

Abstract
Partial inhibition of mitochondrial respiratory complex I by rotenone reproduces aspects of Parkinson's disease in rodents. The hypothesis that rotenone enhancement of neuronal cell death is attributable to oxidative stress was tested in an acute glutamate excitotoxicity model using primary cultures of rat cerebellar granule neurons. As little as 5 nm rotenone increased mitochondrial superoxide (O2·−) levels and potentiated glutamate-induced cytoplasmic Ca2+ deregulation, the first irreversible stage of necrotic cell death. However, the potent cell-permeant O2·− trap manganese tetrakis (N-ethylpyridinium-2yl) porphyrin failed to prevent the effects of the inhibitor. The bioenergetic consequences of rotenone addition were quantified by monitoring cell respiration. Glutamate activation of NMDA receptors used the full respiratory capacity of the in situ mitochondria, and >80% of the glutamate-stimulated respiration was attributable to increased cellular ATP demand. Rotenone at 20 nm inhibited basal and carbonyl cyanide p-trifluoromethoxyphenylhydrazone-stimulated cell respiration and caused respiratory failure in the presence of glutamate. ATP synthase inhibition by oligomycin was also toxic in the presence of glutamate. We conclude that the cell vulnerability in the rotenone model of partial complex I deficiency under these specific conditions is primarily determined by spare respiratory capacity rather than oxidative stress.
Keywords: mitochondria, complex I, rotenone, superoxide, membrane potential, excitotoxicity
Introduction
Mitochondrial dysfunction associated with partial deficiencies in respiratory chain complexes is implicated in aging and many age-related neurodegenerative diseases (for review, see Lin and Beal, 2006; Schapira, 2006). Complex I [nicotinamide adenine dinucleotide (NADH)-ubiquinone oxidoreductase] deficiency in particular is implicated in Parkinson's disease (PD) (Muqit et al., 2006) and underlies the pathology associated with the mitochondrial DNA mutations in Leber's hereditary optic neuropathy (LHON) (Lenaz et al., 2004). Although the exact mechanisms of the pathogenesis attributable to complex I deficiency are not clear, there are two major factors that can lead to a pathogenic outcome, namely bioenergetic deficit and oxidative stress. Endorsement for the oxidative stress hypothesis has come from the rotenone model of PD (Betarbet et al., 2000), in which chronic exposure of rats to low nanomolar concentrations of the complex I inhibitor reproduces many of the pathological features of the disease. The concentration of rotenone in the brain (<30 nm) was deemed insufficient to significantly impair brain mitochondrial respiration, whereas incubation of neural cells in culture with as little as 5 nm inhibitor showed clear evidence of oxidative stress (Betarbet et al., 2000). Similarly, a genetic model for LHON has also suggested the role of oxidative stress in neuronal degeneration because overexpression of superoxide dismutase 2 (SOD2) resulted in decreased optic nerve degeneration (Qi et al., 2004).
Neuronal ATP demand is responsive to changes in electrical activity (Kann and Kovacs, 2006), and, before rejecting a role for energetic insufficiency, it is important to examine the effects of partial complex I deficiency on neuronal function under conditions of high ATP demand. Chronic activation of NMDA-selective glutamate receptors imposes a high ATP load on the neuron (Jekabsons and Nicholls, 2004) and can result in excitotoxicity, one of the key mechanisms of neurodegeneration after stroke, traumatic brain injury, epilepsy, and other pathological conditions (for review, see Krieger and Duchen, 2002). Excitotoxicity is also implicated in the development of Parkinson's disease (Blandini et al., 1996) and is thus a relevant model with which to test the acute effects of rotenone. It is established that energetic insufficiency can exacerbate excitotoxicity (Henneberry et al., 1989), and, in this study, the ability to monitor the respiration of coverslip-attached neurons has allowed us to test the contribution of oxidative stress and respiratory insufficiency to the rotenone-enhanced cell death associated with chronic NMDA receptor activation in primary cultures of rat cerebellar granule neurons (CGNs). It must be emphasized that the conclusions apply specifically in the context of glutamate excitotoxicity and that because oxidative stress can damage ATP generating capacity (Vesce et al., 2005), it should not be assumed that these are mutually opposing concepts.
Materials and Methods
Reagents.
Tetramethylrhodamine methyl ester (TMRM+), MitoSOX, and Fluo-5F AM were from Invitrogen (Carlsbad, CA); rotenone and manganese tetrakis (N-ethylpyridinium-2yl) porphyrin (MnTE-2-PyP) were from Calbiochem (San Diego, CA). (5R,10S)-(+)-5-methyl-10,11-dihydro[a,d]cyclohepten-5,10-imine hydrogen maleate (MK-801) and all other reagents were from Sigma (St. Louis, MO). ATP levels were determined with Sigma FL-AA bioluminescence assay kit.
Preparation of CGNs.
CGNs were prepared from 5- to 7-d-old Wistar rat pups as described previously (Courtney et al., 1990) with modifications. Briefly, cells were plated in coverslip-based one-well (3 × 106 cells) or eight-well (380,000 cells per well) chambers (Lab-Tek, Naperville, IL) that were precoated with 33 μg/ml polyethyleneimine overnight. Cultures were maintained in minimal essential medium supplemented with 10% fetal bovine serum, 30 mm glucose, 20 mm KCl, 2 mm glutamine, 50 U/ml penicillin, and 50 μg/ml streptomycin at 37°C in an incubator with a humidified atmosphere of 5% CO2 and 95% air. Twenty-four hours later, 10 μm cytosine arabinoside was added to inhibit the growth of non-neuronal cells. Neurons were used after 7–12 d in culture as detailed.
In situ respirometry. The respiration rates of intact CGN populations were measured using a “cell respirometer” as described previously (Jekabsons and Nicholls, 2004) but with a modified imaging chamber to simplify the mounting and imaging of cells grown in one-well chambered cover glasses (catalog number 155361; Lab-Tek). Briefly, medium was slowly perfused (∼30 μl/min) over the cells in a thin (250 μm) closed imaging chamber. The deficit in oxygen concentration was monitored downstream by a micro flow-through oxygen electrode (type 16–730; Microelectrodes, Bedford, NH) with output to a chart recorder. Unless otherwise stated, 3 × 106 cells were used in each respirometry experiment. Coverslip-attached cells were washed in respiration buffer containing 120 mm NaCl, 3.5 mm KCl, 1.3 mm CaCl2, 0.4 mm KH2PO4, 20 mm Na–N-Tris-(hydroxymethyl)-methyl-2-amino-ethanesulphonic acid, 5 mm NaHCO3, 1.2 mm Na2SO4, 15 mm d-glucose, and 60 μm fatty acid-free bovine serum albumin and rapidly assembled into the respirometer with a 250 μm gasket (GS-30S/10; Warner Instruments, Hamden, CT), yielding a chamber volume of 95 μl. Respiration buffer that had been equilibrated for at least 90 min at 37°C in air was perfused continuously through the chamber for 20–30 min to allow temperature equilibration. The baseline was set with buffer flowing through a bypass shunt to the electrode for ∼5 min. The electrode response was calibrated with buffers saturated with air, 50% air plus 50% nitrogen, and 100% nitrogen. The entire respirometer assembly was contained within a custom-made enclosure to maintain the temperature at 37°C.
Functional imaging.
Cells cultured in Lab-Tek eight-well chambers and preloaded with appropriate fluorescent indicators were imaged on a Zeiss (Oberkochen, Germany) Pascal Confocal Axiovert 100M microscope with a computer-driven stage. Images were collected using a 20× air objective. A custom-built acrylic chamber enclosed the entire microscope stage, allowing the temperature of the objectives and cells to be maintained at 37°C. For mitochondrial membrane potential (Δψm) determination, cells were preincubated for 2 h with 2.5 nm TMRM+ in the presence of 1 μm tetraphenylboron (TPB−) to facilitate equilibration across the plasma membrane (Nicholls and Budd, 2000). This concentration of TMRM+ is insufficient for aggregation within the mitochondrial matrix (nonquench mode). Subsequently, cells were washed with and incubated in respiration buffer in the presence of 2.5 nm TMRM+ and 1 μm TPB− before starting imaging. TMRM+ fluorescence was excited at 514 nm, and emission was monitored using a 590–650 nm filter.
Mitochondrial O2·− levels were monitored after addition of 0.1–0.2 μm MitoSOX (514 nm excitation, 590–650 nm emission). Fluorescence was monitored for 30 min, rotenone was then added, and recording was continued for another 30–60 min to monitor increases in mitochondrial O2·− levels. The relative levels of O2·− were calculated from the slopes of the MitoSOX fluorescence traces before and after addition of rotenone. Membrane potentials and delayed Ca2+ deregulation (DCD) were monitored in CGNs equilibrated with 50 nm TMRM+ plus 0.5 μm Fluo-5F AM in respiration buffer. The fluorescence of TMRM+ (514 nm excitation, 590–650 nm emission) and Fluo-5F (488 nm excitation, 505–530 nm emission) was monitored. After recording the baseline fluorescence, cells were incubated with different concentrations of rotenone for 60 min and then exposed to 100 μm glutamate in the presence of 10 μm glycine for 60 min, after which cells were treated with the NMDA receptor antagonist MK-801 (10 μm). The cells that failed to show a recovery of free cytoplasmic Ca2+ ([Ca2+]c) to basal levels within 30 min of MK-801 addition were considered to be deregulated.
Cell death.
Neurons were incubated in the presence or absence of 20 nm rotenone. After 1 h, 100 μm glutamate plus 10 μm glycine were added, followed after an additional 1 h by 10 μm MK-801. The original culture medium was restored, and, after 24 h, live/dead cell imaging was performed with Syto 13 and propidium iodide.
Results
Rotenone increases in situ mitochondrial matrix O2·− levels
Experiments with isolated mitochondria have established that partial inhibition of complex I increases the generation of O2·− from NADH-linked substrates (Kudin et al., 2004; Tretter et al., 2004). The rate of oxidation of dihydroethidine to ethidium provides a reliable measure of the intracellular levels of O2·− (Vesce et al., 2004), although the dual localization of the substrate in cytoplasm and matrix makes it difficult to determine the site of origin of the O2·−. To overcome this ambiguity, the recently developed fluorescent mitochondrial O2·− probe MitoSOX (Invitrogen), a positively charged derivative of hydroethidine, was used. Low concentrations of MitoSOX are effectively localized to the matrix in cultured granule neurons (Johnson-Cadwell et al., 2007). Hydroethidine and MitoSOX are relatively inefficient, trapping only a few percent of the generated O2·−, and their rates of oxidation reflect the local levels of O2·− rather than its rate of generation (Johnson-Cadwell et al., 2007). Because the membrane-permeant MitoSOX is positively charged, its uptake (and hence extent of oxidation) is influenced by the mitochondrial and plasma membrane potentials (Johnson-Cadwell et al., 2007), and it was therefore important to establish that any respiratory restriction produced by the suboptimal rotenone concentrations used in this study did not affect either the plasma or mitochondrial membrane potentials. Control experiments monitoring the fluorescence of TMRM+ showed that rotenone concentrations up to 20 nm had no significant effect on the membrane potentials (data not shown), whereas 1 μm rotenone, a concentration sufficient to maximally inhibit complex I and induce ATP synthase reversal, induces a 17 mV mitochondrial depolarization (Johnson-Cadwell et al., 2007).
Even the lowest concentration of rotenone (5 nm) produced a twofold increase in the steady-state level of O2·− in the mitochondria (Fig. 1A), and this was further increased when rotenone was raised to 20 nm (Fig. 1A,C). The potent superoxide dismutase mimetic MnTE-2-PyP (Patel and Day, 1999) decreased the basal rate of MitoSOX oxidation and virtually abolished the increase seen with rotenone (Fig. 1B). It should be noted that interpretation of MitoSOX signals after NMDA receptor activation is not possible because the concentration of the cationic probe in the matrix will be greatly affected by the loss of the plasma membrane potential.
Figure 1.
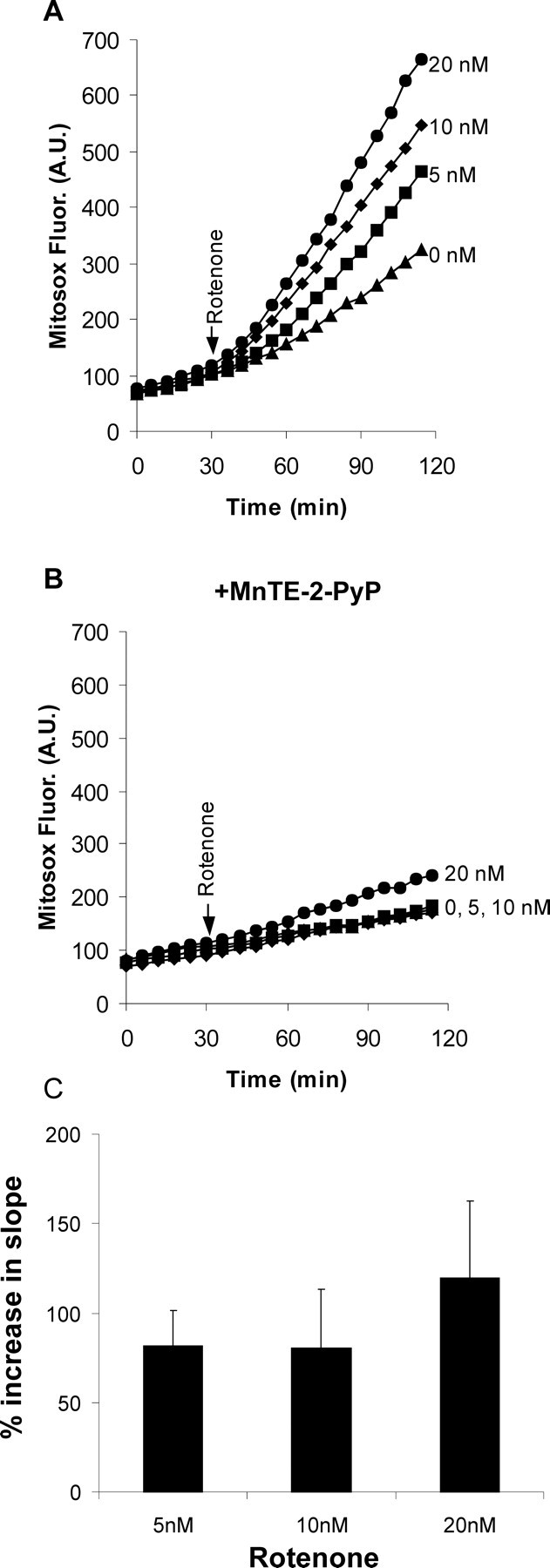
Rotenone-induced mitochondrial O2·− levels in intact granule neurons. Mitochondrial O2·− levels were quantified from the rate of MitoSOX oxidation. A, Rotenone (0–20 nm) was added when indicated. B, The experiment was repeated with CGNs treated with 25 μm MnTE-2-PyP for 45 min before and during imaging. C, Relative levels of matrix O2·− were calculated from the slopes of the fluorescence time courses after rotenone addition, normalized to their corresponding initial slopes. Results show mean and SD of nine independent experiments. Time courses in A and B are representative of nine independent experiments. Reactive oxygen species levels (slopes) in A at all concentrations of rotenone are significantly different from control (p < 0.001). Pooled data for 5 and 10 nm are different from 20 nm. p < 0.05.
Rotenone potentiates glutamate excitotoxicity
Chronic activation of NMDA receptors on CGNs induces plasma membrane depolarization (Johnson-Cadwell et al., 2007), massive Ca2+ entry (Brocard et al., 2001), Ca2+ accumulation by the in situ mitochondria (Budd and Nicholls, 1996), and partial mitochondrial depolarization (Ward et al., 2000) and can ultimately result in mitochondrial bioenergetic collapse and a massive increase in [Ca2+]c (DCD) as the first irreversible stage of necrotic cell death (Tymianski et al., 1993). Glutamate increases [Ca2+]c in the full population of CGNs (Fig. 2A), but deregulating cells can be identified by their failure to restore a low [Ca2+]c and TMRM+ fluorescence after NMDA receptor inhibition by MK-801. Figure 2 shows that 90% of CGNs recovered membrane potentials and Ca2+ homeostasis when MK-801 was added after 60 min of NMDA receptor activation. As little as 5 nm rotenone increased DCD to 40% (Fig. 2C), whereas virtually all cells deregulated after NMDA receptor activation in the presence of 20 nm rotenone.
Figure 2.
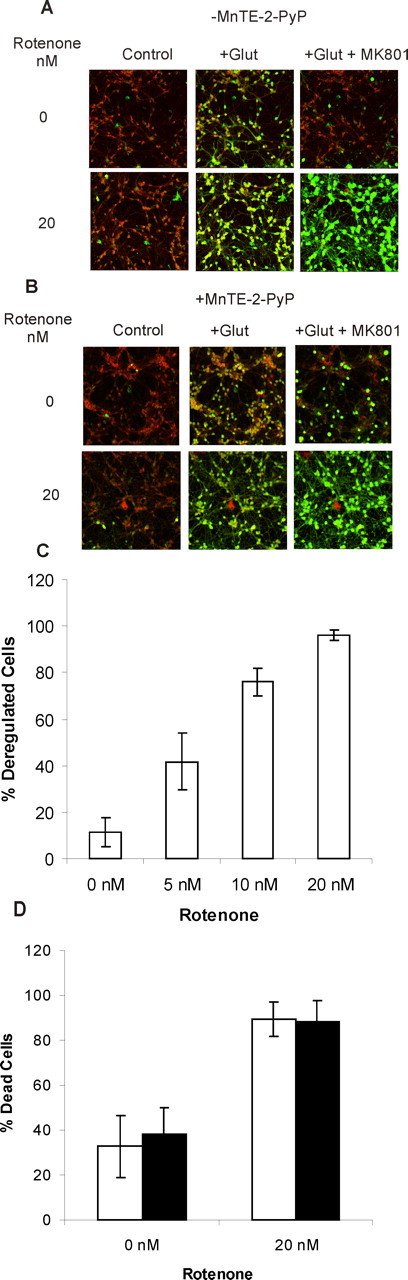
Rotenone-facilitated DCD and failure of MnTE-2-PyP to protect. A, Changes in TMRM+ (red) and Fluo-5F (green) fluorescence before (Control), 6 min after addition of 100 μm glutamate plus 10 μm glycine (+Glut), and 30 min after addition of 10 μm MK-801 after 60 min exposure to glutamate (+Glut + MK801). In the lower sequence, 20 nm rotenone was present throughout. B, Cells were treated with 25 μm MnTE-2-PyP for 45 min before and during imaging. C, The extent of deregulation in the absence of 25 μm Mn-TE-2-PyP as a function of rotenone concentration. Cells in which Fluo-5F fluorescence did not return to basal level after MK-801 treatment were considered deregulated. D, Effect of 20 nm rotenone on the extent of cell death 24 h after 60 min exposure to 100 μm glutamate/10 μm glycine in the presence (filled bars) or absence (open bars) of 25 μm Mn-TE-2-PyP. Results show mean and SD of four independent experiments.
Thus far, the results do not distinguish between an effect of rotenone attributable to inhibition of electron transfer through complex I and that attributable to the observed increase in O2·− levels. However, the inclusion of MnTE-2-PyP failed to protect against DCD (Fig. 2B) or 24 h cell death (Fig. 2D) in the presence of rotenone, suggesting that the enhanced DCD and subsequent cell death may have a more direct energetic basis in this model. Consistent with this, ATP levels 2 min after glutamate addition were reduced to 64 ± 7% in the presence of 20 nm rotenone relative to parallel incubations in the absence of rotenone.
NMDA receptor activation uses the full respiratory capacity of the in situ mitochondria
NMDA receptor activation results in the entry of Na+ and Ca2+ into the cytoplasm. Despite the relative Ca2+ selectivity of most isoforms, the 100-fold excess of Na+ over Ca2+ in the incubation medium means that entry of the monovalent cation is dominant and the cell responds by activating the Na+/K+-ATPase to expel the ion. Previous studies with partially depolarized CGNs in media containing 25 mm KCl (Jekabsons and Nicholls, 2004) showed that NMDA receptor activation used 40–50% of the spare respiratory capacity of the CGNs. In the present study, the cells are incubated in a physiological K+ concentration (3.9 mm), and the respiratory stimulation after glutamate/glycine (Fig. 3B,C) is equal to the maximal respiratory stimulation obtained with optimal concentrations of the protonophore carbonyl cyanide p-trifluoromethoxyphenylhydrazone (FCCP) (Fig. 3A). It should be noted that the absolute values of the respiratory rates are somewhat variable between preparations, which is why it is essential to make sequential additions to the same preparation. Figure 3D shows mean results normalized to either the initial respiration or the maximal FCCP-stimulated respiration.
Figure 3.

Quantitation of the respiratory demand imposed by NMDA receptor activation. A, CGNs were superfused with incubation medium in the respirometer. When indicated the medium was supplemented with FCCP (3 μm F3 or 4 μm F4) for 30 min. Note that 3 μm FCCP is sufficient to attain uncontrolled respiration. B, C, CGNs were superfused with medium containing 100 μm glutamate plus 10 μm glycine (G) or 3 μm FCCP (F3) for the periods indicated. D, Quantitative analysis of the glutamate-stimulated respiration. Respiration rates were normalized relative to the FCCP-stimulated rate and are the mean and SD of four independent experiments. The x-axis is labeled in the same order as the perfusions were made during respirometry in trace C.
Rotenone inhibition of CGN respiration
Basal CGN respiration remains constant for at least 150 min of superfusion with incubation medium (Fig. 4A). In contrast, addition of increasing concentrations of rotenone result in respiratory inhibition. To determine the effect of the inhibitor on maximally stimulated respiration, rotenone additions were made to CGNs superfused with 3 μm FCCP (Fig. 4B,C). Respiratory inhibition was seen with 10 and 20 nm inhibitor. As may be expected, the decrease in respiration produced by each concentration of rotenone is greater when the maximal capacity of the respiratory chain is evoked by the protonophore (Fig. 4D), indicating that the bioenergetic consequences of complex I restriction are most severe under conditions of maximal respiratory demand.
Figure 4.

Rotenone titration of basal and FCCP-stimulated respiration. A, Representative respirometry trace showing inhibition of cellular respiration by successive perfusions with buffer containing 20, 40, and 60 nm rotenone (R20, R40, and R60), respectively. Arrows indicate the times at which perfusion with the indicated buffer was started. Note that respiration in the absence of additions remains stable for the duration of the experiment (broken line). B, C, Representative respirometer traces quantifying the inhibition of uncontrolled respiration by 10 (R10) or 20 nm rotenone (R20) in the presence of 3 μm FCCP (F3). D, Quantitation of the rotenone inhibition in the presence (triangles) or absence (diamonds) of 3 μm FCCP. One-way ANOVA with Tukey's post hoc test for multiple comparisons was used to determine which means differed significantly from the basal respiration rate (for 5–10 nm, n = 6; for 20 nm, n = 7). **p < 0.01; ***p < 0.001.
Rotenone and glutamate-stimulated respiration
The respiratory traces suggest that even a minimal impairment of respiratory capacity could be detrimental to cells under conditions of acute high-energy demand. Under control conditions, CGNs can respond to two sequential periods of NMDA receptor activation with little deterioration of the respiratory rate (Fig. 5A,B). In contrast, if 20 nm rotenone is present during the second cycle, not only is there a decrease in the maximal respiration achieved but also the residual respiration rapidly decays, consistent with the extensive DCD seen in the presence of glutamate with this concentration of rotenone (Fig. 2).
Figure 5.
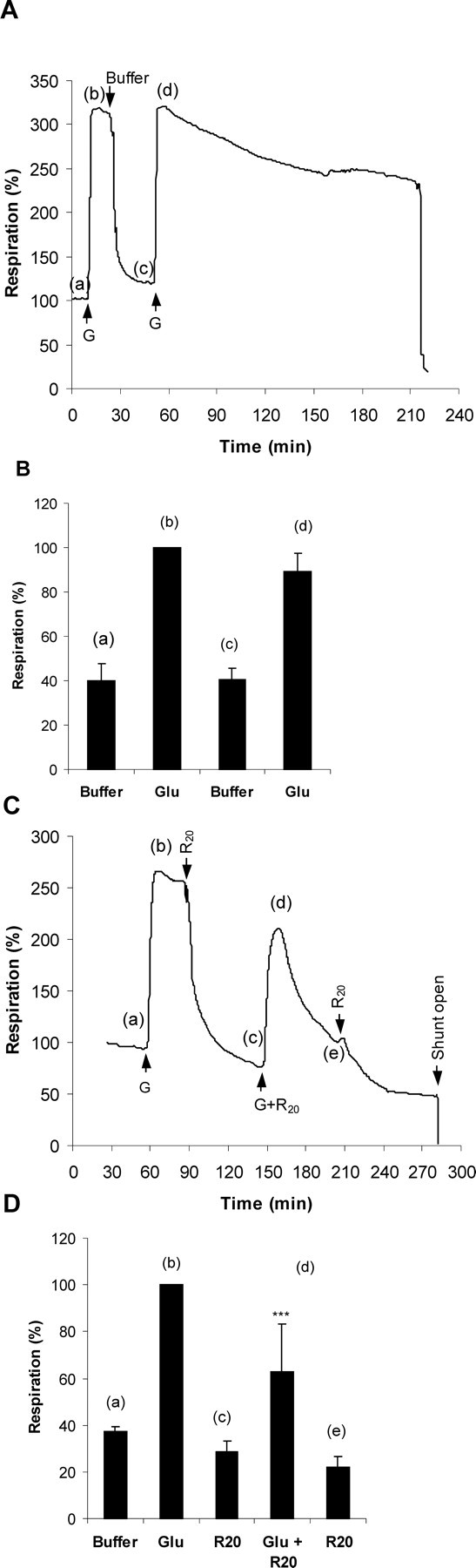
CGN respiration fails rapidly after NMDA receptor activation in the presence of 20 nm rotenone. A, Representative respiratory stimulation by successive glutamate (“G”) exposures. B, Quantitative analysis of the relative respiration rates obtained from replicate respirometer traces as in A. C, A second cycle of NMDA receptor stimulation in the presence of 20 nm rotenone (G+R20) shows decreased maximal stimulation and rapid respiratory failure. D, Quantitative analysis of the relative respiration rates obtained from replicate respirometer traces as in C. Respirations rates are compared with the glutamate-stimulated rate (black bars). ***p < 0.001 first addition versus second addition of glutamate.
If FCCP and glutamate place similar bioenergetic demands on the neuron under these conditions, why is glutamate more toxic to the cell? The answer most likely lies in the Ca2+ influx induced by receptor activation. It is established that much of the Ca2+ entering via the NMDA receptor is accumulated by the mitochondria (Budd and Nicholls, 1996) and that the initial partial depolarization of Δψm is attributable to matrix Ca2+ accumulation rather than ATP demand (Ward et al., 2000). To assess the division of the bioenergetic load after NMDA receptor activation between ATP demand by the plasma membrane ion pumps and enhanced respiration associated with Ca2+ uptake into the mitochondrial matrix, a cycle of glutamate exposure was repeated with the addition of oligomycin simultaneous with the second glutamate addition (Fig. 6A). A limited respiratory stimulation was observed compared with glutamate alone. However, under these conditions, almost all neurons rapidly deregulated as shown in Figure 6B. Extensive DCD was also seen when a single addition of glutamate was made to oligomycin-treated cells (data not shown). This experiment could also be flawed if the action of oligomycin was slower than glutamate. Therefore, we perfused cells first with oligomycin to block the ATP-turnover-driven respiration and then stimulated the cells with glutamate in the continued presence of oligomycin. Data shown in Figure 6C indicates that ∼17% of the glutamate-dependent increase in respiration could be ascribed to the proton current driving net Ca2+ accumulation into the matrix, with the remainder driving enhanced ATP synthesis.
Figure 6.

The ATP-independent component of NMDA receptor-dependent respiratory stimulation. A, Glutamate addition simultaneous with 0.2 μg/ml oligomycin (G+O); the numbers correspond to the images shown in B. B, Simultaneous imaging of TMRM+ and Fluo-5F fluorescence at the time points shown in A. Note the extensive deregulation of the cells after glutamate in the presence of oligomycin (i.e., high [Ca2+]c, low membrane potentials). C, Oligomycin addition (O) before glutamate. Note the respiratory inhibition with oligomycin showing the residual proton leak and the small stimulation with subsequent glutamate.
Discussion
One of the difficulties in modeling neurodegenerative disease is reconciling acute in vitro models with the slowly developing progression of the actual pathology. In the present context, an oxidative stress model would postulate that oxidative damage in vivo slowly accumulates over the years until a threshold was passed at which pathology becomes overt. ATP deficiency, in contrast, causes immediate bioenergetic collapse of the neuron as plasma membrane ion pumps fail. Because the ATP demand depends on the activation state of a neuron, it is possible to explain the pathological time course by assuming that partial complex I restriction gives no phenotype until the neuron is exposed to a maximal ATP demand, a “hundred-year flood”. Neuronal ATP demand in situ is variable, and the present acute in vitro model approximates the maximal bioenergetic demand that could reasonably be imposed on an aerobic neuron. Such maximal stresses may be very rare, accounting for the slow progression of the human disease. Prolonged NMDA receptor activation of individual neurons might rarely be experienced, perhaps as a result of local epileptiform firing, and would result in cell death if ATP generating capacity were compromised.
An early concept of the relationship between oxidative stress and glutamate excitotoxicity originated with the studies of Bockaert and colleagues, who used electron paramagnetic resonance to show that glutamate exposure of CGNs resulted in an increased production of free radicals (Lafon-Cazal et al., 1993). However, the relationship to DCD could not be determined by this technique. Oxidative stress has been subsequently implicated in the number of studies of acute glutamate excitotoxicity. However, when DCD and O2·− levels were determined in real time in individual neurons, it became apparent that the large increase in reactive oxygen species was a consequence rather than a cause of the calcium deregulation (Vesce et al., 2004). Thus, as in the present study, MnTE-2-PyP was an effective O2·− trap but was unable to prevent DCD.
An “energy-linked excitotoxicity” hypothesis is not novel and was proposed some years ago (Henneberry 1989; Henneberry et al., 1989), focusing on the likelihood that bioenergetic insufficiency would potentiate plasma membrane depolarization and hence exacerbate the activation of NMDA receptors by removing the voltage-dependent magnesium block. Glutamate excitotoxicity has been implicated in the pathology of PD (Blandini and Greenamyre, 1998; Greenamyre, 2001), and it is therefore relevant to investigate the synergy between partial complex I inhibition and maximal ATP demand during chronic NMDA receptor activation. Thus, a failure to detect a decrease in ATP in striatal slices after 30 min exposure to 50 nm rotenone (Bao et al., 2005) may not be relevant because the cells are not called on to maximize their ATP generation.
Under the conditions used in this study, <50% of the mitochondrial respiratory capacity is required for the basal ATP demand of the cells and to service the endogenous mitochondrial proton leak. However, maximal activation of NMDA receptors imposes a high energetic demand on the in situ mitochondria that can be quantified by the respirometer. Indeed, it is remarkable that the maximal NMDA receptor activation can use the full respiratory capacity of the mitochondria (Fig. 3). Although both Ca2+ and Na+ enter through the receptor and although most isoforms of the receptor show selectivity for calcium over sodium, the hundredfold excess of the latter cation over Ca2+ means that Na+ entry dominates, leading to activation of the Na+/K+-ATPase. As shown previously with isolated nerve terminals (Scott and Nicholls, 1980), such Na+ pump activation can use virtually all of the spare respiratory capacity of the in situ mitochondria. It should be noted that the present preparations of CGNs appear to possess a considerably lower glycolytic capacity than those investigated in previous studies by the group (Budd and Nicholls, 1996; Ward et al., 2000), in which glycolysis was sufficient to maintain function in the presence of oligomycin and glutamate. We have been unable to determine the reason for this difference.
A previous study from this laboratory (Jekabsons and Nicholls, 2004) showed that NMDA receptor activation did not use the full respiratory capacity of the CGN mitochondria when the cells were incubated in 25 mm KCl, i.e., under conditions of partial plasma membrane depolarization. Independent studies have shown that excitotoxicity is greatly reduced under these circumstances (Kiedrowski, 1999). Because the influx of Na+ and Ca2+ would be decreased as a result of the smaller electrochemical gradient of the ions across the plasma membrane, less ion pump activation, and hence ATP synthesis, will be required under these circumstances.
A limitation with the present study is that, although the respirometer reports the population respiration of the neurons, it cannot distinguish variations in individual cells, to allow correlation with stochastic cell deregulation, or between different intraneuronal locations. Thus, Davey et al. (1997) observed that complex I in presynaptic mitochondria from hippocampus and cortex possessed a lower spare respiratory capacity, deduced from a rotenone titration compared with nonsynaptic mitochondria from the same regions. Thus, bioenergetic failure may initially be restricted to synaptic regions, interestingly correlating with the observation that neurodegeneration in the rat chronic rotenone model may be initiated at striatal dopaminergic nerve terminals (Betarbet et al., 2006).
The concept of “spare respiratory capacity” was reviewed by Fern (2003) who suggested that the relative sensitivity of different neurons to mitochondrial inhibitors correlated with the ratio of the glucose utilization rate to the expression level of respiratory chain complexes. In the present context, we actually found that the spare respiratory capacity of a population of cultured neonatal striatal neurons is actually considerably higher than for CGNs (Oliviera et al., 2007). This does not invalidate the ATP limitation hypothesis for the rotenone model, because it is the nigral dopamineric terminals projecting to the striatum that are selectively vulnerable (Betarbet et al., 2006).
There is incontrovertible evidence that oxidative stress is associated with rotenone-induced complex I restriction in a number of in vivo and in vitro models of PD as well as LHON. Thus, an increase in mitochondria-derived free radicals (Li et al., 2003; Bao et al., 2005; Beretta et al., 2006), lipid peroxidation (Beretta et al., 2006), and oxidative modification of the Parkinson's-associated protein DJ-1 (Betarbet et al., 2006) have been reported, whereas certain antioxidants can reduce the oxidative damage in vitro (Isaev et al., 2004; Moon et al., 2005; Betarbet et al., 2006), although others are ineffective (Moon et al., 2005). Postmortem analysis of mitochondria from individuals with sporadic PD has revealed oxidative damage to complex I (Keeney et al., 2006) and viral delivery of SOD2 to the visual system of a mouse LHON model reduced optic nerve fiber degeneration (Qi et al., 2004). However, the bioenergetic behavior of in situ mitochondria is the resultant of a complex interacting network of functions, and a debate on oxidative stress versus ATP deficiency may be merely semantic. Thus, studies with CGNs have shown that the bioenergetic failure associated with DCD causes a dramatic increase in O2·− levels in the deregulated cells (Vesce et al., 2004). Additionally, oxidative stress induced by acute glutathione depletion restricts CGN ATP generation by inhibiting mitochondrial ATP export to the cytoplasm (Vesce et al., 2005).
As confirmed in Figure 1, oxidative stress, or at least an increase in matrix O2·− levels, occurs in the presence of even low nanomolar concentrations of rotenone. The same range of concentrations results in a dramatic increase in glutamate excitotoxicity under the present experimental conditions (Fig. 2). However, this does not prove a causal relationship, and the inability of the superoxide dismutase mimetic MnTE-2-PyP (Patel and Day, 1999) to protect against DCD clearly demonstrates that the increased O2·− is not causative in this acute excitotoxic model.
In conclusion, the present model shows that low nanomolar concentrations of rotenone enhance mitochondrial O2·− levels, reduce respiratory capacity, and potentiate delayed Ca2+ deregulation in response to chronic NMDA receptor activation. We suggest that bioenergetic insufficiency should be considered together with oxidative stress in animal models of PD.
Footnotes
This work was supported by the National Institute on Aging Grant R01 AG2 1440. The expert technical assistance of Adrienne Wang and Richard Oh is gratefully acknowledged.
References
- Bao L, Avshalumov MV, Rice ME. Partial mitochondrial inhibition causes striatal dopamine release suppression and medium spiny neuron depolarization via H2O2 elevation, not ATP depletion. J Neurosci. 2005;25:10029–10040. doi: 10.1523/JNEUROSCI.2652-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beretta S, Wood JP, Derham B, Sala G, Tremolizzo L, Ferrarese C, Osborne NN. Partial mitochondrial complex I inhibition induces oxidative damage and perturbs glutamate transport in primary retinal cultures. Relevance to Leber hereditary optic neuropathy (LHON) Neurobiol Dis. 2006;24:308–317. doi: 10.1016/j.nbd.2006.07.016. [DOI] [PubMed] [Google Scholar]
- Betarbet R, Sherer TB, MacKenzie G, Garcia-Osuna M, Panov AV, Greenamyre JT. Chronic systemic pesticide exposure reproduces features of Parkinson's disease. Nat Neurosci. 2000;3:1301–1306. doi: 10.1038/81834. [DOI] [PubMed] [Google Scholar]
- Betarbet R, Canet-Aviles RM, Sherer TB, Mastroberardino PG, McLendon C, Kim JH, Lund S, Na HM, Taylor G, Bence NF, Kopito R, Seo BB, Yagi T, Yagi A, Klinefelter G, Cookson MR, Greenamyre JT. Intersecting pathways to neurodegeneration in Parkinson's disease: effects of the pesticide rotenone on DJ-1, alpha-synuclein, and the ubiquitin-proteasome system. Neurobiol Dis. 2006;22:404–420. doi: 10.1016/j.nbd.2005.12.003. [DOI] [PubMed] [Google Scholar]
- Blandini F, Greenamyre JT. Prospects of glutamate antagonists in the therapy of Parkinson's disease. Fundam Clin Pharmacol. 1998;12:4–12. doi: 10.1111/j.1472-8206.1998.tb00918.x. [DOI] [PubMed] [Google Scholar]
- Blandini F, Porter RHP, Greenamyre JT. Glutamate and Parkinson's disease. Mol Neurobiol. 1996;12:73–94. doi: 10.1007/BF02740748. [DOI] [PubMed] [Google Scholar]
- Brocard JB, Tassetto M, Reynolds IJ. Quantitative evaluation of mitochondrial calcium content in rat cortical neurones following a glutamate stimulus. J Physiol (Lond) 2001;531:793–805. doi: 10.1111/j.1469-7793.2001.0793h.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budd SL, Nicholls DG. Mitochondrial calcium regulation and acute glutamate excitotoxicity in cultured cerebellar granule cells. J Neurochem. 1996;67:2282–2291. doi: 10.1046/j.1471-4159.1996.67062282.x. [DOI] [PubMed] [Google Scholar]
- Courtney MJ, Lambert JJ, Nicholls DG. The interactions between plasma membrane depolarization and glutamate receptor activation in the regulation of cytoplasmic free calcium in cultured cerebellar granule cells. J Neurosci. 1990;10:3873–3879. doi: 10.1523/JNEUROSCI.10-12-03873.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davey GP, Canevari L, Clark JB. Threshold effects in synaptosomal and nonsynaptic mitochondria from hippocampal CA1 and paramedian neocortex brain regions. J Neurochem. 1997;69:2564–2570. doi: 10.1046/j.1471-4159.1997.69062564.x. [DOI] [PubMed] [Google Scholar]
- Fern R. Variations in spare electron transport chain capacity: the answer to an old riddle? J Neurosci Res. 2003;71:759–762. doi: 10.1002/jnr.10553. [DOI] [PubMed] [Google Scholar]
- Greenamyre JT. Glutamatergic influences on the basal ganglia. Clin Neuropharmacol. 2001;24:65–70. doi: 10.1097/00002826-200103000-00001. [DOI] [PubMed] [Google Scholar]
- Henneberry RC. The role of neuronal energy in the neurotoxicity of excitatory amino acids. Neurobiol Aging. 1989;10:611–613. doi: 10.1016/0197-4580(89)90149-8. [DOI] [PubMed] [Google Scholar]
- Henneberry RC, Novelli A, Cox JA, Lysko PG. Neurotoxicity at the N-methyl-d-aspartate receptor in energy- compromised neurons. An hypothesis for cell death in aging and disease. Ann NY Acad Sci. 1989;568:225–233. doi: 10.1111/j.1749-6632.1989.tb12512.x. [DOI] [PubMed] [Google Scholar]
- Isaev NK, Stelmashook EV, Ruscher K, Andreeva NA, Zorov DB. Menadione reduces rotenone-induced cell death in cerebellar granule neurons. NeuroReport. 2004;15:2227–2231. doi: 10.1097/00001756-200410050-00017. [DOI] [PubMed] [Google Scholar]
- Jekabsons MB, Nicholls DG. In situ respiration and bioenergetic status of mitochondria in primary cerebellar granule neuronal cultures exposed continuously to glutamate. J Biol Chem. 2004;279:32989–33000. doi: 10.1074/jbc.M401540200. [DOI] [PubMed] [Google Scholar]
- Johnson-Cadwell LI, Jekabsons MB, Wang A, Polster BM, Nicholls DG. “Mild uncoupling” does not decrease mitochondrial superoxide levels in cultured cerebellar granule neurons but decreases spare respiratory capacity and increases toxicity to glutamate and oxidative stress. J Neurochem. 2007;101:1619–1631. doi: 10.1111/j.1471-4159.2007.04516.x. [DOI] [PubMed] [Google Scholar]
- Kann O, Kovacs R. Mitochondria and neuronal activity. Am J Physiol Cell Physiol. 2006;293:C641–D6577. doi: 10.1152/ajpcell.00222.2006. [DOI] [PubMed] [Google Scholar]
- Keeney PM, Xie J, Capaldi RA, Bennett JP., Jr Parkinson's disease brain mitochondrial complex I has oxidatively damaged subunits and is functionally impaired and misassembled. J Neurosci. 2006;26:5256–5264. doi: 10.1523/JNEUROSCI.0984-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiedrowski L. Elevated extracellular K+ concentrations inhibit N-methyl-d-aspartate-induced Ca2+ influx and excitotoxicity. Mol Pharmacol. 1999;56:737–743. [PubMed] [Google Scholar]
- Krieger C, Duchen MR. Mitochondria, Ca2+ and neurodegenerative disease. Eur J Pharmacol. 2002;447:177–188. doi: 10.1016/s0014-2999(02)01842-3. [DOI] [PubMed] [Google Scholar]
- Kudin AP, Bimpong-Buta NYB, Vielhaber S, Elger CE, Kunz WS. Characterization of superoxide-producing sites in isolated brain mitochondria. J Biol Chem. 2004;279:4127–4135. doi: 10.1074/jbc.M310341200. [DOI] [PubMed] [Google Scholar]
- Lafon-Cazal M, Pietri S, Culcasi M, Bockaert J. NMDA-dependent superoxide production and neurotoxicity. Nature. 1993;364:535–537. doi: 10.1038/364535a0. [DOI] [PubMed] [Google Scholar]
- Lenaz G, Baracca A, Carelli V, D'Aurelio M, Sgarbi G, Solaini G. Bioenergetics of mitochondrial diseases associated with mtDNA mutations. Biochim Biophys Acta Bio-Energetics. 2004;1658:89–94. doi: 10.1016/j.bbabio.2004.03.013. [DOI] [PubMed] [Google Scholar]
- Li NY, Ragheb K, Lawler G, Sturgist J, Rajwa B, Melendez JA, Robinson JP. Mitochondrial complex I inhibitor rotenone induces apoptosis through enhancing mitochondrial reactive oxygen species production. J Biol Chem. 2003;278:8516–8525. doi: 10.1074/jbc.M210432200. [DOI] [PubMed] [Google Scholar]
- Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443:787–795. doi: 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- Moon Y, Lee KH, Park JH, Geum D, Kim K. Mitochondrial membrane depolarization and the selective death of dopaminergic neurons by rotenone: protective effect of coenzyme Q10. J Neurochem. 2005;93:1199–1208. doi: 10.1111/j.1471-4159.2005.03112.x. [DOI] [PubMed] [Google Scholar]
- Muqit MM, Gandhi S, Wood NW. Mitochondria in Parkinson disease: back in fashion with a little help from genetics. Arch Neurol. 2006;63:649–654. doi: 10.1001/archneur.63.5.649. [DOI] [PubMed] [Google Scholar]
- Nicholls DG, Budd SL. Mitochondria and neuronal survival. Physiol Rev. 2000;80:315–360. doi: 10.1152/physrev.2000.80.1.315. [DOI] [PubMed] [Google Scholar]
- Oliviera J, Ellerby LM, Rego AC, Nicholls DG. Mitochondrial dysfunction in Huntington's disease: the bioenergetics of isolated and in-situ mitochondria from transgenic mice. J Neurochem. 2007;101:241–249. doi: 10.1111/j.1471-4159.2006.04361.x. [DOI] [PubMed] [Google Scholar]
- Patel M, Day BJ. Metalloporphyrin class of therapeutic catalytic antioxidants. Trends Pharmacol Sci. 1999;20:359–364. doi: 10.1016/s0165-6147(99)01336-x. [DOI] [PubMed] [Google Scholar]
- Qi X, Lewin AS, Sun L, Hauswirth WW, Guy J. SOD2 gene transfer protects against optic neuropathy induced by deficiency of complex I. Ann Neurol. 2004;56:182–191. doi: 10.1002/ana.20175. [DOI] [PubMed] [Google Scholar]
- Schapira AH. Mitochondrial disease. Lancet. 2006;368:70–82. doi: 10.1016/S0140-6736(06)68970-8. [DOI] [PubMed] [Google Scholar]
- Scott ID, Nicholls DG. Energy transduction in intact synaptosomes: influence of plasma- membrane depolarization on the respiration and membrane potential of internal mitochondria determined in situ. Biochem J. 1980;186:21–33. doi: 10.1042/bj1860021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tretter L, Sipos I, Adam-Vizi V. Initiation of neuronal damage by complex I deficiency and oxidative stress in Parkinson's disease. Neurochem Res. 2004;29:569–577. doi: 10.1023/b:nere.0000014827.94562.4b. [DOI] [PubMed] [Google Scholar]
- Tymianski M, Charlton MP, Carlen PL, Tator CH. Secondary Ca2+ overload indicates early neuronal injury which precedes staining with viability indicators. Brain Res. 1993;607:319–323. doi: 10.1016/0006-8993(93)91523-u. [DOI] [PubMed] [Google Scholar]
- Vesce S, Kirk L, Nicholls DG. Relationships between superoxide levels and delayed calcium deregulation in cultured cerebellar granule cells exposed continuously to glutamate. J Neurochem. 2004;90:683–693. doi: 10.1111/j.1471-4159.2004.02516.x. [DOI] [PubMed] [Google Scholar]
- Vesce S, Jekabsons MB, Johnson-Cadwell LI, Nicholls DG. Acute glutathione depletion restricts mitochondrial ATP export in cerebellar granule neurons. J Biol Chem. 2005;280:38720–38728. doi: 10.1074/jbc.M506575200. [DOI] [PubMed] [Google Scholar]
- Ward MW, Rego AC, Frenguelli BG, Nicholls DG. Mitochondrial membrane potential and glutamate excitotoxicity in cultured cerebellar granule cells. J Neurosci. 2000;20:7208–7219. doi: 10.1523/JNEUROSCI.20-19-07208.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]