

Abstract
Background/Aims:
NLRP3 inflammasome activation has been reported to be an early mechanism responsible for glomerular inflammation and injury in obese mice. However, the precise mechanism of obesity-induced NLRP3 inflammasome activation remains unknown. The present study explored whether adipokine visfatin mediates obesity-induced NLRP3 inflammasome activation and consequent podocyte injury.
Methods:
Inflammasome formation and immunofluorescence expressions were quantified by confocal microscopy. Caspase-activity, IL-1β production and VEGF concentrations were measured by ELISA.
Results:
Confocal microscopic analysis showed that visfatin treatment increased the colocalization of Nlrp3 with Asc or Nlrp3 with caspase-1 in podocytes indicating the formation of NLRP3 inflammasomes. This visfatin-induced NLRP3 inflammasome formation was abolished by pretreatment of podocytes with Asc siRNA. Correspondingly, visfatin treatment significantly increased the caspase-1 activity and IL-1β production in podocytes, which was significantly attenuated by Asc siRNA transfection. Further RT-PCR and confocal microscopic analysis demonstrated that visfatin treatment significantly decreased the podocin expression (podocyte damage). Podocytes pretreatment with Asc siRNA or caspase-1 inhibitor, WEHD attenuated this visfatin-induced podocin reduction. Furthermore, Asc siRNA transfection was found to preserve podocyte morphology by maintaining the distinct arrangement of F-actin fibers normally lost in response to visfatin. It also prevented podocyte dysfunction by restoring visfatin-induced suppression of VEGF production and secretion.
Conclusion:
Visfatin induces NLRP3 inflammasome activation in podocytes and thereby resulting in podocyte injury.
Keywords: Obesity, Inflammasomes, Visfatin, Podocytes
Introduction
The prevalence of obesity has led to an increase in the prevalence of related chronic diseases, including chronic kidney disease, diabetes mellitus, hypertension, endothelial dysfunction and cardiovascular diseases. The mechanisms by which increased adiposity leads to renal dysfunction are likely multifactorial, involving alterations in renal haemodynamics, adipokine signaling, insulin resistnace, inflammation, fatty acid metabolism and cholesterol metabolism. Recently we have shown that Nlrp3 inflammasomes activation is an important initiating mechanism responsible for glomerular inflammation and injury in obese mice [1]. In addition, we have also demonstrate that acid sphingomyelinse gene is involved in mediating the obesity-induced NLRP3 inflammasome formation, activation and consequent glomerular injury [2]. However, the precise mechanisms are still unknown, how the obesity-induced Nlrp3 inflammasomes activation and consequent glomerular injury.
Visfatin, a novel adipokine has been identified as a major injurious factor during obesity-associated diseases including diabetes, carotid, coronary atherosclerosis, and chronic kidney disease [3]. It has been postulated that visfatin may play a role in innate immunity during inflammation and obesity (a low-grade inflammatory process). The plasma levels of visfatin were significantly increased in high fat diet fed mice compared to normal chow-fed mice [4] Moreover, recent studies have reported that plasma visfatin level was significantly increased in a large population of patients with CKD [5,6], type 1 and type 2 diabetes, inflammatory bowel disease, rheumatoid arthritis [4, 7-10]. Moschen et al. demonstrated that visfatin treatment dose-dependently upregulated the production of pro or anti- inflammatory cytokines IL-1β, IL1Ra, Il-6, IL-10 and TNF-α in human monocytes [11]. In accordance with these findings, Xia et al. also demonstrated that visfatin-induced the activation of NLRP3 inflammasomes in endothelial cells in vitro and in vivo contributes to endothelial dysfunction and injury, initiating atherosclerosis during obesity [12]. However, it remains unknown whether visfatin-induced NLRP3 inflammasome activation in glomerular podocytes contributes to obesity-induced podocyte injury. The present study was designed to test the hypothesis that activation of NLRP3 inflammasomes is one of the important mechanisms mediating podocyte inflammatory response to visfatin during early stage obesity. Our results for the first time demonstrate that visfatin-induced NLRP3 inflammasome activation in podocytes and contributes to obesity-induced podocyte injury.
Materials and Methods
Cell culture
Kindly provided by Dr. Paul E. Klotman (Division of Nephrology, Department of Medicine, Mount Sinai School of Medicine, New York, USA), a conditionally immortalized mouse podocyte cell line was cultured undifferentiated with 10 U/ml recombinant mouse interferon-γ at 33°C on collagen I-coated flasks in RPMI 1640 medium containing 10% fetal bovine serum, 100 U/ml penicillin and 100 mg/ml streptomycin. Passaged podocytes were allowed to differentiate at 37°C for 10–14 days in the absence of interferon-γ before being ready for use in experiments. Visfatin was used and its concentration and incubation time in cell culture dishes were chosen based on our previous studies [4] and some preliminary experiments.
A conditionally immortalized mouse podocyte cell line was cultured undifferentiated with 10 U/ml recombinant mouse interferon-γ at 33°C on collagen I-coated flasks in RPMI 1640 medium containing 10% fetal bovine serum, 100 U/ml penicillin and 100 mg/ml streptomycin. Passaged podocytes were allowed to differentiate at 37°C for 10–14 days in the absence of interferon-γ before being ready for use in experiments.
ASC siRNA transfection
ASC siRNA was purchased from Qiagen (Valencia, CA, USA), which was confirmed to be effective in silencing the ASC gene in different cells by the company. The scrambled RNA (Qiagen, Valencia, CA, USA) was also confirmed as non-silencing double-strand RNA and was used as a control. Podocytes were serum-starved for 12 h and then transfected with ASC siRNA or scrambled siRNA using siLentFect Lipid Reagent (Bio-Rad, Hercules, CA, USA). After 18 h of incubation at 37 °C, the medium was changed and visfatin (2 μg/ml) added into the medium for indicated time spans in different protocols.
Real-time reverse transcription polymerase chain reaction (RT-PCR)
Total RNA from cultured podocytes or isolated mouse glomeruli was extracted using TRIzol reagent (Invitrogen, Carlsbad, CA. USA) according to the protocol described by the manufacturer. The primers used in this study were synthesized by Operon (Huntsville, AL, USA) and the sequences were: for podocin sense GTGGAAGCTGAGGCACAAAGAC, antisense CAGCGACTGAAGA GTGTGCAAG; and for β-actin sense TCGCTGCGCTGGTCGTC antisense GGCCTCGTCACCCACATAGGA.
Indirect immuno-fluorescent staining and confocal microscopy
For colocalization of inflammasome molecules in podocytes, cultured cells were fixed in 4% PFA for 15 minutes. After being rinsed with phosphate-buffer saline (PBS), the cells were incubated overnight at 4°C with goat anti-NLRP3 (1:200, Abcam, Cambridge, MA, USA) and rabbit anti-ASC (1:50, Enzo, Plymouth Meeting, PA ), or goat anti-NLRP3 (1:200) and anti-caspase-1 (1:100, Abcam, Cambridge, MA, USA) or rabbit anti- podocin or mouse anti-desmin antibodies. After washing, these slides probed with primary antibodies were incubated with Alexa-488- or Alexa-555-labeled secondary antibodies for 1 h at room temperature (Invitrogen, catalog # A11055 or A-31572). After being mounted with DAPI-containing mounting solution, the slides were subjected to examinations using a confocal laser scanning microscope (Fluoview FV1000, Olympus, Japan), with photos being taken and the colocalization of NLRP3 with ASC or caspase-1 analyzed by the Image Pro Plus 6.0 software (Media Cybernetics, Bethesda, MD, USA). The data was expressed as Pearson correlation coefficient (PCC) as we described previously [13].
Direct fluorescent staining of F-actin
To determine the role of NLRP3 inflammasome activation in visfatin-induced cytoskeleton changes, podocytes were cultured in 8-well chambers. After pretreatment with vehicle or transfected with ASC siRNA or scrambled siRNA, the cells were treated with visfatin (2 ug/ml) for 16 h. After washing with PBS, the cells were fixed in 4% paraformaldehyde for 15 min at room temperature, permeabilized with 0.1% Triton X-100, and blocked with 3% bovine serum albumin. F-actin was stained with rhodamine-phalloidin (Invitrogen, Carlsbad, CA, USA) for 15 min at room temperature. After mounting, the slides were examined by a confocal laser scanning microscope.
Assay of the permeability through podocytes monolayer
Podocytes were cultured in 24-well transwell plates and treated as indicated for 24 hr. The transwell inserts were moved into non-used wells with 200 μl fresh media. 100 μl Fluorescein isothiocyanate (FITC)–dextran (10 KDa, Invitrogen) solution was added into each insert and the plate was incubated at 37°C for 2 hours to allow fluorescein molecules flow through the endothelial cell monolayer. The inserts were then removed and fluorescent intensity in each well was determined at excitation/emission of 485/530 nm using a fluorescent microplate reader (FL × 800, BIO-TEK Instruments). The arbitrary fluorescence intensity was used to calculate the relative permeability [14].
ELISA for VEGF-A secretion by podocytes
Podocytes were incubated with different stimulations like visfatin (2 μg/ml) with or without Asc siRNA transfection or WEHD treatment for 12 h. The supernatant was collected for ELISA to measure VEGF-A using a commercially available kit (R&D Systems, Minneapolis, MN, USA).
Caspase-1 activity, IL-1β production
Caspase-1 activity in podocytes (30 μg/30μl) was measured by a commercially available colorimetric assay kit (Biovision, Mountain View, CA). IL-1β production in podocytes supernatant was measured by a commercially available ELISA kit (R&D System, Minneapolis, MN), according to the manufacturer’s instructions [15,16].
Electronic spin resonance (ESR) analysis of O2·− production
For detection of Nox-dependent O2·− production, proteins from cultured podocytes were extracted using sucrose buffer and resuspended with modified Kreb’s–Hepes buffer containing deferoximine (100 mM, Sigma) and diethyldithiocarbamate (5 mM, Sigma). The Nox-dependent O2·− production was examined by addition of 1mM NADPH as a substrate in 50 mg protein and incubated for 15 min at 37 °C in the presence or absence of SOD (200 U/ml), and then supplied with 1mM O2·− specific spin trap 1-hydroxy-3-methoxycarbonyl-2, 2,5, 5-tetramethylpyrrolidine (CMH, Noxygen, Elzach, Germany). The mixture was loaded in glass capillaries and immediately analyzed for O2·− production kinetically for 10 min in a Miniscope MS200 electromagnetic spin resonance (ESR) spectrometer (Magnettech Ltd, Berlin, Germany). The ESR settings were as follows: biofield, 3350; field sweep, 60 G; microwave frequency, 9.78 GHz; microwave power, 20mW; modulation amplitude, 3 G; 4, 096 points of resolution; receiver gain, 50 for cells. The results were expressed as the fold changes of control.
Statistical Analysis
Data are provided as arithmetic means ± SEM; n represents the number of independent experiments. All data were tested for significance using ANOVA or paired and unpaired Student’s t-test as applicable. Only results with p<0.05 were considered statistically significant.
Results
Inhibition of Asc attenuated visfatin-induced NLRP3 inflammasome formation
We tested whether visfatin-induced NLRP3 inflammasome formation and activation in podocytes. Using confocal microscopy, we demonstrated that visfatin (2 μg/ml, 16 hrs) treatment increased colocalization of inflammasome molecules (Nlrp3 with Asc), as shown by yellow spots in podocytes compared to control cells (Fig. 1A), However, prior treatment with Asc siRNA attenuated the visfatin-induced NLRP3 inflammasome formation in podocytes (Fig. 1A and 1B). The summarized data of quantitative co-localization of Nlrp3 with Asc or Nlrp3 with caspase-1 in podocytes were shown in Fig. 1B.
Fig. 1.

Inhibition of ASC attenuate the visfatin-induced inflammasomes formation in podocytes. A. Confocal images representing the colocalization of Nlrp3 (green) with Asc (red) in podocytes (original magnification, ×400). B. Summarized data showing the fold change of Pearson coefficient correlation (PCC) for the colocalization of NLRP3 with Asc and Nlrp3 with Caspase-1. n=5 each group. Ctrl: Control, Vehi: Vehicle, Scram: Scramble, Asc si: Asc siRNA, Casp-1: Caspase-1. * Significant difference (P<0.05) compared to the control group; # Significant difference (P<0.05) compared to the visfatin (vehicle) group.
Inhibition of Asc abrogated visfatin-induced increases in caspase-1 activity and IL-1β secretion
First demonstrated by Srinivasula et al. it is known that the formation of NLRP3 inflammasomes results in downstream caspase-1 activation and subsequent IL-1β maturation [17]. These effects reflect the functionality of the formed inflammasomes and thus were used to determine the effect of Asc inhibition on visfatin-induced inflammasome activation in the current study. Visfatin significantly increased caspase-1 activity and Il–1β production in podocytes compared to control cells, suggesting activation of NLRP3 inflammasomes (Fig. 2A and 2B). Prior treatment with Asc siRNA transfection significantly attenuated visfatin-induced caspase-1 activity and IL-1β production.
Fig. 2.

Inhibition of ASC attenuate the visfatin-induced inflammasomes activation in podocytes. Summarized data showing the fold change of caspase-1 activity (A) and IL-1β production (B) in podocytes with or without stimulation of visfatin and/or Asc siRNA transfection. N=5 each group. Ctrl: Control, Vehi: Vehicle, Scram: Scramble, Asc si: Asc siRNA, Casp-1: Caspase-1. * Significant difference (P<0.05) compared to the control group; # Significant difference (P<0.05) compared to the visfatin (vehicle) group.
Asc inhibition protects the podocytes from visfatin-induced damage
To assess the extent of podocyte damage, the protein expression of slit diaphragm molecules like podocin and desmin was monitored. Podocin, a podocyte-specific marker, decreases in expression during injury, while podocyte damage marker desmin increases during injury. Immunofluorescence analysis demonstrated that visfatin-treated podocytes displayed a dramatic decrease of podocin staining and increase in desmin staining, signifying podocyte damage (Fig. 3A), However, Asc inhibition or WEHD prior treatment resulted in the protection of these podocyte damages as shown by normalized podocin and desmin protein expression to control levels (Fig. 3A and 3B), Moreover, the actin cytoskeleton organization with fibroblast-like stress extending into the foot processes was analyzed because such stress fibers in cultured podocytes correspond to the filamentous actin in podocyte foot processes in vivo, thus representing differentiation of podocytes. Visfatin-induced a dramatic disarrangement of F-actin, which was substantially blocked by prior treatment of Asc siRNA transfection (Fig. 4A), Additionally, VEGF secretion is considered as an indicator of podocyte functionality because glomerular VEGF is primarily produced in podocytes. It was found that visfatin treated podocytes displayed significantly impaired secretion of VEGF, which was restored by prior treatment of these cells with Asc siRNA (Fig. 4B). Taken together these data suggesting that visfatin-induced NLRP3 inflammasome activation contributes to podocyte injury.
Fig. 3.

Podocin and desmin staining in podocytes with or without visfatin and/or Asc siRNA transfection or WEHD treatment. Cultured podocytes were treated with or without visfatin for 16 h. A: Typical fluorescent microscopic images of podocin (upper images) and desmin (lower images) in podocytes with or without visfatin and/or Asc siRNA transfection or WEHD treatment (n=5/group, Original magnification, ×400). C: Values are arithmetic means ± SE (n=5 each group) of summarized data showing the mRNA expression of podocin. Ctrl: Control, Vehi: Vehicle, Scram: Scramble, Asc si: Asc siRNA, * Significant difference (P<0.05) compared to the control group; # Significant difference (P<0.05) compared to the visfatin (vehicle) group.
Fig. 4.

Effects of silencing ASC on visfatin-induced podocyte functional changes. A. Representative microscopic images of F-actin using rhodamine-phalloidin staining (Original magnification, ×400). B. Summarized data showing the fold change of VEGF levels in podocytes with or without visfatin and/or Asc siRNA treatment. N=5 each group. Ctrl: Control, * Significant difference (P<0.05) compared to the control group; # Significant difference (P<0.05) compared to the visfatin (vehicle) group.
Visfatin-induced O2 production in podocytes
Visfatin treatment significantly increased O2·− production compared with control cells (Fig. 5A). However, prior treatment with reactive oxygen species scavenger, Tempol (0.1 mmol/L) for 30 minutes significantly attenuated the visfatin-induced O2·− production. In contrast, caspase-1 inhibitor, WEHD did not alter O2·− production induced by visfatin. These data suggest that visfatin-induced activation of the NLRP3 inflammasomes might be through the NADPH oxidase dependent ROS production. Further, we examined whether the visfatin-induced redox signaling pathway is involved in visfatin-induced NLRP3 inflammasome activation in podocytes. We determined IL-1β production in podocytes with or without prior treatment of Tempol and WEHD. Visfatin treatment significantly increased IL-1β production compared with control cells (Fig. 5B). However, prior treatment with Tempol and caspase-1 inhibitor, WEHD, significantly attenuated visfatin-induced IL-1β production.
Fig. 5.
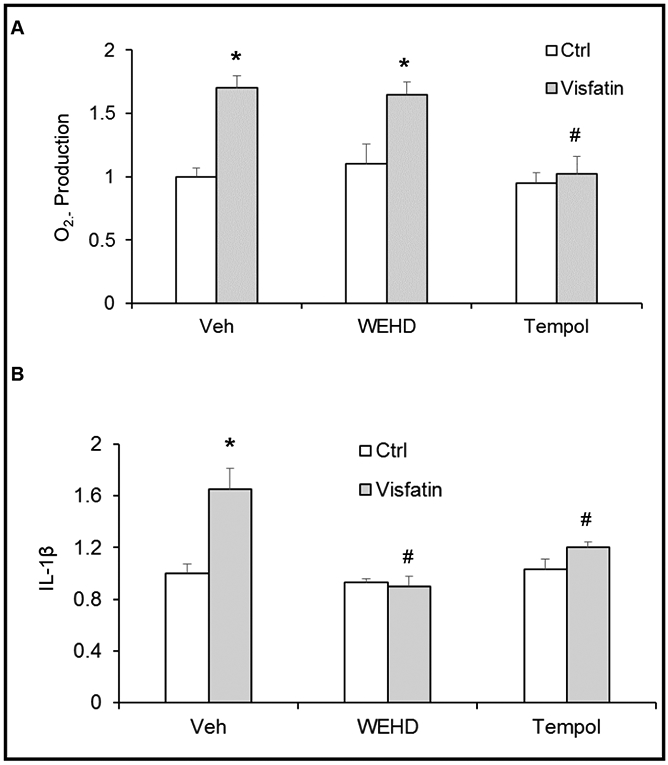
O2·− Production and IL-1β production in podocytes with or without visfatin and/or WEHD or Tempol treatment. Cultured podocytes were treated with or without visfatin for 16 h. Summarized data showing the fold change of O2·− production (A), IL-1β production (B) in podocytes with or without visfatin and/or WEHD or Tempol treatment. N=5 each group. Ctrl: Control, Veh: Vehicle, * Significant difference (P<0.05) compared to the control group; # Significant difference (P<0.05) compared to the visfatin (vehicle) group.
Asc inhibition blocked visfatin-enhanced cell permeability in podocytes
Next, we determined the functional significance of NLRP3 inflammasome activation and examined its influence on visfatin-induced changes in barrier function in podocyte monolayers. As shown in Fig. 6, dextran flux significantly increased in podocytes treated with visfatin. This visfatin-induced increase in permeability of podocytes was markedly reduced in the presence of WEHD or Asc siRNA transfection (Fig. 6).
Fig. 6.
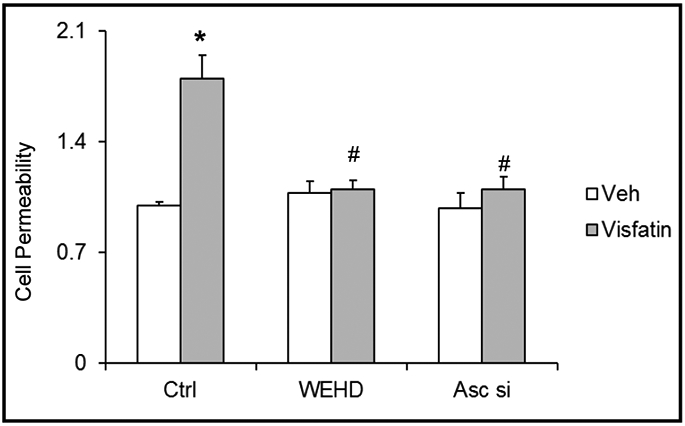
Inhibition of inflammasome attenuates the visfatin-induced cell permeability in podocytes. Cultured podocytes were treated with or without visfatin for 24 h. Summarized data showing the fold change of cell permeability in podocytes with or without visfatin and/or Asc siRNA transfection or WEHD treatment. N=5 each group. Ctrl: Control, Veh: Vehicle, Asc si: Asc siRNA, * Significant difference (P<0.05) compared to the control group; # Significant difference (P<0.05) compared to the visfatin (vehicle) group.
Discussion
The present study was designed to explore whether visfatin-induced NLRP3 inflammasome activation and contributes to the podocyte injury. We first confirmed that visfatin-induced the NLRP3 inflammasome formation and activation in podocytes. However, such inflammasome activation was attenuated in podocytes prior treatment with Asc siRNA transfection. In addition, inhibition of Asc gene or caspase-1 prevented the visfatin-induced podocyte injury and podocyte permeability. These findings for the first time demonstrate the critical role of Nlrp3 inflammasomes activation and subsequent podocyte injury or damage in response to visfatin during obesity.
The inflammasome is a newly identified intracellular machinery of inflammation [18]. Among different kinds of inflammasomes, the Nlrp3 inflammasome is the most fully characterized one in a variety of mammalian cells with high abundance in podocytes of glomeruli [16, 19, 20]. The inflammasome is a proteolytic complex composed of the Nodlike receptor protein 3 (also known as Nlrp3), the adaptor protein apoptosis-associated speck-like protein (Asc), and caspase-1 [21], which is vital for the production of mature IL-1β in response to a variety of agonists or stimuli. It has been reported that IL-1β is an important cytokine with a broad range of biological activities [22, 23] involved in kidney injury or repair [16, 24, 25] and that in glomeruli it is mainly produced by podocytes [26, 27]. The active mature interleukin-1β (IL-1 β) is formed by cleavage of the inactive pro IL-1β precursor by caspase-1, which is activated in a large multiprotein complex, namely, the inflammasome [22, 28-34]. However, little is known about NLRP3 inflammasome contribution to the initiation or development of podocyte dysfunction or damage. Nlrp3 inflammasome has been implicated in the pathogenesis of various metabolic diseases, including obesity, diabetes, gout, silicosis, and acetaminophen-induced liver toxicity [21, 23, 35-40], acute ischemia/reperfusion-induced kidney injury [25], unilateral ureteral obstruction [19, 20], renal biopsies from patients with non-diabetic kidney disease [20] and obesity-induced glomerular injury [2]. The Nlrp3 inflammasome has been reported to be activated by bacterial toxins [18], ATP, monosodium urate crystals [35), β-amyloid [41], muramyldipeptide [42], cholesterol crystals [43] and other stimuli [23]. Moreover, recent studies demonstrated that visfatin has been contributor to endothelial dysfunction through the NLRP3 inflammasome activation in endothelial cells [12]. However, it remains unknown whether visfatin induces NLRP3 inflammasomes activation in podocytes and contributes to podocyte injury or dysfunction and consequent glomerular injury. In the present study, using confocal microscopy, we first confirmed that visfatin-induced the formation and activation of the NLRP3 inflammasome complex in podocytes, as shown by colocalization of NLRP3 with ASC or NLRP3 with caspase-1 and by biochemical analysis of caspase-1 activity and production of IL-1β. However, such inflammasome activation were abolished in podocytes with prior treatment with Asc siRNA or caspase-1 inhibitor, WEHD (Fig. 1 and 2). These results clearly suggest that visfatin-induced NLRP3 inflammasome activation in podocytes, which may contribute to the development of podocyte injury or dysfunction. To our knowledge, the results from the present study provide the first experimental evidence demonstrated that visfatin-induced NLRP3 inflammasome activation in podocytes.
Next we tested a hypothesis that visfatin induces podocyte injury or dysfunction through the activation of Nlrp3 inflammasomes. Podocytes, the epithelial cells lining the outermost layer of the glomeruli, are essential for proper filtration, and injury to podocytes is indicative of impaired glomerular filtration, in time leading to glomerular sclerosis [44-46]. Foot process effacement, considered as the hallmark sign of podocyte injury, is usually accompanied by the destruction of the actin cytoskeleton, increased expression of slit diaphragm molecule and podocyte injury factor desmin, and reduction of the slit diaphragm molecule podocin, which is important for cell polarity and survival. The present study showed that stimulation of podocytes with visfatin decreased the podocin expression and increased the desmin expression. However, inhibition of inflammasome with WEHD or Asc siRNA was able to preserve the morphological structure of podocytes by keeping the distinct arrangement of the F-actin fibers intact and was functionally able to maintain podocin expression and prevent desmin expression increase.
To further explore the mechanism of visfatin-induced Nlrp3 inflammasome activation in podocytes, we determined the NADPH oxidase derived O2·− production in podocytes with or without stimulation of visfatin. It is well documented that several mechanisms underlying inflammasome activation have been reported, including lysosome rupture, ion channel gating, and reactive oxygen species (ROS) activation [15, 46]. Activation of the NLRP3 inflammasome by increased ROS, the most widely accepted and considered to be the most plausible mechanism, suggests that this inflammasome is a general sensor for changes in cellular oxidative stress. Indeed, the present study showed that visfatin significantly increased the NADPH oxidase-dependent O2·− production in podocytes compared to control cells. However, WEHD treatment could not prevent the visfatin-induced increase in superoxide. This suggests that NADPH oxidase activation by visfatin and subsequent production of superoxide are upstream of inflammasome activation, given that inhibition of the inflammasome did not affect the levels of NADPH oxidase-derived superoxide.
Further we examined the role Nlrp3 inflammasomes in visfatin-induced enhancement of podocyte monolayer permeability. There is substantial evidence that glomerular oxidative stress contributes to increases in epithelial monolayer permeability under different pathological conditions such as diabetes, nephritis, hypertension and hHcys [47, 48]. This increased cell permeability importantly participates in the development of glomerular injury and sclerosis [4, 49]. The present study demonstrated that visfatin-induced increase in the podocyte permeability was markedly attenuated by treatment of these cells with Asc siRNA transfection. Moreover, visfatin-induced decrease in the production of VEGF-A, as a glomerular permeability factor, was also reversed by prior treatment with Asc siRNA. These results suggest that visfatin may lead to reduce podocyte function and glomerular barrier integrity and that prior treatment with Asc siRNA prevents such pathological actions of visfatin and protects glomeruli from increased permeability.
Conclusion
In conclusion, the present study demonstrated that adipokine visfatin-induced inflammasome formation, activation and consequent podocyte injury. Therefore, targeting visfatin may be an important therapeutic strategy to prevent inflammasome activation and thereby protect glomeruli from obesity-induced podocyte injury and consequent glomerular injury.
Acknowledgements
This work was supported by National Institutes of Health [grant number DK104031] to K.B.
Footnotes
Disclosure Statement
The authors have no conflicts of interests to declare.
References
- 1.Boini KM, Xia M, Abais JM, Li G, Pitzer AL, Gehr TW, Zhang Y, Li PL: Activation of inflammasomes in podocyte injury of mice on the high fat diet: Effects of ASC gene deletion and silencing. Biochim Biophys Acta 2014;1843:836–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Boini KM, Xia M, Koka S, Gehr TW, Li PL: Instigation of NLRP3 inflammasome activation and glomerular injury in mice on the high fat diet: role of acid sphingomyelinase gene. Oncotarget 2016;7:19031–19044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dahl TB, Yndestad A, Skjelland M, Oie E, Dahl A, Michelsen A, Damas JK, Tunheim SH, Ueland T, Smith C, Bendz B, Tonstad S, Gullestad L, Froland SS, Krohg-Sorensen K, Russell D, Aukrust P, Halvorsen B: Increased expression of visfatin in macrophages of human unstable carotid and coronary atherosclerosis: possible role in inflammation and plaque destabilization. Circulation 2007;115:972–980. [DOI] [PubMed] [Google Scholar]
- 4.Boini KM, Zhang C, Xia M, Han WQ, Brimson C, Poklis JL, Li PL: Visfatin-induced lipid raft redox signaling platforms and dysfunction in glomerular endothelial cells. Biochim Biophys Acta 2010;1801:1294–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Axelsson J, Witasp A, Carrero JJ, Qureshi AR, Suliman ME, Heimburger o, Barany P, Lindholm B, Alvestrand A, Schalling M, Nordfors L, Stenvinkel P: Circulating levels of visfatin/pre-B-cell colony-enhancing factor 1 in relation to genotype, GFR, body composition, and survival in patients with CKD. Am J Kidney Dis 2007;49:237–244. [DOI] [PubMed] [Google Scholar]
- 6.Mahmood N, Junejo AM, Jamal Q, Awan R: Association of visfatin with chronic kidney disease in a cohort of patients with and without diabetes. J Pak Med Assoc 2010;60:922–926. [PubMed] [Google Scholar]
- 7.Brentano F, Schorr O, Ospelt C, Stanczyk J, Gay RE, Gay S, Kyburz D: Pre-B cell colony-enhancing factor/visfatin, a new marker of inflammation in rheumatoid arthritis with proinflammatory and matrix-degrading activities. Arthritis Rheum 2007;56:2829–2839. [DOI] [PubMed] [Google Scholar]
- 8.Malmendal A, Halpain S, Chazin WJ: Nascent structure in the kinase anchoring domain of microtubule-associated protein 2. Biochem Biophys Res Commun 2003;301:136–142. [DOI] [PubMed] [Google Scholar]
- 9.Seo JA, Jang ES, Kim BG, Ryu OH, Kim HY, Lee KW, Kim SG, Choi KM, Baik SH, Choi DS, Kim NH: Plasma visfatin levels are positively associated with circulating interleukin-6 in apparently healthy Korean women. Diabetes Res Clin Pract 2008;79:108–111. [DOI] [PubMed] [Google Scholar]
- 10.Haider DG, Holzer G, Schaller G, Weghuber D, Widhalm K, Wagner O, Kapiotis S, Wolzt M: The adipokine visfatin is markedly elevated in obese children. J Pediatr Gastroenterol Nutr 2006;43:548–549. [DOI] [PubMed] [Google Scholar]
- 11.Moschen AR, Kaser A, Enrich B, Mosheimer B, Theurl M, Niederegger H, Tilg H: Visfatin, an adipocytokine with proinflammatory and immunomodulating properties. J Immunol 2007;178:1748–1758. [DOI] [PubMed] [Google Scholar]
- 12.Xia M, Boini KM, Abais JM, Xu M, Zhang Y, Li PL: Endothelial NLRP3 inflammasome activation and enhanced neointima formation in mice by adipokine visfatin. Am J Pathol 2014;184:1617–1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xia M, Zhang C, Boini KM, Thacker AM, Li PL: Membrane raft-lysosome redox signalling platforms in coronary endothelial dysfunction induced by adipokine visfatin. Cardiovasc Res 2011;89:401–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Boini KM, Hussain T, Li PL, Koka S: Trimethylamine-N-Oxide Instigates NLRP3 Inflammasome Activation and Endothelial Dysfunction. Cell Physiol Biochem 2017;44:152–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Abais JM, Zhang C, Xia M, Liu Q, Gehr T, Boini KM, Li PL: NADPH Oxidase-Mediated Triggering of Inflammasome Activation in Mouse Podocytes and Glomeruli during Hyperhomocysteinemia. Antioxid Redox Signal 2013;18:1537–1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Qin J, Li Y, Cai Z, Li S, Zhu J, Zhang F, Liang S, Zhang W, Guan Y, Shen D, Peng Y, Zhang D, Jie Z, Wu W, Qin Y, Xue W, Li J, Han L, Lu D, Wu P, et al. : A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 2012;490:55–60. [DOI] [PubMed] [Google Scholar]
- 17.Srinivasula SM, Poyet JL, Razmara M, Datta P, Zhang Z, Alnemri ES: The PYRIN-CARD protein ASC is an activating adaptor for caspase-1. J Biol Chem 2002;277:21119–21122. [DOI] [PubMed] [Google Scholar]
- 18.Mariathasan S, Newton K, Monack DM, Vucic D, French DM, Lee WP, Roose-Girma M, Erickson S, Dixit VM: Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature 2004;430:213–218. [DOI] [PubMed] [Google Scholar]
- 19.Anders HJ, Muruve DA: The inflammasomes in kidney disease. J Am Soc Nephrol 2011;22:1007–1018. [DOI] [PubMed] [Google Scholar]
- 20.Vilaysane A, Chun J, Seamone ME, Wang W, Chin R, Hirota S, Li Y, Clark SA, Tschopp J, Trpkov K, Hemmelgarn BR, Beck PL, Muruve DA: The NLRP3 inflammasome promotes renal inflammation and contributes to CKD. J Am Soc Nephrol 2010;21:1732–1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhou R, Tardivel A, Thorens B, Choi I, Tschopp J: Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat Immunol 2010;11:136–140. [DOI] [PubMed] [Google Scholar]
- 22.Davis BK, Wen H, Ting JP: The inflammasome NLRs in immunity, inflammation, and associated diseases. Annu Rev Immunol 2011;29:707–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Imaeda AB, Watanabe A, Sohail MA, Mahmood S, Mohamadnejad M, Sutterwala FS, Flavell RA, Mehal WZ: Acetaminophen-induced hepatotoxicity in mice is dependent on Tlr9 and the Nalp3 inflammasome. J Clin Invest 2009;119:305–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Faul C, Asanuma K, Yanagida-Asanuma E, Kim K, Mundel P: Actin up: regulation of podocyte structure and function by components of the actin cytoskeleton. Trends Cell Biol 2007;17:428–437. [DOI] [PubMed] [Google Scholar]
- 25.Iyer SS, Pulskens WP, Sadler JJ, Butter LM, Teske GJ, Ulland TK, Eisenbarth SC, Florquin S, Flavell RA, Leemans JC, Sutterwala FS: Necrotic cells trigger a sterile inflammatory response through the Nlrp3 inflammasome. Proc Natl Acad Sci U S A 2009;106:20388–20393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Niemir ZI, Stein H, Dworacki G, Mundel P, Koehl N, Koch B, Autschbach F, Andrassy K, Ritz E, Waldherr R, Otto HF: Podocytes are the major source of IL-1 alpha and IL-1 beta in human glomerulonephritides. Kidney Int 1997;52:393–403. [DOI] [PubMed] [Google Scholar]
- 27.Tesch GH, Yang N, Yu H, Lan HY, Foti R, Chadban SJ, Atkins RC, Nikolic-Paterson DJ: Intrinsic renal cells are the major source of interleukin-1 beta synthesis in normal and diseased rat kidney. Nephrol Dial Transplant 1997;12:1109–1115. [DOI] [PubMed] [Google Scholar]
- 28.Horvath GL, Schrum JE, De Nardo CM, Latz E: Intracellular sensing of microbes and danger signals by the inflammasomes. Immunol Rev 2011;243:119–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jin C, Flavell RA: Molecular mechanism of NLRP3 inflammasome activation. J Clin Immunol 2010;30:628–631. [DOI] [PubMed] [Google Scholar]
- 30.Lamkanfi M, Dixit VM: The inflammasomes. PLoS Pathog 2009;5:e1000510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lamkanfi M, Walle LV, Kanneganti TD: Deregulated inflammasome signaling in disease. Immunol Rev 2011;243:163–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schroder K, Tschopp J: The inflammasomes. Cell 2010;140:821–832. [DOI] [PubMed] [Google Scholar]
- 33.Shaw PJ, McDermott MF, Kanneganti TD: Inflammasomes and autoimmunity. Trends Mol Med 2011; 17:57–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wilson SP, Cassel SL: Inflammasome-mediated autoinflammatory disorders. Postgrad Med 2010;122:125–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Martinon F, Petrilli V, Mayor A, Tardivel A, Tschopp J: Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 2006;440:237–241. [DOI] [PubMed] [Google Scholar]
- 36.De Nardo D, Latz E: NLRP3 inflammasomes link inflammation and metabolic disease. Trends Immunol 2011;32:373–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dostert C, Petrilli V, Van Bruggen R, Steele C, Mossman BT, Tschopp J: Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science 2008;320:674–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Horng T, Hotamisligil GS: Linking the inflammasome to obesity-related disease. Nat Med 2011;17:164–165. [DOI] [PubMed] [Google Scholar]
- 39.Stienstra R, Joosten LA, Koenen T, van Tits B, van Diepen JA, van den Berg SA, Rensen PC, Voshol PJ, Fantuzzi G, Hijmans A, Kersten S, Muller M, van den Berg WB, van Rooijen N, Wabitsch M, Kullberg BJ, van der Meer JW, Kanneganti T, Tack CJ, Netea MG: The inflammasome-mediated caspase-1 activation controls adipocyte differentiation and insulin sensitivity. Cell Metab 2010;12:593–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vandanmagsar B, Youm YH, Ravussin A, Galgani JE, Stadler K, Mynatt RL, Ravussin E, Stephens JM, Dixit VD: The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat Med 2011;17:179–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Halle A, Hornung V, Petzold GC, Stewart CR, Monks BG, Reinheckel T, Fitzgerald KA, Latz E, Moore KJ, Golenbock DT: The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat Immunol 2008;9:857–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Martinon F, Agostini L, Meylan E, Tschopp J: Identification of bacterial muramyl dipeptide as activator of the NALP3/cryopyrin inflammasome. Curr Biol 2004;14:1929–1934. [DOI] [PubMed] [Google Scholar]
- 43.Sheedy FJ, Grebe A, Rayner KJ, Kalantari P, Ramkhelawon B, Carpenter SB, Becker CE, Ediriweera HN, Mullick AE, Golenbock DT, Stuart LM, Latz E, Fitzgerald KA, Moore KJ: CD36 coordinates NLRP3 inflammasome activation by facilitating intracellular nucleation of soluble ligands into particulate ligands in sterile inflammation. Nat Immunol 2013;14:812–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kriz W: Podocyte is the major culprit accounting for the progression of chronic renal disease. Microsc Res Tech 2002;57:189–195. [DOI] [PubMed] [Google Scholar]
- 45.Marshall CB, Pippin JW, Krofft RD, Shankland SJ: Puromycin aminonucleoside induces oxidant-dependent DNA damage in podocytes in vitro and in vivo. Kidney Int 2006;70:1962–1973. [DOI] [PubMed] [Google Scholar]
- 46.Abais JM, Xia M, Zhang Y, Boini KM, Li PL: Redox regulation of NLRP3 inflammasomes: ROS as trigger or effector? Antioxid Redox Signal 2015;22:1111–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gonzalez JE, DiGeronimo RJ, Arthur DE, King JM: Remodeling of the tight junction during recovery from exposure to hydrogen peroxide in kidney epithelial cells. Free Radic Biol Med 2009;47:1561–1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Meyer TN, Schwesinger C, Ye J, Denker BM, Nigam SK: Reassembly of the tight junction after oxidative stress depends on tyrosine kinase activity. J Biol Chem 2001;276:22048–22055. [DOI] [PubMed] [Google Scholar]
- 49.Saleh MA, Boesen EI, Pollock JS, Savin VJ, Pollock DM: Endothelin-1 increases glomerular permeability and inflammation independent of blood pressure in the rat. Hypertension 2010;56:942–949. [DOI] [PMC free article] [PubMed] [Google Scholar]