

Abstract
Mitochondria provide ATP, maintain calcium homeostasis, and regulate apoptosis. Neurons, due to their size and complex geometry, are particularly dependent on the proper functioning and distribution of mitochondria. Thus disruptions of these organelles and their transport play a central role in a broad range of neurodegenerative diseases. While in vitro studies have greatly expanded our knowledge of mitochondrial dynamics, our understanding in vivo remains limited. To address this shortcoming, we developed tools to study mitochondrial dynamics in vivo in optically accessible zebrafish. We demonstrate here that our newly generated tools, including transgenic “MitoFish,” can be used to study the in vivo “life cycle” of mitochondria and allows identifying pharmacological and genetic modulators of mitochondrial dynamics. Furthermore we observed profound mitochondrial transport deficits in real time in a zebrafish tauopathy model. By rescuing this phenotype using MARK2 (microtubule-affinity regulating kinase 2), we provide direct in vivo evidence that this kinase regulates axonal transport in a Tau-dependent manner. Thus, our approach allows detailed studies of the dynamics of mitochondria in their natural environment under normal and disease conditions.
Introduction
Mitochondria are vital for aerobic cells. Neurons in particular depend on mitochondrial function, so disrupting these organelles can cause neurological disease (Nicholls and Budd, 2000; Knott et al., 2008; Schon and Przedborski, 2011). This vulnerability results from the high metabolic demands of neurons, their dependence on proper calcium handling and their susceptibility to local proapoptotic and reactive oxygen species signaling, processes in which mitochondria are critically involved. Moreover, neurons need to match the provision, maintenance and removal of mitochondria to the distinct metabolic demands of soma, neurites and synapses. Central to this intracellular homeostasis is microtubule-mediated transport, disturbances of which also manifest as neuropathology (Hirokawa et al., 2010). Cell culture and invertebrate models have begun to reveal the regulators of mitochondrial trafficking (Goldstein et al., 2008; MacAskill and Kittler, 2010; Cai and Sheng, 2012), but many questions remain open, especially about the spatiotemporal characteristics of the mitochondrial life cycle within differentiated vertebrate neurons and during disease.
Tools to address such questions include transgenic mice with fluorescently tagged mitochondria (Chandrasekaran et al., 2006; Misgeld et al., 2007), which allow visualizing mitochondrial dynamics in vivo. The versatility of such models is limited however. Mice, while suitable disease models, have a nervous system difficult to access by microscopy, requiring surgery and preventing visualization of entire neurons. Moreover, delivering drugs or genes to the rodent nervous system can be a challenge. Parallel progress in studying neuronal mitochondria in invertebrates (Stowers et al., 2002; Pilling et al., 2006) is offset by the fact that invertebrate neurons differ from their vertebrate counterparts (e.g., in regeneration or myelination), making modeling neurological diseases difficult. We therefore sought to develop a new set of tools to study mitochondrial dynamics in living zebrafish, an optically and genetically accessible vertebrate organism increasingly used to model neuronal pathology (Kabashi et al., 2010; Paquet et al., 2010).
Here we investigate mitochondrial trafficking in Rohon-Beard (RB) neurons, sensory nerve cells proven to be useful in relating cell biology to neuronal geometry (Sagasti et al., 2005). Our time-lapse imaging studies reveal that mitochondrial transport in RB axons closely resembles transport previously described in mammalian systems. Furthermore, we compare axonal growth and transport as neurons mature. To facilitate efficient imaging of mitochondria, we generated transgenic reporter fish, MitoFish. These fish allowed us to characterize how Tau, a microtubule-binding protein associated with dementia, impacts the distribution of mitochondria. We demonstrate how pharmacological and genetic manipulations can be combined with time-lapse imaging to modulate transport and the Tau-dependent mitochondrial phenotype. This opens up the possibility of “high-content” screens for modulators of mitochondrial disease.
Materials and Methods
Animals.
We used wild-type zebrafish (AB and TLF strains) and the following transgenic fish: memYFP-UAS-mitoCFP (MitoFish, Tg(UAS-E1b:mYFP,mitoCFP)mde6, see below), HuC:Gal4/Tau-UAS-DsRed (TauFish, Tg(elavl3.2:Gal4-VP16)mde4; Tg(UAS-E1b:DsRed,Hsa.MAPT_ P301L)mde3, HuC:Gal4/UAS-DsRed (RedFish, Tg(elavl3.2:Gal4-VP16)mde4; Tg(UAS-E1b:DsRed)mde7) and Huc:Gal4 (Paquet et al., 2009, Tg(elavl3.2:Gal4-VP16)mde4). Fish were maintained, mated, and raised as previously described (Mullins et al., 1994). Embryos were kept in E3 or 0.3× Danieau's solution at 28.5°C and staged as described previously (Kimmel et al., 1995). Animals of either sex were used. All experiments were performed according to local regulations with approval by the Regierung von Oberbayern (Munich, Germany).
Single-cell labeling of RB neurons and their mitochondria.
Sensory:Gal4-VP16 (enhancer element from the islet-1 gene that drives expression of Gal4-VP16 in sensory neurons, gift from A. Sagasti, University of California, Los Angeles), UAS:memYFP (gift from R. Wong, University of Washington, Seattle, Washington) and UAS:mitoCFP were coinjected (each 5 ng/μl) into fertilized eggs of wild-type zebrafish. The UAS:mitoCFP responder construct was generated by conventional blunt-end cloning. Briefly, a fragment from pECFP-mito (Clontech), encoding a fusion of the mitochondrial targeting sequence of human cytochrome c oxidase and enhanced cyan fluorescent protein (ECFP) (mitoCFP), was digested with NheI and NotI. This mitoCFP fragment was blunted and cloned into an upstream activating sequence (UAS) expression vector (cut with EcoRI and NotI), downstream of a 14-mer UAS Gal4 binding sequence fused to a fish basal promoter, E1b (Köster and Fraser, 2001).
Generation of transgenic MitoFish.
To generate transgenic MitoFish with fluorescently labeled mitochondria and cell membranes, we replaced DsRed and the Gateway Cassette GW-R1-R2 in the vector pT2d-DEST_pA_DsRed-T4_E1b_UAS_E1b_GW-R1-R2_pA (Paquet et al., 2009) with memYFP and mitoCFP. In a first step E1b_UAS_E1b was subcloned into pCS2+_memYFP by EcoRI. Then, E1b_UAS_E1b_memYFP_pA was transferred to pT2d-DEST_pA_DsRed-T4_E1b_UAS_E1b_GW-R1-R2_pA by a StuI/PspOMI digest. mitoCFP was amplified by PCR using the following primers: mitoCFP-F Kozak 5′-GCCACCATGTCCGTCCTGACGCCG-3′ and GFP-R BamHI 5′-GGCGGCCGCGGATCCTTACTTGTACAGCTCGTCCATGC-3′, TOPO-cloned into pCR8/TOPO/GW (Invitrogen) and sequenced. mitoCFP was then LR recombined into pT2d-DEST_pA_memYFP_E1b_UAS_E1b_GW-R1-R2_pA, using LR Clonase II (Invitrogen) according to the manufacturer's instructions, which yielded the expression construct pT2d-EXP_pA_memYFP_E1b_UAS_E1b_mitoCFP_pA. Transgenic fish were generated by introducing the expression constructs into the fish genome as previously described (Paquet et al., 2009). Transgenic MitoFish were crossed to the HuC:Gal4 driver line.
Nocodazole treatment.
At 1 day post-fertilization (dpf), embryos were transferred to nocodazole (200–400 nm, Sigma) dissolved in E3 medium containing dimethyl sulfoxide (DMSO; 1%, Sigma) and 1% N-phenylthiourea (PTU, Sigma) and maintained in this solution for 24 h before imaging at 2 dpf. Control embryos were maintained in E3 medium containing DMSO (1%) and PTU (1%) without nocodazole.
In vivo imaging and data analysis.
At 24 h post-fertilization, embryos were transferred to PTU to inhibit pigmentation. At 2 dpf, manually dechorionated embryos were anesthetized using tricaine (Sigma) in medium containing PTU and embedded in low-melting agarose (0.7–0.8%, Sigma) as described by Godinho (2011). For the nocodazole-treated embryos 1% DMSO was added to both the medium and agarose. When cells were to be reimaged on the following day, embryos were unmounted, allowed to recover in PTU-containing medium overnight and remounted in agarose before imaging at 3 dpf.
To investigate mitochondrial dynamics in single cells, agarose-embedded embryos were screened for coexpression of memYFP and mitoCFP in isolated RB neurons. Mitochondrial transport was imaged in the stem axon using a wide-field fluorescence microscope (Olympus BX51W1) equipped with a 100×/numerical aperture (NA) 1.0 water-immersion dipping cone objective, an automated filter wheel (Sutter Instrument Co.), and a cooled CCD camera (either RetigaEXi, QImaging; Photometrix-CoolsnapHQ2, Visitron Systems; or SensiCam, PCO Imaging) and controlled by μManager, an open source microscopy software (www.micro-manager.org; Edelstein et al., 2010). Twelve 10 min movies were acquired with an imaging frequency of 2 s and an exposure time between 200 and 500 ms for each fish. Following transport measurements, the morphology and mitochondrial distribution of the soma, stem axon and entire peripheral axonal arbor of the RB neuron was reconstructed by confocal microscopy (Olympus FV1000, 20×/NA 0.95 water-immersion dipping cone objective).
For the bulk mitochondrial assays, sensory neurons from the caudal part of the fin fold in transgenic MitoFish were imaged using either an Olympus wide-field fluorescence setup (see above) or a Zeiss Axioplan 2 wide-field fluorescence microscope equipped with a 40×/NA 0.8 water-immersion dipping cone objective, an automated filter wheel (Zeiss) and a cooled CCD camera (Retiga 2000R, QImaging) controlled by μManager software. For each specimen, we first took single frames of each fluorescent channel and then acquired a 10 min movie of the mitoCFP channel with an imaging frequency of 1 s. An exposure time of 400 ms was chosen for all images. For all in vivo imaging experiments, neutral density filters were used to attenuate the light when necessary and embryos were maintained at 28°C for the duration of imaging.
MARK2 mRNA synthesis and injection.
To overexpress MARK2 or mutant MARK2 in zebrafish embryos, we generated mRNA using the Ambion mMESSAGE mMACHINE kit (Applied Biosystems) according to the manufacturer's instructions. mRNA was injected at 300 ng/μl into one-cell-stage embryos. Plasmids used encode human wild-type or A208/A212 double-mutant MARK2 (pcDNA3.1_HA-MARK-WT or T208A/S212A, respectively) fused with one or several hemagglutinin (HA) tags for post hoc antibody detection (kind gift from the Mandelkow laboratory; Drewes et al., 1997). Tagging does not alter the expected activity of overexpressed MARK2 (E. M. Mandelkow, DZNE, Bonn, Germany, personal communication). To select MARK2-expressing embryos we coinjected mRNA encoding enhanced green fluorescent protein (GFP; 100 ng/μl) and screened for GFP-positive embryos at 1 dpf. This allows nonspecific effects of mRNA injection to be controlled for. The mRNA-based expression of GFP fades away after 1 dpf, and hence does not interfere with the transport assay.
Western blot.
To analyze MARK2 and Tau levels in TauFish, individual larvae were unmounted after imaging and used for Western blot analysis as previously described (Paquet et al., 2009). The following antibodies were used: K9JA (1:10,000; A0024; Dako), HA-11 (1:1000; Clone 16B12; Eurogentec) and anti-α-Tubulin (1:8000; T6199; Sigma-Aldrich).
Image analysis and processing.
Images were viewed and processed using open-source ImageJ/Fiji software (http://fiji.sc). To represent the spatial location of individual mitochondria over time within the stem axon of RB neurons, we generated kymographs using the Reslice tool with a slice spacing of 1 pixel. To determine the transport flux in single RB neurons, we determined the number of anterogradely and retrogradely moving mitochondria crossing a vertical line drawn across the stem axon.
Mitochondrial volume was calculated under the assumption that a mitochondrion has a cylindrical shape. Half of the mitochondrial width was taken as the radius of the base (r) of the cylinder and the length of the mitochondrion as its height (H). To calculate volume (V) we used the following equation: V = H*π*r2.
To determine the transport characteristics of individual mitochondria, we used the MTrackJ plug-in (ImageJ/Fiji; developed by E. Meijering, Biomedical Imaging Group, Erasmus Medical Center, Rotterdam, The Netherlands). We measured the average and moving speed as well as pause frequency and average pause length. A mitochondrion that moved <0.25 μm between frames was regarded as having paused, while the periods of movement between pauses were regarded as “runs.” “Average speed” was defined as the maximal displacement of a mitochondrion divided by the time over which it was observed, while “moving speed” was defined as displacement during uninterrupted runs divided by the duration of the runs. We divided the number of pauses during a period by the duration of that period to calculate pause frequency and we defined the average time a mitochondrion spent stopping by calculating the average length of pauses during a period of measurement. In our analyses we only included mitochondria that could be tracked for at least 8 frames and that were in motion at the beginning and end of the movies.
To determine mitochondrial density in single neurons, the total number of mitochondria in the peripheral axon was counted. The length of the peripheral axon was measured from maximum intensity projections of confocal stacks using the Simple Neurite Tracer plug-in in Fiji/ImageJ (Longair et al., 2011). The peripheral axon was divided into 3 segments, stem, core and periphery. The axonal segment emanating from the soma but before the first branching point was regarded as the stem. The arbor was divided into a core and periphery, the latter being the very last branches of the arbor that did not branch themselves. Mitochondrial density upon nocodazole treatment was normalized to DMSO control values to allow direct comparisons of drug treatments across compartments.
For the bulk mitochondrial assays in transgenic MitoFish, wide-field images were cropped to a region of interest, in which most neurites were in focus. Only images with a total neurite length >2500 pixels (732 μm) after cropping were used for analysis. To determine mitochondrial density, the number of mitochondria and length of the neurites were measured as described above. Movements of individual mitochondria were analyzed frame by frame by determining the start and end time-points, the location of the movement and the length of the neurite between these two time-points. From these measurements, we calculated the flux density (density of moving mitochondria) per fish. More detailed analysis of mitochondrial movement parameters was performed using the MTrackJ plugin (see above). For all movement events, we set a minimum inclusion criterion of 35 pixels (10 μm) movement distance.
Image panels were assembled with Photoshop software (Adobe) and combined into figures using Illustrator (Adobe). Tiled microscopy images were montaged in Photoshop; the “despeckle” function was used to suppress detector noise for clarity and gamma was adjusted nonlinearly to enhance visibility of low-contrast objects.
Statistics.
Mean values and SEM were calculated using Microsoft Excel. Data series were tested for normal distribution using the D'Agostino-Pearson test. t tests were used to compare datasets that had normal distributions. Anterograde and retrograde fluxes as well as differences between measurements on day 2 and 3 were compared using paired t test. When samples did not follow a normal distribution, we used Mann–Whitney tests, calculated either using the open source software package R and R commander (http://www.r-project.org/) or GraphPad Prism 5. Data are presented as mean ± SEM. p-values <0.05 were considered significant.
Results
Measuring mitochondrial transport in single neurons in vivo
We genetically labeled mitochondria in zebrafish in vivo, which has been shown to be nontoxic (Kim et al., 2008), using the bipartite Gal4-UAS expression system (Köster and Fraser, 2001). We cloned mitoCFP downstream of the Gal4-binding UAS to generate UAS:mitoCFP. By combining this with a second membrane-targeted responder (UAS:memYFP), and a sensory neuron-specific Gal4 driver construct (Sagasti et al., 2005), we could concurrently label sensory neurons and their mitochondria (Fig. 1A,B). Coinjecting the constructs at the one-cell stage (Fig. 1A) lead to mosaic expression patterns, often allowing single RB neurons to be visualized in isolation (Fig. 1B).
Figure 1.

Characterization of mitochondrial transport in RB neurons. A, Schematic of constructs used to label membrane (memYFP) and mitochondria (mitoCFP) in RB neurons. B, RB neuron at 2 dpf (gray box demarcates the area in the fin fold generally imaged). Red box, Area of stem axon imaged in C. Green box, Area of RB peripheral axon magnified to the right, showing mitochondria (top) and membrane (middle); merged image, bottom. C, Kymograph (area in red box in B imaged for 10 min). Arrowheads, Moving mitochondrion (single frames, minutes:seconds). D, Mitochondrial flux in the stem axon at 2 dpf. E, Volumes of anterogradely and retrogradely moving mitochondria. Scale bars: B, 100 μm (magnifications, 20 μm); C, vertical (time) 60 s, horizontal (distance) 5 μm. Data points: D, cells; lines, mean ± SEM.
The superficial location and two-dimensional geometry of RB neurons permitted time-lapse imaging of mitochondrial transport using wide-field microscopy in living zebrafish embryos. We measured mitochondrial flux in single RB neurons at 2 dpf in the “stem axon” (proximal to the first peripheral branch point; Fig. 1B,C). More mitochondria move anterogradely than retrogradely (antero: 0.55 ± 0.04 vs retro: 0.32 ± 0.03 mito/min, n = 26 fish, p < 0.0001; Fig. 1D). This imbalance in numbers likely corresponds to a net translocation of mitochondrial volume into the peripheral arbor, as the mean volume of anterogradely and retrogradely moving mitochondria, as measured by light microscopy, appears to be the same (antero: 0.21 ± 0.01 vs retro: 0.23 ± 0.01 μm3; n = 200 mitochondria from 5 fish in each direction, p > 0.05; Fig. 1E).
Next, we measured several characteristics of mobile mitochondria, namely, average speed (which includes pauses), moving speed, pause frequency and pause length. While mitochondria moving in the anterograde or retrograde direction did not differ in average speed (antero: 0.68 ± 0.02 vs retro: 0.73 ± 0.03 μm/s, n > 140 mitochondria from 12 fish, p > 0.17), retrogradely moving mitochondria displayed significantly faster moving speeds during uninterrupted runs (antero: 0.77 ± 0.01 vs retro: 0.92 ± 0.02 μm/s, n > 140 mitochondria from 12 fish, p < 0.0001). However, since retrogradely moving mitochondria paused more frequently (antero: 2.23 ± 0.18 vs retro: 2.83 ± 0.17 pauses/min, n > 140 mitochondria from 12 fish, p < 0.002) and for longer periods (antero: 3.6 ± 0.6 vs retro: 4.9 ± 0.5 s, n > 140 mitochondria from 12 fish, p < 0.001), there was no difference in average speed.
The sparse labeling allowed us to re-identify and measure mitochondrial transport in the same neuron repetitively over time to explore whether axonal growth and transport correlated. Following time-lapse recording, we reconstructed the soma and entire peripheral arbor of the cell using confocal microscopy (Fig. 2A). While the average size of RB peripheral axons increased from 2 to 3 dpf by ∼30% (3.82 ± 0.32 to 5.02 ± 0.45 mm, n ≥ 19 cells, p < 0.01), neither anterograde nor retrograde flux changed (compare values from 2 dpf described above with those at 3 dpf, antero: 0.53 ± 0.04, retro: 0.27 ± 0.03 mito/min, n = 19 cells, p > 0.4; total flux shown in Fig. 2B). Accordingly, there was no correlation between net flux (anterograde minus retrograde flux) and peripheral arbor growth (Fig. 2C). At the same time, the number of mitochondria in the arbors increased from 2 to 3 dpf proportionally to growth (2 dpf: 225 ± 16; 3 dpf: 334 ± 36, n ≥ 14 cells, p < 0.05), leaving density largely constant, even though we detected a small (< 10%), but statistically significant difference (2 dpf: 65 ± 3 vs 3 dpf: 71 ± 4 mito/mm, n ≥ 14 cells, p < 0.05, Fig. 2C,D).
Figure 2.

Mitochondrial dynamics during cell growth. A, The same RB neuron at 2 and 3 dpf. B, Total mitochondrial flux (anterograde plus retrograde) over time. C, Left, Net mitochondrial flux (anterograde minus retrograde). Right, Number of mitochondria vs arbor size. D, Density of mitochondria at 2 and 3 dpf. E, Nocodazole-treated RB neuron (2 dpf). Red box, Area magnified to the right. F, Normalized mitochondrial densities (total and in subcompartments) in vector (DMSO)- and nocodazole-treated RB neurons. Scales: A, E, 100 μm (magnifications, 10 μm). Data points: Cells; lines, mean ± SEM. Please note that in some of the panels, error bars cannot be seen, as they are hidden behind the data symbols (e.g., C, right panel, and D). The 2 dpf values in B are replotted from Figure 1D.
To investigate whether we could assay changes in transport using our approach, we treated zebrafish embryos with nocodazole, an inhibitor of microtubule assembly (Vasquez et al., 1997). As expected for disruptions of microtubule transport tracks, we observed a reduction in mitochondrial total density (85.8 ± 8.8% of DMSO control), which was even more pronounced in the distal parts of nocodazole-treated axons (73.8 ± 8.8%), while the opposite was seen in the stem axon (127.4 ± 12.2%), suggesting a deficit in mitochondrial trafficking with a resulting central retention of cargos (Fig. 2E,F). However, the changes described here did not reach significance (p > 0.4, n = 9 cells), as obtaining sufficient numbers of cells to overcome the large base-line variation proved difficult due to the need to identify and measure one cell at a time. Therefore we set out to develop a bulk assay of mitochondrial transport in vivo by generating transgenic fish that stably express mitoCFP and memYFP (MitoFish; Fig. 3).
Figure 3.

Transgenic MitoFish. A, Constructs used to generate HuC:Gal4 driver fish and reporter MitoFish. B, Wide-field image of MitoFish at 2 dpf. C, Peripheral arbors of MitoFish RB neurons. D, Transport of mitochondria in MitoFish; superimposition of three pseudo-colored frames of the red box in C (color code as indicated, minutes:seconds). E, Mitochondrial density in RB neurons in MitoFish vs single cells. Scales: B, 1 mm; C, D, 10 μm. Data points: E, fish for MitoFish, cells for transient injections of constructs; lines, mean ± SEM. Please note that in E some error bars cannot be seen, as they are smaller than the size of the data symbols.
Transgenic zebrafish for large-scale transport assays
MitoFish are transgenic responder fish that harbor a bidirectional UAS sequence (Paquet et al., 2009) to concomitantly express mitoCFP and memYFP in a Gal4-dependent manner (Fig. 3A). Given the plethora of Gal4-driver lines available, MitoFish can be used to examine mitochondrial dynamics in different cell-types. Indeed, when we crossed MitoFish to a previously generated HuC:Gal4-driver line (Paquet et al., 2009), we obtained fish coexpressing mitoCFP and memYFP in many neurons (Fig. 3B,C), including RB neurons. We used the mitochondrial dye CMXRos to confirm that mitoCFP was faithfully labeling mitochondria in MitoFish (data not shown).
Next, we imaged mitochondrial transport in the peripheral RB axon arbors in the MitoFish (crossed to the HuC:Gal4-driver; Fig. 3D). Here, in contrast to the single-cell recordings, we imaged several RB axons at the same time in the fin fold, thus recording the transport characteristics of many mitochondria in a short time-frame. The density of resting mitochondria in MitoFish matched the density found in the most distal parts of the axonal arbor in the single-cell assays (MitoFish: 53.1 ± 1.9, n = 10 fish vs single cells: distal 49.2 ± 3.0; proximal 67.0 ± 3.0; stem 97.8 ± 5.9 mitochondria/mm, n = 20 cells). This suggests that in HuC-driven MitoFish we mostly assayed distal axon branches (Fig. 3E), as would be compatible with the imaging position toward the thin rim of the fin fold, chosen to improve image quality. Indeed, in MitoFish we measured a much lower percentage of moving mitochondria (1.03 ± 0.12%, n = 10 fish) than in the stem axons in our single-cell recordings (16.6 ± 1.6%, n = 10 fish). This is expected as flux density drops more steeply (on average by a factor of 2 per branch point, as the cargos are distributed between the branches) than resting mitochondrial density (which we measured to drop roughly by a factor of 2 from stem to distal branches; cf. Fig. 3E). Thus, both of our assays allow generating consistent and complementary datasets. Indeed, when we treated MitoFish with nocodazole, we observed a dramatic decrease in the density of total and moving mitochondria (total density—nocodazole: 28.8 ± 2.1 vs DMSO: 56.8 ± 3.0 mito/mm; flux density—nocodazole: 1.2 ± 0.3 vs DMSO: 7.3 ± 0.6 mito/mm, n = 10 fish, p < 0.001 in both cases, Fig. 4), while transport speed was not significantly affected (data not shown).
Figure 4.

Nocodazole treatment impairs mitochondrial distribution and transport. A, B, Peripheral RB arbors in MitoFish treated with DMSO (A) and nocodazole (B). C, Mitochondrial density (top) and flux density (bottom) in DMSO- and nocodazole-treated MitoFish. Scale bar, 10 μm. Data points: fish; lines, mean ± SEM.
Mitochondrial distribution and transport in a zebrafish tauopathy model
To investigate whether we could detect disease-related deficits in axonal transport with our MitoFish, we crossed them to a zebrafish model of tauopathy, which we previously described (TauFish; Paquet et al., 2009). In these TauFish human Tau bearing a mutation associated with frontotemporal lobar degeneration (P301L) is coexpressed with DsRed to identify TauP301L-bearing neurites. A responder line that expressed DsRed but no Tau (“RedFish”) served as control. We imaged mitochondria in RB neurons expressing mutated Tau (HuC:Gal4; MitoFish × TauFish) and compared these results with the RedFish (Fig. 5A,B). Additionally, in the TauFish, we could identify RB neurites that only expressed memYFP/mitoCFP and not Tau/DsRed (Fig. 5B), which served as internal controls. We observed a profound reduction in the density of total and moving mitochondria in TauFish (density—control RedFish: 59.9 ± 3.3 mitochondria/mm axon, n = 10 fish; TauFish, non-red axons, i.e., “internal control”: 48.1 ± 2.3, n = 14 fish; TauFish, red axons: 14.1 ± 2.3, n = 14 fish; p < 0.0001; flux density—RedFish: 6.8 ± 0.4 moving mitochondria/mm axon; TauFish, internal control: 6.4 ± 0.7; TauFish, red axons: 1.1 ± 0.4; p < 0.0001; Fig. 5C,D). Since DsRed-negative RB neurites in the MitoFish × TauFish had normal transport (Fig. 5D), we could exclude nonspecific transgene integration effects for the observed transport disruption. Transport was also found to be normal in the MitoFish × RedFish, which express DsRed levels comparable to MitoFish × TauFish (Paquet et al., 2009), excluding a major effect of DsRed overexpression on axonal transport (Fig. 5A,D). When comparing the movement behavior of individual mitochondria in MitoFish with or without Tau overexpression (Fig. 5E), we found that the average speed was reduced (RedFish: 0.68 ± 0.03; TauFish: 0.48 ± 0.06 μm/s, n > 20 mitochondria per group; p = 0.002). More detailed analysis, however, revealed that mitochondria in TauFish seemed to move faster when they were in uninterrupted motion (+25.7 ± 9.2% compared with controls, n = 25 mitochondria, p < 0.0001), but showed a trend toward longer pauses between such runs (+137.5 ± 82.7%, p = 0.2), which overcompensated the accelerated moving speed.
Figure 5.
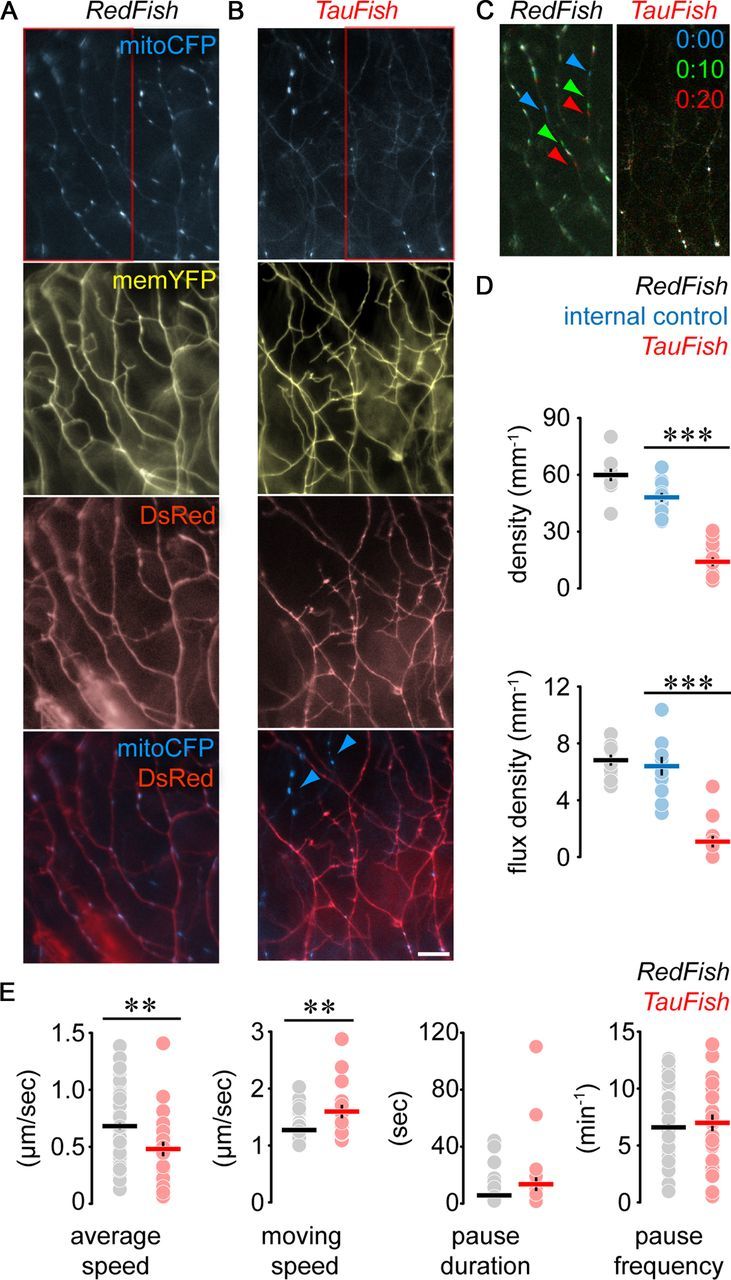
Overexpression of TauP301L reduces mitochondrial density and motility. (A, B) Peripheral RB arbors in DsRed-expressing control (A; RedFish) and TauP301L-expressing (B; TauFish) transgenic lines crossed into the MitoFish. Blue arrowheads, DsRed (and hence TauP301L)-negative neurites (internal control) in B. C, Transport of mitochondria in red boxes in A and B represented as in Figure 3D. D, Mitochondrial density (top) and flux density (bottom) in RedFish vs TauFish (also indicated are internal control neurites). E, Movement behavior of individual mitochondria in RedFish × MitoFish vs TauFish × MitoFish. Scale bar, 10 μm. Data points: D, fish; E: mitochondria; lines, mean ± SEM. Please note that in E some error bars cannot be seen, as they are smaller than the size of the data symbols.
In vitro studies suggest that Tau-induced transport deficits can be reversed by MARK2-mediated phosphorylation of Tau at the microtubule binding domain, which is believed to release Tau from microtubules (Mandelkow et al., 2004). We tested this hypothesis in vivo by expressing MARK2 in the MitoFish × TauFish by mRNA injection. Indeed, MARK2, but not control (GFP) mRNA injections, partially rescued mitochondrial density and transport in TauFish (density—MARK2 in TauFish: 34.3 ± 3.8 mitochondria/mm; GFP in TauFish: 16.8 ± 4.4, n ≥ 7 fish; p = 0.02; flux density—MARK2 6.34 ± 1.4 mitochondria/mm; GFP 0.1 ± 0.1; p = 0.001; Fig. 6). MARK2 overexpression in MitoFish without Tau or in internal control Tau-negative axons (in TauFish) had little effect on mitochondrial density or flux (density—MARK2 in MitoFish: 50.2 ± 2.6 mitochondria/mm; GFP 50.8 ± 2.6; n ≥ 8 fish; p > 0.05; flux density—MARK2: 3.4 ± 0.3 mitochondria/mm; GFP 5.5 ± 0.4; p > 0.05; data for internal control axons not shown). Similarly, overexpression of a mutant form of MARK2 that lacks critical residues to phosphorylate Tau, failed to rescue the Tau-induced mitochondrial phenotype (density—mutant MARK2 13.8 ± 1.7 mito/mm, n = 10 fish; p < 0.000.1; flux density—mutant MARK2 0.9 ± 0.3 mito/mm; p < 0.0001). While this suggests that the effect of MARK2 might be mediated by Tau phosphorylation as shown previously in vitro (Mandelkow et al., 2004), drawing definitive mechanistic conclusions from this dataset is difficult. This is due to the fact that MARK2 can also phosphorylate other microtubule-associated proteins in addition to Tau (Drewes et al., 1997) and the difficulty of ascertaining whether the expression level of MARK2 and mutant MARK2 were precisely matched in RB neurons in our experiments. Still, MARK2 overexpression in TauFish increased the average speed of moving mitochondria (MARK2 0.58 μm/s ± 0.04, n = 58 mitochondria; p > 0.05 compared with TauFish), and partially reversed the Tau-induced changes in the transport behavior of single mitochondria, such as movement speed (data not shown), suggesting that MARK2 might act on cargo-track interactions rather than simply altering the number of mitochondria entering the axon.
Figure 6.
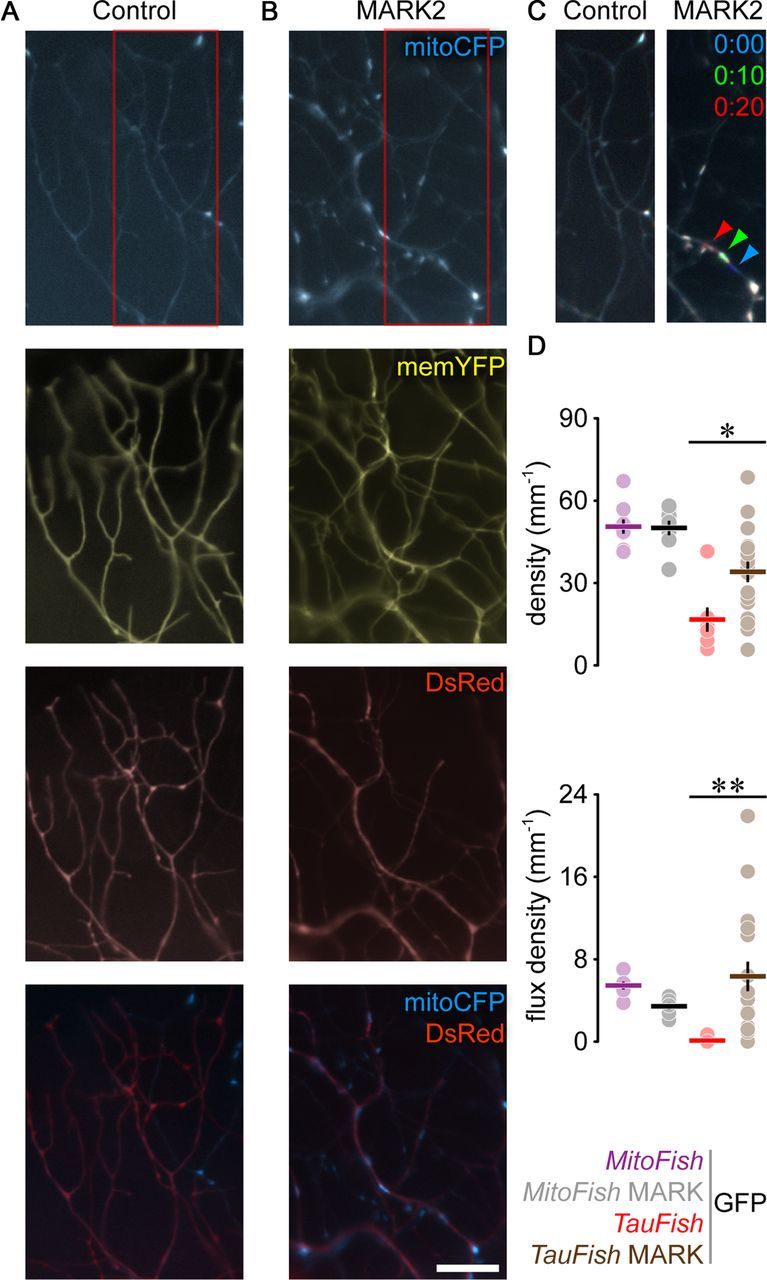
MARK2 overexpression rescues TauFish phenotype. A, B, Peripheral RB arbors in control (GFP-mRNA injected) (A) and MARK2/GFP-expressing (B) TauFish × MitoFish. C, Transport of mitochondria in areas demarcated by red boxes in A and B represented as in Figure 3D. D, Mitochondrial density (top) and flux density (bottom) in GFP- vs MARK2/GFP-expressing MitoFish and TauFish × MitoFish. Scale bar, 10 μm. Data points: fish; lines, mean ± SEM. Please note that in D some error bars cannot be seen, as they are smaller than the size of the data symbols. Also, the values for the flux density in the Tau group in D mostly lie on top of each other (n = 7 data points). The ubiquitous GFP signal that results from mRNA injections fades away after 1 dpf, so it does not interfere with the transgenic labeling.
Discussion
Zebrafish as a model to study transport
The study of neuronal mitochondria and their transport is central to a mechanistic understanding of many neurological diseases. The assays presented here allow quantitative investigations of the life cycle of neuronal mitochondria within their natural milieu in a living vertebrate, zebrafish, and characterization of pharmacologically and genetically induced changes in transport.
Beyond the classical studies using the cytoplasm of large invertebrate axons (Brady et al., 1982; Vale et al., 1985), previous efforts to visualize mitochondria span the phylogenetic spectrum from nematodes to mammals. However, none of these models allows visualizing transport noninvasively, and to our knowledge, no repetitive in vivo measurement of transport in the same axon had been achieved until now. The central advantages of a zebrafish-based assay include: (1) Larvae are transparent allowing for noninvasive imaging with subcellular resolution. (2) Generating transgenic fish is easy. (3) Molecular mechanisms can be probed using mutants, knock-downs or targeted gene disruptions. (4) Myelinated axon tracts in zebrafish, which most invertebrates lack, allow the role of axon-glia interactions in transport to be studied. (5) Imaging zebrafish larvae can be automated for high-content screens (Pardo-Martin et al., 2010). Combined with zebrafish models of human pathology, this might allow for identification of drug candidates that affect subcellular homeostasis (Zon and Peterson, 2005).
Mitochondrial transport and turnover
Our ability to monitor axonal arbor size as well as mitochondrial number and flux in the same neuron over time allows us to deduce important parameters, including the estimated residence time of mitochondria in axons, potential sites of mitochondrial removal and the relationship between transport and axon growth. For instance, the bias toward anterograde translocation of mitochondria we find here has also been reported in Drosophila larvae (Pilling et al., 2006), as well as in embryonic mammalian neurons in culture (Wang et al., 2011). While plausibly being needed for axon growth, our own observation that this bias persists life-long in mice (Misgeld et al., 2007) suggests the alternative of constant distal removal. As disturbed removal of mitochondria plays a central role in Parkinson's disease (Narendra et al., 2008; Geisler et al., 2010; Wang et al., 2011) and given the uncertainty of the site of mitochondrial removal in neurons, distinguishing between these two models—growth-related supply versus peripheral removal—is important. Quantitative analysis of our data shows that the net translocation of individual mitochondria between 2 and 3 dpf exceeds what is needed to maintain a stable mitochondrial density in the growing arbors roughly by a factor of 2 (based on the cylindrical approximation that we used to estimate mitochondrial volume and our flux measurements, ∼60% of the imported mitochondrial volume returns via retrograde transport, ∼20% remains and compensates for growth, another ∼20% are lost; data not shown). This imbalance points to the parallel existence of peripheral removal (Wang et al., 2011) and mitochondrial return (Miller and Sheetz, 2004). Our data also suggest that the average axonal residence time of a mitochondrion is likely shorter than a day (within ∼9 h the same volume of mitochondria is imported as is already present in the arbor at 2 dpf; only ∼20% of this is needed to compensate for growth). This raises the question of whether peripheral residence time is uniform across all mitochondria, or whether subpopulations with different residence times exist (e.g., permanently “resident” vs “cycling”; Wong et al., 2012). Obviously, the potential for fusions and fission complicates this picture considerably (Hoppins et al., 2007; Cai and Sheng, 2012), but we believe that future experiments in zebrafish using optical pulse chase techniques will permit addressing such questions.
Tau-induced changes in transport
Alterations of mitochondrial transport following Tau overexpression have been described in various model systems, including cultured neurons (Ebneth et al., 1998; Mandelkow et al., 2003), flies (Mudher et al., 2004), and transgenic mice (Stokin et al., 2005), although the latter model is fraught with conflicting reports. Importantly, transport alterations in these mouse models were analyzed indirectly, e.g., by examining putative consequences of transport deficits, such as axonal swellings, or by quantifying the levels of axonal proteins. In contrast, we directly visualized mitochondrial transport in vivo and found profound transport disruptions in a zebrafish tauopathy model. Interestingly, while the number of motile mitochondria was greatly reduced in the TauFish, and their average speed was decreased due to increased pauses, the moving speed was increased. This points to an explanation where supernumerary Tau molecules occupy the microtubule surface and interfere with motor protein binding to microtubules (Mandelkow et al., 2003; Dixit et al., 2008), rather than unbound Tau affecting the motors directly. Indeed, a detailed analysis of the behavior of single mitochondria revealed that once moving, mitochondria faced with excessive Tau levels actually appear to move faster than in controls (probably because some short pauses in movement flow are now extended, and can thus be resolved and excluded from a run)—but once mitochondria disengage, they often do so for longer periods of time and might even leave the moving pool. This model is supported by the observation that we could rescue the Tau phenotype by overexpressing MARK2, which regulates Tau's affinity to microtubules by phosphorylating the repeat domain of Tau (Drewes et al., 1997) and thus directly demonstrate the influences of MARK2 on axonal transport in vivo. However, proving the mechanism of such a rescue directly in vivo is challenging, and requires detailed biochemical and biophysical experiments. Still, the ability to swiftly test hypotheses generated from such in vitro experiments in a living vertebrate will be an important addition to our attempts to unravel the mechanisms of mitochondrial transport disturbances. Together, our findings underscore the utility of MitoFish for rapid and sensitive in vivo transport assays and open up the possibility of their use in high-content screening efforts (Zon and Peterson, 2005) for drugs that ameliorate axonal transport deficits common to numerous neurological diseases.
Notes
Supplemental material for this article is available at http://www.misgeld-lab.me.tum.de/new. This URL points to videos illustrating the time-lapse recordings used in this paper to quantify mitochondrial transport.
Video 1: Mitochondrial transport in single RB neurons (cf. Fig. 1; 10 min at 0.5 Hz).
Video 2: Imaging of mitochondrial transport in transgenic MitoFish (cf. Fig. 3; 10 min at 1 Hz). This material has not been peer reviewed.
Footnotes
This work was supported by the Sonderforschungsbereich “Molecular Mechanisms of Neurodegeneration” (SFB 596); the Center for Integrated Protein Sciences (Munich) and the European Community's Seventh Framework Programme (FP7/2007-2013) under Grant agreement no. 200611 (MEMOSAD). C.H. is supported by a research professorship at Ludwig-Maximilians-University. T.M. was supported by the Technische Universität München Institute of Advanced Studies, and the Alexander von Humboldt Foundation. G.P. was supported by the Graduate School of Technische Universität München (TUM-GS). We thank M. Teucke, D. Würzinger, and K. Wullimann for fish husbandry. We thank E.M. and E. Mandelkow for helpful discussions.
References
- Brady ST, Lasek RJ, Allen RD. Fast axonal transport in extruded axoplasm from squid giant axon. Science. 1982;218:1129–1131. doi: 10.1126/science.6183745. [DOI] [PubMed] [Google Scholar]
- Cai Q, Sheng ZH. Mitochondrial transport in neurons: impact on synaptic homeostasis and neurodegeneration. Nat Rev Neurosci. 2012;13:77–93. doi: 10.1038/nrn3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandrasekaran K, Hazelton JL, Wang Y, Fiskum G, Kristian T. Neuron-specific conditional expression of a mitochondrially targeted fluorescent protein in mice. J Neurosci. 2006;26:13123–13127. doi: 10.1523/JNEUROSCI.4191-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixit R, Ross JL, Goldman YE, Holzbaur EL. Differential regulation of dynein and kinesin motor proteins by tau. Science. 2008;319:1086–1089. doi: 10.1126/science.1152993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drewes G, Ebneth A, Preuss U, Mandelkow EM, Mandelkow E. MARK, a novel family of protein kinases that phosphorylate microtubule-associated proteins and trigger microtubule disruption. Cell. 1997;89:297–308. doi: 10.1016/s0092-8674(00)80208-1. [DOI] [PubMed] [Google Scholar]
- Ebneth A, Godemann R, Stamer K, Illenberger S, Trinczek B, Mandelkow E. Overexpression of tau protein inhibits kinesin-dependent trafficking of vesicles, mitochondria, and endoplasmic reticulum: implications for Alzheimer's disease. J Cell Biol. 1998;143:777–794. doi: 10.1083/jcb.143.3.777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edelstein A, Amodaj N, Hoover K, Vale R, Stuurman N. Computer control of microscopes using μManager. Curr Protoc Mol Biol Chapter. 2010;14 doi: 10.1002/0471142727.mb1420s92. Unit14.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geisler S, Holmström KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ, Springer W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol. 2010;12:119–131. doi: 10.1038/ncb2012. [DOI] [PubMed] [Google Scholar]
- Godinho L. Live imaging of zebrafish development. Cold Spring Harb Protoc. 2011;7:770–777. doi: 10.1101/pdb.top119. [DOI] [PubMed] [Google Scholar]
- Goldstein AY, Wang X, Schwarz TL. Axonal transport and the delivery of pre-synaptic components. Curr Opin Neurobiol. 2008;18:495–503. doi: 10.1016/j.conb.2008.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirokawa N, Niwa S, Tanaka Y. Molecular motors in neurons: transport mechanisms and roles in brain function, development, and disease. Neuron. 2010;68:610–638. doi: 10.1016/j.neuron.2010.09.039. [DOI] [PubMed] [Google Scholar]
- Hoppins S, Lackner L, Nunnari J. The machines that divide and fuse mitochondria. Annu Rev Biochem. 2007;76:751–780. doi: 10.1146/annurev.biochem.76.071905.090048. [DOI] [PubMed] [Google Scholar]
- Kabashi E, Champagne N, Brustein E, Drapeau P. In the swim of things: recent insights to neurogenetic disorders from zebrafish. Trends Genet. 2010;26:373–381. doi: 10.1016/j.tig.2010.05.004. [DOI] [PubMed] [Google Scholar]
- Kim MJ, Kang KH, Kim CH, Choi SY. Real-time imaging of mitochondria in transgenic zebrafish expressing mitochondrially targeted GFP. Biotechniques. 2008;45:331–334. doi: 10.2144/000112909. [DOI] [PubMed] [Google Scholar]
- Kimmel CB, Ballard WW, Kimmel SR, Ullmann B, Schilling TF. Stages of embryonic development of the zebrafish. Dev Dyn. 1995;203:253–310. doi: 10.1002/aja.1002030302. [DOI] [PubMed] [Google Scholar]
- Knott AB, Perkins G, Schwarzenbacher R, Bossy-Wetzel E. Mitochondrial fragmentation in neurodegeneration. Nat Rev Neurosci. 2008;9:505–518. doi: 10.1038/nrn2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köster RW, Fraser SE. Tracing transgene expression in living zebrafish embryos. Dev Biol. 2001;233:329–346. doi: 10.1006/dbio.2001.0242. [DOI] [PubMed] [Google Scholar]
- Longair MH, Baker DA, Armstrong JD. Simple Neurite Tracer: open source software for reconstruction, visualization and analysis of neuronal processes. Bioinformatics. 2011;27:2453–2454. doi: 10.1093/bioinformatics/btr390. [DOI] [PubMed] [Google Scholar]
- MacAskill AF, Kittler JT. Control of mitochondrial transport and localization in neurons. Trends Cell Biol. 2010;20:102–112. doi: 10.1016/j.tcb.2009.11.002. [DOI] [PubMed] [Google Scholar]
- Mandelkow EM, Stamer K, Vogel R, Thies E, Mandelkow E. Clogging of axons by tau, inhibition of axonal traffic and starvation of synapses. Neurobiol Aging. 2003;24:1079–1085. doi: 10.1016/j.neurobiolaging.2003.04.007. [DOI] [PubMed] [Google Scholar]
- Mandelkow EM, Thies E, Trinczek B, Biernat J, Mandelkow E. MARK/PAR1 kinase is a regulator of microtubule-dependent transport in axons. J Cell Biol. 2004;167:99–110. doi: 10.1083/jcb.200401085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller KE, Sheetz MP. Axonal mitochondrial transport and potential are correlated. J Cell Sci. 2004;117:2791–2804. doi: 10.1242/jcs.01130. [DOI] [PubMed] [Google Scholar]
- Misgeld T, Kerschensteiner M, Bareyre FM, Burgess RW, Lichtman JW. Imaging axonal transport of mitochondria in vivo. Nat Meth. 2007;4:559–561. doi: 10.1038/nmeth1055. [DOI] [PubMed] [Google Scholar]
- Mudher A, Shepherd D, Newman TA, Mildren P, Jukes JP, Squire A, Mears A, Drummond JA, Berg S, MacKay D, Asuni AA, Bhat R, Lovestone S. GSK-3beta inhibition reverses axonal transport defects and behavioural phenotypes in Drosophila. Mol Psychiatry. 2004;9:522–530. doi: 10.1038/sj.mp.4001483. [DOI] [PubMed] [Google Scholar]
- Mullins MC, Hammerschmidt M, Haffter P, Nüsslein-Volhard C. Large-scale mutagenesis in the zebrafish: in search of genes controlling development in a vertebrate. Curr Biol. 1994;4:189–202. doi: 10.1016/s0960-9822(00)00048-8. [DOI] [PubMed] [Google Scholar]
- Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. 2008;183:795–803. doi: 10.1083/jcb.200809125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls DG, Budd SL. Mitochondria and neuronal survival. Physiol Rev. 2000;80:315–360. doi: 10.1152/physrev.2000.80.1.315. [DOI] [PubMed] [Google Scholar]
- Paquet D, Bhat R, Sydow A, Mandelkow EM, Berg S, Hellberg S, Fälting J, Distel M, Köster RW, Schmid B, Haass C. A zebrafish model of tauopathy allows in vivo imaging of neuronal cell death and drug evaluation. J Clin Invest. 2009;119:1382–1395. doi: 10.1172/JCI37537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paquet D, Schmid B, Haass C. Transgenic zebrafish as a novel animal model to study tauopathies and other neurodegenerative disorders in vivo. Neurodegener Dis. 2010;7:99–102. doi: 10.1159/000285515. [DOI] [PubMed] [Google Scholar]
- Pardo-Martin C, Chang TY, Koo BK, Gilleland CL, Wasserman SC, Yanik MF. High-throughput in vivo vertebrate screening. Nat Methods. 2010;7:634–636. doi: 10.1038/nmeth.1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilling AD, Horiuchi D, Lively CM, Saxton WM. Kinesin-1 and Dynein are the primary motors for fast transport of mitochondria in Drosophila motor axons. Mol Biol Cell. 2006;17:2057–2068. doi: 10.1091/mbc.E05-06-0526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagasti A, Guido MR, Raible DW, Schier AF. Repulsive interactions shape the morphologies and functional arrangement of zebrafish peripheral sensory arbors. Curr Biol. 2005;15:804–814. doi: 10.1016/j.cub.2005.03.048. [DOI] [PubMed] [Google Scholar]
- Schon EA, Przedborski S. Mitochondria: the next (neurode)generation. Neuron. 2011;70:1033–1053. doi: 10.1016/j.neuron.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stokin GB, Lillo C, Falzone TL, Brusch RG, Rockenstein E, Mount SL, Raman R, Davies P, Masliah E, Williams DS, Goldstein LS. Axonopathy and transport deficits early in the pathogenesis of Alzheimer's disease. Science. 2005;307:1282–1288. doi: 10.1126/science.1105681. [DOI] [PubMed] [Google Scholar]
- Stowers RS, Megeath LJ, Górska-Andrzejak J, Meinertzhagen IA, Schwarz TL. Axonal transport of mitochondria to synapses depends on milton, a novel Drosophila protein. Neuron. 2002;36:1063–1077. doi: 10.1016/s0896-6273(02)01094-2. [DOI] [PubMed] [Google Scholar]
- Vale RD, Schnapp BJ, Reese TS, Sheetz MP. Movement of organelles along filaments dissociated from the axoplasm of the squid giant axon. Cell. 1985;40:449–454. doi: 10.1016/0092-8674(85)90159-x. [DOI] [PubMed] [Google Scholar]
- Vasquez RJ, Howell B, Yvon AM, Wadsworth P, Cassimeris L. Nanomolar concentrations of nocodazole alter microtubule dynamic instability in vivo and in vitro. Mol Biol Cell. 1997;8:973–985. doi: 10.1091/mbc.8.6.973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Winter D, Ashrafi G, Schlehe J, Wong YL, Selkoe D, Rice S, Steen J, LaVoie MJ, Schwarz TL. PINK1 and Parkin target Miro for phosphorylation and degradation to arrest mitochondrial motility. Cell. 2011;147:893–906. doi: 10.1016/j.cell.2011.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong MY, Zhou C, Shakiryanova D, Lloyd TE, Deitcher DL, Levitan ES. Neuropeptide delivery to synapses by long-range vesicle circulation and sporadic capture. Cell. 2012;148:1029–1038. doi: 10.1016/j.cell.2011.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zon LI, Peterson RT. In vivo drug discovery in the zebrafish. Nat Rev Drug Discovery. 2005;4:35–44. doi: 10.1038/nrd1606. [DOI] [PubMed] [Google Scholar]