

Abstract
Huntingtin is an antiapoptotic protein that becomes toxic when its polyglutamine stretch is expanded, resulting in Huntington's disease (HD). Protein context and posttranslational modifications regulate huntingtin toxicity. Identifying signaling pathways that act on huntingtin is, therefore, key to understanding huntingtin function in normal and pathological conditions. We show here that huntingtin is phosphorylated by the cyclin-dependent kinase 5 (Cdk5) at serines 1181 and 1201. Phosphorylation can be induced by DNA damage in vitro and in vivo. The state of huntingtin phosphorylation is a crucial regulator of neuronal cell death. Absence of phosphorylation of huntingtin at serines 1181 and 1201 confers toxic properties to wild-type huntingtin in a p53-dependent manner in striatal neurons and accelerates neuronal death induced by DNA damage. In contrast, phosphorylation at serines 1181 and 1201 protects against polyQ-induced toxicity. Finally, we show in late stages of HD a sustained DNA damage that is associated with a decrease in Cdk5/p35 levels. We propose that wild-type huntingtin is a component of the DNA damage response signal in neurons and that the Cdk5/DNA damage pathway is dysregulated in HD.
Keywords: Huntington's disease, polyglutamine, phosphorylation, neuronal survival, neurodegeneration, p53
Introduction
Huntington's disease (HD) is a neurodegenerative disorder characterized by involuntary movements, personality changes, and dementia (Young, 2003). The mutation that causes HD is an abnormal polyglutamine (polyQ) expansion in the protein huntingtin (htt), leading to the selective dysfunction and death of cortical and striatal neurons (MacDonald et al., 2003). The mechanisms by which mutant htt leads to disease involve various pathways (Ross, 2004; Cattaneo et al., 2005; Borrell-Pages et al., 2006; Li and Li, 2006). Late events include the cleavage and translocation of htt into the nucleus and subsequent transcriptional dysregulation (Saudou et al., 1998; Sugars and Rubinsztein, 2003; Graham et al., 2006). Initial events, such as alteration of axonal transport, take place in the cytoplasm (Gunawardena et al., 2003; Gauthier et al., 2004; Trushina et al., 2004). Also, early dysfunctions including alterations in mitochondrial homeostasis lead to the impairment of mitochondrial enzymes, disrupted Ca2+ signaling, and oxidative damage (Sawa, 2001; Panov et al., 2002; Bezprozvanny and Hayden, 2004; Browne and Beal, 2004; Milakovic and Johnson, 2005; Benchoua et al., 2006). Interestingly, the tumor suppressor gene p53 could play a nodal role in disease by linking nuclear and mitochondrial dysfunction and mediating neuronal apoptosis (Steffan et al., 2000; Bae et al., 2005).
Although genetic studies have established that polyQ–htt kills neurons through the gain of a new toxic function, recent evidence supports that the loss of function of wild-type htt contributes to pathogenesis (Cattaneo et al., 2005; Borrell-Pages et al., 2006). Indeed, htt possesses antiapoptotic properties. Mice lacking htt show an increased programmed cell death during development (Zeitlin et al., 1995). Wild-type htt is protective against various stresses in neuronal cultures and promotes neuronal survival in mouse in normal and pathological situations (Dragatsis et al., 2000; Rigamonti et al., 2000; Leavitt et al., 2001). Furthermore, htt enhances transcription and axonal transport of the prosurvival brain derived neurotrophic factor (BDNF). In disease, polyQ expansion in htt leads to a reduction of BDNF transcription and transport, thereby reducing neurotrophic support (Zuccato et al., 2001; Gauthier et al., 2004). Arguments for a key role of the protein context in pathogenesis also come from the observations that cleavage and posttranslational modifications regulate the toxicity of polyQ–htt (Humbert et al., 2002; Steffan et al., 2004; Luo et al., 2005; Graham et al., 2006; Pardo et al., 2006; Schilling et al., 2006). For example, phosphorylation at serine 421 by the prosurvival kinase Akt blocks polyQ–htt-induced toxicity in vitro and in vivo (Humbert et al., 2002; Warby et al., 2005; Pardo et al., 2006).
Another pathway that could be critical in HD involves the cyclin-dependent kinase 5 (Cdk5). Cdk5 is a serine–threonine kinase, the activity of which being mostly restricted to postmitotic and mature neurons, reflecting the localized expression of its required regulatory subunits p35 and p39 (Dhavan and Tsai, 2001). Cdk5 participates to the proper maturation and maintenance of the CNS. In contrast, Cdk5 is also associated with cell death under pathological conditions (Cruz and Tsai, 2004). Interestingly, htt is phosphorylated at serine 434 by Cdk5, which reduces mutant htt cleavage, aggregation, and cell death (Luo et al., 2005). However, the mechanisms leading to Cdk5 regulation in normal and HD pathological situation is unknown. Also, whether Cdk5 phosphorylates htt at other residues and how these phosphorylations impact polyQ–htt-induced toxicity and the function of htt in non-HD conditions remain to be established.
Materials and Methods
Kinase assays.
Kinase assays were performed as described previously (Ko et al., 2001). Neurons were treated at 3 d in vitro with camptothecin (CPT; 10 μm; Sigma, St. Louis, MO) and roscovitine (10 μm, 4 h; Calbiochem, Darmstadt, Germany). Cells were lysed in NP-40 lysis buffer (20 mm Tris-HCl, pH 7.5, 150 mm NaCl, 2 mm EGTA, 1% Nonidet P-40, 10 mm β-glycerophosphate, 5 mm NaF, 1 mm NaPPi, 2 mm DTT, 1 mm sodium vanadate, and 100 μm PMSF), and 150 μg of total protein were used to immunoprecipitate Cdk5 with C8 antibody (1.5 μg) and precipitated with protein A-Sepharose beads (Sigma). Immunoprecipitated complexes were incubated with 1 μg of histone H1 and 2.5 μCi of [32P]ATP in 40 μl of kinase buffer (50 mm HEPES, pH 7.0, 10 mm MgCl2, 5 mm MnCl2, 1 mm DTT, and ATP 100 μm) for 30 min at room temperature.
Glutathione S-transferase (GST)–htt fragments were expressed 1 h at 37°C in 10 ml of BL21 culture after 1 mm isopropyl-β-d-thiogalactopyranoside induction. Peptides were purified on 200 μl of glutathione-agarose beads and stored at −20°C. GST-fused proteins (10 μl) and 5 ng of recombinant Cdk5/p35 or 2.5 ng Cdk5/p25 (Cell Signaling Technology, Boston, MA) were incubated as above.
Cell culture, transfection, and cell lysate.
Primary culture of striatal neurons were prepared from embryonic day 17 Sprague Dawley rats and cultured and transfected at 3 d in vitro by a modified calcium phosphate technique (Saudou et al., 1998). When indicated, neurons were electroporated the day of plating with the rat neuron Nucleofector kit (Amaxa Biosystems, Cologne, Germany). Mouse neuronal cells derived from wild-type htt mouse (neuronal cells, +/+) and from Hdh109Q/109Q knock-in mouse (109Q/109Q) were cultured as described previously (Trettel et al., 2000). Cells were transiently transfected by lipofectamine (Invitrogen, Carlsbad, CA). Neuroblastoma cell line (SH-SY5Y) were grown in RPMI (Roswell Park Memorial Institute) medium containing 10% bovine calf serum, 6 mm l-glutamine, 100 U/ml penicillin G, and 100 μg/ml streptomycin sulfate. Cells were electroporated with cell line Nucleofector kit V (Amaxa Biosystems). HEK 293T cells were grown in DMEM containing 10% bovine calf serum, 6 mm l-glutamine, 100 U/ml penicillin G, and 100 μg/ml streptomycin sulfate and transiently transfected by the calcium phosphate precipitation method (16 μg/10 cm plate DNA). Two days after transfection or treatment, cells were lysed using NP-40 lysis buffer. Western blot analysis was performed on 50 μg of extracts of cells. NIH Image J software was used for quantifications of phospho-huntingtin levels (represented as a percentage of phosphorylation standardized to total htt levels).
Antibodies.
To generate htt-specific phospho-antibodies S1181 and S1201, two phosphopeptides of the sequences TNPPSLpSPIRRKC (S1181; Eurogentec, Seraing, Belgium) and CKEPGEQASVPLpSPKK (S1201; NeoMPS, Strasbourg, France) were synthesized, coupled to KLH, and used to immunize rabbits. Two polyclonal antibodies to the KLH-coupled peptides, anti-P-htt–S1181 and anti-P-htt–S1201, were obtained and purified as described previously (Humbert et al., 2002).
Measurement of neuronal death.
For death assays, striatal neurons were transfected at 3 d in vitro by a modified calcium phosphate technique (Saudou et al., 1998) with the plasmids of interest and the cytomegalovirus–β-galactosidase plasmid (10:1 ratio). Forskolin (10 μm; Sigma) and IBMX (100 μm; Sigma) were added to the cultures 1 h after transfection. To lower the expression levels of htt (see Fig. 6E), neurons were electroporated the day of plating with the rat neuron Nucleofector kit (Amaxa Biosystems). One day after electroporation or transfection, cells were fixed with 4% paraformaldehyde (PFA) for 20 min and immunostained with anti-htt 4C8 and anti-β-galactosidase antibodies. When stated, striatal neurons were treated with 100 nm pifithrin-α (12–16 h; Sigma), or else neurons were electroporated the day of plating with Si-p53 and, 2 d after electroporation, transfected with the different htt constructs. Cells were fixed and immunostained 1 d after transfection as above. 4C8-positive neurons of similar intensities were scored under fluorescence microscopy in a blinded manner. Neuronal degeneration was assessed by neurite loss and nuclear shrinkage (see Fig. 3D). Death induced by htt was deduced by scoring β-galactosidase-positive neurons in control condition (empty vector). For Figure 6, C and F, neurons were treated as stated, at 1 and 3 d in vitro, respectively, fixed, and immunostained, and neuronal degeneration was assessed by nuclear shrinkage. For all experiments, each graph represents three independent experiments performed in duplicates. Data are expressed as the percentage of neuronal cell death. Each bar in a given graph corresponds to the scoring of ∼1200 neurons.
Figure 6.

Phosphorylation of huntingtin occurs early before neuronal death. A, Striatal neurons treated with CPT (10 μm) for varying lengths of time are fixed in methanol and immunostained for P-htt–S1201. Quantification of the nuclear P-htt–S1201 dots intensity reveals an increased phosphorylation at S1201 of htt at 3 h of CPT treatment. At 8 h, S1201 phosphorylation returns to basal level. B, Phosphorylation of htt at S1181 follows the same kinetic during CPT treatment than that of S1201. Primary culture of striatal neurons were treated with DMSO (0.1%, control), roscovitine (10 μm, 4 h), and CPT (10 μm, 3 and 8 h). Protein extracts of neuronal cultures were analyzed by immunoblotting with anti-P-S1181 and anti-htt antibodies. C, Neuronal cell death is induced at 8 h of CPT treatment. Neurons are treated as in A and fixed and scored for neuronal cell death. D, Neurons are treated with CPT (10 μm) for 3 h and washed with culture media for 5 h when indicated (wash), fixed in methanol, and immunostained for P-htt–S1201 or scored for neuronal death. The increased phosphorylation at S1201 of htt at 3 h of CPT treatment is reversible by removal of CPT. As a control, cell death remains constant. E, Unphosphorylated htt accelerates DNA damage-induced cell death. Neurons electroporated with low amount of 1301–17Q, 1301–17Q–AA, and 1301–17Q–DD and treated with DMSO (0.1%, control) and CPT (10 μm, 3 h) were scored for neuronal cell death. F, Reducing Cdk5 activity increases CPT-induced neuronal death. Striatal neurons were treated with DMSO (0.1%, control), CPT (10 μm, 3 h), and roscovitine (10 μm, 4 h) and analyzed as in A. *p < 0.05; **p < 0.01; ***p < 0.001. NS, Not significant.
Figure 3.
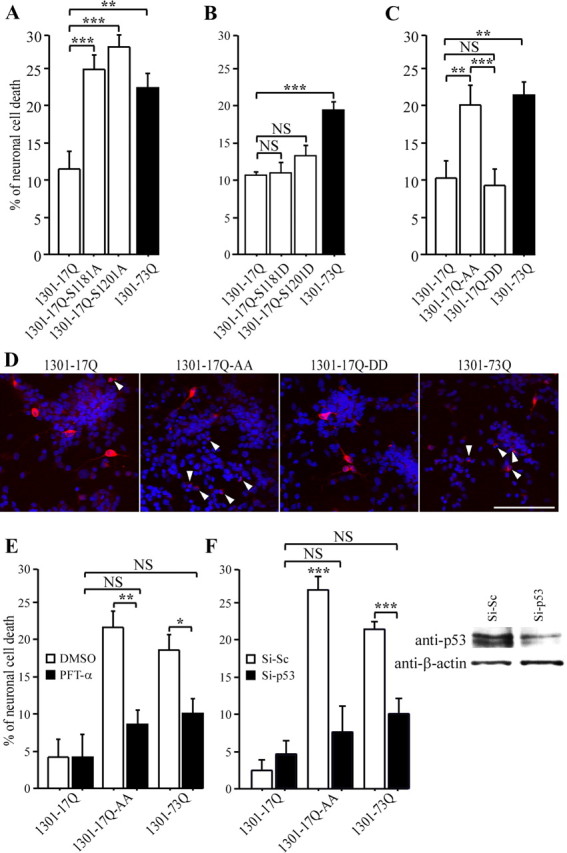
Absence of phosphorylation at serines 1181 and 1201 turns huntingtin into a toxic protein through a p53-dependent mechanism. A–F, Primary cultures of striatal neurons are transfected by a modified phosphate calcium technique with constructs encoding the first 1301 amino acids of human htt containing 17 (wild-type, 1301–17Q) or 73 (1301–73Q) glutamines with intact serines or serines to alanines or aspartic acids mutations. Neurons are scored for neuronal cell death. D, Striatal neurons are immunostained using anti-htt 4C8 antibody (red). Nuclei are stained with Hoechst. White arrows indicate degenerating neurons. Scale bar, 100 μm. E, F, p53 mediates polyQ-induced toxicity and the toxicity of wild-type htt induced by the absence of phosphorylations at serines 1181 and 1201. The activity of p53 is inhibited using pifithrin-α (PTF-α) (E) or RNA interference (F). Although a control siRNA (Si-Sc) had no effect, the p53-directed siRNA (Si-p53) lowers p53 levels as revealed by immunoblotting against p53 and β-actin (loading control). *p < 0.05; **p < 0.01; ***p < 0.001. NS, Not significant.
DNA damage and huntingtin phosphorylation.
To determine the level of DNA damage, cells were fixed in methanol for 5 min at −20°C and immunostained with anti-γ-H2A histone family, member X (H2AX). Pictures of immunostained nuclei (Hoechst 33258; 0.25 μg/ml) were collected using a three-dimensional (3D) microscope and analyzed after subsequent deconvolution as described previously (Gauthier et al., 2004). Analyses of nuclear γ-H2AX intensity were generated using a special developed 3D object counter plugin in NIH Image J software (F. P. Cordelières, Institut Curie, Orsay, France; http://rsb.info.nih.gov/ij/plugins/track/objects.html). Twenty pictures corresponding to 20 cells were taken by condition. The mean of total nuclear γ-H2AX intensity was made per cell. Each graph represents three independent experiments performed. Data are expressed as the nuclear γ-H2AX intensity. Each bar in a given graph corresponds to the scoring of ∼60 cells.
Primary culture of striatal neurons were treated at 3 d in vitro with 50 μm H2O2 for 15 min (Laboratoires Gilbert, Hérouville Saint-Clair, France), 500 μm dopamine for 3 h (Sigma), or 10 μm CPT for 3 h (Sigma). The levels of P-htt–S1181 and γ-H2AX were determined by Western blot, and quantifications were done by NIH ImageJ software (http://rsb.info.nih.gov/ij/), calculated in percentage of phosphorylation standardized to total htt levels.
P-htt–S1201 levels were studied by immunocytochemistry. At indicated times, cells were fixed in methanol for 5 min at − 20°C and immunostained with anti-P-htt–S1201 and anti-γ-H2AX antibodies. Pictures of immunostained nuclei (Hoechst 33258; 0.25 μg/ml) were collected using a 3D microscope and analyzed after subsequent deconvolution. Analyses of nuclear P-htt–S1201 intensity were generated as for γ-H2AX intensity (see above).
Irradiation of mice.
Ten-week-old CD1 mice were irradiated with 5 Gy from a 137 Cs source with a dose rate of ∼1.01 Gy/min (Nowak et al., 2006). Eight hours after irradiation, mice were perfused with 4% PFA, and brains were collected and postfixed in 4% PFA for at least 16 h. OCT (Tissue-Tek)-embedded tissue sections (10 μm) were unmasked in 0.1 m citrate solution, pH 6, and permeabilized with 0.5% Triton X-100. Sections were blocked in PBS, 0.1% Triton X-100, and 10% goat serum (1 h at room temperature) and immunostained with anti-P-htt–S1201 and anti-γ-H2AX antibodies. Pictures were collected using a inverted microscope. For immunoblotting analysis, 8 h after irradiation, mice were killed, and striata were collected and lysed using NP-40 lysis buffer. Western blot analysis was performed on 50 μg of extracts.
Brain tissues.
Tissues were obtained from the Harvard Brain Tissue Resource Center (HBTRC; Belmont, MA): three controls (samples 1–3), one HD grade 4 (HD4, sample 4), and one grade 3 (HD3, sample 5) patients. Samples correspond, respectively, to brain numbers 4741, 4744, 4751, 4680, and 4797 as numbered by HBTRC: 1–3, controls (mean ± SEM; age, 55.3 ± 1.9 years; postmortem delay, 23.8 ± 1.9 h); 4 and 5, HD brains grade 3 and 4 (age, 63.5 ± 18.5 years; postmortem delay, 24.0 ± 2.0 h). Tissues were also obtained from Johns Hopkins Medical Institutions (JHMI; Baltimore, MD): three controls (samples 1–3), two HD grade 3 (samples 4–5), and three grade 4 (samples 6–8) patients. Samples 1–8 correspond, respectively, to brain numbers 0991, 1613, 138, 150, 151, 174, 181 and 184 as numbered by JHMI (age, 58.8 ± 13.4 years; postmortem delay, 10.3 ± 5.9 h). Samples were homogenized in NP-40 lysis buffer and cleared by centrifugation at 6000 × g (15 min; 4°C). Western blot analysis was performed on 50 μg of total extracts.
On-line supplemental material.
Detailed information on antibodies and constructs used and statistical analyses may be found in the supplemental data (available at www.jneurosci.org as supplemental material).
Statview 4.5 software (SAS Institute, Cary, NC) was used for statistical analysis. Data are expressed as mean ± SEM.
Results
Cdk5 phosphorylates huntingtin at serines 1181 and 1201 in vitro
Cdk5 exerts its effects by acting on key substrates (Dhavan and Tsai, 2001). Htt is itself a phosphoprotein with a toxicity when mutated that is regulated by phosphorylation (Humbert et al., 2002; Rangone et al., 2004; Luo et al., 2005; Warby et al., 2005; Pardo et al., 2006; Schilling et al., 2006). Cdk5 phosphorylates htt at S434 and regulates its proteolysis (Luo et al., 2005). We analyzed the htt protein sequence and found four putative Cdk5 consensus phosphorylation sites with high probability scores (http://scansite.mit.edu; S1181, S1201, T1859, and S3126) (Fig. 1A). We initially wondered whether Cdk5 phosphorylates fragments of htt that encompass these sites. Thus, we generated three GST-fused htt peptides, containing serines 1181 and 1201 (GST–htt–1145–1240), threonine 1859 (GST–htt–1792–1899), and serine 3126 (GST–htt–3038–3144) (Fig. 1A). The active Cdk5/p35 and the hyperactive Cdk5/p25 kinase complexes phosphorylated these three peptides of htt (Fig. 1B). We also tested a peptide (GST–htt–384–467) (Fig. 1A) containing serine 434. As described previously, this peptide was phosphorylated by the Cdk5/p25 complex (Luo et al., 2005) (Fig. 1B).
Figure 1.

Cdk5 phosphorylates huntingtin at S1181 and S1201 in vitro. A, The human htt sequence contains four consensus sites for Cdk5 phosphorylation. GST-fused htt and huntingtin 1301 fragments used in this study are shown. B, Several fragments of htt are phosphorylated by Cdk5 in vitro. Kinase assays are performed using recombinant Cdk5/p35 and Cdk5/p25 and GST-fused proteins as substrates. C, The Cdk5 consensus site is defined by S/T-P-X-K/R with phosphorylation on the S/T. It is well conserved in vertebrates for serines 1181 and 1201 of htt sequence. D, Serines 1181 and 1201 are phosphorylated in vitro. GST–htt–1145–1240 fragments with S1181A and S1201A mutations are subjected to phosphorylation by recombinant Cdk5/p35 and Cdk5/p25. YFP, Yellow fluorescent protein.
Serines 1181 and 1201 are highly conserved Cdk5 consensus sites in vertebrates (Fig. 1C), suggesting that they may be important for the function of htt. Also, proteomic analyses have shown that these sites are phosphorylated, further supporting their physiological importance (Schilling et al., 2006). We generated a peptide (GST–htt–1145–1240) containing either intact serines S1181/S1201 or a single or double serine to alanine (that cannot be phosphorylated) mutation to demonstrate that these serines are phosphorylated by Cdk5/p35 or Cdk5/p25 complexes. Phosphorylation by Cdk5 decreased in the singly mutated peptide, whereas phosphorylation by Cdk5 was totally inhibited in the double mutated peptide (Fig. 1D). These results show that S1181 and S1201 of htt are phosphorylated by Cdk5 in vitro.
Cdk5 phosphorylates huntingtin at serines 1181 and 1201 in neurons
To unequivocally identify S1181 and S1201 as Cdk5 phosphorylation sites, we raised two phosphorylation state-specific polyclonal antibodies based on the human htt sequence. Constructs encoding the first 1301 amino acids of htt with 17 glutamines (wild-type, 1301–17Q) were generated with either intact serines or serine to alanine mutations at sites 1181 and/or 1201. In HEK 293T cells transfected with 1301–17Q htt, the antibody against phospho-S1181 (anti-P–htt–1181) recognized wild-type and 1301–17Q–S1201A fragments but not 1301–17Q–S1181A (Fig. 2A). In contrast, the anti-P-htt–S1201 antibody recognized wild-type and 1301–17Q–S1181A fragments but not 1301–17Q–S1201A. These results demonstrate that anti-P-htt–S1181 and anti-P-htt–S1201 specifically recognize htt when phosphorylated at serines 1181 and 1201, respectively, and that these sites are phosphorylated in cells.
Figure 2.

Cdk5 phosphorylates huntingtin at S1181 and S1201 in neurons. A–D, Immunoblotting experiments are performed using the anti-P-htt–S1181, anti-P-htt–S1201, anti-htt (4C8), anti-Cdk5, anti-p35, and anti-β-actin antibodies as indicated. A, To assess the specificity of the phospho-antibodies, 1301–17Q, 1301–17Q–S1181A, and 1301–17Q–S1201A are expressed in HEK 293T cells. B, Cdk5/p35 and Cdk5/p25 are expressed in HEK 293T cells. Quantification reveals a statistically significant increase in phosphorylation at S1181 and S1201 of endogenous htt in the presence of Cdk5/p35 and Cdk5/p25. C, D, Htt phosphorylation at S1181 and S1201 is significantly inhibited by decreasing Cdk5 levels by RNA interference (pSuper–Cdk5) or by roscovitine treatment. SH-SY5Y cells (C) and primary cultures of striatal neurons (D) were transfected with pSuper–Cdk5 or treated with roscovitine (10 μm, 3 h). Quantifications reveal decreased phosphorylations when Cdk5 activity is diminished. *p < 0.05; **p < 0.01; ***p < 0.001.
We next asked whether Cdk5 phosphorylates endogenous htt at serines 1181 and 1201. Therefore, we cotransfected non-neuronal HEK 293T cells that have low Cdk5 activity, with Cdk5 and p35 or p25. Cdk5/p35 and Cdk5/p25 induced a statistically significant increase in the phosphorylations of serines 1181 and 1201 of endogenous htt (Fig. 2B, represented as a percentage of phosphorylation standardized to total htt levels). To further demonstrate that Cdk5 is a bona fide kinase that phosphorylates htt in cells, we then determined the effect of decreasing Cdk5 activity in SH-SY5Y human neuronal cell lines in which Cdk5 activity is high. For this purpose, we used small interfering RNA (siRNA) interference to reduce Cdk5 levels or roscovitine to reduce Cdk5 kinase activity. We observed a statistically significant decrease in S1201 phosphorylation in SH-SY5Y cells transfected with pSuper–Cdk5 or treated with roscovitine (Fig. 2C). Because of to the weak efficacy of the anti-P-htt–S1181 to detect phosphorylated htt at endogenous levels in SHSY-5Y cells, we used primary cultures of rat striatum, in which Cdk5 activity is particularly high. Cdk5 activity was inhibited by roscovitine or by RNA interference (Fig. 2D). We found a statistically significant decrease in S1181 phosphorylation of endogenous htt when Cdk5 activity was inhibited (Fig. 2D). Together, these results show that, although other kinases might be involved as well, serines S1181 and S1201 of endogenous htt are phosphorylated by Cdk5 in cells and neurons.
Phosphorylation of huntingtin at S1181 and S1201 modulates neuronal death through a p53-dependent pathway
To investigate the biological consequence of htt phosphorylation by Cdk5, we tested the effect of mutations at S1181 and S1201 on neuronal survival. We generated constructs encoding the first 1301 amino acids of htt with 17 glutamines (wild-type) with intact serines or with serine to alanine or aspartic acid (mimicking constitutive phosphorylation) mutations at sites 1181 and/or 1201. Primary cultures of striatal neurons were transfected with these constructs, and the percentage of neuronal death was measured by neurite loss and nuclear shrinkage and compared with the toxicity induced by a corresponding polyQ fragment (1301–73Q) (Fig. 3A–D). The different constructs were expressed at similar levels in neurons (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Mutations of each serine to alanine increased the toxicity of wild-type htt to that of polyQ–htt (Fig. 3A). However, serine to aspartic acid mutations had no effect on the wild-type htt (Fig. 3B). We tested whether phosphorylations at S1181/S1201 have synergistic or opposite effects on neuronal death using the same construct (1301–17Q) but with both serines being mutated to either alanine or aspartic acid (Fig. 3C,D). In wild-type htt, double mutations had the same effect as the single mutations. Together, these results indicate that absence of htt phosphorylation at positions S1181 and S1201 makes htt toxic at levels similar to those of htt containing the polyQ expansion. This is to our knowledge the first report of such mutation in wild-type htt.
Having found that absence of phosphorylation at S1181 and S1201 turns htt into a proapoptotic protein, we hypothesized that the pathway activated in response to loss of htt phosphorylation could correspond to apoptotic pathways that are activated in disease, i.e., when htt becomes proapoptotic as a result of the presence of the polyQ-expanded stretch. A good candidate for mediating neuronal death in this situation is the apoptotic protein p53. It was observed previously in cellular and animal models of HD that polyQ-induced toxicity is mediated by p53 (Bae et al., 2005). Also, p53 is a key protein that regulates the apoptotic machinery in cells (Vogelstein et al., 2000). Therefore, we investigated whether the toxic effect on wild-type htt of the absence of phosphorylation at S1181 and S1201 also involves this pathway. We inhibited p53 using either pifithrin-α, a specific p53 inhibitor (Fig. 3E), or RNA interference, which efficiently reduces p53 levels in neurons (Fig. 3F). As expected (Bae et al., 2005), polyQ-induced neuronal cell death was abolished by pifithrin-α (Fig. 3E) or by low p53 levels (Fig. 3F). Likewise, the toxicity induced by the absence of phosphorylation of wild-type htt at S1181 and S1201 (1301–17Q–AA) was completely blocked by inhibiting p53 (Fig. 3E,F). Thus, p53 mediates both polyQ-induced neurotoxicity and the toxicity of wild-type htt induced by the loss of phosphorylation at S1181 and S1201. As recently demonstrated for ataxin-1 (Lam et al., 2006), this supports the notion that the same cellular pathways are used by polyQ-containing proteins in health and disease.
Neuronal DNA damage regulates Cdk5 activity and huntingtin phosphorylation
Having demonstrated that htt phosphorylation at S1181 and S1201 is critical to regulate neuronal death and p53 dependent, we sought to identify the cellular event leading to htt phosphorylation. Among several events, we postulated that DNA damage could be involved. Indeed, the tumor suppressor gene p53 is key to regulate the cell response to cellular stresses and in particular to DNA damage (Vogelstein et al., 2000; Jacobs et al., 2006). Furthermore, Cdk5 may participate in the response to DNA damage in neurons (Lee and Kim, 2007). The dysregulation of Cdk5 occurs with aging and concomitantly with an increase in DNA damage (Lu et al., 2004). The expression and activity of Cdk5 are particularly decreased in the late response to DNA damage (O'Hare et al., 2005). We asked whether phosphorylations of htt that regulate neuronal toxicity could be linked to DNA damage and, thus, set up a neuronal system in which DNA damage is induced. We first analyzed the DNA damage response during oxidative stress in primary culture of striatal neurons because striatal neurons are the most vulnerable in HD (Fig. 4A). For this purpose, we assessed the levels of the well known DNA damage marker γ-H2AX. Histone H2AX is indeed phosphorylated during induction of DNA double-strand breaks. Treatment with H2O2 (50 μm, 15 min) or with dopamine (500 μm, 3 h), which cause oxidative stress, resulted in γ-H2AX foci accumulating in the nuclei of neurons, corresponding to the sites of DNA lesions. We more specifically assessed the effect of DNA damage by using a specific inhibitor of topoisomerase I, CPT (10 μm, 3 h). CPT also led to γ-H2AX foci forming.
Figure 4.
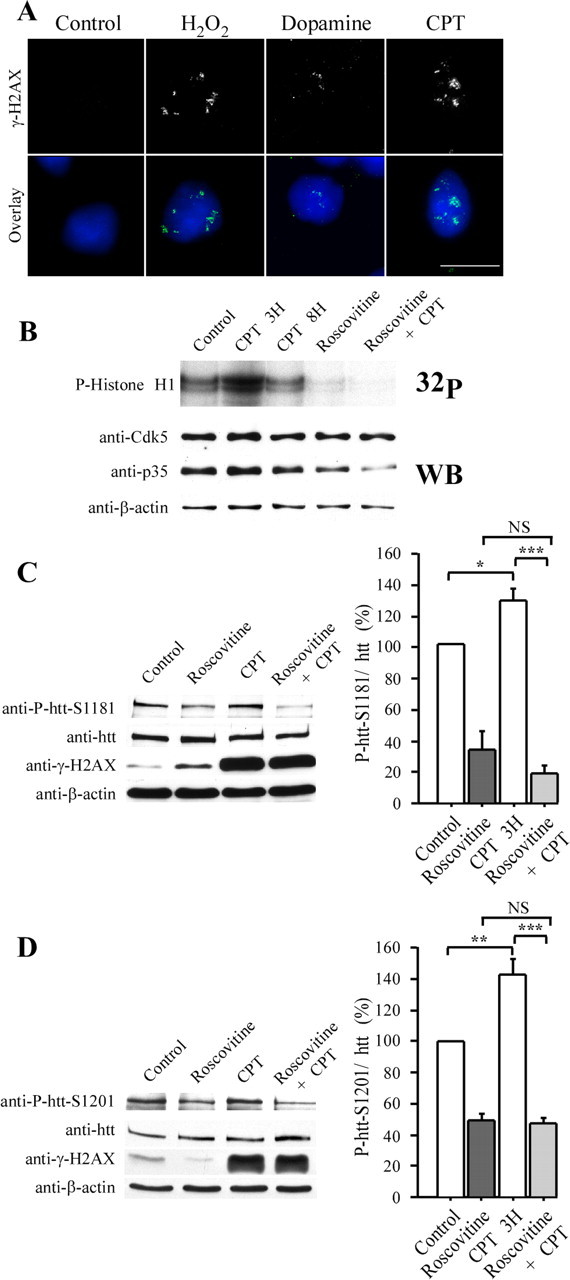
Neuronal DNA damage increases Cdk5 activity and phosphorylations of huntingtin at serines 1181 and 1201. A, Treatment of primary cultures of striatal neurons with DMSO (0.1%, control), 50 μm H2O2, 500 μm dopamine, and 10 μm CPT leads to DNA damage. Cells are immunostained for γ-H2AX. The nuclear staining is shown in blue. Scale bar, 10 μm. B, Cdk5 activity is elevated after 3 h of CPT treatment, although its activity returned to basal level after longer treatment. In vitro kinase assays are performed using immunoprecipitated Cdk5 from primary cultures of striatal neurons treated with DMSO (0.1%, control), 10 μm CPT (3 and 8 h), and, when indicated, 10 μm roscovitine (4 h). Histone H1 is used as a substrate. Analysis of the total extracts by anti-Cdk5, anti-p35, and anti-β-actin immunoblotting reveals that the increase in Cdk5 activity is not linked to an increase in Cdk5 and p35 levels. C, D, Primary cultures of striatal neurons (C) and SH-SY5Y cells (D) are treated with DMSO (0.1%, control), roscovitine (10 μm, 4 h), and 10 μm CPT (3 h). Protein extracts are analyzed by immunoblotting with anti-P-htt–S1181, anti-P-htt–S1201, anti-htt, anti-γ-H2AX, and anti-β-actin antibodies. C, D, Quantifications reveal significantly increased phosphorylations at S1181–S1201 during DNA damage and decreased phosphorylations when Cdk5 activity is diminished by roscovitine treatment. CPT is not able to induce phosphorylation of htt when cells are treated with roscovitine. *p < 0.05; **p < 0.01; ***p < 0.001. WB, Western blot; NS, not significant.
Because primary cultures of striatal neurons showed the classical response to DNA damage, we then investigated Cdk5 activity in these cells (Fig. 4B). Thus, we treated a primary culture of striatal neurons with CPT and examined the activity of Cdk5 during early (10 μm CPT, 3 h) and late (8 h) DNA damage events. After the immunoprecipitation of Cdk5, we performed in vitro kinase assays using histone H1 as a substrate (Fig. 4B). After 3 h of CPT treatment, Cdk5 activity was much higher that in control neurons, although this increase is not linked to an increase in Cdk5 or p35 protein levels (Fig. 4B, immunoblotting). At 8 h, Cdk5 activity returned to basal levels. We treated the cells with roscovitine to determine the specificity of the Cdk5 assay. As expected, in the presence of roscovitine, histone H1 phosphorylation was inhibited. Furthermore, when Cdk5 was inhibited, CPT no longer increased the Cdk5 activity.
Because DNA damage modulates Cdk5 activity in striatal neurons, we investigated whether htt is selectively phosphorylated by Cdk5 during DNA damage. Primary culture of striatal neurons were treated with CPT, and neurons extracts were analyzed by immunoblotting. CPT treatment resulted in an accumulation of γ-H2AX and an increase phosphorylation at S1181 (Fig. 4C). Similar results were obtained using the anti-P-htt–S1201 in human neuroblastoma cells (Fig. 4D). We then decreased Cdk5 activity by roscovitine treatment and found that, in these conditions, CPT had no effect, indicating that CPT-induced phosphorylations at S1181 and S1201 depend on Cdk5. We conclude that in striatal neurons, both Cdk5 activity and huntingtin phosphorylations at S1181 and S1201 increase in response to DNA damage.
Huntingtin is phosphorylated by Cdk5 during DNA damage in nuclei of neurons
We further characterized the phosphorylation of htt at S1201 using the anti-P-htt–S1201 antibody by immunocytochemistry because this antibody works well in immunostaining of fixed cells, therefore allowing a precise analysis of phosphorylation at a single-cell level. The P-htt–S1201-immunopositive staining was observed in neurons as nuclear dots (Fig. 5A), similar to the previously described location of htt in the nucleus (De Rooij et al., 1996; Kegel et al., 2002). The nuclear localization of S1201-phosphorylated huntingtin was further confirmed by cellular fractionation (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). As expected, htt and Cdk5/p35 were found to be present in both the nucleus and the cytoplasm. Because DNA damage occurs in the nucleus, we focused our study on nuclear htt and found by immunoblotting experiments that, during CPT treatment, the level of phosphorylated htt was increased in this compartment.
Figure 5.

Phosphorylation of huntingtin at serine 1201 is induced by Cdk5 during DNA damage in cells and in vivo. A, B, P-htt–S1201-immunopositive nuclear staining is increased by DNA damage. Striatal neurons are untreated or treated with DMSO (0.1%, control), H2O2 (50 μm, 15 min) and CPT (10 μm, 3 h). Fixed neurons are immunostained for the presence of P-htt–S1201 and γ-H2AX. Nuclei are stained with Hoechst. Scale bars, 10 μm. C, D, P-htt–S1201-immunopositive nuclear staining is specific to Cdk5 and htt. Neurons are treated with DMSO (0.1%, control), roscovitine (10 μm, 4 hr), CPT (10 μm, 3 hr), and/or electroporated with control siRNA (Si-Sc), siRNAs directed against htt (Si-htt), or pSuper–Cdk5. Striatal neurons are fixed in methanol and immunostained for P-htt–S1201, and quantification of the nuclear P-htt–S1201 dots intensity is performed. E, Immunoblotting analysis revealed that si-htt and pSuper–Cdk5 reduce htt and Cdk5 levels, respectively, in primary cultures of striatal neurons. F, Radiation induces DNA damage and phosphorylation of htt at serines 1181 and 1201 in mouse. Striatal coronal sections of a wild-type mice 8 h after irradiation (bottom row) and of an unirradiated control (top row) were stained for the presence of P-htt–S1201 and γ-H2AX. Nuclei are stained with Hoechst. Scale bar, 100 μm. Brain extracts of an unirradiated control (Control) and of a wild-type mice 8 h after irradiation (5 Gy) were analyzed by immunoblotting for P-htt–S1181, htt, Cdk5, p35, and β-actin. *p < 0.05; **p < 0.01; ***p < 0.001. NS, Not significant.
We then analyzed the effect of DNA damage by treating primary cultures of striatal neurons with H2O2 or CPT (Fig. 5B). These treatments induced DNA damage, as confirmed by γ-H2AX immunostaining, leading to an increase in the intensity of P-htt–S1201-immunopositive nuclear dots. We quantified this intensity by analyzing deconvoluted pictures of immunostained nuclei from at least 20 independent cells per condition using a 3D microscope (Fig. 5C) (Gauthier et al., 2004). We used a specially developed 3D object counter plugin in NIH Image J to calculate the mean of the total nuclear P-htt–S1201 intensity per cell. The CPT-induced increase in htt phosphorylation at S1201 was found to be statistically significant (Fig. 5C). When treating cells with roscovitine, the CPT-increased phosphorylation at S1201 was completely blocked (Fig. 5C), suggesting that htt phosphorylation at S1201 in response to DNA damage occurs through Cdk5.
To further assess the specificity of the observed phospho-staining and to unequivocally demonstrate that Cdk5 is indeed the kinase involved in this phosphorylation, we used appropriate siRNA to downregulate these proteins. As detected by immunoblotting, this allows to decrease the levels of htt and Cdk5 (Fig. 5E). In both conditions, the intensity of the P-htt–S1201-immunopositive dots was significantly decreased (Fig. 5D, white bars). Furthermore, CPT was no longer able to induce an increased phosphorylation at S1201 when htt and Cdk5 levels were reduced by RNA interference (Fig. 5D, black bars). Therefore, we conclude that htt is phosphorylated by Cdk5 at S1181 and S1201 after DNA damage in primary culture of striatal neurons.
Huntingtin is phosphorylated during DNA damage in the mouse striatum
We next wondered whether phosphorylation of htt during DNA damage occurs in vivo. To this end, 10-week-old wild-type mice were exposed to 5 Gy of γ radiation. Coronal sections of brains collected at 8 h after irradiation were analyzed by immunostaining using anti-P-htt–S1201 and anti-γ-H2AX antibodies (Fig. 5F). Very few striatal neurons were stained by the γ-H2AX antibody in unirradiated control brains (top row). In contrast, 8 h after irradiation, most if not all striatal neurons contained nuclear foci of γ-H2AX bottom row) indicative of DNA damage. We also analyzed phosphorylation state of htt using the P-htt–S1201 antibody. Similar to the observation in cultured neurons (Fig. 5B), under irradiation conditions, P-htt–S1201-immunopositive staining was observed in neurons as nuclear dots. We next aimed to demonstrate phosphorylation on S1181 in vivo. Because the P-htt–S1181 antibody does not work by immunohistochemistry, we immunoblotted brain extracts of control and irradiated mice with this antibody and anti-htt, anti-Cdk5, anti-p35, and anti-β-actin antibodies. Although the total levels of htt, Cdk5, and p35 remained constant, phosphorylation of htt at S1181 was induced by DNA damage in vivo (Fig. 5F, right). These results unequivocally demonstrate that htt is phosphorylated in vitro and in vivo at S1181 and S1201 in response to DNA damage and is directly associated to the DNA damage response pathway in neurons.
Phosphorylation of huntingtin at serines 1181 and 1201 precedes DNA damage-induced neuronal death
During the early phases of DNA damage, Cdk5 activity is increased, whereas aging and the accumulation of DNA damage decrease Cdk5 levels in the brain (Lu et al., 2004). Therefore, we assessed the kinetics of htt phosphorylation during treatment with CPT for varying lengths of time (Fig. 6A). The intensity of P-htt–S1201 spots increased rapidly and significantly, reaching a maximum intensity after 3 h of induced DNA damage. At 8 h, the level of phosphorylation at S1201 in surviving neurons decreased, returning to basal levels. We observed similar statistically significant results when immunoblotting extracts from the same experiment with the anti-P-htt–S1181 (Fig. 6B). Under the same experimental conditions, we examined CPT-induced neuronal death (Fig. 6C). Consistent with a previous report (O'Hare et al., 2005), at early stages of DNA damage (3 h, CPT), we observed no statistically significant neuronal toxicity, whereas a progressive and marked neuronal death was observed after longer CPT treatments (8 h). To further confirm that the event of phosphorylation is an early response to DNA damage, we treated neurons with CPT for 3 h, washed them, and incubated them for another 5 h with standard medium. Interestingly, the short exposure of neurons to CPT did not induce neuronal toxicity at 8 h but still triggered the phosphorylation of htt that was reversible (Fig. 6D). Together, we conclude that htt phosphorylation is an early response to DNA damage when no neuronal death is observed.
Finally, we treated neurons with CPT and examined the effect of htt phosphorylations or not on DNA damage-induced neuronal death. We electroporated neurons with 1301–17Q–AA and 1301–17Q–DD. This allowed a better efficiency of transfection compared with the calcium phosphate method but with a lower amount of the construct of interest in each individual neuron (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). As a consequence, the kinetics of death induced by 1301–73Q or 1301–17Q–AA were slower using electroporation (data not shown). We could thus look at a time point at which the expression of 1301–17Q–AA did not yet induce cell death (Fig. 6E, left bars). In these conditions, CPT treatment drastically increased cell death when htt could not be phosphorylated (Fig. 6E). We also examined the effect of inhibiting Cdk5 on CPT-induced cell death. Consistent with htt and its phosphorylation by Cdk5 playing a role in the DNA damage response, we found that roscovitine treatment of striatal neurons increased cell death, which was further enhanced by CPT treatment (Fig. 6F). These results indicate that htt phosphorylations by Cdk5 participate to the DNA damage-induced neuronal death.
DNA damage and Cdk5 are regulated in HD
Having demonstrated that phosphorylation of wild-type htt regulates DNA damage-induced cell death, we investigated whether the DNA damage pathway could be altered in disease. First, we assessed the levels of γ-H2AX in the genetic HD situation. We used mouse cell lines derived from striatal precursor cells of knock-in mice in which a CAG expansion was inserted into the endogenous mouse htt gene (Trettel et al., 2000). These cell lines carry either two copies of wild-type htt (wild-type cells, +/+) or two copies of mutant htt (homozygous HD mutant cells, 109Q/109Q). They most closely reflect the situation in HD patients because wild-type or polyQ–htt are expressed endogenously. Immunoblotting (Fig. 7A) or immunocytochemistry (Fig. 7B) showed that the levels of γ-H2AX were significantly higher in 109Q/109Q cells than in +/+ cells. The increased of nuclear γ-H2AX foci intensity was found to be statistically significant (2.1-fold increase in 109Q/109Q cells with respect to wild-type cells; unpaired t test, t(97) = −2.85, p = 0.0054).
Figure 7.
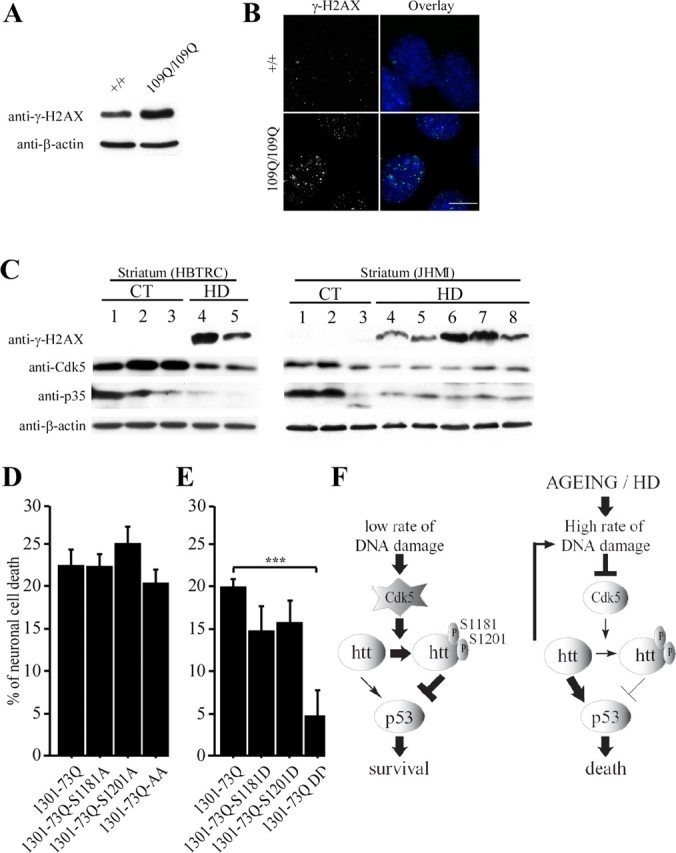
DNA damage, cdk5, and phosphorylations of huntingtin at S1181 and S1201 in HD. A, B, Extracts from wild-type (+/+) and 109Q/109Q mouse striatal cells are analyzed for the presence of γ-H2AX. Immunoblotting with an anti-β-actin antibody is used as a control. Accumulation of γ-H2AX is observed in polyQ cells. B, Immunostaining of γ-H2AX nuclear foci in +/+ and 109Q/109Q cells reveals an increase of foci in the presence of polyQ–htt. Scale bar, 10 μm. C, Whereas γ-H2AX levels are increased in the striatum of HD patients, Cdk5 and p35 levels are reduced. Protein extracts are prepared from whole striatum of control (CT) and HD individuals and analyzed for the presence of γ-H2AX, Cdk5, and p35. Immunoblotting with an anti-β-actin antibody is used as a control. D, E, Phosphorylation of serines 1181 and 1201 in polyQ–htt blocks neuronal death. Primary cultures of striatal neurons are transfected by a modified phosphate calcium method with constructs encoding the first 1301 amino acids of human htt containing 73 glutamines (polyQ, 1301–73Q) with intact serines or serines to alanines or aspartic acids mutations. Neurons are scored for neuronal cell death. F, In normal conditions, DNA damage events occur from spontaneous decay, replication errors, and cellular metabolism. Cdk5 is then activated as a response to DNA damage leading to phosphorylation of serines 1181 and 1201 of htt. Htt phosphorylation blocks polyQ-induced neuronal death in a p53-dependent manner. In contrast, in aging and late stages of HD that correspond to accumulation of DNA damage, the activity of Cdk5 is decreased, and the reduced phosphorylation of wild-type htt turns it into a toxic protein, therefore accelerating DNA damage-induced cell death. ***p < 0.001.
We also analyzed the levels of γ-H2AX in striatal samples from HD patients corresponding to late stages of the disease. It is well established that, in HD, there is a selective atrophy of the brain, with the striatum being affected the most severely. We analyzed human samples from control (CT) and HD grade 3 and 4 (HD) patients from two independent cohorts (HBTRC and JHMI) using an antibody against γ-H2AX (Fig. 7C). Anti-β-actin was used as a control for protein loading. The level of γ-H2AX in HD samples was considerably higher than that in control samples, demonstrating that DNA damage is increased in HD. Consistent with this, DNA damage markers have been observed accumulating in fibroblasts from HD patients (Giuliano et al., 2003) and in a mouse model of HD (Bogdanov et al., 2001).
We next compared by anti-Cdk5 and anti-p35 immunoblotting the level of Cdk5 and its regulatory partner p35 in these HD extracts. In several HD human brain samples, we observed a pronounced downregulation of Cdk5 and its regulatory partner versus that in control human brain. This is in agreement with a previous study (Luo et al., 2005) showing that Cdk5 activity is reduced in HD mouse brains compared with controls.
Phosphorylations of huntingtin at S1181 and S1201 regulate polyQ-induced toxicity
To investigate the consequences of DNA damage-induced regulation of Cdk5 in HD, we assessed the effect of the absence or constitutive phosphorylation at S1181 and S1201 in HD situation on neuronal survival. We used constructs encoding the first 1301 amino acids of htt with 73 glutamines (polyQ, 1301–73Q) with intact serines or with serine to alanine or aspartic acid mutations at sites 1181 and/or 1201. Primary cultures of striatal neurons were transfected with these constructs, and the percentage of neuronal death was measured as in Figure 3. The absence of phosphorylation had no obvious effect on polyQ–htt-induced cell death (Fig. 7D). However, whereas polyQ-induced toxicity was only slightly decreased by single mutations to aspartic acid, double mutations completely rescued polyQ-induced toxicity (Fig. 7E) (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). These data show that phosphorylations at S1181 and S1201 inhibit polyQ–htt-induced cell death in neurons.
Discussion
DNA damage response has been extensively described in cycling cells, in particular regarding its role in cancer, but much less is known in differentiated cells like neurons. We now demonstrate that the increase in the activity of the neuronal kinase Cdk5 in response to DNA damage results in the specific phosphorylation of htt at serines 1181 and 1201 (Fig. 7F). These phosphorylations are crucial to regulate neuronal cell death through the p53 pathway. In HD, phosphorylation of polyQ–htt by Cdk5 completely protects against polyQ-induced p53-mediated cell death. In contrast, in the absence of phosphorylation, wild-type htt becomes toxic and accelerates the DNA damage response. Our data suggest that, whereas transient activation of this pathway is an early neuroprotective response, sustained DNA damage as observed in the pathological situation, results in loss of Cdk5 activity with subsequent htt dephosphorylation and enhanced p53-mediated cell death.
The observation of neurological deficits in human syndromes associated with genetic defects in DNA damage response, including ataxia telangiectasia, have linked DNA damage response to neurodegeneration (Rolig and McKinnon, 2000). In neurodegenerative diseases such as Alzheimer's, Parkinson's, and Huntington's diseases, oxidative stress in neurons may cause numerous DNA lesions to accumulate, resulting in apoptosis (Caldecott, 2004). Although the tumor suppressor gene p53 is more often studied in proliferating cells, it is likely to play a similar role in neurons by either regulating the DNA repair process or inducing apoptosis (Culmsee and Mattson, 2005). Indeed, p53 mediates neuronal death in many cellular or animal models of neurodegenerative disorders and after DNA damage in neurons (Mattson et al., 2001; Jacobs et al., 2006). Our study links Cdk5 and htt to the p53-mediated response to DNA damage in HD. This further emphasizes the importance of increased oxidative stress and DNA damage in neurodegenerative disorders. Dysregulation of this pathway is also central during aging. Comparing two highly diverged animals, Caenorhabditis elegans and Drosophila, shows that aging is characterized by common changes in mitochondrial and DNA repair genes (McCarroll et al., 2004). Furthermore, Lu et al. (2004) defined a set of genes, including Cdk5, having a reduced expression in the human brain after 40 years of age attributable to selective damage of their promoters by oxidative stress. This suggests there are shared mechanisms in the DNA damage response in both normal aging and neurodegenerative disorders. However, in diseases such as HD, this age-related process is accelerated by the presence of the mutation.
Previous reports have established the involvement of p53 in HD pathogenesis (Steffan et al., 2000; Trettel et al., 2000; Bae et al., 2005). PolyQ–htt dysregulates normal transcription in neurons. PolyQ–htt may sequester transcription factors into aggregates, as seen for the CREB (cAMP response element-binding protein)-binding protein (CBP) resulting in the inhibition of CBP- and p53-dependent transcriptions (Steffan et al., 2000; Nucifora et al., 2001). Furthermore, transcriptional dysregulation occurs through the interaction of soluble polyQ–htt with transcription factors, including p53 (Yu et al., 2002; Schaffar et al., 2004; Bae et al., 2005; Cong et al., 2005). In addition, elevated levels of p53 are observed in the 109Q/109Q mouse cell lines derived from striatal precursor cells of knock-in mice (Trettel et al., 2000). These elevated levels of p53 are also observed in neuronal cells, HD mice, and HD patients (Bae et al., 2005). Finally, Bae et al. (2005) demonstrate that the neurodegeneration in HD model of flies and mice is prevented by perturbation of p53 either by pifithrin-α, RNA interference, or genetic deletion. We now show that, in nonpathological conditions, Cdk5 through the phosphorylation of wild-type htt modulates neuronal death through a p53-dependent pathway. A link between p53 and htt in normal and pathological conditions is further supported by the observation that p53 regulates htt expression at the transcriptional level (Feng et al., 2006). Finally, the idea that the same cellular pathways and partners are involved in health and disease may apply to other polyQ disorders, as shown recently for ataxin-1 (Lam et al., 2006).
A recent study has identified several phosphorylation sites on htt by a mass spectrometric characterization (Schilling et al., 2006). Among six novel phosphorylation sites on full-length htt, serines 1181 and 1201 were found to be phosphorylated, supporting their physiological importance. We now identify Cdk5 as the kinase responsible for phosphorylating htt at these sites. The functions of Cdk5 that have been best characterized are functions that dictate the proper maturation and maintenance of the CNS (Cruz and Tsai, 2004; Cheung et al., 2006). However, emerging evidence involves Cdk5 in neurodegeneration. In toxic conditions, Cdk5 activity is dysregulated when its regulators p35 and p39 are cleaved by calpain, therefore transforming Cdk5 into an hyperactive kinase that initiates events associated with neuronal death. Accumulation of p25 has been observed in neurons undergoing toxic conditions, in cases of human Alzheimer brains and in animal models of stroke, Parkinson's disease, and amyotrophic lateral sclerosis (Cruz and Tsai, 2004; Kesavapany et al., 2004; Shelton and Johnson, 2004). In HD, the situation could be different. In agreement with a previous report (Luo et al., 2005), we show that Cdk5 and p35 are downregulated in HD postmortem samples corresponding to late stages of the disease when DNA damage has accumulated. We propose that, as an early response to DNA damage, Cdk5 activity is elevated and contributes to the survival of neurons by phosphorylating htt. The mechanism leading to the elevation of Cdk5 activity remains to be determined, but it is not a consequence of the generation of p25 (data not shown). In late stages of DNA damage, Cdk5 activity decreases, rendering htt less phosphorylated at serines 1201 and 1181 and toxic.
Several studies have shown that wild-type htt has antiapoptotic properties and that these are lost in the presence of an abnormal polyQ expansion (Cattaneo et al., 2005). Some of these properties have been associated with the capacity of htt to promote neuronal survival by stimulating transport of BDNF (Gauthier et al., 2004). When htt contains an abnormal polyQ expansion, the transport and the release of BDNF are reduced. Furthermore, htt binds to the transcriptional repressor REST/NRSF (repressor element-1 transcription/neuron restrictive silencer factors) and sequesters this complex in the cytoplasm away from its nuclear target, the NRSE (neuron restrictive silencer element), which is a consensus sequence found in genes such as the gene encoding BDNF (Zuccato et al., 2003). As for transport, this beneficial activity of htt in transcription is lost when htt is mutated, resulting in a decreased production of BDNF and a decreased neurotrophic support for striatal neurons. Here, we described a new antiapoptotic function for htt that is linked to its capacity to regulate the death response to DNA damage in neurons. Indeed, phosphorylation of htt at serines S1181 and S1201 completely abolishes the toxicity of the pathological polyQ–htt in neurons and slows the death rate during DNA damage. In contrast, the absence of phosphorylation in wild-type htt is sufficient to turn htt into a toxic protein independent of it possessing the pathological polyQ expansion and to accelerate the DNA damage response. Except for the polyQ expansion, this is the first mutation in htt that changes wild-type htt from an antiapoptotic to a proapoptotic protein. Therefore, the antiapoptotic properties of htt can be lost by the presence of an abnormally expanded polyQ but also by modification of the protein context in absence of the polyQ mutation.
Our study highlights the need to understand the normal function of htt and supports previous observations that protein domains outside of the polyQ stretch are key elements that regulate htt function in the normal and pathological condition. A key step during the cascade of events leading to death of striatal neurons is the cleavage of htt into smaller N-terminal fragments. We have shown previously that one consequence of the proteolysis could be to inactivate the positive function of htt in transport (Gauthier et al., 2004). Because proteolysis of normal htt is also observed in HD brain, cleavage of htt by reducing BDNF transport and the neurotrophic supply of striatal neurons could enhance the neurodegeneration induced by the polyQ expansion in the mutant allele. Similarly, during disease progression in the most commonly affected patients, those heterozygous for HD, the increase of DNA damage and subsequent loss of Cdk5 activity attributable to the polyQ expansion in htt may affect the normal htt allele and increase its toxicity. Therefore, these growing list of observations supports that the loss of normal htt functions could contribute to the pathogenesis of HD and is consistent with HD not being simply a gain-of-function disease.
Footnotes
This work was supported by Agence Nationale de la Recherche Grant MRAR-018-01 (F.S.), Association Francaise contre les Myopathies (F.S.), Association pour la Recherche sur le Cancer (ARC) Grant 3665 (S.H.), Fondation pour la Recherche Médicale and Fondation BNP Paribas (F.S.), Fédération pour la Recherche sur le Cerveau (F.S.), HighQ Foundation (F.S., S.H.), and Provital/P. Chevalier (F.S., S.H.). S.L.A. was supported by HighQ Foundation and currently by an ARC doctoral fellowship. F.S. is a recipient of a European Molecular Biology Organization Young Investigator Award, and F.S. and S.H. are Institut National de la Santé et de la Recherche Médicale/Assistance Publique-Hôpitaux de Paris investigators. We thank L.-H. Tsai for Cdk5 constructs; C. A. Ross, J. Troncoso, and Harvard Brain Tissue Resource Center (Belmont, MA), which is supported in part by Public Health Service Grant Number MH/NS 31862, for providing human brain tissue; F. P. Cordelières and the Institut Curie Imaging Facility; L. M. Ellerby, P.-M. Girard, and C. A. Ross for discussions; and members of the Saudou/Humbert laboratory for help and comments.
References
- Bae BI, Xu H, Igarashi S, Fujimuro M, Agrawal N, Taya Y, Hayward SD, Moran TH, Montell C, Ross CA, Snyder SH, Sawa A. p53 mediates cellular dysfunction and behavioral abnormalities in Huntington's disease. Neuron. 2005;47:29–41. doi: 10.1016/j.neuron.2005.06.005. [DOI] [PubMed] [Google Scholar]
- Benchoua A, Trioulier Y, Zala D, Gaillard MC, Lefort N, Dufour N, Saudou F, Elalouf JM, Hirsch E, Hantraye P, Deglon N, Brouillet E. Involvement of mitochondrial complex II defects in neuronal death produced by N-terminus fragment of mutated huntingtin. Mol Biol Cell. 2006;17:1652–1663. doi: 10.1091/mbc.E05-07-0607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezprozvanny I, Hayden MR. Deranged neuronal calcium signaling and Huntington disease. Biochem Biophys Res Commun. 2004;322:1310–1317. doi: 10.1016/j.bbrc.2004.08.035. [DOI] [PubMed] [Google Scholar]
- Bogdanov MB, Andreassen OA, Dedeoglu A, Ferrante RJ, Beal MF. Increased oxidative damage to DNA in a transgenic mouse model of Huntington's disease. J Neurochem. 2001;79:1246–1249. doi: 10.1046/j.1471-4159.2001.00689.x. [DOI] [PubMed] [Google Scholar]
- Borrell-Pages M, Zala D, Humbert S, Saudou F. Huntington's disease: from huntingtin function and dysfunction to therapeutic strategies. Cell Mol Life Sci. 2006;63:2642–2660. doi: 10.1007/s00018-006-6242-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browne SE, Beal MF. The energetics of Huntington's disease. Neurochem Res. 2004;29:531–546. doi: 10.1023/b:nere.0000014824.04728.dd. [DOI] [PubMed] [Google Scholar]
- Caldecott KW. DNA single-strand breaks and neurodegeneration. DNA Repair (Amst) 2004;3:875–882. doi: 10.1016/j.dnarep.2004.04.011. [DOI] [PubMed] [Google Scholar]
- Cattaneo E, Zuccato C, Tartari M. Normal huntingtin function: an alternative approach to Huntington's disease. Nat Rev Neurosci. 2005;6:919–930. doi: 10.1038/nrn1806. [DOI] [PubMed] [Google Scholar]
- Cheung ZH, Fu AK, Ip NY. Synaptic roles of Cdk5: implications in higher cognitive functions and neurodegenerative diseases. Neuron. 2006;50:13–18. doi: 10.1016/j.neuron.2006.02.024. [DOI] [PubMed] [Google Scholar]
- Cong SY, Pepers BA, Evert BO, Rubinsztein DC, Roos RA, van Ommen GJ, Dorsman JC. Mutant huntingtin represses CBP, but not p300, by binding and protein degradation. Mol Cell Neurosci. 2005;4:560–571. [PubMed] [Google Scholar]
- Cruz JC, Tsai LH. A Jekyll and Hyde kinase: roles for Cdk5 in brain development and disease. Curr Opin Neurobiol. 2004;14:390–394. doi: 10.1016/j.conb.2004.05.002. [DOI] [PubMed] [Google Scholar]
- Culmsee C, Mattson MP. p53 in neuronal apoptosis. Biochem Biophys Res Commun. 2005;331:761–777. doi: 10.1016/j.bbrc.2005.03.149. [DOI] [PubMed] [Google Scholar]
- De Rooij KE, Dorsman JC, Smoor MA, Den Dunnen JT, Van Ommen GJ. Subcellular localization of the Huntington's disease gene product in cell lines by immunofluorescence and biochemical subcellular fractionation. Hum Mol Genet. 1996;5:1093–1099. doi: 10.1093/hmg/5.8.1093. [DOI] [PubMed] [Google Scholar]
- Dhavan R, Tsai LH. A decade of CDK5. Nat Rev Mol Cell Biol. 2001;2:749–759. doi: 10.1038/35096019. [DOI] [PubMed] [Google Scholar]
- Dragatsis I, Levine MS, Zeitlin S. Inactivation of hdh in the brain and testis results in progressive neurodegeneration and sterility in mice. Nat Genet. 2000;26:300–306. doi: 10.1038/81593. [DOI] [PubMed] [Google Scholar]
- Feng Z, Jin S, Zupnick A, Hoh J, de Stanchina E, Lowe S, Prives C, Levine AJ. p53 tumor suppressor protein regulates the levels of huntingtin gene expression. Oncogene. 2006;25:1–7. doi: 10.1038/sj.onc.1209021. [DOI] [PubMed] [Google Scholar]
- Gauthier LR, Charrin BC, Borrell-Pages M, Dompierre JP, Rangone H, Cordelieres FP, De Mey J, MacDonald ME, Lessmann V, Humbert S, Saudou F. Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell. 2004;118:127–138. doi: 10.1016/j.cell.2004.06.018. [DOI] [PubMed] [Google Scholar]
- Giuliano P, De Cristofaro T, Affaitati A, Pizzulo GM, Feliciello A, Criscuolo C, De Michele G, Filla A, Avvedimento EV, Varrone S. DNA damage induced by polyglutamine-expanded proteins. Hum Mol Genet. 2003;12:2301–2309. doi: 10.1093/hmg/ddg242. [DOI] [PubMed] [Google Scholar]
- Graham RK, Deng Y, Slow EJ, Haigh B, Bissada N, Lu G, Pearson J, Shehadeh J, Bertram L, Murphy Z, Warby SC, Doty CN, Roy S, Wellington CL, Leavitt BR, Raymond LA, Nicholson DW, Hayden MR. Cleavage at the caspase-6 site is required for neuronal dysfunction and degeneration due to mutant huntingtin. Cell. 2006;125:1179–1191. doi: 10.1016/j.cell.2006.04.026. [DOI] [PubMed] [Google Scholar]
- Gunawardena S, Her LS, Brusch RG, Laymon RA, Niesman IR, Gordesky-Gold B, Sintasath L, Bonini NM, Goldstein LS. Disruption of axonal transport by loss of huntingtin or expression of pathogenic polyQ proteins in Drosophila. Neuron. 2003;40:25–40. doi: 10.1016/s0896-6273(03)00594-4. [DOI] [PubMed] [Google Scholar]
- Humbert S, Bryson EA, Cordelieres FP, Connors NC, Datta SR, Finkbeiner S, Greenberg ME, Saudou F. The IGF-1/Akt pathway is neuroprotective in Huntington's disease and involves Huntingtin phosphorylation by Akt. Dev Cell. 2002;2:831–837. doi: 10.1016/s1534-5807(02)00188-0. [DOI] [PubMed] [Google Scholar]
- Jacobs WB, Kaplan DR, Miller FD. The p53 family in nervous system development and disease. J Neurochem. 2006;97:1571–1584. doi: 10.1111/j.1471-4159.2006.03980.x. [DOI] [PubMed] [Google Scholar]
- Kegel KB, Meloni AR, Yi Y, Kim YJ, Doyle E, Cuiffo BG, Sapp E, Wang Y, Qin ZH, Chen JD, Nevins JR, Aronin N, DiFiglia M. Huntingtin is present in the nucleus, interacts with the transcriptional corepressor C-terminal binding protein, and represses transcription. J Biol Chem. 2002;277:7466–7476. doi: 10.1074/jbc.M103946200. [DOI] [PubMed] [Google Scholar]
- Kesavapany S, Li BS, Amin N, Zheng YL, Grant P, Pant HC. Neuronal cyclin-dependent kinase 5: role in nervous system function and its specific inhibition by the Cdk5 inhibitory peptide. Biochim Biophys Acta. 2004;1697:143–153. doi: 10.1016/j.bbapap.2003.11.020. [DOI] [PubMed] [Google Scholar]
- Ko J, Humbert S, Bronson RT, Takahashi S, Kulkarni AB, Li E, Tsai LH. p35 and p39 are essential for cyclin-dependent kinase 5 function during neurodevelopment. J Neurosci. 2001;21:6758–6771. doi: 10.1523/JNEUROSCI.21-17-06758.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam YC, Bowman AB, Jafar-Nejad P, Lim J, Richman R, Fryer JD, Hyun ED, Duvick LA, Orr HT, Botas J, Zoghbi HY. ATAXIN-1 Interacts with the repressor capicua in its native complex to cause SCA1 neuropathology. Cell. 2006;127:1335–1347. doi: 10.1016/j.cell.2006.11.038. [DOI] [PubMed] [Google Scholar]
- Leavitt BR, Guttman JA, Hodgson JG, Kimel GH, Singaraja R, Vogl AW, Hayden MR. Wild-type huntingtin reduces the cellular toxicity of mutant huntingtin in vivo. Am J Hum Genet. 2001;68:313–324. doi: 10.1086/318207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Kim KT. Regulation of cyclin-dependent kinase 5 and p53 by ERK1/2 pathway in the DNA damage-induced neuronal death. J Cell Physiol. 2007;210:784–797. doi: 10.1002/jcp.20899. [DOI] [PubMed] [Google Scholar]
- Li S, Li XJ. Multiple pathways contribute to the pathogenesis of Huntington disease. Mol Neurodegener. 2006;1:19. doi: 10.1186/1750-1326-1-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu T, Pan Y, Kao SY, Li C, Kohane I, Chan J, Yankner BA. Gene regulation and DNA damage in the ageing human brain. Nature. 2004;429:883–891. doi: 10.1038/nature02661. [DOI] [PubMed] [Google Scholar]
- Luo S, Vacher C, Davies JE, Rubinsztein DC. Cdk5 phosphorylation of huntingtin reduces its cleavage by caspases: implications for mutant huntingtin toxicity. J Cell Biol. 2005;169:647–656. doi: 10.1083/jcb.200412071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald ME, Gines S, Gusella JF, Wheeler VC. Huntington's disease. Neuromolecular Med. 2003;4:7–20. doi: 10.1385/NMM:4:1-2:7. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Duan W, Pedersen WA, Culmsee C. Neurodegenerative disorders and ischemic brain diseases. Apoptosis. 2001;6:69–81. doi: 10.1023/a:1009676112184. [DOI] [PubMed] [Google Scholar]
- McCarroll SA, Murphy CT, Zou S, Pletcher SD, Chin CS, Jan YN, Kenyon C, Bargmann CI, Li H. Comparing genomic expression patterns across species identifies shared transcriptional profile in aging. Nat Genet. 2004;36:197–204. doi: 10.1038/ng1291. [DOI] [PubMed] [Google Scholar]
- Milakovic T, Johnson GV. Mitochondrial respiration and ATP production are significantly impaired in striatal cells expressing mutant huntingtin. J Biol Chem. 2005;280:30773–30782. doi: 10.1074/jbc.M504749200. [DOI] [PubMed] [Google Scholar]
- Nowak E, Etienne O, Millet P, Lages CS, Mathieu C, Mouthon MA, Boussin FD. Radiation-induced H2AX phosphorylation and neural precursor apoptosis in the developing brain of mice. Radiat Res. 2006;165:155–164. doi: 10.1667/rr3496.1. [DOI] [PubMed] [Google Scholar]
- Nucifora FC, Jr, Sasaki M, Peters MF, Huang H, Cooper JK, Yamada M, Takahashi H, Tsuji S, Troncoso J, Dawson VL, Dawson TM, Ross CA. Interference by huntingtin and atrophin-1 with cbp-mediated transcription leading to cellular toxicity. Science. 2001;291:2423–2428. doi: 10.1126/science.1056784. [DOI] [PubMed] [Google Scholar]
- O'Hare MJ, Kushwaha N, Zhang Y, Aleyasin H, Callaghan SM, Slack RS, Albert PR, Vincent I, Park DS. Differential roles of nuclear and cytoplasmic cyclin-dependent kinase 5 in apoptotic and excitotoxic neuronal death. J Neurosci. 2005;25:8954–8966. doi: 10.1523/JNEUROSCI.2899-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panov AV, Gutekunst CA, Leavitt BR, Hayden MR, Burke JR, Strittmatter WJ, Greenamyre JT. Early mitochondrial calcium defects in Huntington's disease are a direct effect of polyglutamines. Nat Neurosci. 2002;5:731–736. doi: 10.1038/nn884. [DOI] [PubMed] [Google Scholar]
- Pardo R, Colin E, Regulier E, Aebischer P, Deglon N, Humbert S, Saudou F. Inhibition of calcineurin by FK506 protects against polyglutamine-huntingtin toxicity through an increase of huntingtin phosphorylation at S421. J Neurosci. 2006;26:1635–1645. doi: 10.1523/JNEUROSCI.3706-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangone H, Poizat G, Troncoso J, Ross CA, MacDonald ME, Saudou F, Humbert S. The serum- and glucocorticoid-induced kinase SGK inhibits mutant huntingtin-induced toxicity by phosphorylating serine 421 of huntingtin. Eur J Neurosci. 2004;19:273–279. doi: 10.1111/j.0953-816x.2003.03131.x. [DOI] [PubMed] [Google Scholar]
- Rigamonti D, Bauer JH, De-Fraja C, Conti L, Sipione S, Sciorati C, Clementi E, Hackam A, Hayden MR, Li Y, Cooper JK, Ross CA, Govoni S, Vincenz C, Cattaneo E. Wild-type huntingtin protects from apoptosis upstream of caspase-3. J Neurosci. 2000;20:3705–3713. doi: 10.1523/JNEUROSCI.20-10-03705.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolig RL, McKinnon PJ. Linking DNA damage and neurodegeneration. Trends Neurosci. 2000;23:417–424. doi: 10.1016/s0166-2236(00)01625-8. [DOI] [PubMed] [Google Scholar]
- Ross CA. Huntington's disease: new paths to pathogenesis. Cell. 2004;118:4–7. doi: 10.1016/j.cell.2004.06.022. [DOI] [PubMed] [Google Scholar]
- Saudou F, Finkbeiner S, Devys D, Greenberg ME. Huntingtin acts in the nucleus to induce apoptosis but death does not correlate with the formation of intranuclear inclusions. Cell. 1998;95:55–66. doi: 10.1016/s0092-8674(00)81782-1. [DOI] [PubMed] [Google Scholar]
- Sawa A. Mechanisms for neuronal cell death and dysfunction in Huntington's disease: pathological cross-talk between the nucleus and the mitochondria? J Mol Med. 2001;79:375–381. doi: 10.1007/s001090100223. [DOI] [PubMed] [Google Scholar]
- Schaffar G, Breuer P, Boteva R, Behrends C, Tzvetkov N, Strippel N, Sakahira H, Siegers K, Hayer-Hartl M, Hartl FU. Cellular toxicity of polyglutamine expansion proteins: mechanism of transcription factor deactivation. Mol Cell. 2004;15:95–105. doi: 10.1016/j.molcel.2004.06.029. [DOI] [PubMed] [Google Scholar]
- Schilling B, Gafni J, Torcassi C, Cong X, Row RH, Lafevre-Bernt MA, Cusack MP, Ratovitski T, Hirschhorn R, Ross CA, Gibson BW, Ellerby LM. Huntingtin phosphorylation sites mapped by mass spectrometry: modulation of cleavage and toxicity. J Biol Chem. 2006;281:23686–23697. doi: 10.1074/jbc.M513507200. [DOI] [PubMed] [Google Scholar]
- Shelton SB, Johnson GV. Cyclin-dependent kinase-5 in neurodegeneration. J Neurochem. 2004;88:1313–1326. doi: 10.1111/j.1471-4159.2003.02328.x. [DOI] [PubMed] [Google Scholar]
- Steffan JS, Kazantsev A, Spasic-Boskovic O, Greenwald M, Zhu YZ, Gohler H, Wanker EE, Bates GP, Housman DE, Thompson LM. The Huntington's disease protein interacts with p53 and CREB-binding protein and represses transcription. Proc Natl Acad Sci USA. 2000;97:6763–6768. doi: 10.1073/pnas.100110097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffan JS, Agrawal N, Pallos J, Rockabrand E, Trotman LC, Slepko N, Illes K, Lukacsovich T, Zhu YZ, Cattaneo E, Pandolfi PP, Thompson LM, Marsh JL. SUMO modification of Huntingtin and Huntington's disease pathology. Science. 2004;304:100–104. doi: 10.1126/science.1092194. [DOI] [PubMed] [Google Scholar]
- Sugars KL, Rubinsztein DC. Transcriptional abnormalities in Huntington's disease. Trends Genet. 2003;19:233–238. doi: 10.1016/S0168-9525(03)00074-X. [DOI] [PubMed] [Google Scholar]
- Trettel F, Rigamonti D, Hilditch-Maguire P, Wheeler VC, Sharp AH, Persichetti F, Cattaneo E, MacDonald ME. Dominant phenotypes produced by the HD mutation in STHdh(Q111) striatal cells. Hum Mol Genet. 2000;9:2799–2809. doi: 10.1093/hmg/9.19.2799. [DOI] [PubMed] [Google Scholar]
- Trushina E, Dyer RB, Badger JD, II, Ure D, Eide L, Tran DD, Vrieze BT, Legendre-Guillemin V, McPherson PS, Mandavilli BS, Van Houten B, Zeitlin S, McNiven M, Aebersold R, Hayden M, Parisi JE, Seeberg E, Dragatsis I, Doyle K, Bender A, Chacko C, McMurray CT. Mutant huntingtin impairs axonal trafficking in mammalian neurons in vivo and in vitro. Mol Cell Biol. 2004;24:8195–8209. doi: 10.1128/MCB.24.18.8195-8209.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000;408:307–310. doi: 10.1038/35042675. [DOI] [PubMed] [Google Scholar]
- Warby SC, Chan EY, Metzler M, Gan L, Singaraja RR, Crocker SF, Robertson HA, Hayden MR. Huntingtin phosphorylation on serine 421 is significantly reduced in the striatum and by polyglutamine expansion in vivo. Hum Mol Genet. 2005;14:1569–1577. doi: 10.1093/hmg/ddi165. [DOI] [PubMed] [Google Scholar]
- Young AB. Huntingtin in health and disease. J Clin Invest. 2003;111:299–302. doi: 10.1172/JCI17742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu ZX, Li SH, Nguyen HP, Li XJ. Huntingtin inclusions do not deplete polyglutamine-containing transcription factors in HD mice. Hum Mol Genet. 2002;11:905–914. doi: 10.1093/hmg/11.8.905. [DOI] [PubMed] [Google Scholar]
- Zeitlin S, Liu JP, Chapman DL, Papaioannou VE, Efstratiadis A. Increased apoptosis and early embryonic lethality in mice nullizygous for the Huntington's disease gene homologue. Nat Genet. 1995;11:155–163. doi: 10.1038/ng1095-155. [DOI] [PubMed] [Google Scholar]
- Zuccato C, Ciammola A, Rigamonti D, Leavitt BR, Goffredo D, Conti L, MacDonald ME, Friedlander RM, Silani V, Hayden MR, Timmusk T, Sipione S, Cattaneo E. Loss of huntingtin-mediated BDNF gene transcription in Huntington's disease. Science. 2001;293:493–498. doi: 10.1126/science.1059581. [DOI] [PubMed] [Google Scholar]
- Zuccato C, Tartari M, Crotti A, Goffredo D, Valenza M, Conti L, Cataudella T, Leavitt BR, Hayden MR, Timmusk T, Rigamonti D, Cattaneo E. Huntingtin interacts with REST/NRSF to modulate the transcription of NRSE-controlled neuronal genes. Nat Genet. 2003;35:76–83. doi: 10.1038/ng1219. [DOI] [PubMed] [Google Scholar]