

Abstract
The Ig superfamily adhesion molecule CHL1, the close homolog of the adhesion molecule L1, promotes neurite outgrowth, neuronal migration, and survival in vitro. We tested whether CHL1, similar to its close homolog L1, has a beneficial impact on recovery from spinal cord injury using adult CHL1-deficient (CHL1−/−) mice and wild-type (CHL1+/+) littermates. In contrast to our hypothesis, we found that functional recovery, assessed by locomotor rating and video-based motion analyses, was improved in CHL1−/− mice compared with wild-type mice at 3–6 weeks after compression of the thoracic spinal cord. Better function was associated with enhanced monoaminergic reinnervation of the lumbar spinal cord and altered pattern of posttraumatic synaptic rearrangements around motoneurons. Restricted recovery of wild-type mice was likely related to early and persistent (3–56 d after lesion) upregulation of CHL1 in GFAP-positive astrocytes at the lesion core. In both the intact spinal cord and cultured astrocytes, enhanced expression of CHL1 and GFAP was induced by application of basic fibroblast growth factor, a cytokine involved in the pathophysiology of spinal cord injury. This upregulation was abolished by inhibitors of FGF receptor-dependent extracellular signal-regulated kinase, calcium/calmodulin-dependent kinase, and phosphoinositide-3 kinase signaling pathways. In homogenotypic and heterogenotypic cocultures of neurons and astrocytes, reduced neurite outgrowth was observed only if CHL1 was simultaneously present on both cell types. These findings and novel in vitro evidence for a homophilic CHL1–CHL1 interaction indicate that CHL1 is a glial scar component that restricts posttraumatic axonal growth and remodeling of spinal circuits by homophilic binding mechanisms.
Keywords: astrocytes, basic fibroblast growth factor, close homolog of L1, homophilic binding, neurite outgrowth, spinal cord regeneration
Introduction
Recognition molecules mediate neural cell proliferation, migration, differentiation, neurite outgrowth and polarization, neural survival, and formation of synapses. Their important roles in synaptic plasticity and regeneration after trauma are becoming increasingly recognized (Loers and Schachner, 2007). Among the recognition molecules of the Ig superfamily, the close homolog of L1 (CHL1) was discovered as a novel member of the L1 family (Holm et al., 1996; Hillenbrand et al., 1999). Like L1, CHL1 promotes neurite outgrowth and neuronal survival, when substrate-coated in vitro (Chen et al., 1999) and, as a coreceptor with integrins, enhances cell migration (Holm et al., 1996; Hillenbrand et al., 1999; Buhusi et al., 2003).
Mutations in the CHL1 gene in humans have been linked to mental retardation (Angeloni et al., 1999; Frints et al., 2003) and schizophrenia (Sakurai et al., 2002; Chen et al., 2005). CHL1-deficient mice have normal appearance and motor abilities, but their social and exploratory behavior, reactivity to novelty, and ability to gate sensorimotor information are altered (Montag-Sallaz et al., 2002; Irintchev et al., 2004; Morellini et al., 2007). Behavioral deficits are possibly related to developmental abnormalities such as misguided projections of hippocampal mossy fibers and olfactory axons (Montag-Sallaz et al., 2002), and positioning and dendrite orientation of pyramidal cells in the visual cortex (Demyanenko et al., 2004). Furthermore, behavioral abnormalities in CHL1-deficient mice may arise from synaptic dysfunctions related to defective clathrin-mediated endocytosis that initiates recycling of synaptic vesicles (Leshchyns'ka et al., 2006), as well as abnormal inhibitory circuitries and inhibitory synaptic transmission in the hippocampus (Nikonenko et al., 2006).
After injuries of the adult mouse CNS and peripheral nervous system, CHL1 is upregulated in its expression in both neurons and astrocytes (Chaisuksunt et al., 2000; Zhang et al., 2000; Rolf et al., 2003). The functional consequences of this posttraumatic response have, however, remained unknown. Here we report enhanced motor recovery in CHL1-deficient mice after spinal cord injury, indicating that CHL1 has a negative impact on spinal cord regeneration. Upregulation of CHL1 expression induced by basic fibroblast growth factor (FGF-2) in glial scar astrocytes was identified as a possible cause of this inhibition. Furthermore, in vitro findings that CHL1 impedes neurite outgrowth when present on both neurons and astrocytes in coculture, and evidence for a previously unrecognized homophilic binding mechanism support the idea that CHL1 has dual functions, conductive and inhibitory, depending on its spatial, temporal, and cell-type-specific expression.
Materials and Methods
Animals.
Female C57BL/6J as well as CHL1-deficient (CHL1−/−) mice and age-matched wild-type (CHL1+/+) littermates from heterozygous breeding [mixed C57BL/6J-129Ola genetic background, five backcrosses into C57BL/6J (Montag-Sallaz et al., 2002)] were used at the ages of 3–4 months. Genotyping of the mice from the CHL1 stock was performed using PCR assays. The animals were kept under standard laboratory conditions. All experiments were conducted in accordance with the German and European Community laws on protection of experimental animals, and the procedures used were approved by the responsible committee of The State of Hamburg. Numbers of animals studied in different experimental groups and at different time periods after surgery are given in the text and figures. All animal treatments, data acquisition, and analyses were performed in a blind manner.
Antibodies and reagents.
The following antibodies were used: goat anti-CHL1 antibody (1:200; R & D Systems, Minneapolis, MN), mouse anti-CHL1 antibody (1:500; R & D Systems), goat anti-choline acetyltransferase antibody (ChAT) (1:100; Millipore, Hofheim, Germany), guinea pig anti-5-hydroxytryptamine transporter (5-HT) (1:1000; Millipore), rabbit anti-tyrosine hydroxylase (TH) (1:800; Millipore), mouse anti-vesicular GABA transporter (VGAT) (1:1000; Synaptic Systems, Göttingen, Germany), and mouse anti-vesicular glutamate transporter 1 (1:500; Synaptic Systems), rabbit anti-ionized binding calcium adapter molecule 1 (Iba-1) (1:1500; Wako Chemicals, Richmond, VA), rabbit anti-S-100 (1:2000; DakoCytomation, Glostrup, Denmark), rabbit anti-glial fibrillary acidic protein (GFAP) (1:1000; DakoCytomation), mouse anti-GFAP (1:1000; Sigma, St. Louis, MO), rabbit anti-fibronectin (1:100; Sigma), rabbit anti-myelin basic protein (MBP) (1:5000; Millipore), rabbit anti-chondroitin proteoglycan sulfate (NG2) (1:500; Millipore), anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (1:1000; Millipore), rabbit anti-neuronal class III β-tubulin (1:2000; Covance, Berkeley, CA), rabbit anti-tau (1:100; Sigma), and mouse anti-microtubule associated protein 2 (MAP2) (1:100; Sigma). The following cyanine 3 (Cy3)-, Cy2-, and Cy5-conjugated secondary antibodies were used for immunofluorescence: goat anti-rabbit, goat anti-mouse, donkey anti-goat, donkey anti-rabbit, donkey anti-mouse, and donkey anti-guinea pig (Jackson ImmunoResearch via Dianova, Hamburg, Germany).
The mouse CHL1–Fc and the mouse L1–Fc fusion proteins were produced and purified as described by Chen et al. (1999) and Haspel et al. (2000). They contain the extracellular domain of the mouse protein (CHL1 or L1) and the Fc region of the human IgG. Human Fc (Athens Research & Technology, Athens, GA) served as a negative control. Recombinant FGF-2 was purchased from Peprotech (Rocky Hill, NJ), and PD98059 [2-(2-amino-3-methyoxyphenyl)-4H-1-benzopyran-4-one], U0126 [1,4-diamino-2,3-dicyano-1,4-bis(o-aminophenylmercapto)butadiene], KN-93 (2-[N-(2-hydroxyethyl)]-N-(4-methoxybenzenesulfonyl)amino-N-(4-chlorocinnamyl)-N-methylbenzylamine), and 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran (LY29400) were obtained from Sigma.
Surgical procedures.
For surgery, the mice were anesthetized by intraperitoneal injections of ketamine and xylazine [100 mg of Ketanest (Parke-Davis/Pfizer, Karlsruhe, Germany) and 5 mg of Rompun (Bayer, Leverkusen, Germany), per kilogram of body weight]. Laminectomy was performed at the T7–T9 level with mouse laminectomy forceps (Fine Science Tools, Heidelberg, Germany). A mouse spinal cord compression device was used to elicit compression injury (Curtis et al., 1993). Compression force (degree of closure of the forceps) and duration were controlled by an electromagnetic device. The spinal cord was maximally compressed (100%, according to the operational definition of Curtis et al., 1993) for 1 s by a time-controlled current flow through the electromagnetic device. The skin was then surgically closed using 6-0 nylon stitches (Ethicon, Norderstedt, Germany). After the operation, mice were kept in a heated room (35°C) for several hours to prevent hypothermia and thereafter singly housed in a temperature-controlled (22°C) room with water and standard food provided ad libitum. During the postoperative time period, the bladders of the animals were manually voided twice daily.
Analysis of motor function.
The recovery of ground locomotion was evaluated using the Basso, Beattie, Bresnahan (BBB) rating scale (Basso et al., 1995), modified for mice (Joshi and Fehlings, 2002), and a novel single-frame motion analysis (Apostolova et al., 2006). This method includes evaluation of four parameters in three different tests: beam walking (foot-stepping angle and rump-height index), voluntary movements without body weight support (extension–flexion ratio), and inclined ladder climbing (number of correct steps). Assessment was performed before and at 1, 3, and 6 weeks after the injury. Values for the left and right extremities were averaged. Recovery indices were used as a measure of functional recovery on an individual animal level as described previously (Apostolova et al., 2006).
Tissue fixation and sectioning.
Mice were anesthetized with 16% solution of sodium pentobarbital (Narcoren, 5 μl/g body weight; Merial, Hallbergmoos, Germany). The animals were transcardially perfused with fixative consisting of 4% formaldehyde and 0.1% CaCl2 in 0.1 m cacodylate buffer, pH 7.3, for 15 min at room temperature (RT). After perfusion, the spinal cords were left in situ for 2 h at RT, after which they were dissected out and postfixed overnight (18–22 h) at 4°C in the same solution used for perfusion. Tissue was then immersed into 15% sucrose solution in 0.1 m cacodylate buffer, pH 7.3, for 2 d at 4°C, embedded in Tissue Tek (Sakura Finetek, Zoeterwoude, The Netherlands), and frozen by 2 min immersion into 2-methyl-butane (isopentane) precooled to −80°C. Serial transverse or longitudinal sections were cut in a cryostat (CM3050; Leica, Nussloch, Germany). Sections, 25 μm thick, were collected on SuperFrost Plus glass slides (Roth, Karlsruhe, Germany). Sampling of sections was always done in a standard sequence so that four sections 250 μm apart were present on each slide.
Immunohistochemistry and in situ hybridization.
Procedures for immunohistochemistry have been described previously (Irintchev et al., 2005). Water-bath antigen demasking was performed in 0.01 m sodium citrate solution, pH 9.0, for 30 min at 80°C (Jiao et al., 1999) for all antigens. Nonspecific binding was blocked using 5% normal serum from the species in which the secondary antibody was produced dissolved in PBS and supplemented with 0.2% Triton X-100, 0.02% sodium azide for 1 h at RT. Incubation with the primary antibody, diluted in PBS containing 0.5% λ-carrageenan (Sigma) and 0.02% sodium azide, was performed for 3 d at 4°C. After washing in PBS (three times for 15 min at RT), the appropriate secondary antibody diluted 1:200 in PBS–carrageenan solution was applied for 2 h at RT. After a subsequent wash in PBS, cell nuclei were stained for 10 min at RT with bis-benzimide solution (Hoechst 33258 dye, 5 μg/ml in PBS; Sigma). Finally, the sections were washed again, mounted in anti-fading medium (Fluoromount G; Southern Biotechnology Associates via Biozol, Eching, Germany) and stored in the dark at 4°C. For double and triple stainings, the primary antibodies were mixed at optimal dilutions and subsequently detected using mixtures of appropriate secondary antibodies. Photographic documentation was made on an LSM 510 confocal microscope (Zeiss, Oberkochen, Germany) or an Axiophot 2 microscope equipped with a digital camera AxioCam HRC and AxioVision software (Zeiss). The images were processed using Adobe Photoshop 7.0 software (Adobe Systems, San Jose, CA).
For in situ hybridization, a digoxigenin (DIG)-labeled antisense cRNA probe corresponding to the extracellular domain of CHL1 was prepared as described previously (Holm et al., 1996). Slides were postfixed with 4% formaldehyde in PBS at 4°C overnight and hybridized with the DIG-labeled probe at 55°C overnight. After extensive washing, alkaline phosphatase-conjugated sheep anti-DIG Fab fragments (1:1000; Roche Diagnostics, Mannheim, Germany) were applied at 4°C overnight. Hybridization signals were detected using nitro blue tetrazolium and 5-bromo-4-chloro-3-indolyl phosphate and observed on a Zeiss Axiophot microscope. Sections incubated with the sense probe, developed in parallel with the antisense probe, did not show any labeling (data not shown).
Motoneuron soma size and quantification of perisomatic terminals.
Estimations of soma areas and perisomatic terminals were performed as described previously (Apostolova et al., 2006). Longitudinal spinal cord sections stained for ChAT, VGAT, or vesicular glutamate transporter 1 (VGLUT1) were examined under a fluorescence microscope to select sections that contained motoneuron cell bodies for a distance of at least 500 μm distal from the lesion scar. Stacks of 1-μm-thick images were obtained on a LSM 510 confocal microscope (Zeiss) using a 63×, 1.5 oil- immersion objective and digital resolution of 512 × 512 pixel. Four adjacent stacks (frame size, 153 × 153 μm) were obtained consecutively in a rostrocaudal direction so that motoneurons located both close and remote to the lesion scar were sampled. One image per cell at the level of the largest cell body cross-sectional area was used to measure soma area, perimeter, and number of perisomatic terminals. Areas and perimeters were measured using the Image Tool 2.0 software program (University of Texas, San Antonio, TX). Linear density was calculated as number of perisomatic terminals per unit length.
Estimation of lesion scar volume.
Spaced serial 25-μm-thick sections 250 μm apart were stained with cresyl violet/Luxol fast blue and used for estimations of the scar volume using the Cavalieri principle. Areas of the scar required for volume estimation were measured directly under the microscope using the Neurolucida software (MicroBrightField, Magdeburg, Germany).
Primary cultures of astrocytes.
Primary cultures of astrocytes were prepared as described previously (Cole and Vellis, 2001). Briefly, cerebral cortices of 1- to 2-d-old C57BL/6J mice were mechanically dissociated, and the obtained mixture of glial cells was seeded on tissue culture flasks in DMEM (Invitrogen, Piscataway, NJ) containing 10% fetal bovine serum (FBS), 2% penicillin, and 2% streptomycin and incubated at 37°C in CO2 enriched (5%) and humidified (95% relative humidity) atmosphere. The culture medium was replaced twice weekly. On days 7 or 8, the flasks were shaken at 200 rpm for 6 h at 37°C to remove the microglia. On culture days 8–10, the cell cultures were again shaken at 200 rpm to remove oligodendroglia. Then, the adherent astrocytes were trypsinized, replated, and grown to confluence. Cells were treated with 100 ng/ml FGF-2 for different time periods or for 3 d with different concentrations of FGF-2. To examine the effects of mitogen-activated protein kinase kinase (MEK), calcium/calmodulin dependent protein kinase II (CaMKII), and phosphoinositide-3 kinase (PI3K) antagonists, astrocytes were pretreated with the indicated inhibitors for 1 h, followed by 3 d of FGF-2 treatment.
Assay for migration.
Astrocytes from CHL1+/+ and CHL1−/− mice were plated in DMEM supplemented with 10% FBS on poly-l-lysine (PLL) (Sigma) coated glass coverslips and grown to confluence. The medium was then removed, and the monolayer was scratched with a sterile 1–10 μl pipette tip. The cells were washed twice with prewarmed (37°C) HBSS (Invitrogen) and maintained in DMEM with 10% FBS. After 6 or 20 h of incubation with or without FGF-2 at 100 ng/ml, cells were fixed with 4% formaldehyde in 0.1 m phosphate buffer, pH 7.3, and immunostained for GFAP. Nuclei were counterstained with 4′-6-diamidino-2-phenylindole.
Assay for proliferation.
Astrocytes from CHL1+/+ and CHL1−/− mice were plated into PLL-coated 96-well plates (20,000 cells per well in 200 μl of DMEM). Cells were cultured with or without FGF-2 (100 ng/ml) for 4 d. Cell proliferation was estimated using an MTT assay kit (Sigma) according to the instructions of the manufacturer.
Cocultures of hippocampal neurons and astrocytes.
Astrocytes were isolated from brains of newborn mice as described previously (Shahar et al., 1989), plated on glass coverslips coated with PLL (100 μg/ml), and maintained in hormonally supplemented culture medium containing 10% horse serum (Sigma) for 24 h, a time period during which they formed a continuous layer. Hippocampal neurons were prepared as described previously (Leshchyns'ka et al., 2003) and seeded on top of the astrocyte monolayer. To avoid extensive neurite outgrowth that hinders subsequent analyses, cultures were fixed only 4 h after plating with 4% formaldehyde in PBS, pH 7.3, for 15 min, washed, and permeabilized with 0.25% Triton X-100 in PBS for 5 min. Cultures were then blocked in 1% bovine serum albumin (BSA) in PBS. Antibodies against tau or MAP2 used to identify neurons were applied in 1% BSA in PBS for 2 h and detected with appropriate secondary antibodies after incubation for 45 min at RT. Coverslips were embedded in Aqua-Poly/Mount (Polysciences, Warrington, PA). Images were acquired on an Axiophot 2 microscope (Zeiss) equipped with an AxioCam HRc digital camera (Zeiss), AxioVision software (version 3.1; Zeiss), and a Plan-Neofluar 40× objective (numerical aperture, 0.75; Zeiss). Lengths of tau or MAP2-labeled neurites were measured using NIH ImageJ software.
Neurite outgrowth assay.
Dissociated cerebellar granule cell cultures were prepared from cerebella of 6- to 8-d-old CHL1+/+ mice and CHL1−/− littermates. The cell suspension was transferred to 12-well plates (105 cells in 1 ml/well) containing one 15 mm round glass coverslip per well coated with PLL followed by mouse CHL1–Fc fusion protein (37.5 nm) or equimolar concentrations of mouse L1–Fc fusion protein, human Fc, or laminin (Sigma). Cells were cultured in serum-free basal medium Eagle (Invitrogen) supplemented with 2 mm l-glutamine (PAA Laboratories, Cölbe, Germany), 1 mg/ml BSA, 12.5 μg/ml insulin, 30 nm sodium selenite, 0.027 TIU/ml aprotinin, 4 nm thyroxine (Sigma), 100 μg/ml transferrin (Merck Biosciences, Schwalbach, Germany), 0.1 mg/ml streptomycin, and 10 U/ml penicillin (PAA Laboratories). After cultivation for 22 h in vitro, cells were fixed with 2.5% glutaraldehyde in PBS, pH 7.4, and stained with toluidine blue/trypan blue. Total neurite length per cell was measured using the KS image analysis system (Kontron/Zeiss, Oberkochen, Germany). In each experiment, at least 50 cells per coverslip were analyzed.
Bead aggregation assay.
Ten microliters of protein A magnetic beads (Dynabeads; Dynal Biotech, Hamburg, Germany) were washed three times with PBS, pH 7.4, and incubated overnight at 4°C in 200 μl of PBS containing 37.5 nm mouse CHL1–Fc or human Fc. After washing the beads three times with PBS, pH 7.3, containing 0.01% Tween 20, the beads were resuspended in PBS and sonicated in ice-cold water for 30 min to ensure bead disaggregation. After centrifugation for 4 min at 10,000 × g, the supernatant was discarded, and the pellet was gently resuspended in 100 μl of 0.1 mg/ml BSA for analysis of aggregation. For inhibition of homophilic interaction of CHL1, rabbit anti-CHL1 and rabbit anti-L1 polyclonal antibodies directed against the extracellular domains of the molecules were added at a final concentration of 600 μg/ml before resuspension of beads. The suspensions were kept for 30 min at normal gravity and RT, and thereafter 10 μl samples were transferred onto microscope glass slides for analysis. Images were digitally captured and analyzed using a Zeiss Axiovert microscope and the Axiovision software (Kontron/Zeiss). The size of each bead aggregate was individually assessed, and aggregates were assigned to three categories: <5 beads, 5–20 beads, and >20 beads per aggregate. Number of aggregates in each category was expressed as percentage of the total bead number.
FGF-2 microinjection into the intact spinal cord.
FGF-2 (0.4 μg/2 μl PBS, per mouse) or PBS alone were injected into the spinal cords of 3-month-old C57BL/6J mice at T9–10 level. After 3 or 7 d, 5-mm-long spinal cord segments including the injection site in the center were dissected for Western blot analysis.
Western blot analysis.
Three 5-mm-long segments from each injured spinal cord were dissected: a segment at the thoracic level with the lesion scar in the center and two adjacent segments rostrally and caudally containing the rostral part of the thoracic spinal cord and the lumbar intumiscence, respectively. Samples were mechanically dissociated and lysed in radioimmunoprecipitation assay (RIPA) buffer (Sigma). Primary cultures of astrocytes grown in 12-well plates at confluence were serum starved for 24 h before treatment at 37°C with selected agents. The medium was then aspirated, and the cells were washed twice with ice-cold PBS and lysed in 100 μl of RIPA buffer. Samples from spinal cords and astrocyte cultures were briefly sonicated, heated at 96°C for 5 min, and centrifuged (20,600 × g) at 4°C for 5 min. The supernatants were electrophoretically separated on 4–12% NuPAGE Novex Bis-Tris gradient gels (Invitrogen) and then transferred to nitrocellulose membranes (Invitrogen). Blots were incubated overnight at 4°C with primary antibodies and washed three times in Tris-buffered saline with 0.05% Tween 20 before probing with horseradish peroxidase-conjugated secondary antibodies (Jackson ImmunoResearch, West Grove, PA) for 1 h at room temperature. Blots were then visualized with an enhanced chemiluminescence reagent (GE Healthcare, Piscataway, NJ) and quantified with a laser-scanning densitometer (Canon, Lake Success, NY).
Statistical analysis.
All numerical data are presented as group mean values with SEM. Parametric or nonparametric tests were used for comparisons, as indicated in the text and figure legends. Analyses were performed using the SYSTAT 9 software package (SPSS, Chicago, IL). The threshold value for acceptance of differences was 5%.
Results
Improved recovery of motor functions in CHL1-deficient mice
Spinal cord compression caused severe disabilities in both CHL1+/+ and CHL1−/− mice, as assessed by the BBB score 1 week after injury (Fig. 1A). Between 1 and 6 weeks, the walking abilities improved more in CHL1−/− mice than in their CHL1+/+ littermates as revealed by analyses of both absolute BBB score values (Fig. 1A) and recovery indices, estimates of gain of function after the first week as a fraction of the functional loss induced by the surgery (Fig. 1E). In addition to the BBB score, we analyzed the plantar stepping ability of the animals using a novel parameter, the foot-stepping angle (Apostolova et al., 2006). This analysis revealed, in agreement with the BBB scores, enhanced recovery in CHL1−/− compared with CHL1+/+ mice at 3 and 6 weeks (Fig. 1B,F).
Figure 1.

Motor recovery in CHL1−/− and CHL1+/+ mice after spinal cord injury. A–D, Time course and degree of functional recovery in CHL1−/− mice and wild-type littermates after spinal cord injury. Shown are mean ± SEM values of open-field locomotion (BBB) scores (A), foot-stepping angles (B), rump-height indices (C), and extension–flexion ratios (D) before surgery (day 0) and at 1, 3, and 6 weeks after injury. *p < 0.05, significant differences between group mean values at a given time period (one-way ANOVA for repeated measurements with Tukey's post hoc test; n = 10 mice per genotype). E–J, Recovery indices (mean + SEM values) calculated from individual animal values for the parameters shown in A–D. I, J, Group mean values (I) and individual values (J) of overall recovery indices calculated as individual means of the recovery indices shown in E–H. *p < 0.05, values different from that of CHL1+/+ mice (one-way ANOVA with Tukey's post hoc test).
We also evaluated more complex motor functions than plantar stepping. We analyzed the rump-height index, a measure of the ability to support body weight during ground locomotion, ability requiring activity and coordination of muscles working at different joints in both extremities. Also for this parameter, recovery at 6 weeks was better in CHL1−/− mice compared with CHL1+/+ littermates (Fig. 1C,G). In addition, CHL1−/− mice performed voluntary movements without body weight support, estimated by the extension–flexion ratio, better than CHL1+/+ mice at both 3 and 6 weeks after injury (Fig. 1D,H). However, numbers of correct steps made by the animals during inclined ladder climbing, reduced to almost 0 in both groups 1 week after injury, did not improve with time in either CHL1+/+ or CHL1−/− mice (data not shown). The ladder-climbing test allows estimation of the ability to perform precise movements requiring a higher degree of supraspinal control compared with the types of movement assessed by the other tests. From the values of the parameters shown in Figure 1E–H, we calculated, on an individual animal basis, overall recovery indices for each animal (Fig. 1J) and group mean values (Fig. 1I). This analysis revealed an overall better functional outcome in CHL1−/− mice compared with their CHL1+/+ littermates.
Because both the BBB score and the numerical parameters used for functional analysis strongly depend on the lesion severity (Kloos et al., 2005; Apostolova et al., 2006), we estimated lesion scar volume in both genotypes at 1 and 6 weeks after injury. The mean volume of the lesion in CHL1−/− and CHL1+/+ mice was similar at both 7 d (1.76 ± 0.16 vs 1.69 ± 0.19 mm3; p > 0.05, t test) and at 6 weeks after injury (1.59 ± 0.13 vs 1.67 ± 0.21 mm3; p > 0.05, t test). Within-genotype comparisons also did not reveal differences between the values at 7 d and 6 weeks. Therefore, better functional recovery in CHL1−/− mice was not related to lower damage susceptibility or tissue scarring.
Enhanced monoaminergic reinnervation of the distal spinal cord in CHL1-deficient mice
Monoaminergic descending pathways, located in the ventral and lateral columns of the spinal cord, originate from the reticular formation, raphe nuclei, locus ceruleus, pretectal area, and the substantia nigra (Kiehn et al., 1992, Shapiro, 1997) and modulate the excitability of spinal cord circuitries capable of initiating and controlling rhythmic coordinated movements (Fouad and Pearson, 2004). To assess whether improved motor functions in CHL1−/− mice may be in part related to enhanced monoaminergic reinnervation of the spinal cord caudal to the injury, we counted numbers of catecholaminergic [TH-positive (TH+)] and serotonergic (5-HT+) axons projecting beyond an arbitrarily selected border 250 μm caudally to the lesion site in spaced serial parasagittal sections at 7 d and 6 weeks after the injury (Fig. 2A,B). Numbers of TH+ and 5-HT+ fibers at 7 d after the injury, considered to be fibers spared after the lesion, were similar in the two groups. Compared with 1 week, numbers of monoaminergic fibers at 6 weeks were increased by a factor of two in CHL1−/− mice but not changed in CHL1+/+ mice. Although it is not clear whether the increase in fiber numbers was attributable to regrowth of axons across the lesion scar or sprouting of spared axons caudal to the lesion, this observation indicates a more vigorous regenerative response of monoaminergic fibers in the injured CHL1−/− spinal cord.
Figure 2.

Monoaminergic innervation caudal to the lesion site. A, B, Mean numbers of TH+ (A) and 5-HT+ (B) fibers crossing an arbitrary border 250 μm caudally from the lesion site at 7 d and 6 weeks after the lesion (*p < 0.05; n = 5 mice per genotype; Wilcoxon–Mann–Whitney test). C, D, Correlations between recovery index calculated for the foot-stepping angle (FSA) and numbers of monoaminergic fibers. Individual recovery index values plotted against numbers of TH+ fibers (C) and 5-HT+ fibers (D). Regression lines are drawn in both panels. The Pearson's correlation coefficient was r = 0.77 (p < 0.01, ANOVA) for the data shown in C and r = 0.93 (p < 0.005, ANOVA) for the data in D.
Regression analysis of data for mice studied both functionally and morphologically revealed high degrees of correlation between the foot-stepping angle on the one hand and numbers of TH+ and 5-HT+ fibers on the other (Fig. 2C,D). Significant positive correlations were also found between BBB score values and numbers of 5-TH+ fibers (r = 0.85; p < 0.01) and TH+ fibers (r = 0.72; p < 0.05). These findings can be interpreted as an indication for a causative relationship between degree of monoaminergic innervation and plantar stepping abilities. At the same time, the rump-height index and the extension-flexion ratio, estimates of functions requiring higher levels of motor control, which were also better in CHL1−/− than in CHL+/+ mice, did not correlate with fiber numbers (data not shown).
Posttraumatic alterations in synaptic inputs to motoneuron somata
To elucidate the possibility that CHL1 deficiency influences the outcome of spinal cord injury also via remodeling of cell circuitries, we estimated densities of synaptic terminals around motoneuron cell bodies. The linear density (number per unit length) of large perisomatic ChAT+ boutons known to form C-type synapses on motoneurons associated with muscarinic type 2 receptors (Davidoff and Irintchev, 1986; Hellstroem et al., 2003) was prominently reduced after injury in mice of both genotypes (Fig. 3B). Coverage of somata by cholinergic synapses was lower in intact spinal cords of CHL1−/− mice compared with CHL1+/+ littermates (Fig. 3B). Perisomatic densities of VGAT+ puncta reflect numbers of GABAergic synapses on the cell bodies (Nikonenko et al., 2006). GABAergic coverage of motoneuron somata, identified by their size and location in the spinal cord, was similar in intact CHL1−/− and CHL1+/+ littermates (Fig. 3C). Spinal cord injury did not affect this coverage in CHL1+/+ mice but caused a decrease in CHL1−/− mice compared with intact mice of the same genotype (Fig. 3C). Glutamatergic (VGLUT1+) puncta around cell bodies are terminals forming either axosomatic or axodendritic synapses. Similar to VGAT+, the density of these perisomatic puncta was not different between genotypes in unlesioned mice (Fig. 3D). In contrast to the GABAergic terminals, however, injury caused a reduction in density of VGLUT1+ boutons in CHL1+/+ mice, whereas the density in CHL1−/− mice remained unchanged (Fig. 3D). We also measured areas of motoneuron cell body profiles in intact and lesioned spinal cords of both CHL1−/− and CHL1+/+ mice and found no differences among the groups (Fig. 3A). Thus, the observed genotype- and lesion-related alterations in synaptic terminal densities reflect changes in total numbers per motoneuron. Overall, the results on perisomatic coverage indicate that, in the intact spinal cord, CHL1 is important for the establishment or maintenance of cholinergic perisomatic synapses on motoneurons. After spinal cord lesion, CHL1 deficiency is associated with attenuated loss of glutamatergic terminals and reduction of the perisomatic GABAergic input.
Figure 3.

Analysis of size and perisomatic terminals of motoneurons. A–D, Soma size of ChAT+ motoneurons (A) and linear densities of ChAT+ (B), VGAT+ (C), and VGLUT1+ (D). Shown are group mean + SEM values calculated from mean values of individual animals. Numbers of injured versus intact CHL1−/− (9 vs 4) and CHL1+/+ (7 vs 4) mice for soma area and ChAT+ puncta. For VGAT+ and VGLUT1+ puncta, numbers of injured versus intact CHL1−/− and CHL1+/+ mice were 5 versus 4. Symbols indicate mean values significantly different between intact and injured animals of the same genotype (*) or between different genotypes (#) (p < 0.05, one-way ANOVA with Tukey's post hoc test). E–G, Correlations between functional parameters and linear densities of perisomatic terminals. Individual values for recovery indices for BBB score (E) and the foot-stepping angle (FSA; F, G) are plotted against numbers of ChAT+ (E, F) and VGLUT1+ (G) terminals. Drawn are regression lines for the data from each genotype group except for CHL1−/− mice in G. The Pearson's correlation coefficients for CHL1+/+ and CHL1−/− mice were r = 0.87 and r = 0.82 (p < 0.01, ANOVA) for E, r = 0.76 and r = 0.77 (p < 0.01, ANOVA) for F, and r = −0.97 (p < 0.01, ANOVA) and r = 0.09 (p > 0.05, ANOVA) for G, respectively.
We also analyzed the degrees of covariation of functional measures of recovery and densities of perisomatic terminals using data from individual animals. No correlations between functional parameters and densities of GABAergic terminals were found, suggesting lack of causative relationships. The same was true for the other two types of terminals studied with three notable exceptions. We found strong positive within-group correlations between numbers of ChAT+ terminals on the one hand and the BBB score values and the foot-stepping angle on the other (Fig. 3E,F). In addition, a negative correlation between VGLUT1 terminal density and plantar stepping ability, as estimated by the foot-stepping angle, was found for CHL1+/+ but not for CHL1−/− mice (Fig. 3G). Although the results on synaptic coverage indicate altered injury-induced posttraumatic remodeling of neuronal circuitries in the absence of CHL1, only alterations in the modulatory cholinergic perisomatic innervation can be considered as potentially linked to functional performance, and this is primarily limited to intrinsic spinal cord functions.
Expression of CHL1 and cell-type-specific markers in the spinal cord before and after the lesion
To assess in which ways CHL1 may be involved in spinal cord regeneration, we examined CHL1 mRNA and protein expression in tissue sections of intact and injured adult wild-type mice. In the intact spinal cord, we found CHL1 mRNA expression in motoneurons (Fig. 4A). After spinal cord injury, CHL1 mRNA was upregulated specifically at the lesion site (Fig. 4B), in contrast to L1, which was not detectable around the lesion site (Fig. 4C). In the intact spinal cord, CHL1 protein was expressed in the ventral horn by motoneurons, by dorsal root axons, as well as in descending tracts traveling through the lateral column, but not in the dorsal corticospinal tract (Fig. 4D). After lesion, CHL1 was upregulated mainly at the lesion site (Fig. 4E–J). This upregulation was detectable by Western blot analysis 3 d after injury and sustained at high levels and even increased during 8 weeks after the injury, the latest time period tested (Fig. 5A). Immunohistochemistry revealed that most of CHL1 protein expression at the lesion site colabeled with the astrocyte markers S-100 and GFAP (Fig. 4E,H,I). CHL1 protein was not expressed by other cell types at the lesion site, namely cells in the fibronectin positive core of the scar (Fig. 4G,J) or Iba-1+ microglia (Fig. 4L). In the spinal cord segments further rostral and caudal to the lesion site, astrocytes did not express CHL1, its expression was limited mainly to axons (Fig. 4K). Finally, we observed that CHL1 was expressed in some (Fig. 4M, arrows) but not all ChAT+ terminals (Fig. 4M, arrowheads) surrounding motoneuron cell bodies.
Figure 4.
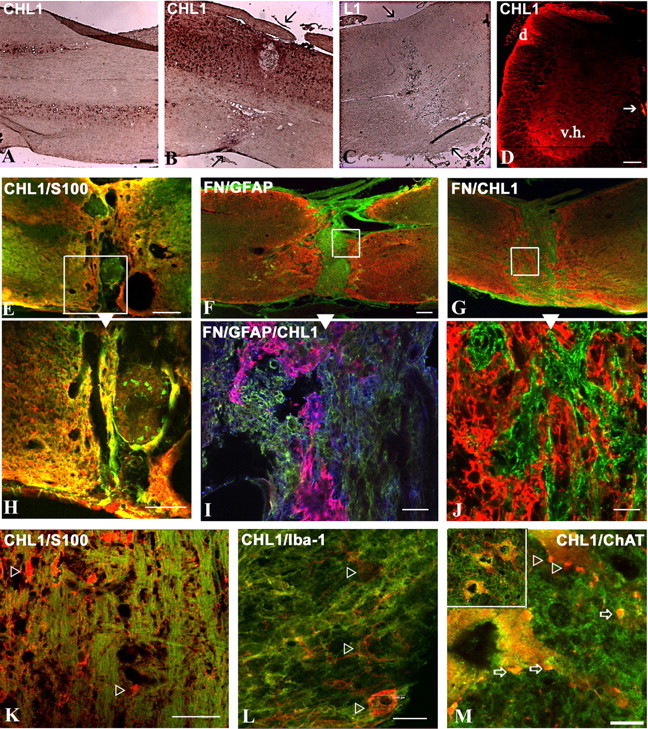
CHL1 expression in the intact and injured CHL1+/+ mouse spinal cord. A–C, In situ hybridization for CHL1 (A, B), but not for L1 (C), is detectable in parasagittal sections of intact (A) and injured (6 weeks after injury; B, C) spinal cords. Arrows point to the lesion site. D, Immunofluorescence for CHL1 on a transverse section of the intact lumbar spinal cord. CHL1 is detectable in the ventral horn gray matter (v.h.), in the dorsal root (d), and the ventral corticospinal tract (arrow). E, H, Double immunofluorescence of a parasagittal section at the lesion site for CHL1 (green) and S-100 (red), with E showing a low-power micrograph and H showing the high-power image of the inset in E. F, Double immunofluorescence of a parasagittal section for GFAP (red) and fibronectin (FN; green) at the lesion site 6 weeks after injury. I, In addition to the fibronectin and GFAP staining seen in F, CHL1 staining is shown in blue. Colocalization of CHL1 and GFAP is seen in purple. G, J, Double immunofluorescence on a parasagittal section for CHL1 (red) and fibronectin (green) at the lesion site 6 weeks after injury. K, Double immunofluorescence of a parasagittal section for CHL1 (green) and S-100 (red) distal from the lesion site 6 weeks after injury. S-100+ astrocytes (arrowheads) do not coexpress CHL1. L, Double immunofluorescence of a parasagittal section at the lesion site 7 d after injury for Iba-1+ microglia (red, arrowheads) and CHL1 (green). M, Double immunofluorescence of a parasagittal section at the lesion site 6 weeks after injury for CHL1 (green) and ChAT+ motoneurons (red). CHL1 is detectable in some (arrows) but not all (arrowheads) perisomatic and dendritic ChAT+ synaptic terminals. Top left corner shows a low-power view of the enlarged image. Scale bars: A–G, 100 μm; H, J, L, 20 μm; I, K, 40 μm; M, 8 μm.
Figure 5.
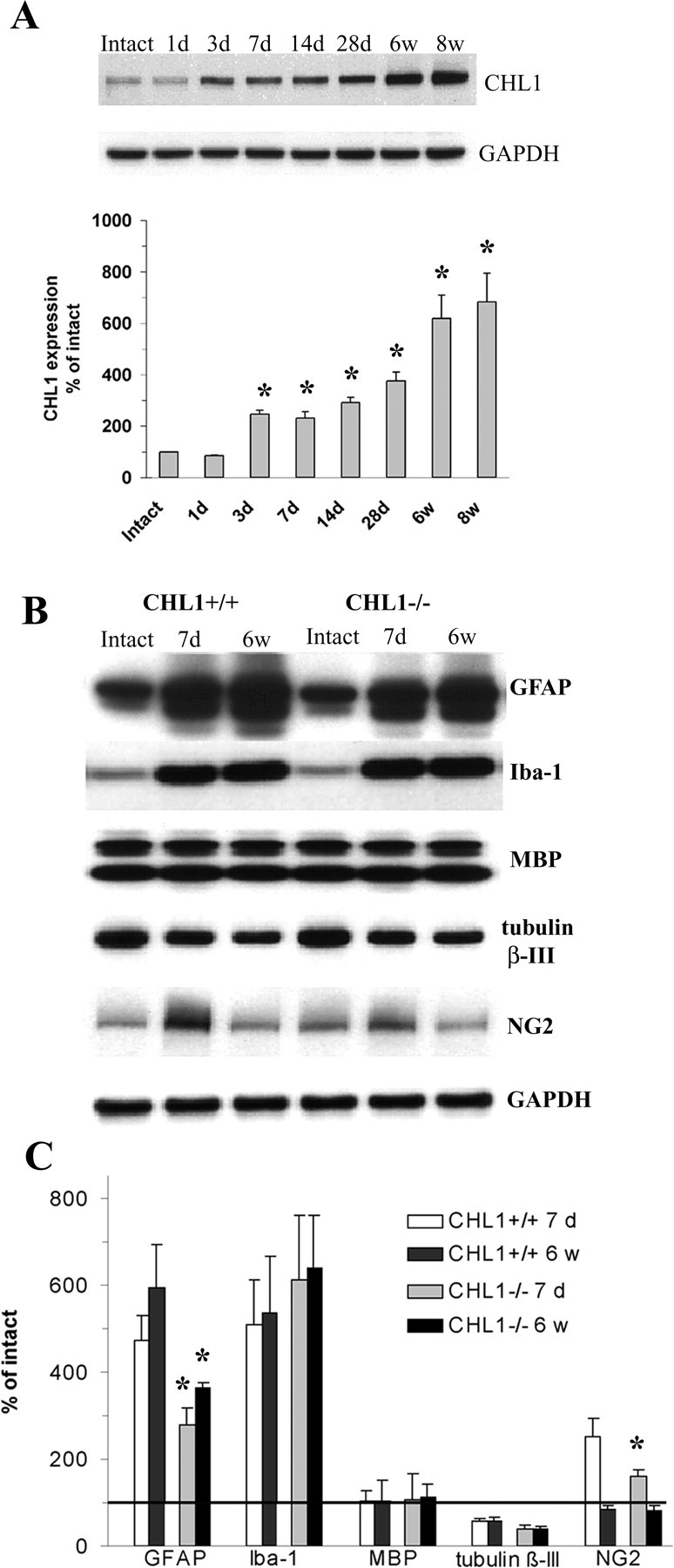
Immunoblot analysis of CHL1 and cell-type-specific markers after injury. A, CHL1 protein expression in intact mice and at different time points after injury at the lesion site. Top, Representative immunoblots for CHL1 (180 kDa band) and the loading control, GAPDH. Bottom, CHL1 expression levels, as estimated by optical density measurements, in the intact spinal cords (set to 100%) and 1 d to 8 weeks after injury as percentage of intact. Mean + SEM values (*p < 0.05 compared with intact, ANOVA with Dunnett's post hoc test; n = 5 mice per time point). B, C, Expression of GFAP, MBP, Iba-1, β-III tubulin, and NG2 in intact and injured spinal cords of CHL1+/+ and CHL1−/− mice at 7 d and 6 weeks after injury at the lesion site. GAPDH was used as a loading control. B, Representative immunoblots. C, Expression levels, normalized to intact (100%, horizontal line), of the marker proteins. Shown are mean + SEM values. *p < 0.05, significant differences between CHL1−/− and CHL1+/+ mice at the same time point after injury (t test; n = 5 mice per group).
We also measured the expression of cell-type-specific markers in intact spinal cords of CHL1+/+ and CHL1−/− mice as well as the expression of these markers 7 d and 6 weeks after lesion in the two genotypes (Fig. 5B,C). The expression of GFAP, Iba-1, the myelin marker MBP, the neuronal marker β-III tubulin, and NG2, a growth-inhibitory chondroitin sulfate proteoglycan produced by a variety of cells after injury, was similar in the intact spinal cords of both genotypes (Fig. 5C). At 7 d and 6 weeks after the lesion, GFAP expression at the lesion site was reduced in CHL1−/− versus CHL1+/+ mice. Also, expression of NG2 was decreased in CHL1−/− mice at 7 d but not at 6 weeks after the lesion. No differences between CHL1−/− and CHL1+/+ mice were seen either at 7 d or at 6 weeks after the lesion for Iba-1, MBP, and β-III tubulin. Differences between CHL1−/− and CHL+/+ mice were not detected in the rostral and caudal parts away from the lesion site at 1 and 6 weeks after the lesion (supplemental Fig. 1, available at www.jneurosci.org as supplemental material).
To address the question as to whether attenuated GFAP expression in CHL1−/− mice results from a spatially restricted astrocytic response, we estimated the volume of the astrocytic scar, as delineated by enhanced GFAP expression in tissue sections and including the fibronectin-positive lesion core, at 7 d and 6 weeks after injury, and found no differences between the genotypes (data not shown). Therefore, lower expression levels of GFAP reflect lower astrocyte densities and/or protein expression in individual cells in CHL1−/− mice compared with CHL1+/+ littermates.
We conclude that CHL1 mRNA and protein are expressed by neurons of the intact adult mouse spinal cord, including motoneurons. CHL1 is present in axonal tracts in the lateral columns of the white matter, as well as in ChAT+ terminals on motoneurons. After injury, CHL1 expression is upregulated by astrocytes surrounding the lesion site but not in its fibronectin-expressing core. Comparisons between the expression of markers for neurons, oligodendrocytes, and macrophages/microglia showed no differences between lesioned spinal cords of CHL1−/− and CHL1+/+ mice, whereas GFAP, a marker for fibrous and reactive astrocytes, was significantly reduced in CHL1−/− mice, and the neurite-repellent proteoglycan NG2 was reduced in expression at 7 d after the lesion. The combined observations suggest that increased expression of CHL1 in lesioned wild-type mice is associated with higher expression of GFAP and NG2 (the latter only at 7 d after lesion) and that the expression of these markers is reduced in CHL1−/− mice, suggesting that the absence of CHL1 is accompanied by reduced astrogliosis.
CHL1 mediates neurite outgrowth and branching on astrocytes in culture
Because the glial scar is a barrier for regrowing axons, we were interested whether expression of CHL1 by astrocytes and/or neurons could contribute to reduced recovery in CHL1+/+ mice. We thus resorted to a prototypical way of measuring neuron to astrocyte interactions in cell culture by reconstituting these interactions through plating of dissociated hippocampal CHL1+/+ neurons on top of CHL1+/+ astrocytes. Indirect immunofluorescence labeling with antibodies against CHL1 showed that CHL1 was expressed by both CHL1+/+ neurons and CHL1+/+ astrocytes in these cocultures (for neurons, see supplemental Fig. 2, available at www.jneurosci.org as supplemental material; for astrocytes, see Fig. 8 and supplemental Fig. 3, available at www.jneurosci.org as supplemental material). CHL1+/+ neurons on CHL1+/+ astrocytes were then compared with CHL1−/− neurons plated on top of CHL1−/− astrocytes. To identify neurites, neurons were labeled with antibodies against the neuronal marker proteins MAP2 and tau, both being in all neurites of these nonpolarized neurons maintained in culture for only 4 h (data not shown). Neurites of CHL1+/+ neurons on CHL1+/+ astrocytes were 44% shorter and contained 31% less branches than neurites of CHL1−/− neurons on CHL1−/− astrocytes (Fig. 6A–C). When CHL1+/+ or CHL1−/− neurons were plated on CHL1−/− or CHL1+/+ astrocytes, respectively, neurite lengths were similar to the level seen for CHL1−/− neurons plated on CHL1−/− astrocytes. In addition to lengths, also numbers of neurite branches were reduced when CHL1 was present in both neurons and astrocytes (Fig. 6C). These observations suggest that CHL1-mediated trans-interactions between neurons and astrocytes inhibit neurite outgrowth and branching.
Figure 8.

Immunoblot analysis of CHL1 expression in cultured astrocytes after FGF-2 treatment. A, B, Time dependence (A) and dose dependence (B) of CHL1 expression during treatment with FGF-2. C, CHL1 upregulation in cultured astrocytes during FGF-2 treatment is completely blocked by MEK (U0126 plus PD98059), CaMKII (KN-93), and PI3K (LY29400) inhibitors. *p < 0.05 compared with zero (paired two-tailed t test in 5 independent experiments).
Figure 6.

Neurite outgrowth and branching in homogenotypic and heterogenotypic cocultures of neurons and astrocytes. The length of the longest neurite, total neurite lengths, and number of branching points were analyzed in CHL1+/+ and CHL1−/− neurons grown on top of the monolayer of CHL1+/+ or CHL1−/− astrocytes. Shown are mean + SEM values. *p < 0.05, difference from CHL1+/+ neurons on CHL1+/+ astrocytes; #p < 0.05, difference from CHL1−/− neurons on CHL1−/− astrocytes (two-tailed t test; n > 200 neurons per genotype in 2 independent experiments).
Bead aggregation assay and neurite outgrowth of cultured cerebellar neurons on substrate-coated CHL1
To investigate the previously unrecognized possibility that CHL1 interacts by homophilic binding mechanisms, a bead aggregation assay was performed, in which beads were coated with CHL1-Fc or Fc (Fig. 7A). Aggregation of CHL1–Fc-coated beads was considerably enhanced compared with Fc-coated beads. Antibodies against extracellular epitopes of CHL1 inhibited bead aggregation (Fig. 7A,B). There were no aggregates of >20 beads when beads were coated with Fc only. Numbers of large aggregates of CHL1–Fc-coated beads were reduced, and those of small aggregates were increased in the presence of CHL1 antibodies, whereas antibodies to L1 did not have these effects (Fig. 7B).
Figure 7.
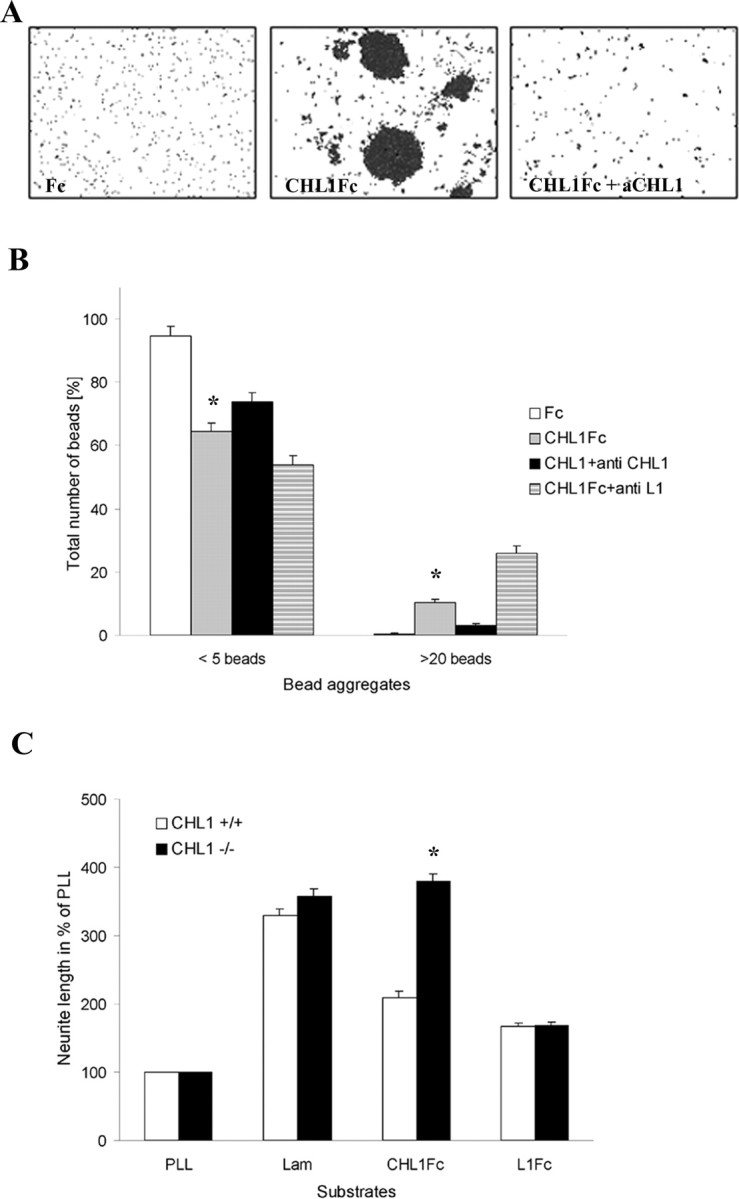
CHL1-mediated bead aggregation assay and neurite outgrowth of cultured cerebellar neurons. A, B, Bead aggregation assay. Beads were coated with CHL1–Fc for estimation of homophilic binding or with Fc as a negative control. Aggregation of CHL1–Fc was disrupted by anti-CHL1 (aCHL1) but not anti-L1 antibody. Diagram (B) shows the percentage of aggregates of less than five beads and large aggregates of >20 beads. Shown are mean + SEM values. *p < 0.05, groups significantly different from all other groups in each category (two-way ANOVA with Newman–Keuls post hoc test in 5 independent experiments). C, Neurite outgrowth assay in cerebellar neuron cultures. CHL1+/+ and CHL1−/− cerebellar neurons were grown on PLL, laminin (Lam), and CHL1–Fc substrate-coated coverslips. Total neurite length on CHL1–Fc-coated coverslips is reduced in CHL1+/+ but strongly enhanced in CHL1−/− cultures. Shown are mean + SEM values. *p < 0.01, significant difference compared with CHL1+/+ cultures (two-way ANOVA with Newman–Keuls post hoc test; n > 100 neurons per genotype in 3 independent experiments).
The possibility that neurite outgrowth depends on homophilic CHL1 interactions was investigated by substrate coating CHL1–Fc and plating cerebellar neurons from CHL1+/+ and CHL1−/− mice onto this substrate (Fig. 7C). When compared with cultures on PLL, in which neurite lengths of CHL1+/+ and CHL1−/− neurons were similar, neurite outgrowth on the CHL1–Fc substrate was by far more enhanced in CHL1−/− neurons (390 ± 10%) than in CHL1+/+ neurons (210 ± 2%). Neurite outgrowth of CHL1+/+ and CHL1−/− mice was similar on substrate-coated laminin and L1–Fc (Fig. 7C). Immunocytochemical analysis revealed that all cultured neurons, similar to granule cells in the developing and adult cerebellum (Holm et al., 1996; Nikonenko et al., 2006), expressed CHL1 (data not shown).
The combined observations indicate that CHL1 binds homophilically and that this homophilic interaction reduces neurite outgrowth, whereas heterophilic interactions enhance neurite outgrowth in vitro.
Expression of CHL1 in cultures of astrocytes exposed to FGF-2
Because FGF-2 expression is enhanced after spinal cord lesion, we were interested whether CHL1 expression level is regulated by FGF-2 and which signal transduction mechanisms are involved. Cultures of astrocytes were exposed to FGF-2 for up to 3 d in vitro, and the effect of different doses of FGF-2 was measured after a 3 d exposure time (Fig. 8A,B, respectively). CHL1 expression was highest after exposure to 100 ng/ml for 3 d when compared with no exposure (Fig. 8A). Doses of 100 ng/ml were saturating when CHL1 expression was measured after 3 d of exposure to FGF-2 (Fig. 8B).
To gain insights into the signal transduction pathways involved in upregulating CHL1 expression, inhibitors of extracellular signal-regulated kinase (U0126 and PD98069), CaMKII (KN-93), and PI3K (LY29400) were applied in culture. They all blocked CHL1 expression in a dose-dependent manner (Fig. 8C). In the absence of FGF-2, the highest doses of the inhibitors, which blocked upregulation of CHL1 expression in the presence of FGF-2, were not toxic to the astrocytes, as could be seen by the robust expression of CHL1. PI3K inhibitor, however, reduced CHL1 expression in the absence of FGF-2, suggesting that PI3K may also be involved in regulating CHL1 levels independent of FGF-2 signaling. Furthermore, expression levels of the control marker GAPDH were not affected in the presence of FGF-2 (Fig. 8A–C). These observations point to known pathways of FGF-2 signaling via its cognate receptors in astrocytes to upregulate CHL1 expression.
Migration of astrocytes from CHL1+/+ and CHL1−/− mice in the absence and presence of FGF-2
Because activated astrocytes migrate more actively than quiescent ones, we assayed the migratory capacity of CHL1−/− versus CHL1+/+ derived astrocytes in vitro in response to FGF-2. Using the scratch assay and GFAP as a marker for astrocytes, we could not detect difference in migration between the two genotypes in the absence of FGF-2 or after 6 h of exposure to the cytokine (Fig. 9A). After 20 h cultivation with FGF-2, CHL1+/+ astrocytes migrated more into the void area than CHL1−/− astrocytes (Fig. 9A). The difference between the genotypes was less pronounced in the absence of the cytokine.
Figure 9.

Migration (A) and proliferation (B) of astrocytes cultured from CHL1−/− and CHL1+/+ mice. A, Scratched monolayers of astrocytes were treated for 6 or 20 h with (+FGF2) or without (−FGF2) FGF-2. B, After 4 d with (+FGF2) or without (−FGF2) FGF-2 treatment, cells were subjected to a proliferation assay using the MTT assay kit. *p < 0.05 compared between cultures without and with FGF-2; #p < 0.01 compared between CHL1−/− and CHL1+/+ mice (two-tailed t test in 5 independent experiments). C, Immunoblot analysis of CHL1 and GFAP expression after injection of FGF-2 into the intact spinal cord. FGF-2 was injected into the T9–T10 segments of mouse spinal cords. PBS as vehicle served as a negative control. Analysis was performed after 3 and 7 d without FGF-2 and with FGF-2 treatment. *p < 0.05 compared with control groups (paired two-tailed t test; n = 5 mice per group).
Proliferation of astrocytes from CHL1+/+ and CHL1−/− mice in the absence and presence of FGF-2
Proliferation is also a hallmark of astrocytes that respond to a lesion in vivo. We, therefore, compared the proliferative activity of astrocytes after 4 d in vitro in the presence or the absence of 100 ng/ml FGF-2 using the MTT assay kit. In the absence of FGF-2, no difference between the two genotypes was seen. With FGF-2, however, astrocytes from CHL1−/− mice proliferated less than astrocytes from CHL1+/+ mice (Fig. 9B).
Expression of CHL1 and GFAP after injection of FGF-2 into the intact spinal cord of adult wild-type mice
To test whether FGF-2 would have effects on the gliotic features of astrocytes and CHL1 expression in vivo, spinal cords of wild-type mice were injected with FGF-2. Western blot analysis revealed similar amounts of CHL1 and GFAP at 3 and 7 d after vehicle injection (Fig. 9C). A time-dependent increase of both CHL1 and GFAP was observed after FGF-2 injection. These results indicate that astrocytes in the spinal cord upregulate expression of CHL1 and GFAP within days after an increase in the local FGF-2 concentration.
Discussion
In this study, we provide novel evidence for a functional role of CHL1 in spinal cord regeneration. We show that adult CHL1-deficient mice recover better than wild-type mice from compression injury of the spinal cord and suggest that functional improvement is related to remodeling of neuronal circuits and enhanced axonal growth. Furthermore, we propose a FGF-2-mediated upregulation of CHL1 in astrocytes and homophilic CHL1 binding as possible cellular and molecular mechanisms underlying the inhibitory effect of CHL1 on regeneration.
Enhanced functional recovery in CHL1-deficient mice
The improved overall outcome of spinal cord injury in CHL1−/− mice reflected improvement of motor functions controlled in different ways. Plantar stepping, evaluated by both the BBB score and the foot-stepping angle, requires low levels of supraspinal control. We found evidence that the measures of this ability strongly correlate with the degree of monoaminergic innervation of the distal spinal cord. A causative relationship is plausible. Aminergic transmitters, in particular serotonin in the mouse, are powerful modulators of intrinsic circuitries capable of initiating and controlling rhythmic and coordinated movements, the spinal pattern generators (Edgerton et al., 2004; Fouad and Pearson, 2004). Monoaminergic fibers provide diffuse innervation, and it can be expected that a more vigorous axonal regrowth, even if not specifically targeted, would be beneficial, in contrast to other systems that require precise reestablishment of synaptic connections. We also found a possible link between cholinergic perisomatic innervation of motoneurons and plantar stepping abilities after injury. The observed correlations, even if reflecting causative relationships, cannot help to explain genotype-specific differences. Still, they suggest that the degree of preservation and reestablishment of the modulatory cholinergic input to motoneuron somata, considerably reduced after spinal cord lesion, may be essential for motor recovery. This notion is supported by recent electrophysiological data showing that, in the intact spinal cord, the cholinergic perisomatic synapses regulate motoneuron excitability during locomotion (Miles et al., 2007). In addition to plantar stepping, improved walking in CHL1-deficient mice was documented by the rump-height index, a parameter estimating the abilities for coordinated and rhythmic activation of muscles working at different joints. Although we did not find correlations of this parameter with structural variables, we can suggest that structural synaptic plasticity, at different locations and possibly of different kind than observed around the motoneuron perikarya, can account for better performance of CHL1−/− mice. Finally, we observed that also the ability to initiate and perform voluntary movements without body weight support, estimated by the flexion–extension ratio, was improved in CHL1−/− mice. This observation can only be explained by enhanced axonal regrowth across the lesion site and reestablishment of connections, direct or indirect, with higher motor control centers, a notion supported by recent observations on tenascin-R-deficient mice after spinal cord compression (Apostolova et al., 2006). In tenascin-R-deficient mice, voluntary movements were not improved compared with wild-type littermates, and no evidence for axonal regrowth was found. From our observations, we can conclude that CHL1 deficiency in the injured spinal cord favors both axonal growth and remodeling of cell connectivity, which lead to a better functional outcome.
CHL1 expression affects the phenotype of astrocytes and inhibits neurite elongation and branching in vitro
Several lines of evidence indicated that the rapid and prolonged upregulation of CHL1 in the injured spinal cord of wild-type mice was specifically associated with activated astrocytes. At the histological level, CHL1 colocalized with astrocyte markers GFAP and S-100 only within the glial scar. Biochemically, CHL1 protein expression was enhanced in parallel with GFAP but not with changes in the expression level of neuronal and oligodendrocyte markers. Therefore, we assume that upregulation of CHL1 by activated astrocytes is instrumental in limiting functional recovery in wild-type animals.
Because the cytokine FGF-2 is implicated in spinal cord regeneration via different mechanisms (Teng et al., 1999; Meijs et al., 2004) and a link between FGF-2 and enhanced CHL1 expression after optic nerve crush has been suggested (Rolf et al., 2003), we investigated its effect on CHL1 expression in cultured astrocytes. We found that, indeed, CHL1 expression is enhanced in a dose- and time-dependent manner by activation of known FGF receptor-dependent signaling pathways, namely MAP kinase, CaMKII, and PI3K. The fact that inhibitors of these enzymes are able to block the FGF-2-induced response shows that conventional triggering pathways regulate CHL1 expression. Not only in vitro, but also in vivo, when injected into the unlesioned spinal cord, FGF-2 enhanced the expression of CHL1 in parallel with a previously described enhancement of GFAP expression (Lewis et al., 1992; Eclancher et al., 1996; Liu et al., 2005). In addition, in vitro assays confirmed that FGF-2 influences CHL1-associated migration and proliferation of astrocytes as indicated by its more potent effects on CHL1+/+ astrocytes than on CHL1−/− astrocytes. Alternative interpretation of these data is that CHL1-deficient mice may have attenuated FGF-2 receptor function, a possibility that warrants additional studies.
To test the idea that CHL1-expressing astrocytes could be inhibitory to wild-type CHL1-expressing neurons, we analyzed homogenotypic and heterogenotypic cocultures of astrocytes and neurons. Both neurite elongation and branching were strongly reduced when CHL1 was expressed on both culture partners as opposed to cocultures in which CHL1 expression was deficient in either neurons or astrocytes or in both. These observations allow the interpretation that homophilic mechanisms are involved in the reduction of neurite outgrowth and suggest that enhanced functional recovery and axonal regrowth in lesioned CHL1−/− mice are related to the absence of CHL1 on astrocytes or neurons or on both.
Homophilic versus heterophilic CHL1 binding mechanisms
The possibility that CHL1 engages in homophilic interactions was excluded in our previous paper (Hillenbrand et al., 1999) on the basis of the inability of CHL1 ectopically overexpressing fibroblast-like cells to aggregate in vitro. However, at the time, it was not realized that these fibroblast-like cells were proteolytically extremely active, and CHL1 was shed into the culture medium as soon as the cells were plated in vitro. Using an acellular assay for testing homophilic binding, we found that recombinant CHL1 in the absence of proteases was able to engage in homophilic binding. In the bead aggregation assay, strong aggregation was seen when beads were coated with CHL1–Fc. This aggregation was specific, because it was not seen when Fc-coated beads were used and because anti-CHL1 antibodies inhibited CHL1-dependent aggregation.
To analyze whether the observed neurite outgrowth inhibition of CHL1+/+ neurons when grown on CHL1+/+ astrocytes could be interpreted in terms of homophilic interactions, we analyzed neurite outgrowth from CHL1+/+ and CHL1−/− neurons on substrate coated with purified CHL1–Fc. As observed previously (Chen et al., 1999), outgrowth of neurites from wild-type neurons was enhanced when compared with the poly-l-lysine substrate. Interestingly, this enhanced neurite outgrowth by wild-type neurons was even more prominent when CHL1−/− neurons were cultured on the CHL1 substrate. Thus, trans-interacting CHL1 enhances neurite outgrowth, most likely via heterophilic binding mechanisms, whereas homophilic trans-interactions between substrate-coated CHL1 and CHL1-expressing neurons inhibit neurite outgrowth. The combined observations point to a dual function of CHL1: it can be neurite outgrowth promoting by heterophilic mechanisms and neurite outgrowth repellent by a homophilic binding mechanism. Because of the inability to grow adult neurons from brainstem nuclei or spinal cord in culture, it is not possible to test heterophilic and homophilic binding mechanisms in vitro for correlation with the in vivo experiments. However, the fact that CHL1 is upregulated after spinal cord injury by astrocytes and that it is expressed by neurons and axon tracts in the adult spinal cord may allow the inference that repellent homophilic trans-interactions between glial scar astrocytes and severed axons are operant also in the adult spinal cord.
The heterophilic binding partners on astrocytes and neurons that contribute to increase of neurite outgrowth both in vitro and axon regrowth in vivo remain to be discovered. Plausible candidates are members of the semaphorin family and the neuropilin/plexin coreceptors, as found for other L1 family members (Castellani et al., 2000, 2002; Falk et al., 2005). It should be mentioned that also integrins may be involved in CHL1-dependent signaling, thus implicating RGD-dependent extracellular matrix ligands in the complex cellular and molecular milieu of the injured spinal cord.
In conclusion, here we show that CHL1 plays an inhibitory role in the pathogenesis of spinal cord injury and provide evidence for some cellular and molecular mechanisms that may underlie the negative impact of CHL1 upregulation during lesion of the spinal cord.
Footnotes
This work was supported by Deutsche Forschungsgemeinschaft and Roechling Stiftung (M.S.). M.S. is a New Jersey Professor for Spinal Cord Research. We are grateful to Eva Kronberg for animal care, Achim Dahlmann for genotyping, Kathrin Hilke-Steen for typing and formatting this manuscript, and Mira Jakovcevski for help with the statistical analysis.
References
- Angeloni D, Lindor NM, Pack S, Latif F, Wei MH, Lerman MI. CALL gene is haploinsufficient in a 3p-syndrome patient. Am J Med Genet. 1999;86:482–485. doi: 10.1002/(sici)1096-8628(19991029)86:5<482::aid-ajmg15>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- Apostolova I, Irintchev A, Schachner M. Tenascin-R restricts posttraumatic remodeling of motoneuron innervation and functional recovery after spinal cord injury in adult mice. J Neurosci. 2006;26:7849–7859. doi: 10.1523/JNEUROSCI.1526-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basso DM, Beattie MS, Bresnahan JC. A sensitive and reliable locomotor rating scale for open field testing in rats. J Neurotrauma. 1995;12:1–21. doi: 10.1089/neu.1995.12.1. [DOI] [PubMed] [Google Scholar]
- Buhusi M, Midkiff BR, Gates AM, Richter M, Schachner M, Maness PF. Close homolog of L1 is an enhancer of integrin-mediated cell migration. J Biol Chem. 2003;278:25024–25031. doi: 10.1074/jbc.M303084200. [DOI] [PubMed] [Google Scholar]
- Castellani V, Chedotal A, Schachner M, Faivre-Sarrailh C, Rougon G. Analysis of the L1-deficient mouse phenotype reveals cross-talk between Sema3A and L1 signaling pathways in axonal guidance. Neuron. 2000;27:237–249. doi: 10.1016/s0896-6273(00)00033-7. [DOI] [PubMed] [Google Scholar]
- Castellani V, De Angelis E, Kenwrick S, Rougon G. Cis and trans interactions of L1 with neuropilin-1 control axonal responses to semaphorin 3A. EMBO J. 2002;21:6348–6357. doi: 10.1093/emboj/cdf645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaisuksunt V, Campbell G, Zhang Y, Schachner M, Lieberman AR, Anderson PN. The cell recognition molecule CHL1 is strongly upregulated by injured and regenerating thalamic neurons. J Comp Neurol. 2000;425:382–392. doi: 10.1002/1096-9861(20000925)425:3<382::aid-cne4>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- Chen QY, Chen Q, Feng GY, Lindpaintner K, Chen Y, Sun X, Chen Z, Gao Z, Tang J, He L. Case-control association study of the close homologue of L1 (CHL1) gene and schizophrenia in the Chinese population. Schizophr Res. 2005;73:269–274. doi: 10.1016/j.schres.2004.06.001. [DOI] [PubMed] [Google Scholar]
- Chen S, Mantei N, Dong L, Schachner M. Prevention of neuronal cell death by neural adhesion molecules L1 and CHL1. J Neurobiol. 1999;38:428–439. doi: 10.1002/(sici)1097-4695(19990215)38:3<428::aid-neu10>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- Cole R, Vellis JD. Preparation of astrocyte, oligodendrocyte, and microglia cultures from primary rat cerebral cultures. In: Fedoroff S, Richardson A, editors. Protocols for neural cell culture. Totowa, NJ: Humana; 2001. pp. 117–127. [Google Scholar]
- Curtis R, Green D, Lindsay RM, Wilkin GP. Up-regulation of GAP-43 and growth of axons in rat spinal cord after compression injury. J Neurocytol. 1993;22:51–64. doi: 10.1007/BF01183975. [DOI] [PubMed] [Google Scholar]
- Davidoff MS, Irintchev AP. Acetylcholinesterase activity and type C synapses in the hypoglossal, facial and spinal-cord motor nuclei of rats. An electron-microscope study. Histochemistry. 1986;84:515–524. doi: 10.1007/BF00482985. [DOI] [PubMed] [Google Scholar]
- Demyanenko GP, Schachner M, Anton E, Schmid R, Feng G, Sanes J, Maness PF. Close homolog of L1 modulates area-specific neuronal positioning and dendrite orientation in the cerebral cortex. Neuron. 2004;44:423–437. doi: 10.1016/j.neuron.2004.10.016. [DOI] [PubMed] [Google Scholar]
- Eclancher F, Kehrli P, Labourdette G, Sensenbrenner M. Basic fibroblast growth factor (bFGF) injection activates the glial reaction in the injured adult rat brain. Brain Res. 1996;737:201–214. doi: 10.1016/0006-8993(96)00732-9. [DOI] [PubMed] [Google Scholar]
- Edgerton VR, Tillakaratne NJ, Bigbee AJ, de Leon RD, Roy RR. Plasticity of the spinal neural circuitry after injury. Annu Rev Neurosci. 2004;27:145–167. doi: 10.1146/annurev.neuro.27.070203.144308. [DOI] [PubMed] [Google Scholar]
- Falk J, Bechara A, Fiore R, Nawabi H, Zhou H, Hoyo-Becerra C, Bozon M, Rougon G, Grumet M, Puschel AW, Sanes JR, Castellani V. Dual functional activity of semaphorin 3B is required for positioning the anterior commissure. Neuron. 2005;48:63–75. doi: 10.1016/j.neuron.2005.08.033. [DOI] [PubMed] [Google Scholar]
- Fouad K, Pearson K. Restoring walking after spinal cord injury. Prog Neurobiol. 2004;73:107–126. doi: 10.1016/j.pneurobio.2004.04.003. [DOI] [PubMed] [Google Scholar]
- Frints SG, Marynen P, Hartmann D, Fryns JP, Steyaert J, Schachner M, Rolf B, Craessaerts K, Snellinx A, Hollanders K, D'Hooge R, De Deyn PP, Froyen G. CALL interrupted in a patient with non-specific mental retardation: gene dosage-dependent alteration of murine brain development and behavior. Hum Mol Genet. 2003;12:1463–1474. doi: 10.1093/hmg/ddg165. [DOI] [PubMed] [Google Scholar]
- Haspel J, Friedlander DR, Ivgy-May N, Chickramane S, Roonprapunt C, Chen S, Schachner M, Grumet M. Critical and optimal Ig domains for promotion of neurite outgrowth by L1/Ng-CAM. J Neurobiol. 2000;42:287–302. [PubMed] [Google Scholar]
- Hellstroem J, Oliveira AL, Meister B, Cullheim S. Large cholinergic nerve terminals on subsets of motoneurons and their relation to muscarinic receptor type 2. J Comp Neurol. 2003;460:476–486. doi: 10.1002/cne.10648. [DOI] [PubMed] [Google Scholar]
- Hillenbrand R, Molthagen M, Montag D, Schachner M. The close homologue of the neural adhesion molecule L1 (CHL1): patterns of expression and promotion of neurite outgrowth by heterophilic interactions. Eur J Neurosci. 1999;11:813–826. doi: 10.1046/j.1460-9568.1999.00496.x. [DOI] [PubMed] [Google Scholar]
- Holm J, Hillenbrand R, Steuber V, Bartsch U, Moos M, Luebbert H, Montag D, Schachner M. Structural features of a close homologue of L1 (CHL1) in the mouse: a new member of the L1 family of neural recognition molecules. Eur J Neurosci. 1996;8:1613–1629. doi: 10.1111/j.1460-9568.1996.tb01306.x. [DOI] [PubMed] [Google Scholar]
- Irintchev A, Koch M, Needham LK, Maness P, Schachner M. Impairment of sensorimotor gating in mice deficient in the cell adhesion molecule L1 or its close homologue, CHL1. Brain Res. 2004;1029:131–134. doi: 10.1016/j.brainres.2004.09.042. [DOI] [PubMed] [Google Scholar]
- Irintchev A, Rollenhagen A, Troncoso E, Kiss JZ, Schachner M. Structural and functional aberrations in the cerebral cortex of tenascin-C deficient mice. Cereb Cortex. 2005;15:950–962. doi: 10.1093/cercor/bhh195. [DOI] [PubMed] [Google Scholar]
- Jiao Y, Sun Z, Lee T, Fusco FR, Kimble TD, Meade CA, Cuthbertson S, Reiner A. A simple and sensitive antigen retrieval method for free-floating and slide-mounted tissue sections. J Neurosci Methods. 1999;93:149–162. doi: 10.1016/s0165-0270(99)00142-9. [DOI] [PubMed] [Google Scholar]
- Joshi M, Fehlings MG. Development and characterization of a novel, graded model of clip compressive spinal cord injury in the mouse. Part 1. Clip design, behavioral outcomes, and histopathology. J Neurotrauma. 2002;19:175–190. doi: 10.1089/08977150252806947. [DOI] [PubMed] [Google Scholar]
- Kiehn O, Rostrup E, Moller M. Monoaminergic systems in the brainstem and spinal cord of the turtle Pseudemys scripta elegans as revealed by antibodies against serotonin and tyrosine hydroxylase. J Comp Neurol. 1992;325:527–547. doi: 10.1002/cne.903250406. [DOI] [PubMed] [Google Scholar]
- Kloos AD, Fisher LC, Detloff MR, Hassenzahl DL, Basso DM. Stepwise motor and all-or-none sensory recovery is associated with nonlinear sparing after incremental spinal cord injury in rats. Exp Neurol. 2005;191:251–265. doi: 10.1016/j.expneurol.2004.09.016. [DOI] [PubMed] [Google Scholar]
- Leshchyns'ka I, Sytnyk V, Morrow JS, Schachner M. Neural cell adhesion molecule (NCAM) association with PKCbeta2 via betaI spectrin is implicated in NCAM-mediated neurite outgrowth. J Cell Biol. 2003;161:625–639. doi: 10.1083/jcb.200303020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leshchyns'ka I, Sytnyk V, Richter M, Andreyeva A, Puchkov D, Schachner M. The adhesion molecule CHL1 regulates uncoating of clathrin-coated synaptic vesicles. Neuron. 2006;52:1011–1025. doi: 10.1016/j.neuron.2006.10.020. [DOI] [PubMed] [Google Scholar]
- Lewis GP, Erickson PA, Guérin CJ, Anderson DH, Fisher SK. Basic fibroblast growth factor: a potential regulator of proliferation and intermediate filament expression in the retina. J Neurosci. 1992;12:3968–3978. doi: 10.1523/JNEUROSCI.12-10-03968.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Lu B, Tu CQ, Chi LT, Wang GL, Pei FX. Effect of basic fibroblast growth factor on the expression of glial fibrillary acidic protein after tractive spinal cord injury in rats. Chin J Traumatol. 2005;8:117–120. [PubMed] [Google Scholar]
- Loers G, Schachner M. Recognition molecules and neural repair. J Neurochem. 2007;101:865–882. doi: 10.1111/j.1471-4159.2006.04409.x. [DOI] [PubMed] [Google Scholar]
- Meijs MF, Timmers L, Pearse DD, Tresco PA, Bates ML, Joosten EA, Bunge MB, Oudega M. Basic fibroblast growth factor promotes neuronal survival but not behavioral recovery in the transected and Schwann cell implanted rat thoracic spinal cord. J Neurotrauma. 2004;21:1415–1430. doi: 10.1089/neu.2004.21.1415. [DOI] [PubMed] [Google Scholar]
- Miles GB, Hartley R, Todd AJ, Brownstone RM. Spinal cholinergic interneurons regulate the excitability of motoneurons during locomotion. Proc Natl Acad Sci USA. 2007;104:2448–2453. doi: 10.1073/pnas.0611134104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montag-Sallaz M, Schachner M, Montag D. Misguided axonal projections, neural cell adhesion molecule 180 mRNA upregulation, and altered behavior in mice deficient for the close homolog of L1. Mol Cell Biol. 2002;22:7967–7981. doi: 10.1128/MCB.22.22.7967-7981.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morellini F, Lepsveridze E, Kahler B, Dityatev A, Schachner M. Reduced reactivity to novelty, impaired social behavior, and enhanced basal synaptic excitatory activity in perforant path projections to the dentate gyrus in young adult mice deficient in the neural cell adhesion molecule CHL1. Mol Cell Neurosci. 2007;34:121–136. doi: 10.1016/j.mcn.2006.10.006. [DOI] [PubMed] [Google Scholar]
- Nikonenko AG, Sun M, Lepsveridze E, Apostolova I, Petrova I, Irintchev A, Dityatev A, Schachner M. Enhanced perisomatic inhibition and impaired long-term potentiation in the CA1 region of juvenile CHL1-deficient mice. Eur J Neurosci. 2006;23:1839–1852. doi: 10.1111/j.1460-9568.2006.04710.x. [DOI] [PubMed] [Google Scholar]
- Rolf B, Lang D, Hillenbrand R, Richter M, Schachner M, Bartsch U. Altered expression of CHL1 by glial cells in response to optic nerve injury and intravitreal application of fibroblast growth factor-2. J Neurosci Res. 2003;71:835–843. doi: 10.1002/jnr.10533. [DOI] [PubMed] [Google Scholar]
- Sakurai K, Migita O, Toru M, Arinami T. An association between a missense polymorphism in the close homologue of L1 (CHL1, CALL) gene and schizophrenia. Mol Psychiatry. 2002;7:412–415. doi: 10.1038/sj.mp.4000973. [DOI] [PubMed] [Google Scholar]
- Shahar A, de Vellis J, Vernadakis A, Haber B, editors. New York: Liss; 1989. A dissection and tissue culture manual of the nervous system. [Google Scholar]
- Shapiro S. Neurotransmission by neurons that use serotonin, noradrenaline, glutamate, glycine and γ-animobutyric acid in the normal and injured spinal cord. Neurosurgery. 1997;40:168–177. doi: 10.1097/00006123-199701000-00037. [DOI] [PubMed] [Google Scholar]
- Teng YD, Mocchetti I, Taveira-DaSilva AM, Gillis RA, Wrathall JR. Basic fibroblast growth factor increases long-term survival of spinal motor neurons and improves respiratory function after experimental spinal cord injury. J Neurosci. 1999;19:7037–7047. doi: 10.1523/JNEUROSCI.19-16-07037.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Roslan R, Lang D, Schachner M, Lieberman AR, Anderson PN. Expression of CHL1 and L1 by neurons and glia following sciatic nerve and dorsal root injury. Mol Cell Neurosci. 2000;16:71–86. doi: 10.1006/mcne.2000.0852. [DOI] [PubMed] [Google Scholar]