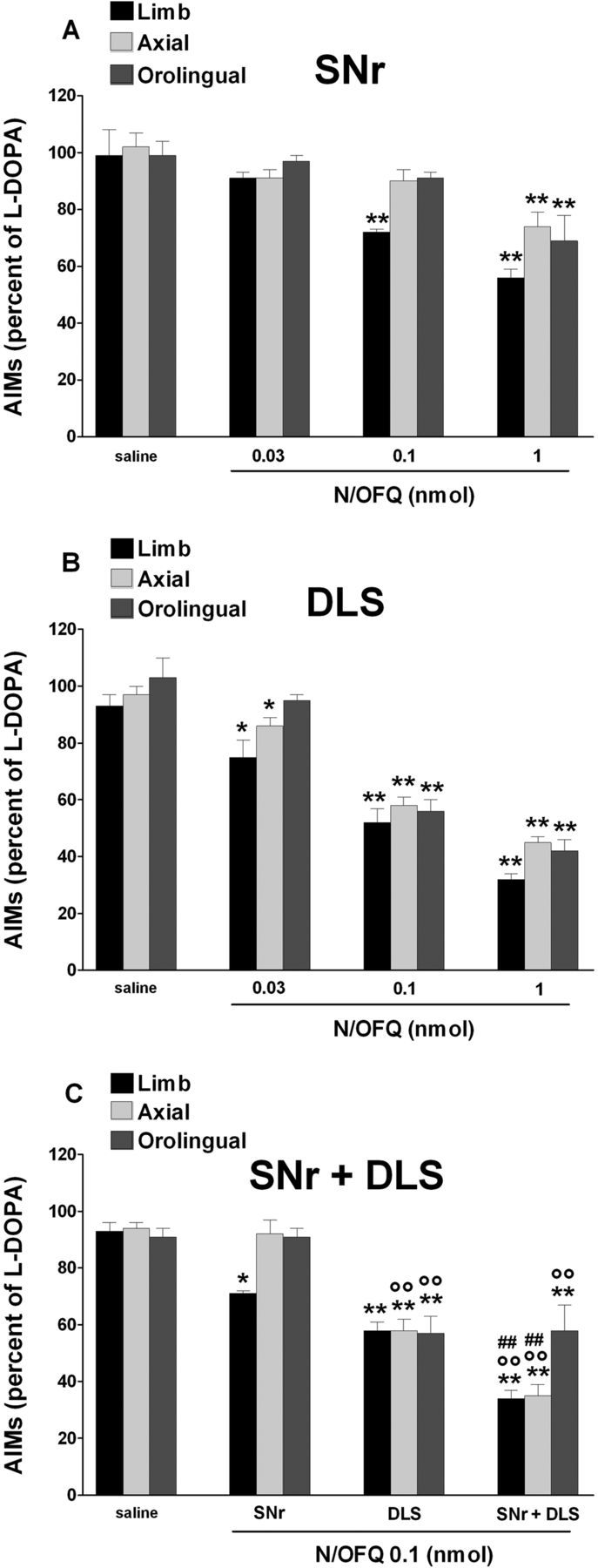
N/OFQ attenuated LID when injected in SNr and DLS. Effect of N/OFQ (0.03–1 nmol, i.c.v.) injected in SNr (A), DLS (B), or both (at 0.1 nmol; C) on ALO AIMs induced by l-DOPA (6 mg/kg plus benserazide 15 mg/kg, i.p.). N/OFQ given 5 min before l-DOPA attenuated the severity of dyskinesias in a dose-related fashion, being more potent when injected in DLS than SNr. Coinjection of N/OFQ in SNr and DLS produced an additive antidyskinetic effect (C). Saline was simultaneously injected in SNr and DLS. Data are expressed as percentage of the l-DOPA effect measured in the same animal in the last training session, and represent the mean ± SEM of 10 determinations. Statistical analysis was performed by one-way (A, B) or two-way (C) ANOVA followed by the Newman–Keuls test for multiple comparisons. A, Significant effect of treatment (F(11,71) = 9.58, p < 0.0001). B, Significant effect of treatment (F(11,71) = 38.8, p < 0.0001). C, Main effect of N/OFQ (F(3,60) = 528.64, p < 0.0001), brain area (F(2,60) = 33.94, p < 0.0001), and their interaction (F(6,60) = 23.76, p < 0.0001). **p < 0.01, *p < 0.05, significantly different from saline. °°p < 0.01, significantly different from SNr injection. ##p < 0.01, significantly different from DLS injection.
Figure 8.
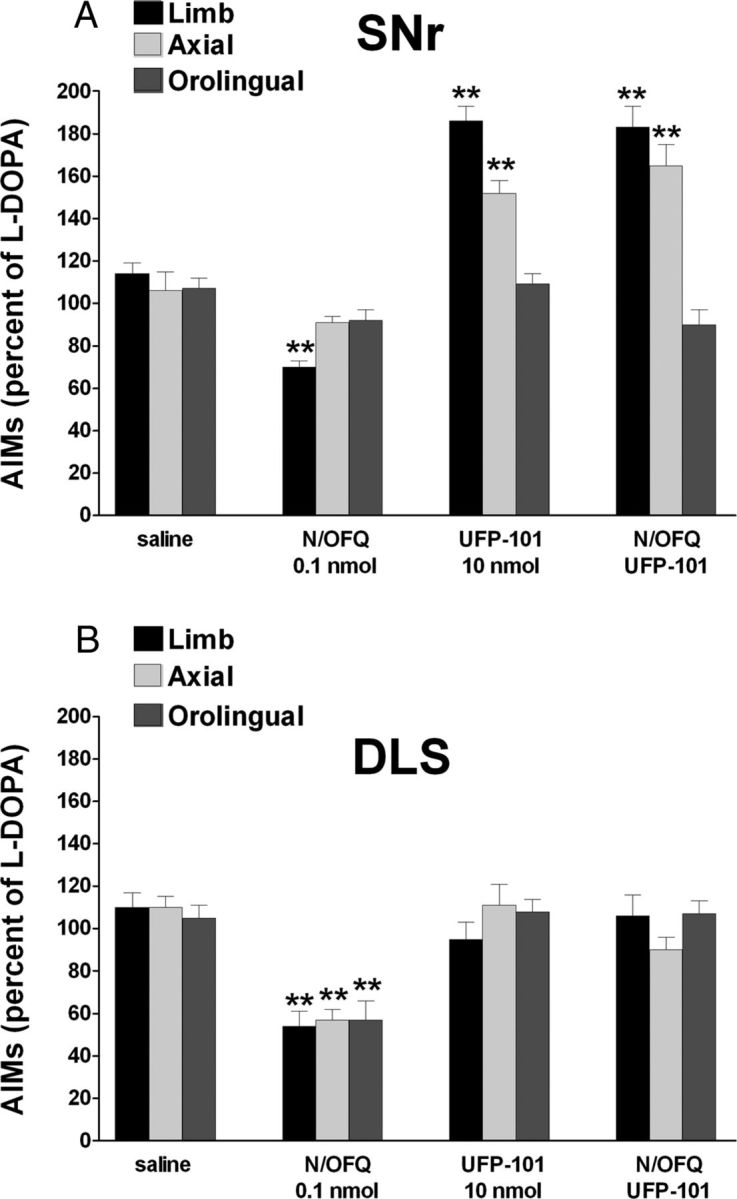
UFP-101 worsened LID when given in SNr but not DLS, preventing N/OFQ action in both regions. Effect of injections of N/OFQ (0.1 nmol, i.c.v.), UFP-101 (10 nmol, i.c.v.), or their combinations in SNr (A) or DLS (B) on ALO AIMs induced by l-DOPA (6 mg/kg plus benserazide 15 mg/kg, i.p.). UFP-101 was administered 5 min before l-DOPA, simultaneously with N/OFQ. Data are expressed as percentage of the l-DOPA effect measured in the same animal in the last training session, and represent the mean ± SEM of 10 determinations. Statistical analysis was performed by two-way ANOVA followed by the Newman–Keuls test for multiple comparisons. A, Limb AIMs, main effect of N/OFQ (F(1,1) = 12.95, p = 0.0012), UFP-101 (F(1,1) = 16.00, p = 0.0004), and their interaction (F(1,28) = 29.18, p < 0.0001); axial AIMs, main effect of N/OFQ (F(1,1) = 13.63, p = 0.0012), UFP-101 (F(1,1) = 45.11, p < 0.0001), and their interaction (F(1,28) = 12.38, p = 0.0015); orolingual AIMs, main effect of N/OFQ (F(1,1) = 26.90, p < 0.0001), UFP-101 (F(1,1) = 24.54, p < 0.0001), and their interaction (F(1,28) = 22.93, p < 0.0001). B, Limb AIMs, main effect of N/OFQ (F(1,1) = 192.51, p < 0.0001), UFP-101 (F(1,1) = 12.64, p = 0.0014), and their interaction (F(1,28) = 9.42, p = 0.0047); axial AIMs, main effect of N/OFQ (F(1,1) = 64.43, p < 0.0001) but not UFP-101 (F(1,1) = 0.01, p = 0.89) or their interaction (F(1,28) = 3.65, p = 0.07); orolingual AIMs, main effect of UFP-101 (F(1,1) = 8.99, p = 0.0056) but not N/OFQ (F(1,1) = 0.02, p = 0.98) or their interaction (F(1,28) = 0.10, p = 0.74). **p < 0.01, significantly different from saline.
Figure 9.

LID was associated with an elevation of NOP receptor binding in the striatum and a reduction in the SN. Representative total binding of 125I[Tyr14]N/OFQ to NOP receptors in cortical and subcortical areas of sham-operated (A, E), hemiparkinsonian (B, F), and dyskinetic (C, G) rats. Twenty micrometer coronal sections were cut at two different brain levels from bregma: AP −0.30 to −0.92 and −3.60 to −5.80. Specific binding values, given in Table 1, were obtained by subtraction of nonspecific from total binding. Binding changes are expressed as percentage of the contralateral hemisphere (D, H) and are means ± SEM of 6–10 animals. Statistical analysis was performed by one-way ANOVA followed by Newman–Keuls test for multiple comparisons. **p < 0.01, significantly different from sham-operated rats.
Experiments in nonhuman primates.
Five macaques rendered stably parkinsonian with 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) and dyskinetic with l-DOPA (see below)(see Fig. 10).
Figure 10.

Ro 65–6570 attenuated LID in MPTP-treated macaques. Five stably parkinsonian macaques were made dyskinetic by chronic l-DOPA treatment, and then challenged with l-DOPA combined with Ro 65–6570 (0.001 and 0.01 mg/kg, i.m.) or saline (A). Dyskinetic conditions were assessed on a Dyskinesia Disability Scale (see Materials and Methods) evaluating choreiform and dystonic components (A, B). The time courses of these motor responses are shown in C and D. Data are expressed as absolute dyskinesia scores in each animal (A), medians with lower and upper quartiles (B), or means ± SEM (C, D). Statistical analysis was performed by one-way nonparametric RM ANOVA (Friedman test) followed by the Dunn's test for multiple comparisons. *p < 0.05, significantly different from saline.
Unilateral lesion with 6-OHDA
Unilateral lesion of DA neurons was induced in isoflurane-anesthetized Sprague Dawley male rats by stereotaxically injecting 8 μg of 6-OHDA (dissolved in 4 μl of saline containing 0.02% ascorbic acid) into the medial forebrain bundle (Marti et al., 2005), according to the following coordinates from bregma: AP −4.4 mm, mediolateral (ML) −1.2 mm, and dorsoventral (DV) −7.8 mm below dura (Paxinos and Watson, 1986). Sham-operated animals were injected with 4 μl of saline (containing 0.02% ascorbic acid). Two weeks after 6-OHDA injection, denervation was evaluated with a test dose of amphetamine (5 mg/kg, i.p.). Rats showing ipsilateral turning >7 turns/min were enrolled in the study (Marti et al., 2002a, 2007).
l-DOPA treatment and Abnormal Involuntary Movement Scale rating
Two weeks after amphetamine testing, DA-depleted rats were made dyskinetic by a 21 d course of l-DOPA treatment (6 mg/kg + benserazide 15 mg/kg, i.p., once daily). Quantification of abnormal involuntary movements (Abnormal Involuntary Movement Scale; AIMs), a correlate of dyskinesia (Cenci et al., 1998), was performed as extensively described in previous papers (Lundblad et al., 2002; Muñoz et al., 2008; Berthet et al., 2009; Mela et al., 2010). Briefly, rats were observed individually for 1 min every 20 min during the 3 h that followed l-DOPA injection. Dyskinetic movements were classified based on their topographic distribution into three subtypes: (1) axial AIMs, i.e., twisted posture or choreiform twisting of the neck and upper body toward the side contralateral to the lesion; (2) forelimb AIMs, i.e., jerky or dystonic movements of the contralateral forelimb and/or purposeless grabbing movement of the contralateral paw; and (3) orolingual AIMs, i.e., orofacial muscle twitching, empty masticatory movements and contralateral tongue protrusion. Each AIM subtype was rated on a frequency scale from 0 to 4 (1, occasional; 2, frequent; 3, continuous but interrupted by sensory distraction; and 4, continuous, severe, and not interrupted by sensory distraction) on each monitoring period (Cenci et al., 1998). In addition, the amplitude of these AIMs was scored based on a scale from 0 to 4 as described by (Cenci and Lundblad, 2007). Axial, forelimb, and orolingual (ALO) AIMs were calculated as the sum of the product of amplitude and frequency scores from all monitoring periods (Cenci and Lundblad, 2007), and presented either separately or together (global score). AIMs scoring was performed five times during l-DOPA treatment; dyskinetic rats showing total AIMs score > 100 in the last session were enrolled in the study. The theoretical maximal total AIMs score for each animal is 432 (48 each of the nine 20 minute sessions).
Tyrosine immunohistochemistry analysis revealed that dyskinetic rats were fully DA denervated since the ratio of optical density in the ipsilateral versus contralateral side was 99.1 ± 0.2% in sham-operated and 0.1 ± 0.01% in dyskinetic rats (n = 6 both).
Rotarod experiments
The rotarod test was used to quantify the degree of motor impairment both ON and OFF l-DOPA. Dyskinetic rats were tested before (control session) and 20, 60, and 120 min after acute drug treatment (l-DOPA, NOP ligands or their combination). These windows were selected based on the AIMs time course (data not shown), showing a peak after 60–80 min and extinction after 180 min from l-DOPA administration. The speed of rotating cylinder was stepwise increased (180 s each step) from 5 rpm until animals fell off the rod (Marti et al., 2004). Dyskinesias negatively correlated with rotarod performance (r = −0.75, Spearman's nonparametric correlation test, p < 0.001), with a maximal reduction of ∼85% at peak (data not shown).
Drug microinjections in dyskinetic rats
Under isoflurane anesthesia, a stainless steel infusion cannula (15 mm long, 24 gauge) was stereotaxically lowered over the lateral ventricle [intracerebroventricular: AP = −0.9 mm; ML = −1.4 mm; ventrodorsal (VD) = −2.0 mm; DLS: AP = +0.7 mm; ML = +3.5 mm; VD = −3.2 mm; and/or SNr: AP = −5.5 mm; ML = −2.2 mm; VD = −7.3 mm] ipsilateral to the lesioned side. A stainless steel stylet was inserted into the cannula to avoid its occlusion. Animals were allowed 6 d to recover, during which they were handled to familiarize with the operator and injection procedures. On the day of experiment, the rat was gently restrained by the operator, the stylet was removed and 0.5 μl of saline (control rats) or saline containing NOP receptor peptide ligands (N/OFQ and UFP-101) was injected without the use of anesthesia. Alternatively, systemic (i.p.) administration of saline, Ro 65–6570 or J-113397, was performed. In combination studies, NOP ligands were given 5 min (N/OFQ and UFP-101), 15 min (J-113397), or 30 min (Ro 65–6570) before l-DOPA.
Microdialysis
Two microdialysis probes (1 mm dialyzing membrane, AN69; Hospal) were implanted in the DA-lesioned SNr (AP −5.5, ML −2.2, VD −8.3) and ipsilateral VMTh (AP −2.3, ML −1.4, VD −7.4) of isoflurane-anesthetized dyskinetic rats, as previously described (Marti et al., 2007, 2008). In the same surgery session, animals were also implanted with an intracerebroventricular cannula (coordinated as above). Twenty-four hours after surgery, probes were perfused with a modified Ringer's solution (1.2 mm CaCl2, 2.7 mm MgCl2, 148 mm NaCl, and 0.85 mm MgCl2) at a 3 μl/min flow rate and, after 6 h rinsing, samples were collected every 15 min. l-DOPA (6 mg/kg plus benserazide 15 mg/kg) was administered systemically (i.p.), whereas N/OFQ 0.1 nmol was injected (i.c.v., 5 min before l-DOPA). In control rats, saline was injected (i.c.v.) and, 5 min later, administered intraperitoneally. AIMs monitoring was performed every 15 (for 1 min). At the end of the experiments, animals were killed and the correct placement of the probes was verified histologically.
Endogenous Glu and GABA analysis
Glu and GABA were measured by HPLC coupled with fluorometric detection following o-phthaldialdehyde/mercaptoethanol derivatization (Marti et al., 2007). Glu and GABA were separated in a 5-C18 Chromsep analytical column (Chrompack) perfused at 0.48 ml/min with a mobile phase containing 0.1 m sodium acetate, 10% methanol and 2.2% tetrahydrofuran, pH 6.5). Glu and GABA were detected by a FP-2020 Plus spectrophotometer (Jasco) with the excitation and the emission wavelengths set at 370 and 450 nm, respectively. The limits of detection for Glu and GABA were ∼1 and ∼0.5 nm, and their retention times were ∼3.5 and ∼ 18.0 min, respectively.
In vivo experiments in mice
To investigate the in vivo selectivity of Ro 65–6570, motor activity was evaluated in NOP+/+ and NOP−/− naive mice (25–30 g; 12–15 weeks old) (Nishi et al., 1997) by means of the drag test. This test measures the ability of the animal to balance its body posture with forelimbs in response to an externally imposed dynamic stimulus (backward dragging) (Marti et al., 2005). The animal was gently lifted from the tail allowing the forepaws to rest on the table, and dragged backwards at a constant speed (∼20 cm/s) for a fixed distance (100 cm). The number of steps made with each paw was recorded by two distinct observers.
In vivo experiments in nonhuman primates
Experiments were conducted according to previously published procedures and methods (Bézard et al., 2003; Aubert et al., 2005; Fasano et al., 2010). Five cynomolgus monkeys (Macaca fascicularis; SAH/Xierxin) were housed in individual primate cages under controlled conditions of humidity (50 ± 5%), temperature (24 ± 1°C), and light (12 h light/dark cycles, time lights on 8:00 A.M.) with food and water ad libitum. Experiments were performed in accordance with European Communities Council Directive of November 24, 1986 (86/609/EEC) for care of laboratory animals in an Association for Assessment and Accreditation of Laboratory Animal Care -accredited facility following acceptance of study design by the Institute of Lab Animal Science (Chinese Academy of Science, Beijing, China) Institutional Animal Care and Use Committee. Veterinarians skilled in the healthcare and maintenance of nonhuman primates supervised animal care.
Animals were treated daily (9:00 A.M.) with MPTP hydrochloride (0.2 mg/kg, i.v.) dissolved in saline (Bezard et al., 2001a). Following stabilization of the MPTP-induced syndrome (2 months), animals received a tailored dose (16–19 mg/kg) of an l-DOPA/carbidopa combination (100/25 mg) designed to optimally reverse the parkinsonian features of each animal, twice daily for 3 months. Animals developed severe and reproducible dyskinesias, presenting choreic-athetoid (characterized by constant writhing and jerking motions), dystonic, and sometimes ballistic movements (large-amplitude flinging and flailing movements). These animals were not terminated after completion of experiments, but we demonstrated in several occasions that animals intoxicated with this regimen display a dramatic reduction of DAergic nigrostriatal markers (>95%) compared with control animals (Bezard et al., 2001b; Guigoni et al., 2005; Fernagut et al., 2010).
Behavioral experiments
Macaque's behavior was first recorded in the OFF state for 0.5 h in an observation cage (dimensions: 1.1 × 1.5 × 1.1 m). Drugs were then administered, and behavior was recorded for at least a further 4 h in the observation cage. The parkinsonian condition (and its reversal) was assessed on a validated parkinsonian monkey rating scale by post hoc analysis of video recordings by observers blinded to treatment. The parkinsonian disability score is a combination of the range of movement, bradykinesia, posture, and tremor scores according to the following formula: range of movement + bradykinesia + postural abnormality + tremor (Bézard et al., 2003; Aubert et al., 2005; Fasano et al., 2010). The severity of dyskinesia is rated using the Dyskinesia Disability Scale: 0, dyskinesia absent; 1, mild, fleeting, and rare dyskinetic postures and movements; 2, moderate, more prominent abnormal movements, but not interfering significantly with normal behavior; 3, marked, frequent and, at times, continuous dyskinesia intruding on the normal repertoire of activity; or, 4, severe, virtually continuous dyskinetic activity replacing normal behavior and disabling to the animal. Locomotor activity is concomitantly monitored with infrared activity monitors, providing a mobility count every 5 min (Bézard et al., 2003; Aubert et al., 2005; Fasano et al., 2010).
Data presentation and statistical analysis
Behavior (Figs. 3, 4, 6) and autoradiography (Fig. 9) data were analyzed using conventional or repeated measure (RM) one-way ANOVA as appropriate, followed by the Newman–Keuls test for multiple comparisons. Conventional two-way ANOVA with a 2 × 2 factorial design was used to analyze drug interactions (Figs. 5, 7, 8). Nonparametric one-way RM ANOVA followed by the Dunn's test for multiple comparisons was used to treat behavioral scores in nonhuman primates (Fig. 10). p values <0.05 were considered to be statistically significant.
Figure 4.

In vivo specificity of NOP receptor agonists. A, B, Ro 65–6570 (0.01–1 mg/kg, i.p.) inhibited stepping activity in the drag test in NOP+/+ mice, being ineffective in NOP−/− mice. Each experiment consisted of three different sessions: a control session followed by two other sessions performed 15 min (A) and 90 min (B) after saline or drug administration. Data are expressed as percentages of motor performance in the control session and are means ± SEM 6–7 determinations per group. C, D, NOP receptor antagonists prevent the antidyskinetic effect of NOP receptor agonists. UFP-101 (10 nmol, i.c.v.) was administered simultaneously with N/OFQ (0.1 nmol, i.c.v.; C) or 5 min before Ro 65–6570 (0.1 mg/kg, i.p.; D). J-113397 (3 mg/kg, i.p.) was administered 15 min before NOP agonists. Data are expressed as percentages of l-DOPA effect, and are means ± SEM of 10–12 determinations per group. Statistical analysis was performed by one-way ANOVA followed by the Newman–Keuls test for multiple comparisons. A, Significant effect of treatment in NOP+/+ mice (F(3,23) = 17.61, p < 0.0001). B, Significant effect of treatment in NOP+/+ mice (F(3,23) = 19.89, p < 0.0001). *p < 0.05, **p < 0.01, different from saline. °°p < 0.01, different from N/OFQ or Ro 65–6570 alone.
Materials
d-Amphetamine sulfate, benserazide hydrochloride, l-DOPA methylester hydrochloride, MPTP hydrochloride, 6-OHDA hydrobromide, and SKF38393 were purchased from Sigma. N/OFQ, UFP-101, J-113397, and Ro 65–6570 were synthesized in our laboratory. All drugs were readily dissolved in saline.
Results
Effects of N/OFQ on D1 receptor-induced ERK phosphorylation and loss of synaptic depotentiation
Upregulation of striatal D1 signaling in LID is associated with an increased activity along the Ras/MEK/ERK kinase pathway (Valjent et al., 2005; Feyder et al., 2011) and a loss of neuron capability to depotentiate striatal synaptic response after LTP induction (Picconi et al., 2003). To investigate the effect of NOP receptor activation on these D1 receptor-dependent biochemical correlates of LID, we explored whether N/OFQ could prevent ERK phosphorylation and restore a physiological depotentiation of synaptic plasticity in striatal slices of naive animals stimulated with the D1 receptor agonist SKF38393.
To quantify ERK phosphorylation, we used a recently established ex vivo system, in which brain slices prepared from adult mice can be stimulated with appropriate agonists and antagonists. For ERK signaling activation we monitored the ribosomal protein S6 phosphorylation at the Thr235/236 site (Orellana et al., 2012). D1 receptor activation by 100 μm SKF38393 application dramatically elevated ERK signaling by ∼10-fold (Fig. 1A,B). N/OFQ 1 μm was ineffective alone but prevented the D1 agonist effect (Fig. 1A,B).
Figure 1.

N/OFQ inhibited D1 receptor-mediated activation of ERK signaling in mouse striatal slices. A, Representative photomicrographs showing ERK-dependent ribosomal protein S6 phosphorylation (pS6 Thr235/236) (green) and NeuN (red) immunofluorescence in mouse striatal slices in response to SKF38393 (100 μm), N/OFQ (1 μm), or their combination. B, Phospho-S6 levels were significantly increased in striatal neurons stimulated with SKF38393 (Student's t test, Control vs SKF38393, t(10) = −6.452, *p = 0.0006). N/OFQ, ineffective by itself, prevented the effect of SKF38393 (Student's t test, Control vs SKF38393+N/OFQ, t(12) = 0.947, p = 0.363). One-way ANOVA revealed a significant effect of treatment F(5,31) = 27.235, p > 0.0001. Bonferroni's post hoc, SKF38393 vs SKF38393+N/OFQ, #p < 0.0001. Data are expressed as number ± SEM of pS6 positive cells with respect to NeuN positive cells. *p < 0.05 different from control. #p < 0.05 different from SKF38393 alone.
Cortically evoked EPSPs were measured in electrophysiologically identified striatal MSNs (Fig. 2). In a first group of control slices, after acquiring EPSPs of stable amplitudes for 10 min, LTP of synaptic transmission was induced by HFS protocol (EPSP amplitude post-HFS, 162.7 ± 6.7%). A subsequent application of LFS protocol in these recorded MSNs produced a depotentiation of the synaptic response to pre-HFS conditions (EPSP amplitude post-LFS, 93.9 ± 7.9%; Fig. 2A,B). Bath application of 3 μm D1 receptor agonist SKF38393 prevented depotentiation of MSN synaptic plasticity (EPSP amplitude post-LFS, 160.8 ± 4.3%; Fig. 2C,D). Bath application of 0.1 μm N/OFQ did not affect LTP or depotentiation in MSNs (EPSP amplitude post-HFS, 152.6 ± 6.5%; EPSP amplitude post-LFS 106.7 ± 6.0%; Fig. 2A,B). However, when 3 μm SKF38393 was bath applied for 20 min in the presence of 0.1 μm N/OFQ, depotentiation of MSN synaptic transmission was fully restored (EPSP amplitude, post-LFS 97.3 ± 5.9%, SKF38393 vs SKF38393 + N/OFQ, p < 0.001; Fig. 2C,D). To verify the selectivity of N/OFQ action, we bath applied 3 μm SKF38393 and 0.1 μm N/OFQ with 1 μm NOP receptor antagonist UFP-101. In these conditions, LFS protocol did not produce depotentiation, confirming that N/OFQ action was accomplished through the NOP receptor.
Figure 2.

N/OFQ restored synaptic depotentiation of MSNs in the presence of a D1 receptor agonist. A, Time course of the HFS-induced LTP followed by the LFS-induced depotentiation of MSN EPSPs in control conditions (n = 3, open circles) and in the presence of 0.1 μm N/OFQ (n = 3, filled circles). B, EPSP traces recorded in representative MSNs before HFS (left), and 20 min post-HFS (middle) or LFS (right) in control conditions and in the presence of 0.1 μm N/OFQ. C, Time course of HFS followed by LFS in the presence of 3 μm D1 receptor agonist SKF38393 applied for 20 min (white bar). SKF38393 alone prevented depotentiation in MSNs (control, n = 3, open circles) whereas N/OFQ reversed this effect (n = 5, filled circles). The NOP receptor antagonist UFP-101 (0.1 μm) counteracted the effect of N/OFQ (n = 4, diamonds). D, EPSP traces acquired before HFS and after HFS or LFS in the presence of SKF38393 applied 10 min after HFS (top), N/OFQ + SKF38393 (middle), and N/OFQ + SKF38393 in the presence of UFP-101 (bottom). ***p < 0.001 different from control.
Effect of NOP receptor agonists on dyskinesias and rotarod performance in rats
Data obtained in vitro predict that NOP receptor agonists might selectively target LID pathways and attenuate LID. We therefore administered exogenous N/OFQ and the NOP receptor synthetic agonist Ro 65–6570 (Röver et al., 2000) to dyskinetic rats challenged with l-DOPA. N/OFQ (i.c.v.) dose-dependently attenuated LID appearance (Fig. 3A) and rescued rotarod performance (Fig. 3B) in the 0.03–1 nmol range. The rescue effect at 1 nmol was, however, less pronounced than at lower doses, consistent with the finding that N/OFQ caused primary hypolocomotive effect OFF l-DOPA at this dose (Fig. 3C). To prove the antidyskinetic potential of NOP receptor agonists, the small molecule Ro 65–6570 was used at doses selective for NOP receptor stimulation (0.01–1 mg/kg, i.p.). Indeed, in this dose range Ro 65–6570 reduced stepping activity in NOP+/+ mice but was ineffective in NOP−/− mice (Fig. 4A,B). Ro 65–6570, given 30 min before l-DOPA, attenuated AIMs appearance at 0.01 mg/kg (Fig. 3D), and maximally at 1 mg/kg. This antidyskinetic effect was accompanied by restoration of rotarod performance (Fig. 3E). Conversely, Ro 65–6570 markedly impaired rotarod performance OFF l-DOPA at 1 mg/kg (Fig. 3F). To confirm the pharmacological specificity of the antidyskinetic action of NOP agonists, UFP-101 and J-113397 prevented the antidyskinetic effect of N/OFQ and Ro 65–6570 (Fig. 4C,D).
Effects of intracerebroventricular injections of N/OFQ on LID and neurochemical correlates in SNr and VMTh
Microdialysis was used to investigate whether the antidyskinetic action of N/OFQ was accompanied by changes of the nigral output. AIMs expression (Fig. 5A) was associated with a ∼2-fold elevation of GABA and Glu in SNr (Fig. 5B,C) as well as a ∼40% reduction of GABA and a ∼60% elevation of Glu in VMTh (Fig. 5D,E). Injection of 0.1 nmol N/OFQ (i.c.v.) attenuated (∼60%) LID severity (Fig. 5A) and simultaneously blunted the surge in nigral amino acids (Fig. 5B,C). In addition, N/OFQ prevented the GABA inhibition and attenuated the Glu surge in VMTh (Fig. 5D,E).
Effect of NOP receptor antagonists on dyskinesias and rotarod performance in rats
To investigate whether not only exogenous but also endogenous N/OFQ affected LID, peptidic (UFP-101) (Calo et al., 2002) and nonpeptidic (J-113397) (Kawamoto et al., 1999) NOP receptor antagonists were used at maximal antiparkinsonian doses (Marti et al., 2005) against a prodyskinetic dose of l-DOPA. UFP-101 (10 nmol, i.c.v.; Fig. 6A) or J-113397 (3 mg/kg, i.p.; Fig. 6D) worsened limb without affecting axial and orolingual AIMs. Consistently, neither antagonist attenuated the impairment of rotarod performance associated with LID (ON l-DOPA; Fig. 6B,E). Conversely, UFP-101 (Fig. 6C) and J-113397 (Fig. 6F) facilitated rotarod performance OFF l-DOPA.
Effect of regional injections of N/OFQ in the SNr and DLS of dyskinetic rats
To investigate the brain areas involved in the antidyskinetic and prodyskinetic effects of NOP receptor ligands, N/OFQ and UFP-101 were microinjected in SNr and/or DLS. Intranigral N/OFQ slightly reduced limb AIMs at 0.1 nmol providing a greater and more generalized effect at 1 nmol (Fig. 7A). Conversely, intrastriatal N/OFQ caused a marked reduction of limb and axial AIMs yet at 0.03 nmol (Fig. 7B). Combined intranigral and intrastriatal injections of 0.1 nmol N/OFQ (Fig. 7C) caused greater AIMs attenuation than each injection alone. Contrary to N/OFQ, UFP-101 (10 nmol) worsened limb and axial AIMs when injected in SNr (Fig. 8A) but not striatum (Fig. 8B). In both cases, however, UFP-101 prevented the antidyskinetic effect of locally injected N/OFQ.
NOP receptor binding associated with parkinsonism and dyskinesias
To investigate whether LID is associated with changes of NOP receptor binding, quantitative autoradiography was used (Fig. 9). In sham-operated rats, binding appeared highest in motor cortex (M1 and M2), intermediate in striatum and SN, and lowest in the subthalamic nucleus of (Fig. 9A,E) (Table 1). 6-OHDA lesioning (Fig. 9B,F) caused an increase of ipsilateral binding throughout the striatum and a reduction in both SN subdivisions (Fig. 9D,H). Similar patterns were observed in the brain of dyskinetic rats (Fig. 9C,D,G–H).
Table 1.
Autoradiographical binding of 125I[Tyr14]N/OFQ to the NOP receptor in cortical and subcortical areas of sham-operated, untreated 6-OHDA lesioned (hemiparkinsonian), and dyskinetic rats
| Sham operated | Hemiparkinsonian | Dyskinetic | |
|---|---|---|---|
| Level AP −0.30 to −0.92 mm | |||
| M1 | |||
| Ipsi | 71.6 ± 6.7 | 89.8 ± 14.9 | 94.6 ± 6.1 |
| C/L | 77.2 ± 7.6 | 83.8 ± 14.1 | 90.6 ± 6.2 |
| M2 | |||
| Ipsi | 80.8 ± 8.6 | 90.9 ± 14.9 | 100.3 ± 6.8 |
| C/L | 81.1 ± 6.1 | 90.0 ± 13.7 | 97.5 ± 6.9 |
| CPuDL | |||
| Ipsi | 22.7 ± 4.8 | 30.6 ± 6.2 | 31.5 ± 3.5 |
| C/L | 23.0 ± 4.9 | 20.9 ± 4.5 | 21.5 ± 3.0 |
| CPuVL | |||
| Ipsi | 24.8 ± 4.7 | 30.1 ± 3.9 | 34.3 ± 3.1 |
| C/L | 25.9 ± 4.7 | 22.5 ± 3.8 | 25.9 ± 2.8 |
| CPuDM | |||
| Ipsi | 24.8 ± 4.9 | 28.6 ± 3.0 | 30.4 ± 3.4 |
| C/L | 26.1 ± 4.7 | 19.2 ± 1.5 | 21.6 ± 3.1 |
| CPuCe | |||
| Ipsi | 27.0 ± 6.4 | 27.7 ± 4.2 | 30.6 ± 3.7 |
| C/L | 27.7 ± 6.5 | 20.7 ± 3.4 | 23.1 ± 3.6 |
| CPuVM | |||
| Ipsi | 29.7 ± 3.0 | 39.5 ± 6.3 | 36.9 ± 3.1 |
| C/L | 29.5 ± 3.1 | 30.0 ± 5.3 | 27.1 ± 3.0 |
| Level AP −3.60 to −5.80 mm | |||
| M1 | |||
| Ipsi | 72.4 ± 6.8 | 89.7 ± 13.9 | 96.6 ± 7.2 |
| C/L | 75.4 ± 8.6 | 84.8 ± 12.1 | 94.6 ± 7.3 |
| M2 | |||
| Ipsi | 78.4 ± 5.0 | 85.7 ± 14.0 | 90.1 ± 7.5 |
| C/L | 76.4 ± 8.6 | 84.8 ± 13.0 | 91.0 ± 7.8 |
| STN | |||
| Ipsi | 17.4 ± 1.7 | 32.3 ± 6.1 | 35.4 ± 7.0 |
| C/L | 18.5 ± 4.1 | 30.9 ± 6.0 | 38.5 ± 7.4 |
| SNr | |||
| Ipsi | 25.5 ± 1.3 | 16.0 ± 4.6 | 16.6 ± 4.4 |
| C/L | 25.1 ± 2.7 | 28.1 ± 4.9 | 26.8 ± 4.7 |
| SNc | |||
| Ipsi | 22.9 ± 0.6 | 17.7 ± 3.9 | 19.9 ± 3.4 |
| C/L | 24.6 ± 1.8 | 34.7 ± 3.2 | 32.1 ± 4.8 |
Twenty micrometer coronal sections were cut at two different brain levels from bregma: AP −0.30 to −0.92 and −3.60 to −5.80. Absolute binding values are given as fmol/mg tissue. Ce, Cerebellar; CPu, caudate-putamen; DL, dorsolateral; DM, dorsomedial; VL, ventrolateral; VM, ventromedial.
NOP receptor agonists in dyskinetic nonhuman primates
To confirm the antidyskinetic potential of NOP receptor agonists, Ro 65–6570 was systemically injected in MPTP-lesioned dyskinetic macaques (Fig. 10A). Ro 65–6570 significantly reduced dyskinetic movements at 0.01 mg/kg (i.m.) but not lower dose (Fig. 9B). Ro 65–6570 was effective against dystonia, although a trend for inhibition of chorea was clearly observed (Fig. 10B). Time-course analysis (Fig. 10C,D) revealed that Ro 65–6570 attenuated the peak and shortened the duration of dyskinetic movements. Ro 65–6570 did not affect the therapeutic effects of l-DOPA on movement range, bradykinesia, and tremor, and even improved posture at the higher dose (Table 2).
Table 2.
Motor behavior of MPTP-treated macaques before (OFF score) and after (ON score) l-DOPA and Ro 65–6570 administration
| OFF score | ON score |
|||
|---|---|---|---|---|
| Saline | Ro 65–6570 0.001 mg/kg | Ro 65–6570 0.01 mg/kg | ||
| Chorea | 0.0 ± 0.0 | 23.8 ± 3.3 | 16.8 ± 3.5 | 15.4 ± 4.1 |
| Dystonia | 0.0 ± 0.0 | 23.8 ± 3.3 | 15.8 ± 3.6 | 16.0 ± 4.4* |
| Total dyskinesia | 0.0 ± 0.0 | 47.6 ± 6.6 | 32.8 ± 7.1 | 31.4 ± 8.4* |
| Movement | 2.4 ± 2.4 | 39.4 ± 1.9 | 39.4 ± 1.5 | 40.0 ± 1.7 |
| Bradykinesia | 36.0 ± 0.0 | 9.4 ± 1.7 | 8.6 ± 2.2 | 8.6 ± 3.0 |
| Posture | 19.2 ± 2.9 | 7.2 ± 1.5 | 3.6 ± 1.6 | 1.8 ± 1.1* |
| Tremor | 2.4 ± 2.4 | 0.6 ± 0.6 | 1.0 ± 0.5 | 3.2 ± 1.4 |
| Total parkinsonian disability | 103.2 ± 6.1 | 25.8 ± 4.7 | 21.8 ± 4.9 | 21.4 ± 6.2 |
Five stably parkinsonian macaques were made dyskinetic by chronic l-DOPA treatment. Macaques were then challenged with l-DOPA combined with Ro 65–6570 (0.001 and 0.01 mg/kg, i.m.) or saline. Parkinsonian condition was assessed on a parkinsonian nonhuman primate rating scale scoring movement range, bradykinesia, posture, tremor, and total parkinsonian disability (see Materials and Methods). Dyskinetic condition was assessed on a Dyskinesia Disability Scale evaluating choreiform, dystonic, and total dyskinesia movements (see Materials and Methods). Data are means ± means from five animals. Statistical analysis was performed by nonparametric one-way RM ANOVA followed by Dunn's test for multiple comparisons.
*p < 0.05, versus saline.
Discussion
Opposite adaptive changes of N/OFQ transmission have been observed in the basal ganglia of the parkinsonian rodent brain: elevation of N/OFQ expression (and release) accompanied by reduction of NOP receptor binding in SNr, and reduction of N/OFQ expression accompanied by elevation of NOP receptor binding in striatum. These changes underlie the different behavioral responses to NOP receptor ligands in PD models. Indeed, we have previously reported that NOP receptor antagonists exert a symptomatic antiparkinsonian effect (Marti et al., 2005, 2008; Viaro et al., 2008, 2010; Visanji et al., 2008; Volta et al., 2010), and that such effect is mediated by blockade of SNr NOP receptors (Marti et al., 2005, 2010). We now report that NOP receptor agonists provide an antidyskinetic effect, which is faithfully replicated by intrastriatal N/OFQ injections, suggesting the involvement of striatal NOP receptors. LIDs are associated with hyperesponsiveness of upregulated D1 receptors on striatonigral MSNs (Aubert et al., 2005; Berthet et al., 2009) leading to an increased activity along the Ras/MEK/ERK kinase pathway (Valjent et al., 2005; Feyder et al., 2011) and a loss of neuron capability to depotentiate striatal synaptic response after LTP induction (Picconi et al., 2003). In vitro and in vivo data revealed that NOP receptor activation opposes D1 signaling in striatum. Indeed, N/OFQ prevented the D1 receptor-mediated increase in ERK phosphorylation and loss of striatal depotentiation of striatal plasticity in striatal MSNs, without being per se effective. These data add to the previous finding that N/OFQ inhibited the D1 receptor stimulated cAMP accumulation in striatal slices (Olianas et al., 2008) and suggest the existence of a negative interaction between the NOP and D1 receptor on the membranes of striatal MSNs. The NOP receptor is expressed not only presynaptically on nigrostriatal DA terminals (Norton et al., 2002) but also postsynaptically on striatal GABAergic MSNs neurons (Olianas et al., 2008). The postsynaptic action of N/OFQ might be favored under dyskinetic conditions, since in the striatum of l-DOPA primed and unprimed 6-OHDA hemilesioned rats, NOP receptor binding is upregulated, probably to compensate for the reduction of N/OFQ expression following DA denervation (Marti et al., 2010). Thus, NOP receptor agonists would reinstate an inhibitory control mediated by endogenous N/OFQ over striatal D1 signaling, which has been lost following DA denervation.
Indirect evidence that the negative NOP receptor modulation of D1 signaling is relevant for in vivo conditions was provided by microdialysis experiments showing that centrally administered N/OFQ attenuated LID simultaneously with the surge of GABA levels in SNr. In fact, the same responses were observed in dyskinetic rats by pairing systemic l-DOPA with intrastriatal perfusion of a D1 receptor antagonist (Mela et al., 2012). This endorses the view that the antidyskinetic effect of N/OFQ is possibly mediated by upregulated postsynaptic NOP receptors opposing the overactivation of the direct striatonigral pathway induced by l-DOPA via D1 receptors. Consistently, microdialysis also revealed that LID was associated with inhibition of thalamic GABA release, and that the antidyskinetic action of N/OFQ was accompanied by blockade of this response. Indeed, inhibition of thalamic GABA might reflect overinhibition of nigrothalamic neurons by direct striatonigral pathway activation, as previously demonstrated for the anti-akinetic effect of l-DOPA in hemiparkinsonian rats (Marti et al., 2007). This lends neurochemical support to the notion that overinhibition of nigrothalamic GABA neurons by striatonigral direct pathway activation underlies both the motor promoting and prodyskinetic actions of l-DOPA.
Remarkably, the antidyskinetic action of N/OFQ was not accompanied by motor inhibition. Indeed, it is well known that one of the main consequences of central NOP receptor stimulation is motor impairment (Reinscheid et al., 1995; Devine et al., 1996; Jenck et al., 1997; Marti et al., 2004, 2009). However, the antidyskinetic effect of N/OFQ and Ro 65–6570 was observed at doses approximately 30- to 100-fold lower than those causing hypolocomotion, suggesting it was not related to motor impairment. Thus, NOP receptor agonists might behave as functional DA antagonists that can be dosed in a way to improve LID without affecting movement in the OFF state.
N/OFQ also attenuated dyskinesia when injected into SNr. Indeed, SNr is involved in LID (Meissner et al., 2006; Westin et al., 2006; Lindgren et al., 2010). In particular, conversion of l-DOPA to DA in SNr (Sarre et al., 1998) generates abnormal nigral DA levels (Lindgren et al., 2010) thereby triggering dyskinetic movements through activation of both D1 and D2 nigral receptors (Mela et al., 2012). However, different from striatum, the hypolocomotive and antidyskinetic doses of N/OFQ given intranigrally largely overlapped, suggesting that attenuation of LID is due to unspecific motor inhibition. Indeed, exogenous N/OFQ microinjected in the SNr of naive rats inhibits movement (Marti et al., 2004, 2009), possibly through elevation of the excitatory Glu (Marti et al., 2002b) and attenuation of the inhibitory GABA inputs (Marti et al., 2007; Mabrouk et al., 2010) impinging over nigrothalamic neurons. Alternatively, we could speculate that dyskinesia pathways in SNr are less sensitive to N/OFQ, or even that N/OFQ itself is less active in the DA-depleted SNr. Indeed, the enhancement of the N/OFQ tone in SNr along with the reduction of the NOP receptor binding (present study) and expression following DA depletion (Marti et al., 2005, 2010) predict a higher NOP receptor saturation by endogenous N/OFQ, which favors the motor promoting and antiparkinsonian action of NOP receptor antagonists (Marti et al., 2007, 2008; Volta et al., 2011).
Previous studies have demonstrated the potential therapeutic benefit of a combination of subthreshold doses of an NOP receptor antagonist with l-DOPA in parkinsonian rodents (Marti et al., 2007; Viaro et al., 2010). We now report that an NOP receptor antagonist given in SNr worsens dyskinesia severity when combined with dyskinesiogenic doses of l-DOPA. This corroborates the view that NOP receptor blockade in SNr potentiates the antiparkinsonian action of l-DOPA at the cost of inducing dyskinesia, as also observed in marmosets (Visanji et al., 2008). Although the prodyskinetic effect induced by intracerebroventricular UFP-101 and systemic J-113397 was mild, limited to limb dyskinesia and observed at maximal antagonist doses, this finding warns of the potential motor side effects of overdosing NOP antagonists as an adjunct to l-DOPA therapy.
The antidyskinetic effect of Ro 65–6570 was also replicated in the gold-standard model of LID, the dyskinetic macaque. The effect of Ro 65–6570 was milder than in rats, but occurred in the same dose range. Axial symptoms such as gait and balance disturbances are very common and disabling in PD patients (Jankovic, 2008). The finding that an NOP agonist improved posture in l-DOPA-treated dyskinetic macaques might contrast with previous findings that an NOP antagonist improved balance in MPTP-treated l-DOPA-unprimed macaques (Viaro et al., 2008). The apparent contradiction that an NOP agonist and an antagonist have the same impact on axial symptoms might be explained on the basis of an action at NOP receptors (possibly located in different brain areas/circuits) that are differently saturated by endogenous N/OFQ. Moreover, we should recall that the models and timing of administration were different in the two studies. Indeed, the overall behavioral patterns were different since the NOP antagonist also improved bradykinesia, tremor, hypokinesia, and rigidity, whereas the NOP agonist did not.
Concluding remarks
A major side effect of long-term therapy with l-DOPA is the development of dyskinesias (Nutt and Gancher, 1994; Bezard et al., 2001a; Obeso et al., 2004; Calabresi et al., 2010), which represent a clinical challenge due to the poor therapeutic armamentarium available to neurologists, essentially limited to amantadine (Verhagen Metman et al., 1998). Here we report that exogenous N/OFQ and the small molecule NOP receptor agonist Ro 65–6570 inhibited LID expression in dyskinetic rats and macaques without attenuating the antiparkinsonian effect of l-DOPA. We also provide evidence that N/OFQ inhibited biochemical, electrophysiological, and neurochemical correlates of striatal D1 receptor activation in the naive or dyskinetic striatum, which suggests that NOP receptor agonists exert a selective antidyskinetic effect by activating striatal NOP receptors that oppose D1 signaling. However, further mechanistic studies in 6-OHDA lesioned dyskinetic and nondyskinetic rats need to be performed to confirm this hypothesis. The possibility that NOP receptor agonists exert an antidyskinetic effect through striatal NOP receptor does not contradict our previous finding that NOP receptor antagonists exert antiparkinsonian effects through blockade of NOP receptors in SNr. Indeed, an NOP receptor antagonist is expected to act primarily where the N/OFQ tone is elevated (i.e., in SNr), whereas an NOP receptor agonist where N/OFQ is low or absent (i.e., in striatum).
In conclusion, NOP receptor agonists have been proven effective for treating pain (Ko et al., 2009), anxiety (Jenck et al., 1997), and drug dependence (Zaveri, 2011). The concordance of the antidyskinetic effects in rats and nonhuman primates suggests that NOP receptor agonists may also prove effective in attenuating LID and rescuing motor activity in dyskinetic PD patients.
Footnotes
This work was supported by the Italian Ministry of the University (FIRB n. RBIN047W33, PRIN 2008 n. 2008EJ85RS to M.M.) and the Italian Ministry of Health (Progetto Giovani Ricercatori 2008).
The authors declare no competing financial interests.
References
- Andersson M, Hilbertson A, Cenci MA. Striatal fosB expression is causally linked with l-DOPA-induced abnormal involuntary movements and the associated upregulation of striatal prodynorphin mRNA in a rat model of Parkinson's disease. Neurobiol Dis. 1999;6:461–474. doi: 10.1006/nbdi.1999.0259. [DOI] [PubMed] [Google Scholar]
- Aubert I, Guigoni C, Håkansson K, Li Q, Dovero S, Barthe N, Bioulac BH, Gross CE, Fisone G, Bloch B, Bezard E. Increased D1 dopamine receptor signaling in levodopa-induced dyskinesia. Ann Neurol. 2005;57:17–26. doi: 10.1002/ana.20296. [DOI] [PubMed] [Google Scholar]
- Berthele A, Platzer S, Dworzak D, Schadrack J, Mahal B, Büttner A, Assmus HP, Wurster K, Zieglgänsberger W, Conrad B, Tölle TR. [3H]-nociceptin ligand-binding and nociceptin opioid receptor mrna expression in the human brain. Neuroscience. 2003;121:629–640. doi: 10.1016/s0306-4522(03)00484-6. [DOI] [PubMed] [Google Scholar]
- Berthet A, Porras G, Doudnikoff E, Stark H, Cador M, Bezard E, Bloch B. Pharmacological analysis demonstrates dramatic alteration of D1 dopamine receptor neuronal distribution in the rat analog of L-DOPA-induced dyskinesia. J Neurosci. 2009;29:4829–4835. doi: 10.1523/JNEUROSCI.5884-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezard E, Brotchie JM, Gross CE. Pathophysiology of levodopa-induced dyskinesia: potential for new therapies. Nat Rev Neurosci. 2001a;2:577–588. doi: 10.1038/35086062. [DOI] [PubMed] [Google Scholar]
- Bezard E, Boraud T, Chalon S, Brotchie JM, Guilloteau D, Gross CE. Pallidal border cells: an anatomical and electrophysiological study in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-treated monkey. Neuroscience. 2001b;103:117–123. doi: 10.1016/s0306-4522(00)00546-7. [DOI] [PubMed] [Google Scholar]
- Bézard E, Ferry S, Mach U, Stark H, Leriche L, Boraud T, Gross C, Sokoloff P. Attenuation of levodopa-induced dyskinesia by normalizing dopamine D3 receptor function. Nat Med. 2003;9:762–767. doi: 10.1038/nm875. [DOI] [PubMed] [Google Scholar]
- Bridge KE, Wainwright A, Reilly K, Oliver KR. Autoradiographic localization of (125)i[Tyr(14)] nociceptin/orphanin FQ binding sites in macaque primate CNS. Neuroscience. 2003;118:513–523. doi: 10.1016/s0306-4522(02)00927-2. [DOI] [PubMed] [Google Scholar]
- Calabresi P, Pisani A, Mercuri NB, Bernardi G. Long-term potentiation in the striatum is unmasked by removing the voltage-dependent magnesium block of NMDA receptor channels. Eur J Neurosci. 1992;4:929–935. doi: 10.1111/j.1460-9568.1992.tb00119.x. [DOI] [PubMed] [Google Scholar]
- Calabresi P, Centonze D, Pisani A, Sancesario G, Gubellini P, Marfia GA, Bernardi G. Striatal spiny neurons and cholinergic interneurons express differential ionotropic glutamatergic responses and vulnerability: implications for ischemia and Huntington's disease. Ann Neurol. 1998;43:586–597. doi: 10.1002/ana.410430506. [DOI] [PubMed] [Google Scholar]
- Calabresi P, Di Filippo M, Ghiglieri V, Tambasco N, Picconi B. Levodopa-induced dyskinesias in patients with Parkinson's disease: filling the bench-to-bedside gap. Lancet Neurol. 2010;9:1106–1117. doi: 10.1016/S1474-4422(10)70218-0. [DOI] [PubMed] [Google Scholar]
- Calò G, Guerrini R, Rizzi A, Salvadori S, Regoli D. Pharmacology of nociceptin and its receptor: a novel therapeutic target. Br J Pharmacol. 2000;129:1261–1283. doi: 10.1038/sj.bjp.0703219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calo G, Rizzi A, Rizzi D, Bigoni R, Guerrini R, Marzola G, Marti M, McDonald J, Morari M, Lambert DG, Salvadori S, Regoli D. [Nphe1,Arg14,Lys15]nociceptin-NH2, a novel potent and selective antagonist of the nociceptin/orphanin FQ receptor. Br J Pharmacol. 2002;136:303–311. doi: 10.1038/sj.bjp.0704706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carta M, Carlsson T, Kirik D, Björklund A. Dopamine released from 5-HT terminals is the cause of L-DOPA-induced dyskinesia in parkinsonian rats. Brain. 2007;130:1819–1833. doi: 10.1093/brain/awm082. [DOI] [PubMed] [Google Scholar]
- Cenci MA, Lundblad M. Ratings of L-DOPA-induced dyskinesia in the unilateral 6-OHDA lesion model of Parkinson's disease in rats and mice. Curr Protoc Neurosci Chapter. 2007;9 doi: 10.1002/0471142301.ns0925s41. Unit 9.25. [DOI] [PubMed] [Google Scholar]
- Cenci MA, Lee CS, Björklund A. L-DOPA-induced dyskinesia in the rat is associated with striatal overexpression of prodynorphin- and glutamic acid decarboxylase mRNA. Eur J Neurosci. 1998;10:2694–2706. [PubMed] [Google Scholar]
- Devine DP, Reinscheid RK, Monsma FJ, Jr, Civelli O, Akil H. The novel neuropeptide orphanin FQ fails to produce conditioned place preference or aversion. Brain Res. 1996;727:225–229. doi: 10.1016/0006-8993(96)00476-3. [DOI] [PubMed] [Google Scholar]
- Fasano S, Bezard E, D'Antoni A, Francardo V, Indrigo M, Qin L, Doveró S, Cerovic M, Cenci MA, Brambilla R. Inhibition of Ras-guanine nucleotide-releasing factor 1 (Ras-GRF1) signaling in the striatum reverts motor symptoms associated with L-dopa-induced dyskinesia. Proc Natl Acad Sci U S A. 2010;107:21824–21829. doi: 10.1073/pnas.1012071107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernagut PO, Li Q, Dovero S, Chan P, Wu T, Ravenscroft P, Hill M, Chen Z, Bezard E. Dopamine transporter binding is unaffected by L-DOPA administration in normal and MPTP-treated monkeys. PLoS One. 2010;5:e14053. doi: 10.1371/journal.pone.0014053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feyder M, Bonito-Oliva A, Fisone G. L-DOPA-induced dyskinesia and abnormal signaling in striatal medium spiny neurons: focus on dopamine D1 receptor-mediated transmission. Front Behav Neurosci. 2011;5:71. doi: 10.3389/fnbeh.2011.00071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flau K, Redmer A, Liedtke S, Kathmann M, Schlicker E. Inhibition of striatal and retinal dopamine release via nociceptin/orphanin FQ receptors. Br J Pharmacol. 2002;137:1355–1361. doi: 10.1038/sj.bjp.0704998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouty S, Brown JM, Rosenberger J, Cox BM. MPTP treatment increases expression of pre-pro-nociceptin/orphanin FQ mRNA in a subset of substantia nigra reticulata neurons. Neuroscience. 2010;169:269–278. doi: 10.1016/j.neuroscience.2010.04.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guigoni C, Dovero S, Aubert I, Li Q, Bioulac BH, Bloch B, Gurevich EV, Gross CE, Bezard E. Levodopa-induced dyskinesia in MPTP-treated macaques is not dependent on the extent and pattern of nigrostrial lesioning. Eur J Neurosci. 2005;22:283–287. doi: 10.1111/j.1460-9568.2005.04196.x. [DOI] [PubMed] [Google Scholar]
- Hunter WM, Greenwood FC. Preparation of iodine-131 labelled human growth hormone of high specific activity. Nature. 1962;194:495–496. doi: 10.1038/194495a0. [DOI] [PubMed] [Google Scholar]
- Jankovic J. Parkinson's disease: clinical features and diagnosis. J Neurol Neurosurg Psychiatry. 2008;79:368–376. doi: 10.1136/jnnp.2007.131045. [DOI] [PubMed] [Google Scholar]
- Jenck F, Moreau JL, Martin JR, Kilpatrick GJ, Reinscheid RK, Monsma FJ, Jr, Nothacker HP, Civelli O. Orphanin FQ acts as an anxiolytic to attenuate behavioral responses to stress. Proc Natl Acad Sci U S A. 1997;94:14854–14858. doi: 10.1073/pnas.94.26.14854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamoto H, Ozaki S, Itoh Y, Miyaji M, Arai S, Nakashima H, Kato T, Ohta H, Iwasawa Y. Discovery of the first potent and selective small molecule opioid receptor-like (ORL1) antagonist: 1-[(3R,4R)-1-cyclooctylmethyl-3-hydroxymethyl-4-piperidyl]-3-ethyl-1, 3-dihydro-2H-benzimidazol-2-one (J-113397) J Med Chem. 1999;42:5061–5063. doi: 10.1021/jm990517p. [DOI] [PubMed] [Google Scholar]
- Ko MC, Woods JH, Fantegrossi WE, Galuska CM, Wichmann J, Prinssen EP. Behavioral effects of a synthetic agonist selective for nociceptin/orphanin FQ peptide receptors in monkeys. Neuropsychopharmacology. 2009;34:2088–2096. doi: 10.1038/npp.2009.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert DG. The nociceptin/orphanin FQ receptor: a target with broad therapeutic potential. Nat Rev Drug Discov. 2008;7:694–710. doi: 10.1038/nrd2572. [DOI] [PubMed] [Google Scholar]
- Lindgren HS, Andersson DR, Lagerkvist S, Nissbrandt H, Cenci MA. L-DOPA-induced dopamine efflux in the striatum and the substantia nigra in a rat model of Parkinson's disease: temporal and quantitative relationship to the expression of dyskinesia. J Neurochem. 2010;112:1465–1476. doi: 10.1111/j.1471-4159.2009.06556.x. [DOI] [PubMed] [Google Scholar]
- Lundblad M, Andersson M, Winkler C, Kirik D, Wierup N, Cenci MA. Pharmacological validation of behavioural measures of akinesia and dyskinesia in a rat model of Parkinson's disease. Eur J Neurosci. 2002;15:120–132. doi: 10.1046/j.0953-816x.2001.01843.x. [DOI] [PubMed] [Google Scholar]
- Mabrouk OS, Marti M, Morari M. Endogenous nociceptin/orphanin FQ (N/OFQ) contributes to haloperidol-induced changes of nigral amino acid transmission and parkinsonism: a combined microdialysis and behavioral study in naive and nociceptin/orphanin FQ receptor knockout mice. Neuroscience. 2010;166:40–48. doi: 10.1016/j.neuroscience.2009.12.006. [DOI] [PubMed] [Google Scholar]
- Marti M, Mela F, Bianchi C, Beani L, Morari M. Striatal dopamine-NMDA receptor interactions in the modulation of glutamate release in the substantia nigra pars reticulata in vivo: opposite role for D1 and D2 receptors. J Neurochem. 2002a;83:635–644. doi: 10.1046/j.1471-4159.2002.01169.x. [DOI] [PubMed] [Google Scholar]
- Marti M, Guerrini R, Beani L, Bianchi C, Morari M. Nociceptin/orphanin FQ receptors modulate glutamate extracellular levels in the substantia nigra pars reticulata. A microdialysis study in the awake freely moving rat. Neuroscience. 2002b;112:153–160. doi: 10.1016/s0306-4522(02)00050-7. [DOI] [PubMed] [Google Scholar]
- Marti M, Mela F, Veronesi C, Guerrini R, Salvadori S, Federici M, Mercuri NB, Rizzi A, Franchi G, Beani L, Bianchi C, Morari M. Blockade of nociceptin/orphanin FQ receptor signaling in rat substantia nigra pars reticulata stimulates nigrostriatal dopaminergic transmission and motor behavior. J Neurosci. 2004;24:6659–6666. doi: 10.1523/JNEUROSCI.0987-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marti M, Mela F, Fantin M, Zucchini S, Brown JM, Witta J, Di Benedetto M, Buzas B, Reinscheid RK, Salvadori S, Guerrini R, Romualdi P, Candeletti S, Simonato M, Cox BM, Morari M. Blockade of nociceptin/orphanin FQ transmission attenuates symptoms and neurodegeneration associated with Parkinson's disease. J Neurosci. 2005;25:9591–9601. doi: 10.1523/JNEUROSCI.2546-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marti M, Trapella C, Viaro R, Morari M. The nociceptin/orphanin FQ receptor antagonist J-113397 and L-DOPA additively attenuate experimental parkinsonism through overinhibition of the nigrothalamic pathway. J Neurosci. 2007;27:1297–1307. doi: 10.1523/JNEUROSCI.4346-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marti M, Trapella C, Morari M. The novel nociceptin/orphanin FQ receptor antagonist Trap-101 alleviates experimental parkinsonism through inhibition of the nigro-thalamic pathway: positive interaction with L-DOPA. J Neurochem. 2008;107:1683–1696. doi: 10.1111/j.1471-4159.2008.05735.x. [DOI] [PubMed] [Google Scholar]
- Marti M, Viaro R, Guerrini R, Franchi G, Morari M. Nociceptin/orphanin FQ modulates motor behavior and primary motor cortex output through receptors located in substantia nigra reticulata. Neuropsychopharmacology. 2009;34:341–355. doi: 10.1038/npp.2008.56. [DOI] [PubMed] [Google Scholar]
- Marti M, Sarubbo S, Latini F, Cavallo M, Eleopra R, Biguzzi S, Lettieri C, Conti C, Simonato M, Zucchini S, Quatrale R, Sensi M, Candeletti S, Romualdi P, Morari M. Brain interstitial nociceptin/orphanin FQ levels are elevated in Parkinson's disease. Mov Disord. 2010;25:1723–1732. doi: 10.1002/mds.23271. [DOI] [PubMed] [Google Scholar]
- Meissner W, Ravenscroft P, Reese R, Harnack D, Morgenstern R, Kupsch A, Klitgaard H, Bioulac B, Gross CE, Bezard E, Boraud T. Increased slow oscillatory activity in substantia nigra pars reticulata triggers abnormal involuntary movements in the 6-OHDA-lesioned rat in the presence of excessive extracellular striatal dopamine. Neurobiol Dis. 2006;22:586–598. doi: 10.1016/j.nbd.2006.01.009. [DOI] [PubMed] [Google Scholar]
- Mela F, Millan MJ, Brocco M, Morari M. The selective D(3) receptor antagonist, S33084, improves parkinsonian-like motor dysfunction but does not affect L-DOPA-induced dyskinesia in 6-hydroxydopamine hemi-lesioned rats. Neuropharmacology. 2010;58:528–536. doi: 10.1016/j.neuropharm.2009.08.017. [DOI] [PubMed] [Google Scholar]
- Mela F, Marti M, Bido S, Cenci MA, Morari M. In vivo evidence for a differential contribution of striatal and nigral D1 and D2 receptors to l-DOPA induced dyskinesia and the accompanying surge of nigral amino acid levels. Neurobiol Dis. 2012;45:573–582. doi: 10.1016/j.nbd.2011.09.015. [DOI] [PubMed] [Google Scholar]
- Meunier JC, Mollereau C, Toll L, Suaudeau C, Moisand C, Alvinerie P, Butour JL, Guillemot JC, Ferrara P, Monsarrat B. Isolation and structure of the endogenous agonist of opioid receptor-like ORL1 receptor. Nature. 1995;377:532–535. doi: 10.1038/377532a0. [DOI] [PubMed] [Google Scholar]
- Mogil JS, Pasternak GW. The molecular and behavioral pharmacology of the orphanin FQ/nociceptin peptide and receptor family. Pharmacol Rev. 2001;53:381–415. [PubMed] [Google Scholar]
- Muñoz A, Li Q, Gardoni F, Marcello E, Qin C, Carlsson T, Kirik D, Di Luca M, Björklund A, Bezard E, Carta M. Combined 5-HT1A and 5-HT1B receptor agonists for the treatment of L-DOPA-induced dyskinesia. Brain. 2008;131:3380–3394. doi: 10.1093/brain/awn235. [DOI] [PubMed] [Google Scholar]
- Navailles S, Bioulac B, Gross C, De Deurwaerdère P. Serotonergic neurons mediate ectopic release of dopamine induced by L-DOPA in a rat model of Parkinson's disease. Neurobiol Dis. 2010;38:136–143. doi: 10.1016/j.nbd.2010.01.012. [DOI] [PubMed] [Google Scholar]
- Nazzaro C, Marino S, Barbieri M, Siniscalchi A. Inhibition of serotonin outflow by nociceptin/orphaninFQ in dorsal raphe nucleus slices from normal and stressed rats: role of corticotropin releasing factor. Neurochem Int. 2009;54:378–384. doi: 10.1016/j.neuint.2009.01.004. [DOI] [PubMed] [Google Scholar]
- Neal CR, Jr, Mansour A, Reinscheid R, Nothacker HP, Civelli O, Watson SJ., Jr Localization of orphanin FQ (nociceptin) peptide and messenger RNA in the central nervous system of the rat. J Comp Neurol. 1999a;406:503–547. [PubMed] [Google Scholar]
- Neal CR, Jr, Mansour A, Reinscheid R, Nothacker HP, Civelli O, Akil H, Watson SJ., Jr Opioid receptor-like (ORL1) receptor distribution in the rat central nervous system: comparison of ORL1 receptor mRNA expression with (125)I-[(14)Tyr]-orphanin FQ binding. J Comp Neurol. 1999b;412:563–605. [PubMed] [Google Scholar]
- Nishi M, Houtani T, Noda Y, Mamiya T, Sato K, Doi T, Kuno J, Takeshima H, Nukada T, Nabeshima T, Yamashita T, Noda T, Sugimoto T. Unrestrained nociceptive response and dysregulation of hearing ability in mice lacking the nociceptin/orphaninFQ receptor. EMBO J. 1997;16:1858–1864. doi: 10.1093/emboj/16.8.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norton CS, Neal CR, Kumar S, Akil H, Watson SJ. Nociceptin/orphanin FQ and opioid receptor-like receptor mRNA expression in dopamine systems. J Comp Neurol. 2002;444:358–368. doi: 10.1002/cne.10154. [DOI] [PubMed] [Google Scholar]
- Nutt JG, Gancher ST. Parkinson's disease dyskinesias. Neurology. 1994;44:1187. doi: 10.1212/wnl.44.6.1187. author reply 1187–1188. [DOI] [PubMed] [Google Scholar]
- Obeso JA, Rodriguez-Oroz M, Marin C, Alonso F, Zamarbide I, Lanciego JL, Rodriguez-Diaz M. The origin of motor fluctuations in Parkinson's disease: importance of dopaminergic innervation and basal ganglia circuits. Neurology. 2004;62(Suppl 1):S17–S30. doi: 10.1212/wnl.62.1_suppl_1.s17. [DOI] [PubMed] [Google Scholar]
- Olianas MC, Dedoni S, Boi M, Onali P. Activation of nociceptin/orphanin FQ-NOP receptor system inhibits tyrosine hydroxylase phosphorylation, dopamine synthesis, and dopamine D(1) receptor signaling in rat nucleus accumbens and dorsal striatum. J Neurochem. 2008;107:544–556. doi: 10.1111/j.1471-4159.2008.05629.x. [DOI] [PubMed] [Google Scholar]
- Orellana D, Morella I, Indrigo M, Papale A, Brambilla R. The Ras-ERK pathway in neuronal cell signalling. In: Mukai H, editor. Protein kinase technologies. New York: Springer; 2012. [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. Ed 2. Orlando, FL: Academic; 1986. [DOI] [PubMed] [Google Scholar]
- Picconi B, Centonze D, Håkansson K, Bernardi G, Greengard P, Fisone G, Cenci MA, Calabresi P. Loss of bidirectional striatal synaptic plasticity in L-DOPA-induced dyskinesia. Nat Neurosci. 2003;6:501–506. doi: 10.1038/nn1040. [DOI] [PubMed] [Google Scholar]
- Reinscheid RK, Nothacker HP, Bourson A, Ardati A, Henningsen RA, Bunzow JR, Grandy DK, Langen H, Monsma FJ, Jr, Civelli O. Orphanin-Fq— a neuropeptide that activates an opioid-like g-protein-coupled receptor. Science. 1995;270:792–794. doi: 10.1126/science.270.5237.792. [DOI] [PubMed] [Google Scholar]
- Röver S, Adam G, Cesura AM, Galley G, Jenck F, Monsma FJ, Jr, Wichmann J, Dautzenberg FM. High-affinity, non-peptide agonists for the ORL1 (orphanin FQ/nociceptin) receptor. J Med Chem. 2000;43:1329–1338. doi: 10.1021/jm991129q. [DOI] [PubMed] [Google Scholar]
- Sarre S, Vandeneede D, Ebinger G, Michotte Y. Biotransformation of L-DOPA to dopamine in the substantia nigra of freely moving rats: effect of dopamine receptor agonists and antagonists. J Neurochem. 1998;70:1730–1739. doi: 10.1046/j.1471-4159.1998.70041730.x. [DOI] [PubMed] [Google Scholar]
- Sbrenna S, Marti M, Morari M, Calò G, Guerrini R, Beani L, Bianchi C. Modulation of 5-hydroxytryptamine efflux from rat cortical synaptosomes by opioids and nociceptin. Br J Pharmacol. 2000;130:425–433. doi: 10.1038/sj.bjp.0703321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao R, Ma Z, Thakkar MM, McCarley RW, Auerbach SB. Nociceptin/orphanin FQ decreases serotonin efflux in the rat brain but in contrast to a kappa-opioid has no antagonistic effect on mu-opioid-induced increases in serotonin efflux. Neuroscience. 2007;147:106–116. doi: 10.1016/j.neuroscience.2007.02.011. [DOI] [PubMed] [Google Scholar]
- Valjent E, Pascoli V, Svenningsson P, Paul S, Enslen H, Corvol JC, Stipanovich A, Caboche J, Lombroso PJ, Nairn AC, Greengard P, Hervé D, Girault JA. Regulation of a protein phosphatase cascade allows convergent dopamine and glutamate signals to activate ERK in the striatum. Proc Natl Acad Sci U S A. 2005;102:491–496. doi: 10.1073/pnas.0408305102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhagen Metman L, Del Dotto P, van den Munckhof P, Fang J, Mouradian MM, Chase TN. Amantadine as treatment for dyskinesias and motor fluctuations in Parkinson's disease. Neurology. 1998;50:1323–1326. doi: 10.1212/wnl.50.5.1323. [DOI] [PubMed] [Google Scholar]
- Viaro R, Sanchez-Pernaute R, Marti M, Trapella C, Isacson O, Morari M. Nociceptin/orphanin FQ receptor blockade attenuates MPTP-induced parkinsonism. Neurobiol Dis. 2008;30:430–438. doi: 10.1016/j.nbd.2008.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viaro R, Marti M, Morari M. Dual motor response to l-dopa and nociceptin/orphanin FQ receptor antagonists in 1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine (MPTP) treated mice: paradoxical inhibition is relieved by D(2)/D(3) receptor blockade. Exp Neurol. 2010;223:473–484. doi: 10.1016/j.expneurol.2010.01.014. [DOI] [PubMed] [Google Scholar]
- Visanji NP, de Bie RM, Johnston TH, McCreary AC, Brotchie JM, Fox SH. The nociceptin/orphanin FQ (NOP) receptor antagonist J-113397 enhances the effects of levodopa in the MPTP-lesioned nonhuman primate model of Parkinson's disease. Mov Disord. 2008;23:1922–1925. doi: 10.1002/mds.22086. [DOI] [PubMed] [Google Scholar]
- Volta M, Mabrouk OS, Bido S, Marti M, Morari M. Further evidence for an involvement of nociceptin/orphanin FQ in the pathophysiology of Parkinson's disease: a behavioral and neurochemical study in reserpinized mice. J Neurochem. 2010;115:1543–1555. doi: 10.1111/j.1471-4159.2010.07061.x. [DOI] [PubMed] [Google Scholar]
- Volta M, Viaro R, Trapella C, Marti M, Morari M. Dopamine-nociceptin/orphanin FQ interactions in the substantia nigra reticulata of hemiparkinsonian rats: involvement of D2/D3 receptors and impact on nigro-thalamic neurons and motor activity. Exp Neurol. 2011;228:126–137. doi: 10.1016/j.expneurol.2010.12.024. [DOI] [PubMed] [Google Scholar]
- Westin JE, Lindgren HS, Gardi J, Nyengaard JR, Brundin P, Mohapel P, Cenci MA. Endothelial proliferation and increased blood-brain barrier permeability in the basal ganglia in a rat model of 3,4-dihydroxyphenyl-L-alanine-induced dyskinesia. J Neurosci. 2006;26:9448–9461. doi: 10.1523/JNEUROSCI.0944-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaveri NT. The nociceptin/orphanin FQ receptor (NOP) as a target for drug abuse medications. Curr Top Med Chem. 2011;11:1151–1156. doi: 10.2174/156802611795371341. [DOI] [PMC free article] [PubMed] [Google Scholar]