

Abstract
Gait and balance disorders unresponsive to dopaminergic drugs in Parkinson's disease (PD) are secondary to lesions located outside the dopaminergic system. However, available animal models of PD fail to display l-3,4-dihydroxyphenylalanine (DOPA)-responsive parkinsonism and drug-resistant gait and balance disorders, and this lack of appropriate model could account for the deficit of efficient treatments. Because the pedunculopontine nucleus (PPN) plays an important role in locomotion control, we conducted the present study to investigate the consequences of combined dopaminergic and PPN lesions in a same animal. We used macaques that received first 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) intoxication to render them parkinsonian and then local stereotaxic lesion of the PPN. Adding bilateral PPN lesions in MPTP-lesioned macaques induced dopamine-resistant gait and balance disorders but unexpectedly improved hypokinesia. Additional MPTP injections resulted in the association of a severe DOPA-responsive parkinsonism together with DOPA-unresponsive gait disorders. Histological examination assessed a severe dopaminergic degeneration and a significant loss of PPN cholinergic neurons. We observed similar results in aged monkeys intoxicated with MPTP: they developed severe DOPA-responsive hypokinesia and tremor together with unresponsive gait and balance disorders and displayed dopaminergic lesion and a weak but significant cholinergic PPN lesion. Our results highlight the complex role of the cholinergic PPN neurons in the pathophysiology of PD because its lesion induces a dual effect with an improvement of hypokinesia contrasting with a worsening of DOPA-unresponsive gait and balance disorders. Thus, we obtained a primate model of PD that could be useful to test symptomatic treatments for these heavily disabling symptoms.
Introduction
Gait and balance disorders represent a major burden in the elderly population and are commonly observed in advanced forms of Parkinson's disease (PD) (Bloem et al., 2004). The lack of efficiency of dopaminergic (DA) treatment on these axial symptoms suggests that the underlying lesions involve non-DA systems. Several studies have shown that the cholinergic neurons of the pedunculopontine nucleus (PPN) degenerates in PD (Hirsch et al., 1987; Zweig et al., 1987; Jellinger, 1988). Growing evidences suggest that the PPN is crucial in the development of gait and balance disorders: (1) experimental manipulation of the PPN in mammals highlights the involvement of this region in posture and locomotion (for review, see Nutt et al., 2011); we showed recently that lesion of PPN cholinergic neurons in monkeys was sufficient to induce gait and balance disorders resistant to DA agents (Karachi et al., 2010); (2) cholinergic PPN neurons are lost in faller but not in non-faller PD patients (Karachi et al., 2010); and (3) low-frequency stimulation of the PPN mildly alleviates gait and falls in advanced PD patients (Stefani et al., 2007; Ferraye et al., 2010; Moro et al., 2010).
Monkeys intoxicated with 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) are widely used as a model of parkinsonism. It has been demonstrated that MPTP injected in young animals induced degeneration of the nigrostriatal pathway without any loss of cholinergic PPN neurons (Herrero et al., 1993; Heise et al., 2005) associated with classical parkinsonian symptoms. Because all these symptoms are reversed by DA treatment (Karachi et al., 2010), this model does not recapitulate entirely the clinical features of advanced PD patients who suffer from gait and balance disorders resistant to DA treatment.
To gain additional insights in the understanding of gait disorders in PD and to better reproduce the symptomatology of PD, our goal was to determine whether PPN lesions in MPTP-lesioned monkeys will add gait and balance disorders resistant to DA treatment, to classic parkinsonian symptoms. To test this hypothesis, we choose to combine in the same animal both DA and cholinergic PPN lesions using two different approaches.
The first approach was to determine whether a PPN lesion in young MPTP-lesioned monkeys will add gait and balance disorders resistant to DA treatment associated with parkinsonian l-3,4-dihydroxyphenylalanine (DOPA)-sensitive symptoms. We also wanted to test whether a specific lesion involving only PPN cholinergic neurons (Clark et al., 2007; Karachi et al., 2010) will have the same behavioral effect than a lesion affecting both cholinergic and noncholinergic neurons.
The second approach was to use aged MPTP-lesioned monkeys because they have been shown to display balance disorders (Ovadia et al., 1995) associated with a loss of PPN cholinergic neurons (Karachi et al., 2010). However, axial symptoms as well as their sensitivity to DA treatment have never been fully characterized.
Using these approaches, our aim was to develop a new monkey model of advanced PD. This model would be pertinent to better understand the pathophysiology of the disease and to test new therapeutic approaches for the end stage of PD.
Materials and Methods
Animals.
All experiments were performed in accordance with the recommendations contained in the European Community Council Directives of 1986 (86/609/EEC) and the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the French Animal Ethics Committee of the National Institute of Health and Medical Research. The animals were housed under conditions of constant temperature (22 ± 1°C), humidity (55 ± 5%), and air replacement (16 times per hour) on a 12 h light/dark cycle with access ad libitum to food and water. The experimental protocol was designed to minimize the number of animals used, and everything was done to minimize their suffering.
The study was performed on 10 macaques (Macaca fascicularis). The first group was composed of six aged female macaques, estimated to be ∼25–30 years old according to their dentition and their hair appearance. Three were MPTP intoxicated (A MPTP group) and were maintained for 6–10 weeks to observe the motor disorders obtained, including gait and posture. Three other aged monkeys were used as a control group (A control group). The second group of four young male macaques, 3–5 years old and weighing 3–5 kg, received MPTP intoxication and then stereotactic lesions of the PPN, followed by a few of MPTP injections (Y double-lesioned group). We also used brain sections obtained from five control young male macaques (Y control group) used previously in another study (Karachi et al. 2010) to quantify the number of cholinergic PPN and DA nigral neurons.
MPTP treatment.
We used a previously described regimen of MPTP intoxication (Karachi et al., 2010). Injections were performed intramuscularly under anesthesia (5 mg/kg ketamine) every 3–5 d until a stable parkinsonian state was reached. A lower dose of MPTP was administered in aged macaques than in young ones (0.3 vs 0.4 mg/kg, respectively) because of the increased susceptibility of aged animals to MPTP (Ovadia et al., 1995). The animals were examined every 2 d, and parkinsonian symptoms were scored on a disability scale of 0–25 (Luquin et al., 1999).
PPN lesions.
We performed PPN lesions stereotactically on young MPTP-lesioned monkeys using the procedure described previously (Karachi et al., 2010). We used injections of urotensin II-conjugated diphtheria toxin (10 μl, 20%) into the PPN of two macaques and ibotenic acid (5 μl, 10 μg/μl) into the PPN of the two others. Urotensin toxin has been developed to specifically kill PPN cholinergic neurons, which have been shown previously to express specifically the urotensin II receptor in rodents (Clark et al., 2007). Lesions were made unilaterally first in all four animals and then, 3–4 weeks later, contralaterally in three animals. The fourth animal only received unilateral PPN lesion because it was to be involved in a subsequent experimental protocol.
Behavioral analyses.
The global motor activity of the animals was assessed as they freely moved in their home cage by means of a video image analyzer system (Vigie Primates, Lyon, France). This system determined the level of activity over the 20 min period of observation from changes in gray level in pixels from one image to the next counted every 80 ms.
Rigidity and tremor were assessed on a semiquantitative scale (from 0 to 3) when the monkey sat quietly in a primate chair. Rigidity was assessed qualitatively to differentiate plastic parkinsonian hypertonia with cogwheel from other types of hypertonia. To better characterize tremor episodes, we used TremAn software (Uhrikova et al., 2011) that was designed to perform frequency analysis from video recordings. Gait and posture parameters were measured while the animals were walking in a hallway after a training period. These parameters included speed and length of steps, back curve, knee angle, height of the pelvis, and tail position and were quantified as described previously (Karachi et al., 2010). Balance deficit was also rated between 0 and 3 based on the frequency of disequilibrium in the hallways (number of disequilibrium occurring during 10 min, averaged on five sessions). We considered that the global activity in the home cage, the length and speed of steps, reflected hypokinesia. Back curve, knee angle, height of the pelvis, and balance deficits were used as indicators of postural parameters.
Old (n = 3) and young (n = 4) monkeys were assessed at each step of the experimental paradigm started by a control condition. They were tested without and with apomorphine treatment (Figs. 1, 2).
Figure 1.

A, Comparison of MPTP-induced symptoms in young macaques and aged animals. Parkinsonian and postural parameters were evaluated in each animal under control conditions (Ctrl) after MPTP intoxication in young (Y MPTP) and aged (A MPTP) macaques, and after apomorphine treatment in young (Y Apo) and aged (A apo) macaques. All values are expressed in percentage of control data, except for tremor scaled from 0 to 3. *p < 0.05, Kruskall–Wallis test followed by Mann–Whitney U test in the event of statistically significant differences. B, Spectral analysis of tremor recorded at the left elbow in one aged macaque.
Figure 2.
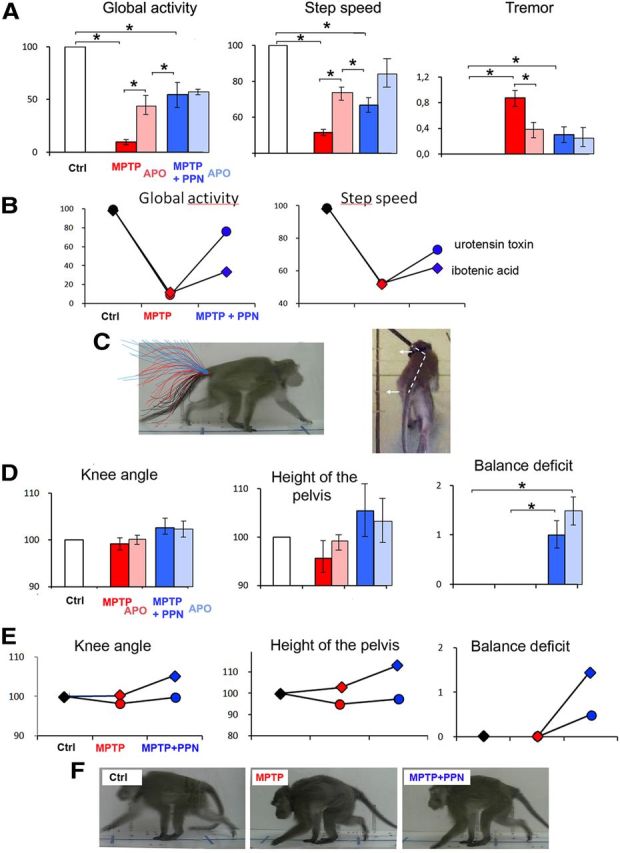
Effect of PPN lesion on parkinsonian symptoms and gait parameters in young MPTP macaques (n = 4). Parkinsonian and postural parameters were evaluated under control conditions (Ctrl) after MPTP intoxication (MPTP), after a subsequent PPN lesion (MPTP + PPN), and after apomorphine treatment (Apo) in the two MPTP states. A, PPN lesion resulted in an improvement of hypokinesia (global activity and step speed) and tremor. Step length and knee angle were also improved but at a lesser degree. B, Improvement of hypokinesia was less after ibotenic acid lesions than after urotensin toxin lesions. C, Worsening of posture and locomotion after PPN lesion assessed by the position of the tail and body, which was turned contralaterally to the lesion. Knee angle, height of the pelvis, and balance deficit were also worsened. D, Postural disorders shown in the same monkey under control condition (Ctrl), after intoxication by MPTP, after a subsequent PPN lesion, and after apomorphine treatment (Apo) in the two MPTP states. PPN lesion resulted in a worsening of these postural parameters. E, Worsening of postural parameters after PPN lesion was more important after ibotenic acid lesions than after urotensin toxin lesions. All values are expressed in percentage of control data, except for the tremor and balance deficits scaled from 0 to 3. *p < 0.05, Kruskall–Wallis test followed by Mann–Whitney U test in the event of statistically significant differences. F, Photographs of the same animal in the control state (Ctrl), after MPTP administration, and then after PPN lesion (MPTP + PPN).
Apomorphine treatment.
Intramuscular injections of apomorphine (Aguettant) at doses of 60–120 μg/kg were given at each step of the experiment (after MPTP intoxication and PPN lesion) when symptoms reached a maximum level of severity. The therapeutic dose required to release the symptoms has been determined for each individual after MPTP treatment and did not change over the whole experiment.
Tissue processing.
All animals were killed 3–4 weeks after the last MPTP injection. The aged monkeys received a lethal overdose of anesthesia and were perfused intracardially with 0.9% NaCl as fresh tissue was needed for other studies. The brains were hemisected along the midline, and each hemisphere was cut into blocks along the frontal plane. Blocks from one hemisphere were postfixed in 4% paraformaldehyde in PBS for 9 d, rinsed in PBS, and then immersed in 30% sucrose for 4 d. Free-floating serial 40 μm transverse sections were then cut on a freezing microtome. Young animals were deeply anesthetized and perfused transcardially with 400 ml of saline, followed by 5 L of 4% paraformaldehyde (in 0.1 m PBS, pH 7.4, at 4°C) and 1 L of PBS with 5% sucrose. The brains were removed from the skull, rinsed in PBS complemented with 10% sucrose for 1 d and 20% sucrose for 1 d, and then frozen and cut into 50-mm-thick sections transversally.
Histological analyses.
Series of regularly interspaced (500 μm apart) fresh-frozen sections were processed for NADPH histochemistry as described previously (Hirsch et al., 1987). Some sections were counterstained with neural red or cresyl violet. Other series of free-floating sections used for tyrosine hydroxylase (TH) immunohistochemistry (François et al., 1999).
The number of neurons immunoreactive for NADPH (NADPH+) was counted on five regularly interspaced PPN sections (every 400 μm in aged animals and 500 μm in young macaques) covering the anteroposterior extent of the structure. The extent of DA denervation was assessed by counting TH+ neurons on eight regularly interspaced nigral sections (every 1440 μm) using the same method. Estimation of the total number of cholinergic and DA neurons was performed using a semiautomatic stereology system with a computer-based system (Mercator; ExploraNova). The number of non-NADPH+, Nissl-stained neurons in the PPN and in the adjacent cuneiform nucleus (CuN) was also counted on NADPH counterstained sections using the same method and was expressed as the number of cells per cubic millimeters.
Statistical analyses.
Behavioral parameters were quantified (1) at the control state, (2) after MPTP intoxication for all the monkeys (n = 7), (3) after unilateral PPN lesion (n = 4 young monkeys) and bilateral PPN lesion (n = 3 young animals), (4) after a subsequent MPTP intoxication, and (5) after apomorphine treatment at each step of the experiment.
Mann–Whitney rank-sum test was performed to determine the loss of TH+, NADPH+, and non-NADPH+ neurons between two groups of individuals. The relative effects of MPTP on TH+ nigral neurons were assessed using aged-matched controls: young macaques treated with MPTP (Y double-lesioned group) were compared with a control population of young healthy macaques (Y control group). Conversely, a control population of old healthy macaques (A control group) was used to assess the loss of TH+ nigral cell in older macaques after MPTP intoxication (A MPTP group). A Kruskall–Wallis test, followed by a Mann–Whitney U test in the event of statistically significant differences, was used to analyze the variations in parkinsonian symptoms and postural parameters in the different conditions. Results with p ≤ 0.05 were considered significant for all the analyses. All data are presented as mean ± SEM.
Results
Behavioral effects of PPN lesions performed after MPTP intoxication in young macaques
Parkinsonian symptoms
After MPTP intoxication, macaques displayed severe hypokinesia, characterized by a dramatic decrease in global activity and in the speed and length of steps, plastic hypertonia with limb cogwheel rigidity, and episodes of postural and action tremor. Postural parameters were also affected after MPTP intoxication, characterized by an increase in the height of the back and a decrease in the angle of the knee and the height of the pelvis. Apomorphine injections resulted in an improvement of all parameters (50%). No balance deficit was detectable either with or without apomorphine treatment. Therefore, young MPTP-lesioned macaques displayed DOPA-sensitive parkinsonian symptoms and gait disorders but no balance deficit.
PPN lesions in MPTP-lesioned macaques
PPN lesions were then performed in the four young MPTP-lesioned macaques using either a toxin specific for cholinergic neurons (two animals) or a nonspecific toxin (ibotenic acid) (the other two animals). A unilateral and especially a bilateral PPN lesion performed with either specific or nonspecific toxin for cholinergic neurons resulted in an improvement of tremor and hypokinesia in the four monkeys (Fig. 2A; Table 1). The PPN lesions performed with the specific toxin resulted in a greater improvement of hypokinesia than the injections performed with ibotenic acid (Fig. 2B). Conversely, muscle tone of limbs was markedly decreased on the side contralateral to the lesion but was increased on the ipsilateral side, extending to the neck.
Table 1.
Summary of the effect of MPTP intoxication and then PPN lesion on parkinsonian and gait and postural parameters
| Treatment | Global activity (%) | Step speed (%) | Step length (%) | Tremor (0–3) | Rigidity (0–3) | Height of the back (%) | Angle of the knee (%) | Pelvis height (%) | Balance deficit (0–3) |
|---|---|---|---|---|---|---|---|---|---|
| Control | 100 | 100 | 100 | 0 | 0 | 100 | 100 | 100 | 0 |
| A MPTP (apo) (n = 3) | 3 (21) | 48 (66) | 75 (80) | 2 (1.2) | 1.7 (1) | 125 (114) | 91 (96) | 91 (98) | 0 (1.7) |
| Y MPTP (apo) (n = 4) | 10* (45**) | 52* (73**) | 81* (89) | 0.9* (0.4**) | 1.8* (0.5**) | 133* (114) | 97 (99) | 96 (99) | 0 |
| Y PPN (apo) (n = 4) | 52*,*** (58) | 67*,*** (80) | 83* (87) | 0.3*** (0.3) | 0.5*, ***(0.5) | 123 (116) | 103 (102) | 106 (102) | 1*,*** (1.5) |
| Y MPTP2 (apo) (n = 4) | 8* (17) | 46* (66) | 76* (79) | 1* (0.5) | 1.7* (0.8) | 150* (129) | 95 (95) | 98 (100) | 0.8 (1) |
Mean values of parkinsonian and gait and postural parameters were calculated in aged macaques in the control state and after MPTP intoxication (A MPTP). They were also calculated in young macaques in the control state and after MPTP intoxication (Y MPTP) and then after PPN lesion (Y PPN) and after an additional series of MPTP injections performed after PPN lesion (Y MPTP2). Values are expressed as a percentage of corresponding values in the control state, except for tremor, rigidity, and balance deficit, expressed on a scale of 0–3. Values in parentheses correspond to values in a given state after apomorphine (apo) treatment.
*p < 0.05 versus control.
**p < 0.05 versus respective treatment before apomorphine.
***p < 0.05 versus MPTP. Kruskall–Wallis test followed by Mann–Whitney U test in the event of statistically significant differences.
The PPN lesion, performed with either specific or nonspecific toxin, impaired gait and worsened postural parameters: a flexed trunk deviated toward the side contralateral to the lesion and erect tail (Fig. 2C), an increase of knee angle and height of the pelvis, and weak but persistent balance deficits that induced falls (Fig. 2D). These symptoms were maximal immediately after the lesion and progressively regressed over 3–4 weeks but without returning to baseline. Compared with a specific cholinergic PPN lesion, a nonspecific lesion resulted in more severe balance deficits and changes in muscle tone (Fig. 2E).
At the same dose as that given after MPTP intoxication, apomorphine injection improved hypokinesia but did not result in a significant improvement of postural and gait parameters (Fig. 2A,D; Table 1).
Monkey model of advanced PD in young animals
To obtain robust gait deficits and classical parkinsonian symptoms in all our young monkeys, some additional MPTP injections were performed after the PPN lesions because of the improvement of hypokinesia.
Additional doses of MPTP induced a dramatic worsening of hypokinesia. Finally, hypokinesia reached a more severe plateau than the one observed at the peak of the first MPTP intoxication period. Global activity in the home cage was nearly suppressed, and both the length and speed of steps were strongly decreased. The postural parameters were severely affected (increase in the height of the back and decrease in knee angle). All the animals displayed balance deficits during walking. None of these symptoms improved over a 3 week observation period (Fig. 3A,B; Table 1). Interestingly, the effect was particularly drastic after only one MPTP injection in the two macaques with cholinergic PPN lesions. After the second series of MPTP injections, the percentage improvement after apomorphine treatment was intermediate (35%) (Fig. 3B).
Figure 3.

Drastic worsening of all symptoms including balance deficits after additional doses of MPTP in double-lesioned macaques. Apomorphine (Apo) was also administered to analyze the symptoms restored by DA treatment. A, Photographs of the same animal in control state (Ctrl), after MPTP administration, and then after PPN lesion and additional MPTP injections (MPTP + PPN + MPTP). B, Examples of parkinsonian and postural and gait parameters that were evaluated under control conditions (Ctrl) after MPTP intoxication (MPTP), after a subsequent PPN lesion and additional MPTP injections (MPTP + PPN + MPTP), and after apomorphine treatment (Apo) in the two MPTP states. All values are expressed in percentage except for balance deficits scaled from 0 to 3. #p < 0.05 for comparison between pre- and post-apomorphine conditions; *p < 0.05 for comparisons with control state. C, Photographs of balance deficit observed after additional doses of MPTP in a monkey. *p < 0.05, Kruskall–Wallis test followed by Mann–Whitney U test in the event of statistically significant differences.
Simultaneous lesion of both cholinergic and DA neurons in aged MPTP-lesioned macaques
We take advantage of aged parkinsonian monkeys to determine whether simultaneous DA and cholinergic lesions provoked by MPTP intoxication induced DOPA-resistant gait and balance disorders.
After MPTP intoxication, aged macaques displayed more severe hypokinesia, hypertonia, and episodes of postural and action tremor than in young monkeys (Table 1). The mean frequency of the tremor obtained from 15 measurements on the four limbs in the most severely impaired aged macaque was 5.84 ± 0.33 Hz (Fig. 1B). Postural parameters measured during walking were also more severely affected in aged than in young monkeys after MPTP intoxication (Table 1). Apomorphine injections resulted in an improvement of hypokinesia, rigidity, tremor, gait, and posture, which was greater in young (50%) than in aged (37%) macaques (Fig. 1A; Table 1). Disequilibrium and falls during walking in the hallway after apomorphine were observed in all aged MPTP-lesioned macaques.
Assessment of lesions
We quantified nigral DA neurons and cholinergic and noncholinergic neurons of the PPN and CuN (Table 2). As expected, a severe decrease in TH immunoreactivity was observed in the putamen and caudate nucleus of all MPTP-lesioned macaques. There was a loss of 74% of TH-positive cells in the substantia nigra pars compacta of young macaques (lesioned, 22,594 ± 743 neurons, n = 4; control, 84,150 ± 4154 neurons, n = 5; p < 0.005, Mann–Whitney U test) and a loss of 73% in aged macaques (lesioned, 20,588 ± 4037 neurons, n = 3; control, 76,121 ± 1125, n = 3; p < 0.05, Mann–Whitney U test) (Fig. 4A).
Table 2.
Extent of the lesion after MPTP intoxication and PPN lesion
| Groups of macaques | TH+ neurons | NADPH+ neurons | PPN non-NADPH+ neurons | CuN non-NADPH+ neurons |
|---|---|---|---|---|
| A control (n = 3) | 76,121 ± 1125 | 8128 ± 81 | 2893 ± 227 | 19,544 ± 774 |
| A MPTP (n = 3) | 20,588 ± 4037 | 6360 ± 80 | 2786 ± 284 | 17,683 ± 547 |
| Loss | 73%* | 22%* | ||
| Y control (n = 5) | 84,150 ± 4154 | 8178 ± 198 | 3551 ± 117 | 25,142 ± 118 |
| Y double-lesioned (n = 4) | 22,594 ± 743 | 4808 ± 370 | 3341 ± 53 | 23,882 ± 919 |
| Loss | 74%* | 41%* | 6% | 7% |
The numbers of TH+ neurons in the substantia nigra pars compacta, NADPH+ neurons in the PPN, and non-NADPH+ neurons in the PPN and CuN were stereologically quantified in aged control (A control) and MPTP-lesioned (A MPTP) macaques and in young control (Y control) and double-lesioned (Y double-lesioned) macaques. The neuronal loss is expressed as a percentage, taking the respective controls as 0%.
*p < 0.05 versus MPTP, Mann–Whitney U test.
Figure 4.
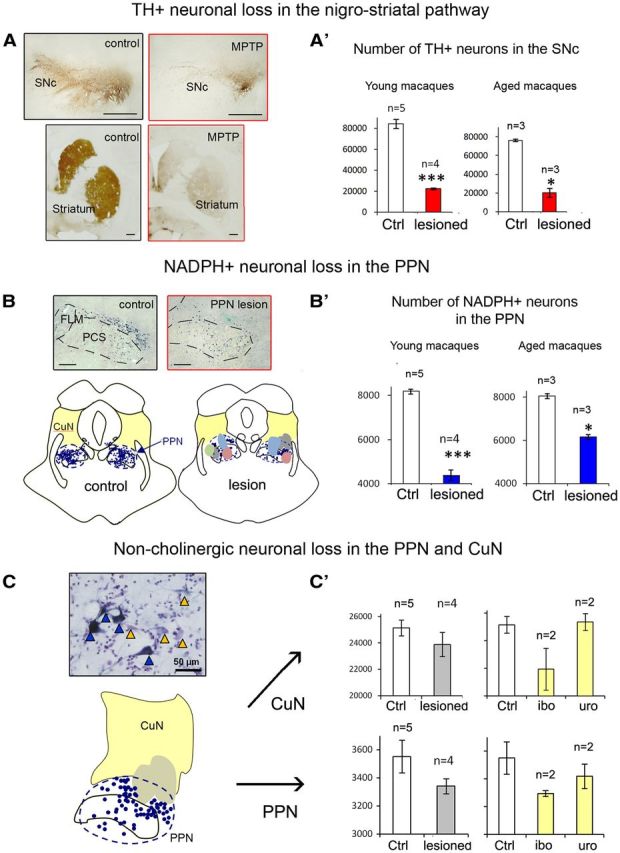
Assessment of DA lesion within the nigrostriatal pathway and of cholinergic and noncholinergic lesion in the PPN and the CuN. A, Coronal sections immunostained with TH showing a severe decrease in TH+ fibers in the striatum (putamen and caudate nucleus) and in TH+ neurons in the substantia nigra pars compacta (SNc) in a young double-lesioned macaque (MPTP) compared with a young control. A′, Graphic representation of the number of nigral TH+ neurons in young double-lesioned macaques and their controls and in aged MPTP-lesioned macaques and their controls. B, Top, Photomicrographs of PPN sections labeled for NADPH histochemistry showing the toxin injection site into the PPN compared with a control. Bottom, NADPH+ neurons (blue) were mapped in a control and in lesioned animals. All injection sites of the four macaques were transferred onto the corresponding brainstem map. Each individual is represented by a different color. Note that the cholinergic part of the PPN was lesioned in all animals. Each dot represents an NADPH+ neuron; yellow areas represent the extent of the CuN. FLM, Medial longitudinal fasciculus; PCS, superior cerebellar peduncle. B′, Graphic representation of the number of NADPH+ neurons in the PPN in young double-lesioned macaques and their controls and in aged MPTP-lesioned macaques and their controls. C, Section immunostained with NADPH and counterstained with Nissl showing the presence of non-NADPH+ neurons (yellow triangles) and NADPH+ neurons (blue triangles). Computer-generated map showing that the injection site (gray area) involved not only the PPN but also the CuN more dorsally located (yellow area). C′, Graphic representation of the distribution of noncholinergic neurons in the CuN (top) and PPN (bottom) in animals that received toxin lesions. Left, The number of noncholinergic neurons in lesioned macaques (n = 4) were compared with controls (n = 5). Right, Distribution of noncholinergic neurons in macaques after ibotenic acid lesion (n = 2) and after urotensin toxin lesion (n = 2). Ctrl, Control; ibo, ibotenic acid; uro, urotensin toxin. Scale bars: A, B, 2 mm; C, 50 μm. *p < 0.01, ***p < 0.001, Mann–Whitney U test.
Whereas there was no loss of NADPH+ neurons in the PPN of young MPTP-lesioned macaques, the loss reached 22% in aged MPTP-lesioned monkeys (lesioned, 6360 ± 80 neurons, n = 3; control, 8128 ± 81, n = 3; p < 0.05, Mann–Whitney U test) (Table 2). Toxin PPN injections in the four young MPTP-lesioned macaques resulted in a bilateral neuronal loss of 41% (range, 24–57%) (lesioned, 4808 ± 370 neurons, n = 4; control, 8178 ± 198 neurons, n = 5; p < 0.005, Mann–Whitney U test) (Fig. 4B; Table 2).
We also estimated the loss of Nissl-stained noncholinergic neurons in both the PPN and the CuN (Fig. 4C, photograph). Whatever the toxin injected, the number of noncholinergic neurons was reduced by only 6% (range, 2–19%) in the whole PPN of the MPTP-lesioned group compared with controls (lesioned, 3341 ± 53 neurons, n = 4; control, 3551 ± 117 neurons, n = 5; p = 0.394, Mann–Whitney U test) (Fig. 4B; Table 2). The lesion also reached the CuN, with a loss of 7% of noncholinergic neurons (range, 1–26%), which was not statistically significant (lesioned, 23,882 ± 919 neurons, n = 4; control, 25,142 ± 118 neurons, n = 5; p < 0.05, Mann–Whitney U test) (Fig. 4B; Table 2). Ibotenic acid injections induced a higher loss of noncholinergic neurons in the PPN and above all in the CuN than the urotensin toxin (10 and 13% in the PPN and CuN vs 3 and 1%, respectively). There was no loss of noncholinergic neurons in either the PPN or the CuN of aged MPTP-lesioned macaques (Table 2).
Discussion
In this study, using two different approaches, we showed that a PPN lesion together with a DA lesion in monkeys induced gait disorders unresponsive to dopamine agonist in addition to classical parkinsonian symptoms. To our knowledge, this is the first attempt to combine both DA and PPN lesions in monkeys that allowed reproducing a close model of the advanced PD.
In aged macaques, MPTP intoxication induced severe parkinsonism with mild midline symptoms, such as balance disorders, as described previously (Ovadia et al., 1995). Compared with young MPTP-lesioned macaques, hypokinesia in aged monkeys was less DOPA responsive. All three aged macaques displayed postural deficits, but only one developed marked balance disorders during gait, worsened by apomorphine. These clinical findings are consistent with the histological examination that showed a severe TH neuronal cell loss in the substantia nigra in both young and aged MPTP animal groups, whereas the loss of PPN cholinergic neurons was significant in aged monkeys only. However, the mild severity of this loss (22%) compared with the loss of 44% induced by stereotaxic PPN lesion could explain why the axial symptoms were not predominant. Moreover, lesions or pathological changes outside the substantia nigra and PPN probably existed in these animals and may also explain the existence of DA-resistant symptoms. Thus, because both PPN cholinergic loss and dopamine-unresponsive gait disorders were mild in aged MPTP-lesioned monkeys, we considered that this approach could not be a convincing candidate for a model of advanced PD. In contrast, young MPTP-lesioned monkeys with subsequent stereotaxic lesion of the PPN developed robust midline signs, including gait and balance disorders, independently of apomorphine treatment. The extent of the non-DA lesions was fully controlled, involving the PPN area without any other extranigral neuronal loss. In addition, according to the toxin used (specific to cholinergic neurons or not), we could dissect the specific contribution of these different populations of PPN neurons in the pathophysiology of parkinsonian symptoms. As demonstrated previously, diphtheria toxin conjugated with urotensin II resulted in a lesion mainly restricted to the PPN and almost specific to cholinergic neurons (Clark et al., 2007; Karachi et al., 2010) with no extension in the adjacent CuN. In comparison, the nonspecific ibotenic acid injected into the cholinergic part of the PPN induced a larger lesion, which extended to the noncholinergic neurons of the CuN.
As hypothesized and in line with our previous results (Karachi et al., 2010), PPN lesions in young MPTP-lesioned monkeys resulted in the additional occurrence of DA-unresponsive alteration of gait, balance, and posture. The postural changes, as assessed by tail position and knee angle, were strictly similar to those observed in the animals that received PPN bilateral lesion but were not intoxicated with MPTP in our previous study and were not improved by DA agonist treatment (Karachi et al., 2010). In addition, they remained unchanged after the second series of MPTP injections, whereas hypokinesia was dramatically worsened. Thus, postural changes induced by PPN lesion seem to be essentially independent of the relationship between the PPN and the DA/basal ganglia system. It is possible that postural changes related to PPN lesions are mediated through complex effects on axial and limb muscle tone because the PPN belongs to the mesencephalic reticular formation, known to control muscle tone (Rye et al., 1988; Lai and Siegel 1990; Takakusaki et al., 2004) via its descending input to the reticulospinal tract. The occurrence of alterations in muscle tone after PPN lesions is in line with experiments in decerebrated cats showing that electrical modulation of the PPN area resulted in a dramatic decrease of muscle tone (Takakusaki et al., 2003). Moreover, it has been reported recently that cholinergic inputs to reticulospinal neurons, possibly arising from the PPN, were capable of amplifying and extending the duration of locomotor output, depending on axial muscle tone, in lampreys (Smetana et al., 2010). Hypertonia has been reported previously after bilateral specific cholinergic lesions of the PPN (Karachi et al., 2010). A larger PPN lesion performed unilaterally in monkeys using kainic acid also produced flexed posture in the limbs but on the contralateral side (Kojima et al., 1997). Although a plausible explanation is not easy to find, differences in the location and extent of the lesions might account for this discrepancy regarding the side of hypertonia.
In young monkeys, balance deficits and falls were observed only with the combination of DA and PPN lesions, never with DA or PPN lesions alone (Karachi et al., 2010). This observation is crucial because it highlights the fact that neither DA nor PPN degeneration alone is sufficient to induce falls and that balance failure depends on lesions within multiple neuronal systems. This is highly consistent with our previous finding in PD patients that fallers had a greater neuronal loss in the PPN than non-fallers (Karachi et al., 2010). Interestingly, in that study, we focused on the PPN cholinergic neurons. In the present work, PPN lesion performed with a toxin specific for cholinergic neurons in two MPTP-lesioned monkeys systematically resulted in the occurrence of falls, suggesting that cholinergic neurons are involved in balance control. In humans, the results of a positron emission tomography study with a cholinergic tracer also support this hypothesis, because faller PD patients had a lower cholinergic innervation of the thalamus arising from the PPN than patients non-fallers (Bohnen et al., 2009). However, our monkeys with nonspecific cholinergic lesions displayed more severe balance deficits than those with specific lesions, indicating that noncholinergic neurons of the PPN and above all of the CuN are also probably involved in balance control, in addition to cholinergic and DA systems. In all the monkeys, balance deficits and falls were increased under apomorphine. Our results are consistent with the observation that the response of gait and balance disorders to levodopa decreases over time in PD patients and that some forms of freezing of gait may be worsened by prolonged levodopa treatment or high-frequency stimulation of the subthalamic nucleus (Giladi et al., 2001). Some reports also suggest that falls may be more frequent under DA agonist in older patients with advanced PD than in younger patients (Elmer et al., 2012).
Several hypotheses might be considered to explain the improvement of hypokinesia that we observed in our MPTP-lesioned monkeys after PPN lesion. (1) One could argue that parkinsonian symptoms recovered over time. However, this is unlikely to account for the improvement of hypokinesia, because the delay between the end of MPTP intoxication and PPN lesion was 9 weeks in the most severely affected MPTP-lesioned monkey. In a previous study, we showed that, after the interruption of MPTP intoxication, parkinsonism either recovered or stabilized within 2–5 weeks (Mounayar et al., 2007). (2) Another possible explanation is that a partial cholinergic PPN lesion performed with urotensin II-conjugated diphtheria toxin, which is ∼50% and thus similar to that observed in PD postmortem (Hirsch et al., 1987; Karachi et al., 2010), induces an increased activity of the remaining cholinergic neurons. Accordingly, hyperactive cholinergic terminals were observed at the level of DA nigral neurons in PD patients (Anglade et al., 1993). It is possible that, when lesions are more severe and nonspecific to cholinergic neurons, the remaining neurons might no longer be able to activate their nigral and thalamic targets. That could explain why a larger PPN lesion using kainic acid in intact nonhuman primates induced typical parkinsonism with hypokinesia and hypertonia (Kojima et al., 1997; Munro-Davies et al., 1999; Matsumura and Kojima, 2001), whereas this was not the case after partial lesion specific to cholinergic neurons (Karachi et al., 2010). This may also explain why, in the current study, nonspecific PPN lesions were less effective at improving hypokinesia than a cholinergic-specific lesion. However, although the excitatory PPN projection to the remaining DA nigrostriatal neurons and to thalamocortical neurons might explain an improvement of hypokinesia after partial PPN lesion, its excitatory drive to the subthalamic nucleus and the zona incerta would, on the contrary, contribute to an excessive overactivity of these nuclei and would thus result in a worsening of hypokinesia. (3) It is also possible that PPN neurons display decreased activity after partial PPN lesion in MPTP-lesioned monkeys, as reported after MPTP intoxication in macaques (Gomez-Gallego et al., 2007). Consequently, PPN neurons would reduce their excitatory drive toward the subthalamic nucleus, resulting in an improvement of hypokinesia. Improvement of tremor is another interesting point. Because there is a cerebellar pathway to the PPN (Hazrati and Parent, 1992), a loss of the target for cerebellar input may in turn modulate the severity of tremor. This is in accordance with recent functional MRI work in tremor-predominant PD patients showing that tremor amplitude is related to a dysfunction within the cerebello-thalamo-cortical pathway (Helmich et al., 2012). In summary, the symptoms ultimately obtained in double-lesioned macaques would result from the balance between the severity of the DA cell loss and the complex effects attributable to the PPN lesion. Double-lesioned macaques could also be useful to study other non-DA symptoms of PD, possibly related to PPN dysfunction, such as sleep disorders (Arnulf et al., 2010) or cognitive deficits (Winn, 2006).
In conclusion, the combination of PPN and DA lesions in young MPTP-lesioned monkeys is technically difficult to perform because stereotaxic injections of neurotoxin within the brainstem are needed. Even if the examination of gait and posture is different in monkeys and humans and difficult to compare, these monkeys had consistent gait and balance disorders resistant to DA associated with classical parkinsonian symptoms. These results shed new light on the complex role of PPN degeneration in the pathophysiology of parkinsonian axial symptoms and pave the way toward a closer model of advanced PD.
Footnotes
Part of this work was funded by the National Institute of Health and Medical Research of France and the Michael J. Fox Foundation.
The authors declare no competing financial interests.
References
- Anglade P, Tsuji S, Hirsch EC, Javoy-Agid F, Agid Y. Ultrastructural relations between nigrostriatal dopaminergic neurons and cholinergic nerve endings in the human brain. Histol Histopathol. 1993;8:501–504. [PubMed] [Google Scholar]
- Arnulf I, Ferraye M, Fraix V, Benabid AL, Chabardès S, Goetz L, Pollak P, Debû B. Sleep induced by stimulation in the human pedunculopontine nucleus area. Ann Neurol. 2010;67:546–549. doi: 10.1002/ana.21912. [DOI] [PubMed] [Google Scholar]
- Bloem BR, Hausdorff JM, Visser JE, Giladi N. Falls and freezing of gait in Parkinson's disease: a review of two interconnected, episodic phenomena. Mov Disord. 2004;19:871–884. doi: 10.1002/mds.20115. [DOI] [PubMed] [Google Scholar]
- Bohnen NI, Müller ML, Koeppe RA, Studenski SA, Kilbourn MA, Frey KA, Albin RL. History of falls in Parkinson disease is associated with reduced cholinergic activity. Neurology. 2009;73:1670–1676. doi: 10.1212/WNL.0b013e3181c1ded6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark SD, Alderson HL, Winn P, Latimer MP, Nothacker HP, Civelli O. Fusion of diphtheria toxin and urotensin II produces a neurotoxin selective for cholinergic neurons in the rat mesopontine tegmentum. J Neurochem. 2007;102:112–120. doi: 10.1111/j.1471-4159.2007.04529.x. [DOI] [PubMed] [Google Scholar]
- Elmer LW, Surmann E, Boroojerdi B, Jankovic J. Long-term safety and tolerability of rotigotine transdermal system in patients with early-stage idiopathic Parkinson's disease: a prospective, open-label extension study. Parkinsonism Relat Disord. 2012;18:488–493. doi: 10.1016/j.parkreldis.2012.01.008. [DOI] [PubMed] [Google Scholar]
- Ferraye MU, Deb û B, Fraix V, Goetz L, Ardouin C, Yelnik J, Henry-Lagrange C, Seigneuret E, Piallat B, Krack P, Le Bas JF, Benabid AL, Chabardès S, Pollak P. Effects of pedunculopontine nucleus area stimulation on gait disorders in Parkinson's disease. Brain. 2010;133:205–214. doi: 10.1093/brain/awp229. [DOI] [PubMed] [Google Scholar]
- François C, Yelnik J, Tand é D, Agid Y, Hirsch EC. Dopaminergic cell group A8 in the monkey: anatomical organization and projections to the striatum. J Comp Neurol. 1999;414:334–347. doi: 10.1002/(SICI)1096-9861(19991122)414:3<334::AID-CNE4>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- Giladi N, McDermott MP, Fahn S, Przedborski S, Jankovic J, Stern M, Tanner C, Parkinson Study Group Freezing of gait in PD: prospective assessment in the DATATOP cohort. Neurology. 2001;56:1712–1721. doi: 10.1212/WNL.56.12.1712. [DOI] [PubMed] [Google Scholar]
- Gomez-Gallego M, Fernandez-Villalba E, Fernandez-Barreiro A, Herrero MT. Changes in the neuronal activity in the pedunculopontine nucleus in chronic MPTP-treated primates: an in-situ hybridization study of cytochrome oxidase subunit I, choline acetyl transferase and substance P mRNA expression. J Neural Transm. 2007;114:319–326. doi: 10.1007/s00702-006-0547-x. [DOI] [PubMed] [Google Scholar]
- Hazrati LN, Parent A. Projection from the deep cerebellar nuclei to the pedunculopontine nucleus in the squirrel monkey. Brain Res. 1992;585:267–271. doi: 10.1016/0006-8993(92)91216-2. [DOI] [PubMed] [Google Scholar]
- Heise CE, Teo ZC, Wallace BA, Ashkan K, Benabid AL, Mitrofanis J. Cell survival patterns in the pedunculopontine tegmental nucleus of methyl-4-phenyl-1,2,3,6-tetrahydropyridine-treated monkeys and 6OHDA-lesioned rats: evidence for differences to idiopathic Parkinson disease patients? Anat Embryol (Berl) 2005;210:287–302. doi: 10.1007/s00429-005-0053-1. [DOI] [PubMed] [Google Scholar]
- Helmich RC, Hallett M, Deuschl G, Toni I, Bloem BR. Cerebral causes and consequences of parkinsonian resting tremor: a tale of two circuits? Brain. 2012;135:3206–3226. doi: 10.1093/brain/aws023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrero MT, Hirsch EC, Javoy-Agid F, Obeso JA, Agid Y. Differential vulnerability to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine of dopaminergic and cholinergic neurons in the monkey mesopontine tegmentum. Brain Res. 1993;624:281–285. doi: 10.1016/0006-8993(93)90088-5. [DOI] [PubMed] [Google Scholar]
- Hirsch EC, Graybiel AM, Duyckaerts C, Javoy-Agid F. Neuronal loss in the pedunculopontine tegmental nucleus in Parkinson disease and in progressive supranuclear palsy. Proc Natl Acad Sci U S A. 1987;84:5976–5980. doi: 10.1073/pnas.84.16.5976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jellinger K. The pedunculopontine nucleus in Parkinson's disease, progressive supranuclear palsy and Alzheimer's disease. J Neurol Neurosurg Psychiatry. 1988;51:540–543. doi: 10.1136/jnnp.51.4.540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karachi C, Grabli D, Bernard FA, Tand é D, Wattiez N, Belaid H, Bardinet E, Prigent A, Nothacker HP, Hunot S, Hartmann A, Lehéricy S, Hirsch EC, François C. Cholinergic mesencephalic neurons are involved in gait and postural disorders in Parkinson disease. J Clin Invest. 2010;120:2745–2754. doi: 10.1172/JCI42642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojima J, Yamaji Y, Matsumura M, Nambu A, Inase M, Tokuno H, Takada M, Imai H. Excitotoxic lesions of the pedunculopontine tegmental nucleus produce contralateral hemiparkinsonism in the monkey. Neurosci Lett. 1997;226:111–114. doi: 10.1016/S0304-3940(97)00254-1. [DOI] [PubMed] [Google Scholar]
- Lai YY, Siegel JM. Muscle tone suppression and stepping produced by stimulation of midbrain and rostral pontine reticular formation. J Neurosci. 1990;10:2727–2734. doi: 10.1523/JNEUROSCI.10-08-02727.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luquin MR, Montoro RJ, Guillén J, Saldise L, Insausti R, Del Río J, López-Barneo J. Recovery of chronic parkinsonian monkeys by autotransplants of carotid body cell aggregates into putamen. Neuron. 1999;22:743–750. doi: 10.1016/S0896-6273(00)80733-3. [DOI] [PubMed] [Google Scholar]
- Matsumura M, Kojima J. The role of the pedunculopontine tegmental nucleus in experimental parkinsonism in primates. Stereotact Funct Neurosurg. 2001;77:108–115. doi: 10.1159/000064614. [DOI] [PubMed] [Google Scholar]
- Moro E, Hamani C, Poon YY, Al-Khairallah T, Dostrovsky JO, Hutchison WD, Lozano AM. Unilateral pedunculopontine stimulation improves falls in Parkinson's disease. Brain. 2010;133:215–224. doi: 10.1093/brain/awp261. [DOI] [PubMed] [Google Scholar]
- Mounayar S, Boulet S, Tand é D, Jan C, Pessiglione M, Hirsch EC, Féger J, Savasta M, François C, Tremblay L. A new model to study compensatory mechanisms in MPTP-treated monkeys exhibiting recovery. Brain. 2007;130:2898–2914. doi: 10.1093/brain/awm208. [DOI] [PubMed] [Google Scholar]
- Munro-Davies LE, Winter J, Aziz TZ, Stein JF. The role of the pedunculopontine region in basal ganglia mechanisms of akinesia. Exp Brain Res. 1999;129:511–517. doi: 10.1007/s002210050921. [DOI] [PubMed] [Google Scholar]
- Nutt JG, Bloem BR, Giladi N, Hallett M, Horak FB, Nieuwboer A. Freezing of gait: moving forward on a mysterious clinical phenomenon. Lancet Neurol. 2011;10:734–744. doi: 10.1016/S1474-4422(11)70143-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ovadia A, Zhang Z, Gash DM. Increased susceptibility to MPTP toxicity in middle-aged rhesus monkeys. Neurobiol Aging. 1995;16:931–937. doi: 10.1016/0197-4580(95)02012-8. [DOI] [PubMed] [Google Scholar]
- Rye DB, Lee HJ, Saper CB, Wainer BH. Medullary and spinal efferents of the pedunculopontine tegmental nucleus and adjacent mesopontine tegmentum in the rat. J Comp Neurol. 1988;269:315–341. doi: 10.1002/cne.902690302. [DOI] [PubMed] [Google Scholar]
- Smetana R, Juvin L, Dubuc R, Alford S. A parallel cholinergic brainstem pathway for enhancing locomotor drive. Nat Neurosci. 2010;13:731–738. doi: 10.1038/nn.2548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefani A, Lozano AM, Peppe A, Stanzione P, Galati S, Tropepi D, Pierantozzi M, Brusa L, Scarnati E, Mazzone P. Bilateral deep brain stimulation of the pedunculopontine and subthalamic nuclei in severe Parkinson's disease. Brain. 2007;130:1596–1607. doi: 10.1093/brain/awl346. [DOI] [PubMed] [Google Scholar]
- Takakusaki K, Habaguchi T, Ohtinata-Sugimoto J, Saitoh K, Sakamoto T. Basal ganglia efferents to the brainstem centers controlling postural muscle tone and locomotion: a new concept for understanding motor disorders in basal ganglia dysfunction. Neuroscience. 2003;119:293–308. doi: 10.1016/S0306-4522(03)00095-2. [DOI] [PubMed] [Google Scholar]
- Takakusaki K, Habaguchi T, Saitoh K, Kohyama J. Changes in the excitability of hind limb motoneurons during muscular atonia induced by stimulating the pedunculopontine tegmental nucleus in cats. Neuroscience. 2004;124:467–480. doi: 10.1016/j.neuroscience.2003.12.016. [DOI] [PubMed] [Google Scholar]
- Uhríkova Z, Sprdlík O, Hoskovcov á M, Komárek A, Ulmanov á O, Hlaváč V, Nugent CD, Růžička E. Validation of a new tool for automatic assessment of tremor frequency from video recordings. J Neurosci Methods. 2011;198:110–113. doi: 10.1016/j.jneumeth.2011.02.033. [DOI] [PubMed] [Google Scholar]
- Winn P. How best to consider the structure and function of the pedunculopontine tegmental nucleus: evidence from animal studies. J Neurol Sci. 2006;248:234–250. doi: 10.1016/j.jns.2006.05.036. [DOI] [PubMed] [Google Scholar]
- Zweig RM, Whitehouse PJ, Casanova MF, Walker LC, Jankel WR, Price DL. Loss of pedunculopontine neurons in progressive supranuclear palsy. Ann Neurol. 1987;22:18–25. doi: 10.1002/ana.410220107. [DOI] [PubMed] [Google Scholar]