

Abstract
Alzheimer's disease (AD) involves activation of apoptotic pathways that may be regulated through signaling cascades initiated by the amyloid precursor protein (APP). Enlarged endosomes have been observed in postmortem AD brains at very early stages of the disease. We show here that exogenous expression of a familial AD (FAD) mutant of APP or of the APP binding protein APP–BP1 in neurons causes enlargement of early endosomes, increased receptor-mediated endocytosis via a pathway dependent on APP–BP1 binding to APP, and apoptosis. Levels of both APP–BP1 and Rab5 are elevated in early endosomes in cortical embryonic neurons expressing APP(V642I) or APP–BP1, in cultured skin fibroblast cells from Down syndrome subjects, and in postmortem hippocampal tissue of individuals with AD. Indeed, Rab5 was found to bind specifically to APP–BP1, between amino acids 443 and 479. Inhibition of Rab5 or dynamin activity, but not of Eps15 (epidermal growth factor receptor pathway substrate 15) activity, rescued neurons from apoptosis induced by either APP(V642I) or APP–BP1, without affecting levels of intracellular or secreted amyloid-β (Aβ). Induction of Rab5 activity via expression of a constitutively active mutant led to an increase in neuronal apoptosis more than twice that attributable to induction of endosome enlargement via a Rab5-independent mechanism, regardless of Aβ production. Together, these findings suggest that Rab5 activation via an APP/APP–BP1-initiated signaling pathway mediates neuronal apoptosis caused by FAD mutants of APP and that, within this pathway, Rab5 has a specific role in signaling that is distinct from, although not independent of, its role in trafficking.
Keywords: Alzheimer's disease, endosomes, amyloid precursor protein, apoptosis, APP–BP1, Rab5
Introduction
The amyloid precursor protein (APP) is the source of the β-amyloid peptides that accumulate in brains of patients with Alzheimer's disease (AD). APP primacy in the etiology of AD is underscored by the fact that virtually all individuals with Down syndrome (DS), who as a result of chromosome 21 trisomy overexpress APP, develop AD if they live past the age of 40 years. This, combined with the fact that specific mutations in APP or duplication of the APP gene locus (Rovelet-Lecrux et al., 2006; Sleegers et al., 2006) cause some forms of familial AD (FAD), raises the possibility that an alteration of normal APP function may contribute to AD neuropathology. Kang et al. (1987) were the first to propose that APP might act as a signaling receptor. The first direct evidence in support of this notion was the finding that the APP cytodomain interacts with the heterotrimeric G-protein Go (Nishimoto et al., 1993; Brouillet et al., 1999). Subsequently, we described a second binding protein for the APP cytodomain, APP–BP1 (Chow et al., 1996). APP–BP1 is a cell cycle protein that drives the S to M transition in dividing cells (Y. Chen et al., 2000). Exogenous expression of APP–BP1 in neurons has been shown to cause DNA synthesis and consequent apoptosis via a signaling pathway that is dependent on APP–BP1 binding to APP (Chen et al., 2003).
During our characterization of this signaling pathway, we addressed the possibility that it may be at least partly routed through endosomes. Endosomal abnormalities, including enlarged Rab5-positive endosomes, comprise one of the earliest neuropathological alterations in sporadic and familial AD (Cataldo et al., 2000, 2001). Threonine-668 phosphorylated APP cofractionates with endosome markers and is present in enlarged endosomes of AD hippocampal neurons (Lee et al., 2003), and C-terminal fragments of APP localize to endocytic compartments (Grbovic et al., 2003). Interestingly, upregulation of the endocytic pathway by overexpression of the early endosome protein Rab5 increases the level of C-terminal fragments of APP and the secretion of amyloid-β (Aβ) (Grbovic et al., 2003).
We showed previously that exogenous expression of FAD APP mutations in primary neurons causes the same intracellular accumulation of C-terminal fragments of APP and increased Aβ secretion, with subsequent apoptosis (McPhie et al., 1997, 2001). Given the similarities between the consequences of overexpression of Rab5 and expression of FAD APP, we asked whether expression of FAD APP in primary neurons could lead to abnormal endosomal activation. We show that herpes simplex virus (HSV)-mediated expression of APP(V642I) (numbering according to that of APP-695) or of the APP–BP1 increases both Rab5 levels and the number and size of Rab5-positive structures and enhances receptor-mediated endocytosis in a pathway dependent on APP–BP1 binding to APP. It also causes increased levels of both APP–BP1 and Rab5 in early endosomes, a phenomenon also seen in skin fibroblasts from DS subjects and in postmortem hippocampal tissue of individuals with AD. Rab5 specifically binds to a region between amino acids 443 and 479 in APP–BP1. Finally, we find that dominant-negative mutants of Rab5 and dynamin, but not of Eps15 (epidermal growth factor receptor pathway substrate 15), a protein that mediates a Rab5-independent component of clathrin-mediated endocytosis, inhibits the neuronal apoptosis caused by APP(V642I) or APP–BP1. Conversely, expression of constitutively active Rab5 leads to an increase in neuronal apoptosis that is twice that induced by endosome enlargement via a Rab5-independent mechanism. These data suggest that Rab5 activation mediates neuronal apoptosis caused by FAD mutants of APP and that this Rab5-mediated pathway has a specific role in signaling that is distinct from, but likely linked to, trafficking endocytosis.
Materials and Methods
Primary rat embryonic cortical culture and HSV infection
Primary cortical cultures from embryonic day 17 rat embryos were plated at a density of 2.5 × 105 cells per 17 mm well on glass coverslips coated with poly-d-lysine. Four days after plating, neurons were infected with the appropriate viruses at a multiplicity of infection (MOI) of 1 per virus. At this MOI, 70% or more of the neurons in the cultures express the exogenous cDNAs. HSV expressing Escherichia coli β-galactosidase (HSV–LacZ) was used as a control. In experiments in which two different viruses were coexpressed, HSV–LacZ was added to the conditions involving expression of single viruses to equalize the amount of virus in all conditions. The cells were processed for analysis 16 h after infection.
Culture of skin fibroblasts from Down syndrome subjects and controls
Skin fibroblasts from a 5-month-old Down syndrome subject and matched control skin fibroblasts (Coriell Institute for Medical Research, Camden, NJ) were grown at 37°C in the presence of 5% CO2. The growth medium, MEM with 2 mm l-glutamine and non-inactivated 10% fetal bovine serum, was changed every 2 d.
Plasmid construction
All plasmid constructs were made in the pHSVPrpUC vector using standard techniques. The APP–BP1 and dominant-negative hUbc12(C111S) HSV vectors have been described previously (Y. Chen et al., 2000), as has the APP(V642I) HSV vector (McPhie et al., 1997). The HSV vectors expressing FAD presenilin 1 (PS1) mutants were constructed in the same manner as described for the HSV vectors expressing wild-type PS1 and PS1 A246E by Bursztajn et al. (1998). cDNAs for green fluorescent protein (GFP)–Rab4 and GFP–Rab7 and for dominant-negative mutants of these cDNAs were subcloned into HSVPrpUC from plasmids obtained from Dr. Angela Wandinger-Ness (University of New Mexico, Albuquerque, NM). Rab5 and Rab5(S34N) cDNAs were subcloned into HSVPrpUC from plasmids provided by Dr. Marianne Wessling-Resnick (Harvard School of Public Health, Boston, MA). Dominant-negative Eps15 and hepatocyte growth factor-regulated kinase substrate (Hrs) cDNAs were provided by Drs. Alexandre Benmerah (Université Paris Descartes, Paris, France) and Masayuki Komada (Tokyo Institute of Technology, Yokohama, Japan), respectively.
Immunocytochemistry of cultured cells
Primary cortical neurons were infected with the appropriate viruses at an MOI of 1 and, 16 h later, were fixed for 20 min with 4% paraformaldehyde (PFA). Cultures were washed three times with 5% normal goat serum (NGS) in PBS and were incubated overnight with the appropriate antibody made up in 5% NGS containing 0.3% Triton X-100. Cells were then washed three times with PBS and incubated for 1 h with a fluorescent-tagged secondary antibody. After the incubation, cells were washed once with PBS, and the coverslips were mounted on slides using Gel/Mount (Biomeda, Foster City, CA). The antibodies used were rabbit anti-Rab5A (1:400; Santa Cruz Biotechnology, Santa Cruz, CA), rabbit anti-Rab7 (1:250; Santa Cruz Biotechnology), rabbit ant-early endosomal antigen 1 (EEA1) (1:250; Abcam, Cambridge, MA), and mouse anti-APP–BP1 (1:100; BD Transduction Laboratories, San Jose, CA).
Immunocytochemistry of postmortem tissue
Forty-micrometer-thick postmortem brain tissues from the prefrontal cortex of subjects diagnosed with neuropathological evidence of early stage AD (described in detail by Cataldo et al., 2000), fixed in PFA, were immersed in TBS (20 mm Tris, pH 7.4, and 8.75% NaCl), rinsed three times with PBS, and rinsed twice with diluting buffer (10 mm phosphate containing 2% BSA, 0.4% Triton X-100, and 1% normal goat serum). The tissue sections were incubated in 20% normal goat serum in 10 mm phosphate buffer for 10 min, after which they were incubated with rabbit anti-APP–BP1, BP339, and mouse anti-Rab5 at room temperature for 40 h. Sections were washed once in 10 mm phosphate buffer and twice in diluting buffer. The sections were incubated with Alexa Fluor 488 goat anti-mouse and Alexa Fluor 555 goat anti-rabbit overnight and washed before they were mounted onto slides.
Immunoblots
Sixteen hours after infection of the cultures with HSV vectors, primary neurons were homogenized in lysis buffer [100 mm Tris-HCl, 20 mm NaCl, 10 mm EDTA, 10 mm EGTA, 1% SDS with 10 μg/ml leupeptin, 10 μg/ml aprotinin, 1 mm Na3VO4, 1 mm phenylmethylsulfonyl fluoride (PMSF), 1 mm benzamidine, and 10 mm β-glycerol phosphate], and proteins in the lysates were separated by SDS-PAGE. Proteins were then transferred to polyvinylidene difluoride (Millipore, Bedford, MA) membranes for immunoblot analysis (Western Star immunodetection system; Tropix Applied Biosystems, Foster City, CA). The antibodies used for the immunoblots were rabbit anti-Rab5A (1:1000; Santa Cruz Biotechnology), mouse anti-Rab11 (1:1000; BD Transduction Laboratories), mouse anti-APP–BP1 (1:1000; BD Transduction Laboratories), rabbit anti-EEA1 (1:1000; Abcam), and mouse anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (1:1000; Abcam). Densitometry analysis was performed with NIH ImageJ software, and levels were standardized to GAPDH.
Apoptosis assays
Bisbenzimide staining.
For analysis of condensed nuclei, primary cortical cultures were infected with the appropriate viruses at an MOI of 1, and 16 h later the cells were fixed for 20 min with 4% paraformaldehyde and stained with bisbenzimide as described previously (McPhie et al., 2001). Ten random fields of 150–250 cells were analyzed for each condition. The number of cells with condensed nuclei relative to the total number of cells per field was calculated and expressed as a percentage. All experiments were repeated three to four times. The statistical analysis has been described in detail by Chen et al. (2003).
Terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling assay.
Apoptosis was independently assessed by the ApopTag Plus Apoptosis Detection kit (Chemicon Temecula, CA). The assay was applied according to the instructions of the manufacturer. In brief, neurons were fixed in 1% paraformaldehyde, pH 7.4, washed, and then incubated in ethanol/acetic acid 2:1 for 5 min. After equilibration with buffer, cells were incubated at 37°C for 1 h with TdT enzyme. The reaction was stopped and the cells were incubated for 30 min at room temperature with anti-digoxigenin conjugate, after which they were washed, counterstained with propidium iodide, and mounted.
Receptor-mediated endocytosis
Receptor-mediated uptake was examined according to Grbovic et al. (2003) using fluorescent tagged transferrin–Alexa 588 (Invitrogen, Carlsbad, CA). Primary neurons grown in 24-well plates were washed and incubated for 30 min at 37°C in PBS containing 0.1% bovine serum albumin and 20 mm HEPES. Cells were then incubated with transferrin (50 μg/ml) on ice for 1 h, after which they were washed three times before being warmed to 37°C for 5 min (to initiate uptake specifically by early endosomes). After this incubation, cells were fixed with 4% paraformaldehyde for 20 min.
Quantitative transferrin uptake assay
Quantitation of transferrin uptake was based on the method of Fukumori et al. (2006). Briefly, internalized transferrin was assessed by washing cells three times in HBSS (Sigma, St. Louis, MO), pH 7.4. Cells were incubated 30 min on ice with 8 μg/ml biotin–transferrin (Sigma) in conditioned medium, after which they were incubated for 7 min at 37°C. Cells were then washed three times with HBSS, pH 4.0, and harvested for immunoblot analysis. To determine levels of surface-bound transferrin, cells were incubated for 30 min with biotin–transferrin at 4°C and washed three times with HBSS, pH 7.4. Cells were then harvested for immunoblot analysis.
Endosome fractionation
Fractionation of endosomes was based on the method of Seemann et al. (1997) and was described previously (McPhie et al., 2003). Briefly, 100 mg of frozen hippocampal postmortem tissue (Harvard Brain Tissue Resource Center) (Table 1) was lysed in 2 ml of homogenization buffer (HB) plus protease inhibitors. The lysate was passed 10 times through a 26 gauge needle. After a 1300 × g spin at 4°C, the postnuclear supernatant was adjusted to 40.6% sucrose and 3 mm imidazole, pH 7.4, and transferred to an SW41 tube (Beckman Coulter, Fullerton, CA). The sample was overlaid successively with the following: 4 ml of 35% sucrose, 3 mm imidazole, pH 7.4; 3 ml of 25% sucrose, 3 mm imidazole, pH 7.4; and 2 ml of HB. This sucrose gradient was centrifuged at 160,000 × g for 90 min. The late endosome fraction (at the 25% sucrose–HB interface), early endosome fraction (at the 25–35% sucrose interface), and heavy membrane fraction (at the 35–40.6% sucrose interface) were taken. Twenty micrograms of each sample, in triplicate, were then electrophoresed on an SDS-PAGE gel.
Table 1.
Demographic data for postmortem hippocampal tissue
| Tissue number | Group | Age (years) | Sex | PMI |
|---|---|---|---|---|
| B5095 | Control | 82 | M | 17.16 |
| B5140 | Control | 74 | F | 23.00 |
| B5803 | Control | 74 | M | 14.33 |
| B5919 | Control | 80 | M | 18.68 |
| B5985 | Control | 77 | M | 24.58 |
| B5033 | AD/Braak 5 | 74 | M | 14.75 |
| B5480 | AD/Braak 5 | 80 | M | 17.08 |
| B5929 | AD/Braak 6 | 74 | F | 17.50 |
| B5939 | AD/Braak 5/6 | 82 | M | 17.92 |
M, Male; F, female; PMI, postmortem interval.
Coimmunoprecipitation
Primary cortical neurons were seeded at 5 × 106 cells per dish in 60 mm poly-d-lysine-coated dishes. Neurons were infected with HSV–myc–APP–BP1 and HSV–Rab5 for 16 h. Coimmunoprecipitation was performed at 4°C throughout and was based on the procedure described by Enouf et al. (2003). In brief, cells were washed in Dulbecco's PBS (in g/liter: 0.1 anhydrous CaCl2, 0.2 KCl, 0.2 KH2PO4, 0.1 MgCl2·6H2O, 8.0 NaCl, and 2.16 Na2HPO4·7H2O) and lysed with 10 mm potassium acetate/10 mm HEPES, pH 7.3, containing 50 mm NaF, 1 mm PMSF, 10 μg/ml aprotinin, 10 μg/ml leupeptin, 1 mm Na3VO4, 1 mm iodoacetamide, 1 mm benzamidine, and 10 mm β-glycerol phosphate. The lysates were subjected to 10 passages through a 26 gauge needle, after which the buffer was adjusted to 25 mm potassium acetate/125 mm HEPES, pH 7.3. The homogenate was centrifuged at 2000 × g for 20 min to remove nuclei, followed by centrifugation at 18,600 × g for 15 min. Lysates were precleared with either protein G [for monoclonal antibody (mAb)] or protein A (for polyclonal antibody) beads for 1 h, after which they were incubated with 3 μg of 9E10 anti-myc antibody (Upstate Biotechnology, Lake Placid, NY), anti-hemagglutinin (HA) antibody (Roche Diagnostics, Indianapolis, IN), or anti-APP–BP1 antibody (BD Transduction Laboratories) overnight at 4°C. Subsequently, protein G was added for 2 h, after which the beads were washed four times in 25 mm potassium acetate/125 mm HEPES, pH 7.3, with protease inhibitors. The proteins in the immune complexes were separated by SDS-PAGE.
Analysis of Aβ levels
Secreted and intracellular Aβ levels were analyzed by sandwich ELISA as described previously (Murphy et al., 2000). Media were collected 16 h after infection, and cell debris was removed by centrifugation. Cells were lysed with 0.1% Triton X-100 and pelleted at 20,000 × g. Complete protease inhibitor cocktail (Roche Diagnostics) was added to the media and to the cell lysate. Aβ40 and Aβ42 were quantified by mAb9/mAb40.1–HRP and mAb42.2/mAb9–HRP ELISAs, respectively. All measurements were performed in triplicate.
Confocal microscopy
Confocal images were obtained using a Leica (Nussloch, Germany) TCS-NT laser confocal microscope.
Results
Exogenous expression of APP(V642I) or APP–BP1 increases the number and size of Rab5-positive structures in neurons
Abnormalities in the endosomal pathway represent one of the earliest manifestations of AD pathology. Therefore, we examined the morphology of early endosomes in primary neuronal cultures infected with an HSV vector expressing the FAD APP mutant V642I (McPhie et al., 1997). Immunocytochemical analysis of HSV vector-infected cortical neurons with an antibody to the early endosomal protein Rab5 revealed that, whereas HSV–LacZ-infected control cells displayed small uniform vesicular compartments immunoreactive with anti-Rab5, cells infected with HSV–APP(V642I) contained more numerous and atypically large Rab5-positive structures (Fig. 1). Structures positive for Rab7, a late endosomal marker, did not differ in size between experimental and control cultures. These data suggest that an FAD mutant of APP can cause enlargement of early endosomes in primary neuronal cultures.
Figure 1.

Exogenous expression of APP–BP1 or APP(V642I) increases the number and size of Rab5-positive structures in neurons. Primary neurons were infected with HSV vectors expressing APP–BP1, APP(V642I), or LacZ control. Sixteen hours later, the infected cells were harvested, and their proteins were analyzed by immunoblot to confirm expression of the transgene (data not shown). Cells were fixed and stained with rabbit polyclonal anti-Rab5 plus Alexa 555-conjugated secondary (red). Cells expressing APP–BP1 (B) or APP(V642I) (C) present larger and more numerous Rab5-positive structures than do the control HSV–LacZ-infected cells (A). D, E, and F are the corresponding differential interference contrast images for A, B, and C, respectively. Scale bar, 5 μm.
We showed previously that APP interacts at its C terminus with the cell cycle protein APP–BP1 (Chow et al., 1996; Y. Chen et al., 2000) and that expression of APP(V642I) in neurons causes an increase in the levels of APP–BP1 (Chen et al., 2003). Therefore, we ectopically increased the levels of APP–BP1 in primary neuronal cultures by infecting them with an HSV vector encoding APP–BP1 (HSV–APP–BP1). Immunoblots confirmed overexpression of APP–BP1 (data not shown). Immunocytochemical analysis of the cultures with the antibody to Rab5 demonstrated that, similar to HSV–APP(V642I)-infected cells, cells infected with HSV–APP–BP1 contained more numerous and larger Rab5-positive structures than control HSV–LacZ-infected cells (Fig. 1), whereas Rab7-positive structures remained unchanged.
HSV-mediated APP–BP1 or APP(V642I) expression enhances receptor-mediated endocytosis
As described above, HSV-mediated expression of APP–BP1 or of APP(V642I) in neurons in vitro causes increases in the number and size of Rab5-positive structures, which are probably early endosomes. To examine whether APP–BP1 or APP(V642I) expression not only causes structural changes in early endosomes but also alters endosomal function, we analyzed the effects of exogenous APP–BP1 and APP(V642I) expression on receptor-mediated endocytosis. Primary neuronal cultures infected with HSV–APP–BP1 or HSV–APP(V642I) were incubated with fluorophore Alexa 588-conjugated transferrin for 5 min. Cells infected with HSV–APP–BP1 or with HSV–APP(V642I) showed a qualitative increase in the number of transferrin-positive vesicles relative to control HSV–LacZ-infected cells, indicating an increase in constitutive receptor-mediated endocytosis (Fig. 2A). We quantified the increases in receptor-mediated endocytosis by measuring the ratio of internalized versus bound transferrin in APP–BP1- and APP(V642I)-expressing cells. Primary cortical neurons infected with HSV–APP–BP1 or with HSV–APP(V642I) showed a quantitative and significant increase in the ratio of internalized to bound transferrin relative to controls (Fig. 2B).
Figure 2.

Exogenous expression of APP–BP1 or APP(V642I) causes a quantitative increase in receptor-mediated endocytosis. A, Primary neurons expressing APP–BP1, APP(V642I), or the LacZ control were incubated on ice with fluorescent-tagged transferrin for 1 h before being incubated for 5 min at 37°C and then fixed. Cells expressing APP–BP1 or APP(V642I) show higher levels of receptor-mediated endocytosis, as indicated by the higher level of transferrin uptake at 5 min compared with control LacZ-expressing cells. Scale bar, 10 μm. B, Cells infected with HSV–APP–BP1, HSV–APP(V642I), or HSV–LacZ were incubated for 30 min on ice with biotin transferring (Tf), after which they were harvested to assess bound transferring, or incubated at 37°C for 5 min to assess internalized transferrin. ANOVA showed that neurons expressing APP–BP1 or APP(V642I) present a significantly higher ratio of internalized to bound transferrin compared with control. *p < 0.01. The inset shows a representative blot depicting internalized transferrin in the different conditions.
Exogenous expression of APP–BP1 or APP(V642I) in neurons causes increases of Rab5 and EEA1 protein levels but not of Rab11 levels
To confirm our immunocytochemical results at the biochemical level, we infected primary neuronal cultures with HSV–APP–BP1 or HSV–APP(V642I). The cell lysates were immunoblotted with antibodies to specific proteins involved in the endocytic pathway. As shown in Figure 3, the levels of Rab5 and of another marker of early endosomes, EEA1, are increased in cultures infected with HSV–APP–BP1 or HSV–APP(V642I) relative to control cultures. However, the levels of Rab11, involved in endosome recycling, are not increased in these same cultures. This result suggests that the endosome abnormalities caused by exogenous expression of APP–BP1 or of APP(V642I) are specific for early endosomes.
Figure 3.

Exogenous expression of APP(V642I) or APP–BP1 in neurons causes increases of Rab5 and EEA1 protein levels but not of Rab11 levels. Primary neurons were infected with HSV vectors expressing APP–BP1, APP(V642I), or LacZ; 16 h later, the cells were lysed. Equal amounts of protein were separated by SDS-PAGE on three separate gels, and each was transferred to a polyvinylidene difluoride (PVDF) membrane. Blots were probed with rabbit polyclonal anti-Rab5, rabbit polyclonal anti-EEA1, or mouse monoclonal anti-Rab11. Each blot was also probed with mouse monoclonal anti-GAPDH to standardize for loading differences between samples. ANOVA revealed that neurons expressing APP–BP1 or APP(V642I) presented a significant increase in Rab5 and EEA1 protein levels, but not in Rab11 levels, compared with mock- or HSV–LacZ-infected control neurons. *p < 0.01. Values shown are densitometry values standardized to GAPDH. Insets show a representative gel for each condition.
APP–BP1- and APP(V642I)-mediated changes in the morphology and the function of early endosomes are dependent on the binding of APP–BP1 to APP but are independent of the neddylation pathway
We showed previously that APP–BP1 interacts directly with APP (Chow et al., 1996). To test whether this interaction was necessary for APP–BP1- or APP(V642I)-mediated changes in the structure and function of early endosomes, we used HSV vectors to coexpress in neurons a peptide representing the domain of APP–BP1 that binds to APP (Chow et al., 1996; Y. Chen et al., 2000), together with APP–BP1 or APP(V642I). Expression of this peptide, APP–BP1(145–251), blocks both the enlargement of endosomes (Fig. 4A) and the enhanced receptor-mediated endocytosis (Fig. 5A) caused by exogenous expression of APP–BP1 or APP(V642I), suggesting that interaction of APP with APP–BP1 is required for the endosomal changes caused by these proteins. We cannot, however, rule out the possibility that expression of APP–BP1(145–251) blocks interactions of the C terminus of APP with other proteins that bind to it as well and that these disruptions contribute to the abnormal endocytosis.
Figure 4.

APP–BP1- and APP(V642I)-mediated changes in the morphology of early endosomes are dependent on the binding of APP–BP1 to APP but are independent of the neddylation pathway. Primary neurons expressing exogenous APP–BP1 or APP(V642I) were coinfected with either APP–BP1(145–251), the region to which APP binds, or Ubc12(C111S), a dominant-negative mutant in the neddylation pathway. Neurons were then fixed and stained with mouse anti-EEA1 (red) and rabbit anti-Rab5 (green). The right side of each image shows the merged image of EEA1 and Rab5, depicting early endosomes. The insets are enlarged images demonstrating discrete endosomes in single neurons. A, Coexpression of APP–BP1(145–251) with APP–BP1 or APP(V642I) in primary neurons blocks the increase in number and size of early endosomes seen in neurons expressing APP–BP1 or APP(V642I) alone. B, In contrast, coexpression of Ubc12(C111S) does not block the increase in number and size of early endosomes seen in neurons expressing APP–BP1 or APP(V642I) alone. Scale bar, 5 μm.
Figure 5.

Enhanced receptor-mediated endocytosis caused by exogenous expression of APP–BP1 or APP(V642I) in neurons is inhibited when APP binding to APP–BP1 is blocked but not when the neddylation pathway is blocked. Primary neurons coexpressing APP–BP1 (145–251) together with APP–BP1 or APP(V642I) do not show an increase in receptor-mediated endocytosis, as indexed by transferrin uptake, which is observed in neurons expressing APP–BP1 or APP(V642I) alone. In contrast, neurons coexpressing Ubc12(C111S) together with APP–BP1 or APP(V642I) retain the enhanced receptor-mediated endocytosis caused by these proteins. Scale bar, 10 μm.
We have shown that one of the pathways initiated by APP–BP1 is the neddylation pathway (Y. Chen et al., 2000). This pathway includes hUbc12, the NEDD8-conjugating enzyme in the neddylation pathway. To determine whether the neddylation pathway mediates the effects of APP–BP1 or APP(V642I) expression on endosome structure and function, we coexpressed in neurons a dominant-negative mutant (C111S) of hUbc12 with APP–BP1 or APP(V642I). As shown in Figures 4B and 5B, inhibition of the neddylation pathway by expression of hUbc12(C111S), which blocks apoptosis caused by exogenous expression of APP–BP1 or APP(V642I) (Chen et al., 2003), does not block either the enlargement of endosomes (Fig. 4B) or the enhanced receptor-mediated endocytosis (Fig. 5B) caused by APP–BP1 or APP(V642I). These data suggest that the endosomal changes caused by these proteins are independent of neddylation, which may occur downstream of Rab5 activation or in an independent APP–BP1-mediated pathway.
Rab5 binds to a subregion of APP–BP1 between amino acids 443 and 479
Given that the effects on endosomes of exogenous expression of APP–BP1 are independent of the neddylation pathway, we addressed the possibility that direct interaction of APP–BP1 with Rab5 might mediate the endosomal abnormalities seen in neurons infected with HSV–APP–BP1. To determine whether APP–BP1 and Rab5 interact directly in neurons, we infected primary rat embryonic cortical neurons with HSV vectors expressing myc epitope-tagged APP–BP1 and Rab5, or myc-APP–BP1 and Rab7. Myc-APP–BP1 was immunoprecipitated from the neuronal cell lysates with 9E10, an antibody specific for the myc epitope. The immunoprecipitated material was blotted with antibodies to Rab5 or to Rab7. As shown in Figure 6, the antibody to Rab5 immunodetected a specific protein band in the immunoprecipitated fraction that comigrates with myc–Rab5 in the lysate from a culture infected with HSV–myc–Rab5 (Fig. 6A). The Rab7 antibody did not immunodetect a protein in the immunoprecipitated fraction (Fig. 6B). These data suggest that APP–BP1 interacts specifically with Rab5 in cells in rat primary cortical cultures.
Figure 6.

Rab5 binds to a subregion of APP–BP1 within amino acids 443–479. Primary neurons were infected with HSV vectors expressing myc–APP–BP1 and Rab5 or myc–APP–BP1 and Rab7. myc–APP–BP1 was precipitated from the cell lysate with the anti-myc antibody 9E10 or with an irrelevant anti-HA antibody. A, B, The blots were probed with anti-Rab5 (A) or anti-Rab7 (B). Rab5, but not Rab7, coprecipitated with myc–APP–BP1. The lane labeled “whole cell” represents 4% of the lysate used for immunoprecipitation (i.p.). C, To determine the subregion of APP–BP1 to which Rab5 binds, neurons were coinfected with HSV–Rab5 and HSV vectors expressing APP–BP1(Δ401–479), HA–APP–BP1(443–479), or HA–APP–BP1(145–251). Immunoprecipitation was performed with anti-APP–BP1 or anti-HA, and the immunoprecipitate was blotted with an antibody to Rab5. A specific protein band was detected in cells coexpressing APP–BP1 or APP–BP1(443–479) with Rab5 but not in those coexpressing APP–BP1(Δ401–479) or APP–BP1(145–225), suggesting that Rab5 binds to a region within amino acids 443–479 in APP–BP1.
To define the subregion of APP–BP1 to which Rab5 binds, we coinfected cortical neurons with HSV–Rab5 and one of three vectors expressing three different APP–BP1 peptides: APP–BP1(Δ401–479), HA-tagged APP–BP1(443–479), or HA-tagged APP–BP1(145–225). APP–BP1(Δ401–479) was immunoprecipitated with anti-APP–BP1, and HA–APP–BP1(443–479) or HA–APP–BP1(145–225) was immunoprecipitated with anti-HA. The immunoprecipitated material was blotted with an antibody to Rab5. As shown in Figure 6C, the antibody to Rab5 immunodetected a specific protein band in cells overexpressing full-length APP–BP1, as well as in those expressing APP–BP1(443–479), but not in those expressing APP–BP1(Δ401–479) or APP–BP1(145–225). This suggests that Rab5 binds to a region within amino acids 443–479 in APP–BP1.
APP–BP1 and Rab5 proteins colocalize in cultured neurons and in neurons in control and AD frontal cortex
To determine whether APP–BP1 and Rab5 were present in the same subcellular compartments, we infected primary cultures of rat cortical neurons with an HSV vector expressing APP–BP1. Immunocytochemical analysis of the cultures with antibodies to APP–BP1 and Rab5 revealed overlap in their subcellular distributions, primarily in punctuate endosome-like structures (Fig. 7A). To determine whether these in vitro data are reflected in vivo, postmortem prefrontal cortex tissue from AD and control human brains was stained with antibodies to APP–BP1 and Rab5. The staining confirmed the existence of enlarged Rab5-positive endosomes in AD brain compared with control and revealed a significant overlap between APP–BP1 and Rab5, in both control and AD brain, that was particularly pronounced in the enlarged endosomes in AD brain (Fig. 7B).
Figure 7.

APP–BP1 and Rab5 colocalize in cultured neurons overexpressing APP–BP1 and in neurons in control and AD frontal cortex, as shown by confocal microscopy. A, Immunocytochemical analysis of neuronal cultures expressing exogenous APP–BP1 revealed close to a 63% overlap in their subcellular distributions, primarily in punctuate endosome-like structures. Scale bar, 5 μm. B, To determine whether these data are reflected in vivo, we stained postmortem prefrontal cortex tissue from AD and control human brains with antibodies to APP–BP1 and Rab5. The staining reveals an average of 82% overlap between APP–BP1 and Rab5 in control and AD brain, particularly in the enlarged endosomes in AD (arrows). Scale bar, 5 μm. Overlap was determined by comparing the number of costained cells with the total number of stained cells.
APP–BP1 and Rab5 levels are increased in early endosomes in primary neurons expressing exogenous APP–BP1 or APP(V642I), in DS skin fibroblasts, and in AD hippocampus
Subcellular fractionation of rat brain tissue (Fig. 8A) was performed to confirm the localization of APP–BP1 in a fraction enriched in Rab5-containing endosomes. Whole rat brain tissue was fractionated into heavy membranes, early endosomes, and late endosomes (Seemann et al., 1997). Equal amounts of protein from the three fractions were subjected to immunoblot analysis with antibodies to APP–BP1 and Rab5. Indeed, APP–BP1 was enriched in the Rab5-positive early endosome fraction.
Figure 8.

APP–BP1 and Rab5 levels are increased relative to controls in early endosomes in primary neurons expressing exogenous APP–BP1 or APP(V642I), in DS skin fibroblasts, and in AD hippocampus. A, Subcellular endosomal fractionation of rat brain tissue shows increased levels of APP–BP1 specifically in the Rab5-positive early endosome fraction. H, Heavy membranes; L, late endosomes; E, early endosomes. B, Primary cortical rat embryonic neurons expressing APP–BP1 or APP(V642I) were fractionated and subjected to immunoblot analysis. Equal amounts of protein were loaded for all samples, and densitometry values were standardized to levels of GAPDH. Neurons expressing APP–BP1 or APP(V642I) show an increase in standardized APP–BP1 and Rab5 levels in early endosomes (EE) relative to controls (neurons expressing LacZ). C, D, Lysates of skin fibroblasts from DS subjects (C) and from postmortem hippocampal tissue from control and AD brains (D) were similarly fractionated and subjected to immunoblot analysis. In D, each lane represents a postmortem tissue sample from a distinct control (con) or AD subject (see Table 1). ANOVA showed significant increases in standardized levels of APP–BP1 and Rab5 in early endosomes in DS cells and in AD brain relative to controls. *p < 0.001.
Levels of both APP–BP1 and Rab5 were assessed specifically in early endosomes in primary neurons expressing exogenous APP–BP1 or APP(V642I), in DS skin fibroblasts, or in AD hippocampus relative to controls (Table 1). Rat primary cortical embryonic neurons infected with HSV–APP–BP1 or with HSV–APP(V642I) were fractionated and subjected to immunoblot analysis with antibodies to APP–BP1 and Rab5. As shown in Figure 8B, APP–BP1 and Rab5 levels are increased specifically in early endosomes after exogenous expression of either APP–BP1 or APP(V642I). To determine whether this elevation of APP–BP1 and Rab5 in early endosomes is also seen in DS and AD, lysates of skin fibroblasts from DS subjects, as well as from postmortem hippocampal tissue from AD brains, were similarly fractionated and subjected to immunoblot analysis. Figure 8 demonstrates a significant increase in levels of APP–BP1 and Rab5 in early endosomes in DS cells (Fig. 8C) as well as in AD brain (Fig. 8D) relative to controls, confirming the relevance to DS and AD of these alterations in APP–BP1 and Rab5.
Exogenous expression of FAD mutants of presenilin does not affect the number or size of Rab5-positive structures in neurons
Cataldo et al. (2001) reported that endocytic pathway abnormalities are present in the brains of individuals with Alzheimer's disease caused by certain FAD APP mutations but are not detectable in the brains of individuals with Alzheimer's disease caused by FAD mutants of PS1. To test whether the same was true in our in vitro model, we infected primary neuronal cultures with HSV–APP(V642I), HSV–PS1(A280E), or HSV–PS1(M146L) and analyzed the cultures immunocytochemically with an antibody to Rab5 (Fig. 9). Consistent with the observations of Cataldo et al. (2001), we detected numerous enlarged Rab5-positive structures in neurons infected with HSV–APP(V642I) but not in neurons infected with HSV vectors expressing either of the FAD PS1 mutations.
Figure 9.

Expression of FAD mutants of presenilin does not affect the number or size of Rab5-positive structures in neurons. Primary neurons were infected with HSV vectors expressing either of two presenilin 1 mutations (M146L or A280E) or APP(V642I), fixed, and stained with rabbit polyclonal anti-Rab5. The cells expressing either of the two presenilin 1 mutations did not show an increase in the number or size of Rab5-positive structures relative to those observed in cells expressing APP(V642I). Each panel shows a single neuron. Scale bar, 5 μm.
Dominant-negative Rab5 blocks apoptosis caused by either APP–BP1 or APP(V642I)
We have shown here that primary neurons expressing APP(V642I) or APP–BP1 manifest endosomal abnormalities similar to those observed in AD brain. We showed previously that exogenous expression of APP(V642I) or APP–BP1 in primary neurons in culture caused the neurons to enter apoptosis (Chen et al., 2003). To determine whether the endosomal abnormalities caused by expression of these proteins in neurons is related to neuronal apoptosis, we used HSV vectors to coexpress a dominant-negative mutant of Rab5 (S34N) with APP–BP1 or APP(V642I) in primary neuronal cultures. Immunocytochemical analysis of the infected cultures with an antibody to the early endosomal antigen EEA1 showed that coexpression of the dominant-negative mutant of Rab5 with either APP–BP1 or APP(V642I) inhibited the endosome enlargement caused by expression of either of these proteins alone (Fig. 10A). In addition, incubation of neurons coexpressing the Rab5 mutant with APP–BP1 or APP(V642I) with fluorophore Alexa 588-conjugated transferrin for 5 min showed an inhibition of receptor-mediated uptake compared with neurons expressing APP–BP1 or APP(V642I) alone (Fig. 10B).
Figure 10.

Dominant-negative Rab5 blocks apoptosis caused by exogenous expression of either APP–BP1 or APP(V642I). A, Coexpression of a dominant-negative mutant of Rab5 (S34N) with either APP–BP1 or APP(V642I) inhibits the early endosome enlargement caused by expression of either of these proteins alone. Primary neurons were infected with HSV vectors expressing APP–BP1 and APP(V642I) alone or together with Rab5(S34N), fixed, and stained with the rabbit polyclonal anti-EEA1. Neurons coexpressing Rab5(S34N) did not show the increase in amount and size of early endosome staining that is observed in neurons expressing either APP–BP1 or APP(V642I) alone. Each panel presents endosomes in a single neuron. Scale bar, 5 μm. B, Coexpression of a dominant-negative mutant of Rab5, Rab5(S34N), with either APP–BP1 or APP(V642I) inhibits enhancement of receptor-mediated endocytosis that is caused by expression of either of these proteins alone. Primary neurons were infected with HSV vectors expressing APP–BP1 or APP(V642I), alone or together with Rab5(S34N), and incubated for 5 min with transferrin at 37°C. Cells coexpressing Rab5(S34N) present more membrane-bound transferrin and less uptake of transferrin than do cells expressing APP–BP1 or APP(V642I) alone. Each panel presents endosomes in a single neuron. Scale bar, 5 μm. C, Dominant-negative Rab5 blocks neuronal apoptosis caused by exogenous expression of either APP–BP1 or APP(V642I). Primary neurons were infected with HSV vectors expressing APP–BP1 or APP(V642I), alone or together with Rab5(S34N), Rab7(N125I), or Rab11(S27N), fixed, and stained with bisbenzimide to assess apoptosis. Apoptosis induced by exogenous expression of APP–BP1 or APP(V642I) was blocked by coexpression of Rab5(S34N) but not by coexpression of either Rab7(N125I) or Rab11(S27N). ANOVA for differences between expression of APP–BP1 alone or with the dominant-negative mutants by ANOVA revealed significant differences only between APP–BP1 versus APP–BP1/Rab5(S34N). The analysis for differences between expression of APP(V642I) alone or with the dominant-negative mutants showed a significant difference only between APP(V642I) versus APP(V642I)/Rab5(S34N). *p < 0.001. Inset shows bisbenzimide staining of condensed nuclei in representative fields for each condition.
As shown in Figure 10C, apoptosis caused by exogenous expression of APP–BP1 or APP V642I also is inhibited when Rab5 function is blocked by expression of a Rab5 dominant-negative mutant (S34N). In contrast, coexpression of dominant-negative mutants of Rab7 (N125I) or of Rab11 (S27N) with APP–BP1 or APP(V642I) did not inhibit neuronal apoptosis caused by these proteins. The apoptosis data were obtained by bisbenzimide staining and were independently confirmed by terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling assay.
Dominant-negative Eps15 does not rescue neurons from apoptosis caused by APP–BP1 or APP(V642I) but dominant-negative dynamin does
Rab5 has been shown to play direct signaling roles that are intimately tied to the endocytosis machinery but are distinct from its housekeeping role in endocytosis (Lanzetti et al., 2004; Miaczynska et al., 2004). We asked whether the rescue effect of rab5(S34N) on APP–BP1- or APP(V642I)-induced apoptosis was the consequence of inhibiting endocytosis or of inhibiting a direct Rab5-mediated signaling pathway within the endo-somal compartment. Eps15 is a constitutive component of clathrin-coated pits, and the dominant-negative Eps15 mutant, Eps15Δ95–295, inhibits both ligand-induced and constitutive endocytosis by sequestering the endocytic adaptor protein AP2 (Benmerah et al., 1999), without directly affecting Rab5. Coexpression of Eps15Δ95–295 with APP–BP1 or APP(V642I) in primary cortical embryonic neurons did not rescue neurons from their apoptotic fate (Fig. 11A), suggesting that the involvement of Rab5 in the APP signal transduction pathway may be distinct from its role in endocytosis.
Figure 11.
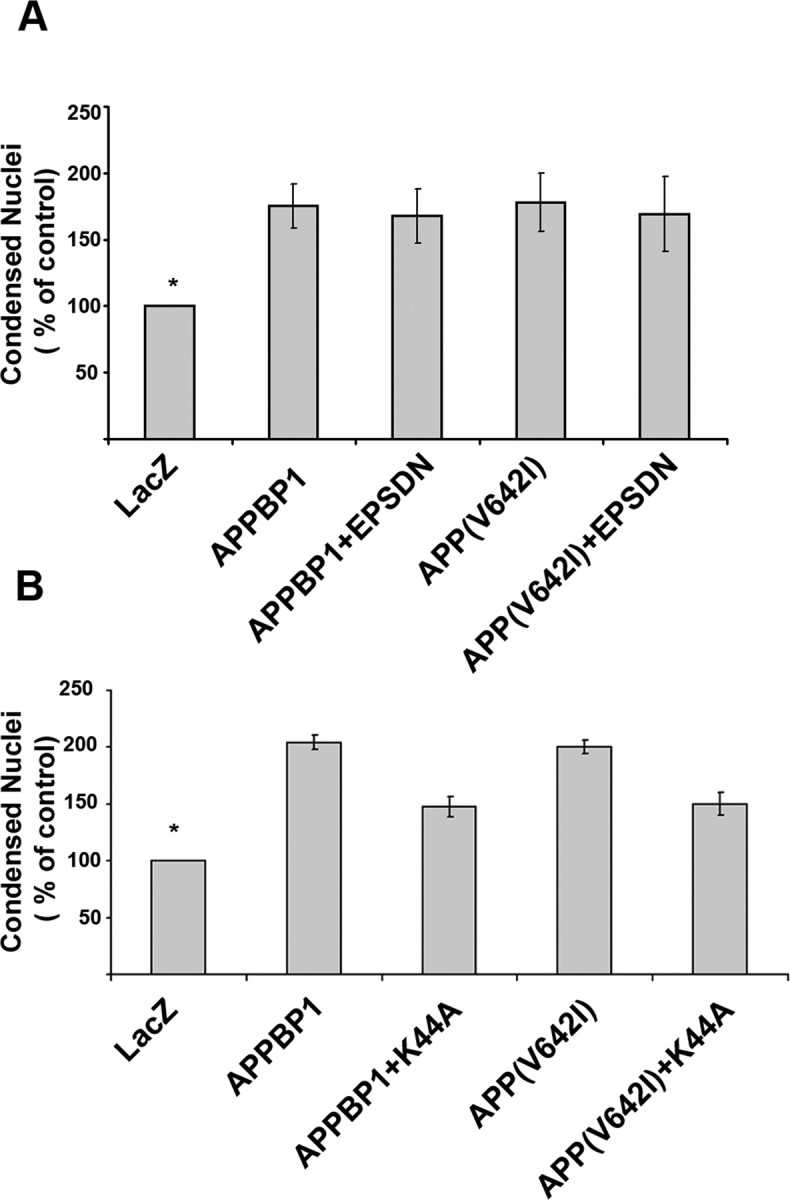
Dominant-negative Eps15 fails to rescue, and dominant-negative dynamin partially rescues, neurons from apoptosis caused by exogenous expression of APP–BP1 or APP(V642I). We coexpressed Eps15Δ95–295 (labeled EPSDN in the figure) or dynamin(K44A) with APP–BP1 or APP(V642I) in primary cortical embryonic neurons and stained the neurons with bisbenzimide to assess apoptosis. A, ANOVA for differences between apoptosis levels in neurons coexpressing Eps15Δ95–295 revealed no significant differences between levels of apoptosis caused by APP–BP1, APP–BP1/Eps15Δ95–295, APP(V642I), or APP(V642I)/Eps15Δ95–295. *p < 0.001 compared with control. B, In contrast, coexpression of dynamin K44A significantly reduced apoptosis caused by APP–BP1 or APP(V642I) (p < 0.01). *p < 0.001, APP–BP1 or APP(V642I) versus lacZ control.
However, despite the fact that Eps15Δ95–295 appeared to inhibit the increase in endosome size caused by exogenous expression of APP–BP1 or APP(V642I) in neurons, it remained possible that a subpopulation of endosomes outside of the clathrin-dependent endocytic pathway remained enlarged in the presence of Eps15Δ95–295 and that this subpopulation mediated apoptosis caused by APP–BP1 or APP(V642I). To test this hypothesis, we coexpressed dominant-negative dynamin(K44A) with APP–BP1 or APP(V642I). Dynamin is a large GTPase that plays an essential role in vesicle scission during endocytosis. Whereas Eps15 participates strictly in clathrin-mediated endocytosis, dynamin participates not only in clathrin-mediated endocytosis but also in calveolin-mediated endocytosis and may play a role in mitogenic signaling that is distinct from its role in receptor-mediated endocytosis (for review, see Sever, 2002). As shown in Figure 11B, dynamin(K44A) partially inhibits neuronal apoptosis caused by APP–BP1 or APP(V642I), suggesting that the apoptosis may be attributable in part to either calveolin-mediated endocytosis and/or rab5 signaling that is distinct from, but not independent of, endocytosis.
Induction of enlarged endosomes through a Rab5-dependent mechanism leads to an increase in neuronal apoptosis greater than that induced by enlargement of endosomes via a Rab5-independent mechanism
The data from the previous experiment suggested that at least part of the function of Rab5 in the apoptotic pathway initiated by APP may be a specialized endocytic event distinct from its housekeeping role in endocytosis, because inhibiting clathrin-dependent endocytosis did not in itself result in neuronal rescue. We therefore asked whether using a Rab5-independent mechanism to enlarge endosomes in cortical neurons would have a different effect on apoptosis from that seen by expressing a constitutively active mutant of Rab5(Q79L). Expression of Rab5(Q79L) causes endosome enlargement (Stenmark et al., 1994) and regulates specific mitogenic signaling (Dinneen and Ceresa, 2004) in cultured cells. Hrs is an early endosomal protein that, when overexpressed, also causes enlargement of endosomes (Komada et al., 1997). Hrs does not interact with Rab5 (Hayakawa and Kitamura, 2000), suggesting that Hrs-induced enlargement of endosomes proceeds by a pathway independent of Rab5. Figure 12 shows that, although either expression of Rab5(Q79L) or overexpression of Hrs in primary neurons in culture leads to apoptosis, the increase in neuronal apoptosis induced by Rab5(Q79L) is more than twofold greater than that observed in Hrs-overexpressing cells.
Figure 12.

Exogenous expression of a constitutively active Rab5(Q79L) leads to an increase in neuronal apoptosis that is more than twofold greater than that induced by overexpression of Hrs. Primary rat cortical embryonic neurons infected with HSV vectors expressing Rab5(Q79L), Hrs, or LacZ (control) were stained with bisbenzimide to assess apoptosis. ANOVA for differences between levels of apoptosis revealed that neurons infected with Rab5(Q79L) showed an increase in the level of apoptosis that was more than two times greater (140 vs 60%) than that observed in neurons infected with Hrs relative to control. *p < 0.001 compared with control. **p < 0.001 compared with Hrs.
Levels of Aβ do not correlate with effects on neuronal apoptosis
The endocytic pathway is thought to be involved in APP metabolism, and endosomes have been shown to be an intracellular site of Aβ generation (Perez et al., 1999; Soriano et al., 1999). To determine whether APP–BP1- or APP(V642I)-mediated neuronal apoptosis correlates with intracellular or secreted Aβ levels, we assessed Aβ levels after blocking APP–BP1- or APP(V642I)-induced enlargement of endosomes by coexpression of dominant-negative mutants of Rab5 or Eps15. Although intracellular and secreted Aβ levels were increased in cells expressing APP(V642I) and intracellular levels were increased, to a lesser degree, by APP–BP1, no difference in either intracellular or secreted Aβ levels was found consequent to coexpression of either of the dominant-negative mutants. Secreted Aβ levels were increased to a similar extent consequent to the induction of enlarged endosomes by expression of either constitutively active Rab5 or overexpressed Hrs. However, the former resulted in an increase in neuronal apoptosis more than twice that caused by overexpression of Hrs, demonstrating a lack of correlation between Aβ levels and endocytic effects on apoptosis (data not shown).
Discussion
We show here that exogenous expression of APP(V642I) or the APP binding protein APP–BP1 increases the number and size of Rab5-positive early endosomes in neurons and enhances receptor-mediated endocytosis. We further demonstrate that APP–BP1 or APP(V642I) exogenous expression increases protein levels of Rab5 in neurons, suggesting that this may be the mechanism by which exogenous expression of these cDNAs causes endosomal enlargement and activation. The activation of endosomes by APP(V642I) or APP–BP1 did not occur when APP binding to APP–BP1 was inhibited or when a dominant-negative mutant of Rab5 was coexpressed. These data confirm the involvement of APP and APP–BP1 in a Rab5-dependent signaling pathway linked to endosomal enlargement and activation.
Endocytosis has been linked to the cell cycle (Sager et al., 1984; Warren et al., 1984; Warren, 1989; Polo et al., 2004), leading us to focus in the current study on APP–BP1, an APP binding partner and a cell cycle protein (Y. Chen et al., 2000), as a potential candidate mediating the FAD APP-initiated signaling pathway that regulates Rab5 activity. Exogenous expression of APP–BP1 in neurons has been shown to cause DNA synthesis and consequent apoptosis via a signaling pathway that is dependent on APP–BP1 binding to APP (Chen et al., 2003). We show here that APP–BP1 binds not only to APP but also to Rab5 in neurons. Indeed, we were able to pinpoint the Rab5 binding site within APP–BP1 to amino acids 443–479, the same region to which hUba3, the APP–BP1 partner in the neddylation pathway, binds.
The functional role of APP–BP1 in modulating Rab5 activity is not known. However, it is possible that APP–BP1 acts as a guanine nucleotide exchange factor (GEF) for Rab5. GEFs stimulate the exchange of GDP for GTP, thereby generating the active forms of small GTPases such as Rab5. It is interesting that exogenous expression of another Rab5 GEF, ALS2 (amyotrophic lateral sclerosis 2), which is mutated in a number of motor neuron diseases, causes the protein to colocalize with Rab5 and EEA1 in early endosomal compartments and stimulates the enlargement of endosomes in cultured cortical neurons (Otomo et al., 2003). We have not shown that APP, APP–BP1, and Rab5 exist as a tripartite complex, but if they do, APP–BP1, acting as a GEF, could actually initiate the recruitment of Rab5 to the membranes.
Although dominant-negative mutants in the neddylation pathway prevent neuronal apoptosis mediated by APP–BP1 or FAD APP, they did not prevent endosome enlargement and activation by these same proteins. These data suggest that the neddylation cascade lies downstream of endocytic activation in the apoptotic pathway induced by APP–BP1 or APP(V642I) or that it represents a separate pathway. Our finding that inhibition of neddylation by expression of either Ubc12(C111S) or the deneddylating enzyme NEDP1 does not alter Rab5(Q79L)- or Hrs-mediated apoptosis (D.Laifenfeld, D. L. McPhie, and R. L. Neve, unpublished data) favors the latter interpretation.
The data presented in this paper support the idea that APP–BP1 links APP signaling to Rab5 and endocytosis in vitro, but is this pathway mechanistically relevant to AD? We demonstrated previously that the increase in APP–BP1 protein in neurons expressing exogenous APP(V642I) is mirrored in neurons in affected regions of AD brain and that NEDD8 is translocated to the cytoplasm in both cultured neurons overexpressing APP–BP1 and hippocampal neurons in AD brain (Chen et al., 2003). Here, we extend these findings by showing that APP–BP1 and Rab5 colocalize in punctuate structures in both cultured neurons and human postmortem brain. Moreover, this colocalization is enhanced in enlarged endosomes in both cultured neurons expressing exogenous APP–BP1 or APP(V642I) and in AD prefrontal cortex relative to controls. We further demonstrate a significant increase in the expression of both APP–BP1 and Rab5 in early endosomes of primary neurons expressing exogenous APP–BP1 or APP(V642I), skin fibroblasts from DS subjects, and neurons in AD hippocampus. These data suggest that this pathway, which links APP signaling directly to endosomes, may play a role in the development of neuropathology in both DS and AD.
FAD can be caused by mutations in the APP gene or in the presenilin genes PS1 and PS2. We reported previously that, in contrast to FAD mutant APPs, neither wild-type nor FAD mutant PS1 enhances apoptosis in neurons (Bursztajn et al., 1998). Similarly, in the present study, we showed that neurons expressing exogenous wild-type or FAD PS1 mutations differed from those expressing exogenous FAD APP in that they did not display an increase in the number or size of Rab5-positive structures. Thus, the APP-APP–BP1–Rab5 signaling pathway is distinct from presenilin signaling pathways or γ-secretase activity. This finding is consistent with the report by Cataldo et al. (2001) that the presence of enlarged endosomes distinguished FAD APP-caused AD from FAD PS1-caused AD. Notably, expression of FAD APP leads to increased levels of secreted α-APP and β-APP and to elevation of levels of specific C-terminal fragments of APP in addition to Aβ, whereas the only known effect of FAD presenilins on APP processing is alteration of the ratio of Aβ42 to Aβ40. These data suggest that fragments of APP other than Aβ may play a role in endocytic abnormalities in AD.
Neuronal loss is associated with AD. There is considerable evidence that one mechanism by which neurons die in AD is apoptosis (for review, see LeBlanc, 2005). Indeed, several groups have reported the presence of activated caspases in the postmortem brains of individuals with AD (Selznick et al., 1999; Stadelmann et al., 1999; Raina et al., 2001) and have described apoptotic neurons in the brains of transgenic mouse models for AD (LaFerla et al., 1995; Chui et al., 1999; Xie et al., 2001; Hwang et al., 2004). We showed previously that exogenous expression of FAD APP mutations or of APP–BP1 in primary neurons causes cell cycle entry followed by apoptosis (McPhie et al., 1997, 2001).
We now demonstrate that the endosomal enlargement induced by exogenous expression of APP(V642I) or APP–BP1 is linked to the induction of apoptosis in these neurons, in that coexpression of a dominant-negative mutant of Rab5, which blocks the endosomal enlargement, inhibits the apoptosis. In contrast, suppression of clathrin-dependent endocytosis without directly affecting Rab5, by coexpressing dominant-negative Eps15, did not affect either APP–BP1- or APP(V642I)-induced apoptosis. However, inhibition of more global endocytosis by coexpression of dominant-negative dynamin partially inhibited both APP–BP1- and APP(V642I)-mediated apoptosis. Conversely, induction of enlarged endosomes via expression of a constitutively active form of Rab5 led to an increase in neuronal apoptosis that was more than twice that induced via a Rab5-independent pathway, by overexpression of the early endosomal protein Hrs. These data suggest that the signaling cascade induced by APP–BP1 or APP(V642I) that leads to apoptosis is only partly caused by endosomal enlargement and may also involve a Rab5 signaling function that is distinct from, but not independent of, trafficking endocytosis.
Notably, the role of Rab5 in apoptosis induced by the APP pathway was shown to be independent of endosomal effects on Aβ levels. Inhibiting APP–BP1- or APP(V642I)- induced endosome enlargement by either dominant-negative Rab5 or dominant-negative Eps15 (with only the former leading to rescue from neuronal apoptosis) had no affect on levels of Aβ in cells expressing exogenous APP–BP1 or APP(V642I). Similarly, although increasing endosome size by either expression of constitutively active Rab5 or overexpression of Hrs led to a similar increase in levels of Aβ, Rab5 activation led to a greater than twofold greater increase in neuronal apoptosis relative to overexpression of Hrs.
Together, our findings point to a pathway initiated by exogenous expression of APP or expression of FAD mutants of APP that is routed through APP–BP1 and Rab5 and that culminates in apoptosis. Recent compelling evidence suggests that, in addition to playing a role in endocytosis, Rab5 can serve as a signaling molecule in other pathways (Barbieri et al., 2004; Lanzetti et al., 2000; Tall et al., 2001; Miaczynska et al., 2004). The present finding that apoptosis induced by APP–BP1 or APP(V642I) is differentially affected by Rab5-dependent versus Rab5-independent inhibition of enlarged endosomes may point to the importance of Rab5 in a signaling pathway separate from its role in trafficking endocytosis, in this pathway. It has been suggested that endosomes may serve as intermediates within signaling pathways that regulate apoptosis (Nixon, 2005). In this regard, proteins such as Alix/AIP (apoptosis-linked gene 2 interacting protein X/apoptosis-linked gene 2 interacting protein) that indirectly regulate endosome formation, shape, and function (for review, see Katzmann et al., 2002) have been found to modulate apoptosis in diverse ways (Missotten et al., 1999; B. Chen et al., 2000). By analogy, APP may serve a similar role. Recently, Miaczynska et al. (2004) identified APPL (adaptor protein containing PH domain, PTB domain, and leucine zipper motif) proteins as Rab5 effectors that transduce signals between the plasma membrane and the nucleus, whereas Lanzetti et al. (2004) identified RN-tre (related to the N terminus of the tre oncogene product) as a Rab5 effector that transduces signals that cause actin remodeling, suggesting additional pathways through which APP/APP–BP1 could propagate Rab5-mediated downstream signaling.
An APP(V642I)- or APP–BP1-induced increase in Rab5 levels affecting Rab5 signaling could lead to a secondary effect on endocytosis, which could result in a cumulative stimulation of apoptosis by the two pathways. Indeed, our data demonstrate that activating the endocytic machinery by overexpression of Hrs led to a low level of neuronal apoptosis. Modulation of the endocytosis machinery by signaling pathways has been shown previously. For example, the epidermal growth factor (EGF) regulates clathrin-coated membrane formation and trafficking and subsequent endocytosis of EGF, via activation of src kinase (Wilde et al., 1999). Such signaling pathways appear to intersect specifically with Rab5 (Cavalli et al., 2001). Thus, EGF activates Rab5 (Barbieri et al., 2004) as well as the Rab5 GTPase-activating protein RN-tre (Lanzetti et al., 2000).
Altered endocytosis can promote neurodegeneration in several ways. For example, neurons rely on endocytosis to communicate with peripheral tissues (Sofroniew et al., 2001), and signaling endosomes, possessing the characteristics of early endosomes, participate in the transduction of signals that promote neuronal survival and differentiation (Delcroix et al., 2003). These properties of endosomes may explain the failure of retrograde signaling by NGF that has been observed in a mouse model of Down syndrome (Cooper et al., 2001). Alternatively, endosome dysfunction could lead to altered APP processing (Grbovic et al., 2003) or to disturbed vesicular transport in AD (Burke et al., 1990; Stokin et al., 2005).
We have shown previously (Chen et al., 2003) that APP–BP1 binds to the C-terminal 31 amino acids of APP (APP–C31). APP–C31 has physiological significance: APP is cleaved by caspases intracytoplasmically at Asp-664 to generate APP–C31, which is cytotoxic (Lu et al., 2000). Both our laboratory and others have reported that exogenous expression of APP–C31 specifically in neurons causes apoptosis (Bertrand et al., 2001; McPhie et al., 2003). In vivo, cleavage of FAD APPs at Asp-664 in transgenic mice appears to be necessary for these genes to cause Alzheimer's-like pathology and defects in synaptic transmission and learning (Galvan et al., 2006; Saganich et al., 2006). It will be interesting to find out whether generation of APP–C31 by caspase cleavage of FAD APPs causes also the endosome abnormalities described in the current study.
Footnotes
This work was supported by National Institutes of Health Grants AG12954 and AG021185 (R.L.N.). We thank Dr. Angela Wandinger-Ness for the Rab4 and Rab7 cDNAs, Dr. Marianne Wessling-Resnick for the Rab5 cDNAs, Dr. Alexandre Benmerah for the dominant-negative Eps15 cDNA, Dr. Richard Vallee for the dominant-negative dynamin cDNA, and Dr. Masayuki Komada for the Hrs cDNA.
References
- Barbieri MA, Fernandez-Pol S, Hunder C, Horazdovsky BH, Stahl PD. Role of rab5 in EGF receptor-mediated signal transduction. Eur J Cell Biol. 2004;83:305–314. doi: 10.1078/0171-9335-00381. [DOI] [PubMed] [Google Scholar]
- Benmerah A, Bayrou M, Cerf-Bensussan N, Dautry-Varsat A. Inhibition of clathrin-coated pit assembly by an Eps15 mutant. J Cell Sci. 1999;112:1303–1311. doi: 10.1242/jcs.112.9.1303. [DOI] [PubMed] [Google Scholar]
- Bertrand E, Brouillet E, Caille I, Bouillot C, Cole GM, Prochiantz A, Allinquant B. A short cytoplasmic domain of the amyloid precursor protein induces apoptosis in vitro and in vivo. Mol Cell Neurosci. 2001;18:503–511. doi: 10.1006/mcne.2001.1030. [DOI] [PubMed] [Google Scholar]
- Brouillet E, Trembleau A, Galanaud D, Volovitch M, Bouillot C, Valenza C, Prochiantz A, Allinquant B. The amyloid precursor protein interacts with Go heterotrimeric protein within a cell compartment specialized in signal transduction. J Neurosci. 1999;19:1717–1727. doi: 10.1523/JNEUROSCI.19-05-01717.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke WJ, Park DH, Chung HD, Marshall GL, Haring JH, Joh TH. Evidence for decreased transport of tryptophan hydroxylase in Alzheimer's disease. Brain Res. 1990;537:83–87. doi: 10.1016/0006-8993(90)90342-9. [DOI] [PubMed] [Google Scholar]
- Bursztajn S, DeSouza R, McPhie DL, Berman SA, Shioi J, Robakis NK, Neve RL. Overexpression in neurons of human presenilin-1 or a presenilin-1 familial Alzheimer disease mutant does not enhance apoptosis. J Neurosci. 1998;18:9790–9799. doi: 10.1523/JNEUROSCI.18-23-09790.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cataldo A, Rebeck GW, Ghetri BG, Hulette C, Lippa C, Van Broeckhoven C, van Duijn C, Cras P, Bogdanovic N, Bird T, Peterhoff C, Nixon R. Endocytic disturbances distinguish among subtypes of Alzheimer's disease and related disorders. Ann Neurol. 2001;50:661–665. [PubMed] [Google Scholar]
- Cataldo AM, Peterhoff CM, Troncoso JC, Gomez-Isla T, Hyman BT, Nixon RA. Endocytic pathway abnormalities preceded amyloid β deposition in sporadic Alzheimer's disease and Down syndrome. Am J Pathol. 2000;157:277–286. doi: 10.1016/s0002-9440(10)64538-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavalli V, Corti M, Gruenberg J. Endocytosis and signaling cascades: a close encounter. FEBS Lett. 2001;498:190–196. doi: 10.1016/s0014-5793(01)02484-x. [DOI] [PubMed] [Google Scholar]
- Chen B, Borinstein SC, Gillis J, Sykes VW, Bogler O. The glioma-associated protein SETA interacts with AIP1/Alix and ALG-2 and modulates apoptosis in astrocytes. J Biol Chem. 2000;275:19275–19281. doi: 10.1074/jbc.M908994199. [DOI] [PubMed] [Google Scholar]
- Chen Y, McPhie DL, Hirschberg J, Neve RL. The amyloid precursor protein-binding protein APP–BP1 drives the cell cycle through the S-M checkpoint and causes apoptosis in neurons. J Biol Chem. 2000;275:8929–8935. doi: 10.1074/jbc.275.12.8929. [DOI] [PubMed] [Google Scholar]
- Chen Y, Liu W, McPhie DL, Hassinger L, Neve RL. APP–BP1 mediates APP-induced apoptosis and DNA synthesis and is increased in Alzheimer's disease brain. J Cell Biol. 2003;163:27–33. doi: 10.1083/jcb.200304003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow N, Korenberg JR, Chen XN, Neve RL. APP–BP1, a novel protein that binds to the carboxyl-terminal region of the amyloid precursor protein. J Biol Chem. 1996;271:11339–11346. doi: 10.1074/jbc.271.19.11339. [DOI] [PubMed] [Google Scholar]
- Chui DH, Tanahashi H Ozawa K, Ikeda S, Checler F, Ueda O, Suzuki H, Araki W, Inoue H, Shirotani K, Takahashi K, Gallyas F, Tabira T. Transgenic mice with Alzheimer presenilin 1 mutations show accelerated neurodegeneration without amyloid plaque formation. Nat Med. 1999;5:560–564. doi: 10.1038/8438. [DOI] [PubMed] [Google Scholar]
- Cooper JD, Salehi A, Delcroix JD, Howe CL, Belichenko PV, Chua-Couzens J, Kilbridge JF, Carlson EJ, Epstein CJ, Mobley WC. Failed retrograde transport of NGF in a mouse model of Down's syndrome: reversal of cholinergic neurodegenerative phenotypes following NGF infusion. Proc Natl Acad Sci USA. 2001;98:10439–10444. doi: 10.1073/pnas.181219298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delcroix JD, Valletta JS, Wu C, Hunt SJ, Kowal AS, Mobley WC. NGF signaling in sensory neurons: evidence that early endosomes carry NGF retrograde signals. Neuron. 2003;39:69–84. doi: 10.1016/s0896-6273(03)00397-0. [DOI] [PubMed] [Google Scholar]
- Dinneen JL, Ceresa BP. Continual expression of Rab5(Q79L) causes a ligand-independent EGFR internalization and diminishes EGFR activity. Traffic. 2004;5:606–615. doi: 10.1111/j.1398-9219.2004.00204.x. [DOI] [PubMed] [Google Scholar]
- Enouf V, Chwetzoff S, Trugnan G, Cohen J. Interactions of rotavirus VP4 spike protein with the endosomal protein Rab5 and the prenylated Rab acceptor PRA1. J Virol. 2003;77:7041–7047. doi: 10.1128/JVI.77.12.7041-7047.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukumori A, Okochi M, Tagami S, Jiang J, Itoh N, Nakayama T, Yanagida K, Ishizuka-Katsura Y, Morihara T, Kamino K, Tanaka T, Kudo T, Tanji H, Ikuta A, Haass C, Takeda M. Presenilin-dependent γ-secretase on plasma membrane and endosomes is functionally distinct. Biochemistry. 2006;45:4907–4914. doi: 10.1021/bi052412w. [DOI] [PubMed] [Google Scholar]
- Galvan V, Gorostiza OF, Banwait S, Ataie M, Logvinova AV, Sitaraman S, Carlson E, Sagi SA, Chevallier N, Jin K, Greenberg DA, Bredesen DE. Reversal of Alzheimer's-like pathology and behavior in human APP transgenic mice by mutation of Asp664. Proc Natl Acad Sci USA. 2006;103:7130–7135. doi: 10.1073/pnas.0509695103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grbovic OM, Mathews PM, Jiang Y, Schnidt SD, Dinakar R, Summers-Terio NB, Ceresa BP, Nixon RA, Cataldo AM. Rab5-stimulated up-regulation of the endocytic pathway increases intracellular β-cleaved amyloid precursor protein carboxyl-terminal fragment levels and Aβ production. J Biol Chem. 2003;278:31261–31268. doi: 10.1074/jbc.M304122200. [DOI] [PubMed] [Google Scholar]
- Hayakawa A, Kitamura N. Early endosomal localization of Hrs requires a sequence within the proline- and glutamine-rich region but not the FYVE finger. J Biol Chem. 2000;275:29636–29642. doi: 10.1074/jbc.M002696200. [DOI] [PubMed] [Google Scholar]
- Hwang DY, Cho JS, Lee SH, Chae KR, Lim HJ, Min SH, Seo SJ, Song YS, Song CS, Paik SG, Sheen YY, Kim YK. Aberrant expressions of pathogenic phenotype in Alzheimer's diseased transgenic mice carrying NSE-controlled APPsw. Exp Neurol. 2004;186:20–32. doi: 10.1016/j.expneurol.2003.09.021. [DOI] [PubMed] [Google Scholar]
- Kang J, Lemaire HG, Unterbeck A, Salbaum JM, Masters CL, Grzeschik KH, Beyreuther K, Muller-Hill B. The precursor of Alzheimer's disease amyloid A4 protein resembles a cell-surface receptor. Nature. 1987;325:733–736. doi: 10.1038/325733a0. [DOI] [PubMed] [Google Scholar]
- Katzmann DJ, Odorizzi G, Emr SD. Receptor downregulation and multivesicular-body sorting. Nat Rev Mol Cell Biol. 2002;3:893–905. doi: 10.1038/nrm973. [DOI] [PubMed] [Google Scholar]
- Komada M, Masaki R, Yamamoto A, Kitamura N. Hrs, a tyrosine kinase substrate with a conserved double zinc finger domain, is localized to the cytoplasmic surface of early endosomes. J Biol Chem. 1997;272:20538–20544. doi: 10.1074/jbc.272.33.20538. [DOI] [PubMed] [Google Scholar]
- LaFerla FM, Tinkle BT, Bieberich CJ, Haudenschild CC, Jay G. The Alzheimer's Aβ peptide induces neurodegeneration and apoptotic cell death in transgenic mice. Nat Genet. 1995;9:21–30. doi: 10.1038/ng0195-21. [DOI] [PubMed] [Google Scholar]
- Lanzetti L, Rybin V, Malabarba MG, Christoforidis S, Scita G, Zerial M, Di Fiore PP. The Eps8 protein coordinates EGF receptor signaling through Rac and trafficking through Rab5. Nature. 2000;408:374–377. doi: 10.1038/35042605. [DOI] [PubMed] [Google Scholar]
- Lanzetti L, Palamidessi A, Areces L, Scita G, Di Fiore PP. Rab5 is a signalling GTPase involved in actin remodeling by receptor tyrosine kinases. Nature. 2004;429:309–314. doi: 10.1038/nature02542. [DOI] [PubMed] [Google Scholar]
- LeBlanc AC. The role of apoptotic pathways in Alzheimer's disease neurodegeneration and cell death. Curr Alzheimer Res. 2005;2:389–402. doi: 10.2174/156720505774330573. [DOI] [PubMed] [Google Scholar]
- Lee M-S, Kao S-C, Lemere CA, Xia W, Tseng H-C, Zhou Y, Neve R, Ahlijanian MK, Tsai L-H. APP processing is regulated by cytoplasmic phosphorylation. J Cell Biol. 2003;163:83–95. doi: 10.1083/jcb.200301115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu DC, Rabizadeh S, Chandra S, Shayya RF, Ellerby LM, Ye X, Salvesen GS, Koo EH, Bredesen DE. A second cytotoxic proteolytic peptide derived from amyloid β-protein precursor. Nat Med. 2000;6:397–404. doi: 10.1038/74656. [DOI] [PubMed] [Google Scholar]
- McPhie DL, Lee RKK, Eckman CB, Olstein DH, Durham SP, Yager D, Younkin SG, Wurtman RJ, Neve RL. Neuronal expression of β-amyloid precursor protein Alzheimer mutations causes intracellular accumulation of a C-terminal fragment containing both the amyloid β and cytoplasmic domains. J Biol Chem. 1997;272:24743–24746. doi: 10.1074/jbc.272.40.24743. [DOI] [PubMed] [Google Scholar]
- McPhie DL, Golde T, Eckman CB, Yager D, Younkin SG, Neve RL. The β-secretase cleavage product of the amyloid precursor protein mediates neuronal apoptosis caused by familial Alzheimer's disease mutations. Mol Brain Res. 2001;97:103–113. doi: 10.1016/s0169-328x(01)00294-7. [DOI] [PubMed] [Google Scholar]
- McPhie DL, Coopersmith R, Hines-Peralta A, Chen Y, Ivins KJ, Manly SP, Kozlowski MR, Neve KA, Neve RL. DNA synthesis and neuronal apoptosis caused by familial Alzheimer disease mutants of the amyloid precursor protein are mediated by the p21 activated kinase PAK3. J Neurosci. 2003;23:6914–6927. doi: 10.1523/JNEUROSCI.23-17-06914.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miaczynska M, Christoforidis S, Giner A, Shevchenko A, Uttenweiler-Joseph S, Habermann B, Wilm M, Parton RG, Zerial M. APPL proteins link rab5 to nuclear signal transduction via an endosomal compartment. Cell. 2004;116:445–456. doi: 10.1016/s0092-8674(04)00117-5. [DOI] [PubMed] [Google Scholar]
- Missotten M, Nichols A, Rieger K, Sadoul R. Alix, a novel mouse protein undergoing calcium-dependent interaction with the apoptosis-linked-gene 2 (ALG-2) protein. Cell Death Differ. 1999;6:124–129. doi: 10.1038/sj.cdd.4400456. [DOI] [PubMed] [Google Scholar]
- Murphy MP, Uljon SN, Fraser PE, Fauq A, Lookingbill HA, Findlay KA, Smith TE, Lewis PA, McLendon DC, Wang R, Golde TE. Presenilin 1 regulates pharmacologically distinct γ-secretase activities. Implication for the role of presenilin in γ-secretase cleavage. J Biol Chem. 2000;275:26277–26284. doi: 10.1074/jbc.M002812200. [DOI] [PubMed] [Google Scholar]
- Nishimoto I, Okamoto T, Matsuura Y, Okamoto T, Murayama Y, Ogata E. Alzheimer amyloid protein precursor complexes with brain GTP-binding protein G(o) Nature. 1993;362:75–79. doi: 10.1038/362075a0. [DOI] [PubMed] [Google Scholar]
- Nixon RA. Endosome function and dysfunction in Alzheimer's disease and other neurodegenerative diseases. Neurobiol Aging. 2005;26:373–382. doi: 10.1016/j.neurobiolaging.2004.09.018. [DOI] [PubMed] [Google Scholar]
- Otomo A, Hadano S, Okada T, Mizumura H, Kunita R, Nishijima H, Showguchi-Miyata J, Yanagisawa Y, Kohiki E, Suga E, Yasuda M, Osuga H, Nishimoto T, Narumiya S, Ikeda JE. ALS2, a novel guanine nucleotide exchange factor for the small GTPase Rab5, is implicated in endosomal dynamics. Hum Mol Genet. 2003;12:1671–1687. doi: 10.1093/hmg/ddg184. [DOI] [PubMed] [Google Scholar]
- Perez RG, Soriano S, Hayes JD, Ostaszewski B, Xia W, Selkoe DJ, Chen X, Stokin GB, Koo EH. Mutagenesis identifies new signals for β-amyloid precursor protein endocytosis, turnover, and the generation of secreted fragments, including Aβ42. J Biol Chem. 1999;274:18851–18856. doi: 10.1074/jbc.274.27.18851. [DOI] [PubMed] [Google Scholar]
- Polo S, Pece S, Di Fiore PP. Endocytosis and cancer. Curr Opin Cell Biol. 2004;16:156–161. doi: 10.1016/j.ceb.2004.02.003. [DOI] [PubMed] [Google Scholar]
- Raina AK, Hochman A, Zhi X, Rottkamp CA, Nunomura A, Siedlak SL, Boux H, Castellani RJ, Perry G, Smith MA. Abortive apoptosis in Alzheimer's disease. Acta Neuropathol. 2001;101:305–310. doi: 10.1007/s004010100378. [DOI] [PubMed] [Google Scholar]
- Rovelet-Lecrux A, Hannequin D, Raux F, Le Meur N, Laquerriere A, Vital A, Dumanchin C, Feuillette S, Brice A, Vercelletto M, Dubas F, Frebourg T, Campion D. APP locus duplication causes autosomal dominant early-onset Alzheimer disease with cerebral amyloid angiopathy. Nat Genet. 2006;38:24–26. doi: 10.1038/ng1718. [DOI] [PubMed] [Google Scholar]
- Saganich MJ, Schroeder BE, Galvan V, Bredesen DE, Koo EH, Heinemann SF. Deficits in synaptic transmission and learning in amyloid precursor protein (APP) transgenic mice require C-terminal cleavage of APP. J Neurosci. 2006;26:13428–13436. doi: 10.1523/JNEUROSCI.4180-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sager PR, Brown PA, Berlin RD. Analysis of transferrin recycling in mitotic and interphase HeLa cells by quantitative fluorescence microscopy. Cell. 1984;39:275–282. doi: 10.1016/0092-8674(84)90005-9. [DOI] [PubMed] [Google Scholar]
- Seemann J, Weber K, Gerke V. Annexin I targets S100C to early endosomes. FEBS Lett. 1997;413:185–190. doi: 10.1016/s0014-5793(97)00911-3. [DOI] [PubMed] [Google Scholar]
- Selznick LA, Holtzman DM, Han BH, Gokden M, Srinivasan AN, Johnson EM, Jr, Roth KA. In situ immunodetection of neuronal caspase-3 activation in Alzheimer disease. J Neuropathol Exp Neurol. 1999;58:1020–1026. doi: 10.1097/00005072-199909000-00012. [DOI] [PubMed] [Google Scholar]
- Sever S. Dynamin and endocytosis. Curr Opin Cell Biol. 2002;14:463–467. doi: 10.1016/s0955-0674(02)00347-2. [DOI] [PubMed] [Google Scholar]
- Sleegers K, Brouwers N, Gijselinck I, Theuns J, Goossens D, Wauters J, Del-Favero J, Cruts M, van Duijn CM, Van Broeckhoven C. APP duplication is sufficient to cause early onset Alzheimer's dementia with cerebral amyloid angiopathy. Brain. 2006;129:2977–2983. doi: 10.1093/brain/awl203. [DOI] [PubMed] [Google Scholar]
- Sofroniew MV, Howe CL, Mobley WC. Nerve growth factor signaling, neuroprotection, and neural repair. Annu Rev Neurosci. 2001;24:1217–1281. doi: 10.1146/annurev.neuro.24.1.1217. [DOI] [PubMed] [Google Scholar]
- Soriano S, Chyung AS, Chen X, Stokin GB, Lee VM, Koo EG. Expression of β-amyloid precursor protein-CD3γ chimeras to demonstrate the selective generation of amyloid β(1–40) and amyloid β(1–42) peptides within secretory and endocytic compartments. J Biol Chem. 1999;274:32295–32300. doi: 10.1074/jbc.274.45.32295. [DOI] [PubMed] [Google Scholar]
- Stadelmann C, Deckwerth TL, Srinivasan A, Bancher C, Bruck W, Jellinger K, Lassmann H. Activation of caspase-3 in single neurons and autophagic granules of granulovacuolar degeneration in Alzheimer's disease. Am J Pathol. 1999;155:1459–1466. doi: 10.1016/S0002-9440(10)65460-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenmark H, Parton RG, Steele-Mortimer O, Lutcke A, Gruenberg J, Zerial M. Inhibition of rab5 GTPase activity stimulates membrane fusion in endocytosis. EMBO J. 1994;13:1287–1296. doi: 10.1002/j.1460-2075.1994.tb06381.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stokin GB, Lillo C, Falzone TL, Brusch RG, Rockenstein E, Mount SL, Raman R, Davies P, Masliah E, Williams DS, Goldstein LS. Axonopathy and transport deficits early in the pathogenesis of Alzheimer's disease. Science. 2005;307:1282–1288. doi: 10.1126/science.1105681. [DOI] [PubMed] [Google Scholar]
- Tall GG, Barbieri MA, Stahl PD, Horazdovsky BF. Ras-activated endocytosis is mediated by the Rab5 guanine nucleotide exchange activity of RIN1. Dev Cell. 2001;1:73–82. doi: 10.1016/s1534-5807(01)00008-9. [DOI] [PubMed] [Google Scholar]
- Warren G. Cell biology: mitosis and membranes. Nature. 1989;342:857–858. doi: 10.1038/342857a0. [DOI] [PubMed] [Google Scholar]
- Warren G, Davoust J, Cockcroft A. Recycling of transferrin receptors in A431 cells is inhibited during mitosis. EMBO J. 1984;3:2217–2225. doi: 10.1002/j.1460-2075.1984.tb02119.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilde A, Beattie EC, Lem L, Riethof DA, Liu SH, Mobley WC, Soriano P, Brodsky FM. EGF receptor signaling stimulates SRC kinase phosphorylation of clathrin, influencing clathrin redistribution and EGF uptake. Cell. 1999;96:677–687. doi: 10.1016/s0092-8674(00)80578-4. [DOI] [PubMed] [Google Scholar]
- Xie J, Chang X, Zhang X, Guo Q. Aberrant induction of Par-4 is involved in apoptosis of hippocampal neurons in presenilin-1 M146V mutant knock-in mice. Brain Res. 2001;915:1–10. doi: 10.1016/s0006-8993(01)02803-7. [DOI] [PubMed] [Google Scholar]