

Abstract
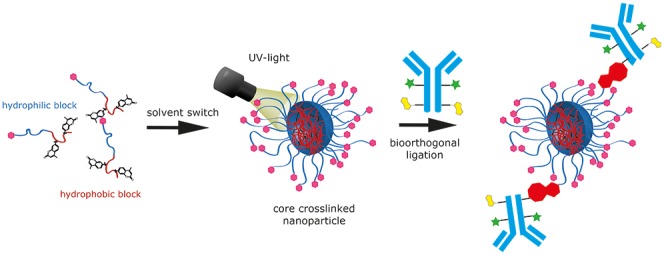
Fast and bioorthogonally reacting nanoparticles are attractive tools for biomedical applications such as tumor pretargeting. In this study, we designed an amphiphilic block copolymer system based on HPMA using different strategies to introduce the highly reactive click units 1,2,4,5-tetrazines (Tz) either at the chain end (Tz-CTA) or statistical into the hydrophobic block. This reactive group undergoes a rapid, bioorthogonal inverse electron-demand Diels–Alder reaction (iEDDA) with trans-cyclooctenes (TCO). Subsequently, this polymer platform was used for the preparation of different Tz-covered nanoparticles, such as micelles and colloids. Thereby it was found that the reactivity of the polymeric micelles is comparable to that of the low molar mass tetrazines. The core-cross-linked micelles can be successfully conjugated at rather low concentrations to large biomacromolecules like antibodies, not only in physiological buffer, but also in human blood plasma, which was confirmed by fluorescence correlation spectroscopy (FCS).
1. Introduction
1.1. Nanoparticles and Imaging
Nanomedicine became of large interest during the last decades.1−3 Nanoparticles such as polymeric micelles, liposomes, dendrimers, antibodies, and colloids can be applied as drug delivery systems.4,5 They show great potential in the treatment of different diseases6 like cancer,7 tuberculosis,8 or neuro diseases.9 Due to their tunable size, constitution, and surface-modification, nanoparticles can reach the desired target sites often preferentially. The advantages of nanomedicine often are improved biodistribution, the protection of the payload against degradation and the ability to accumulate at specific sites.10
Independent of their role as carriers, nanoparticles can also serve as imaging agents, if labeled with a suited marker such as a fluorescent dye, magnetic elements or radionuclides.11 For cancer imaging, the so-called enhanced permeability and retention (EPR) effect often comes into play. This effect was first described in the 1980s by Maeda and co-workers.12 The EPR effect relies on the modified nature of the tumor tissue, which has a leaky vasculature and a reduced lymphatic drainage. In contrast to normal tissue, nanosized drug delivery systems can diffuse into tumor tissue due to the leaky vasculature. Additionally, nanoparticles show an enhanced retention in the tumor based on the decreased lymphatic drainage.13−16 This so-called passive tumor accumulation (targeting) can be improved by an active targeting by adding antibodies or other selective binders to the nanoparticle. The resulting accumulation of the nanoparticles in the tumor can be used, besides the localized delivery of a drug, for imaging and diagnosis.17 In cancer diseases, imaging is crucial to understanding the development and the treatment of the disease.5,18 Typical techniques deployed are magnetic resonance imaging (MRI), computed tomography (CT), optical imaging (OI), single-photon emission computed tomography (SPECT), or positron emission tomography (PET).19,20 During recent years, different combinations of those methods have been developed like PET/MRT or PET/CT and show a great potential for imaging of the main tumor.21,22
1.2. Pretargeting
Click chemistry can be applied to efficiently labeled nanoparticles with various functional groups for active targeting or with radioactive labels for imaging.23−25 In a rapid labeling approach, this is done ex vivo before administration of the nanomedicine. However, in the case of radiolabeling, this can be disadvantageous, considering that the most commonly utilized radionuclides have a short half-life time like 18F or 68Ga with 110 and 68 min, respectively. Thus, only a time window of a few hours can be used.5 However, many applications that are based on the accumulation of nanoparticles within the tumor tissue through the EPR-effect require a longer time frame of more than 24 h before imaging. Thus, long-lived radionuclides such as 111In or 89Zr with half-life times of 2.8 and 3.3 days would be necessary.26,27 This would, however, lead to a higher radiation dose for the patient.23 To circumvent this problem, a pretargeting approach based on bioorthogonal in vivo reactions28 can be used to employ short-lived radionuclides for slowly accumulating targeting vectors such as nanoparticles. In this approach, a targeting vehicle is administered first. This vehicle can be a tumor specific antibody or a nanoparticle, which is modified with reactive moieties for bioorthogonal reactions. After application, a longer time period to accumulate at the target site, either by active or passive targeting, can be provided. Subsequently, a second system is applied bearing the opposite reacting moiety and the radiolabel for imaging. This so-called pull down reagent usually consists of a small molecule to provide fast diffusion into the target site and fast clearance through the kidney in order to reduce the background signal.29−31
1.3. Bioorthogonal Reactions
Highly selective and fast reactions are required to link two moieties within a living system. This class of biocompatible reactions is commonly described as bioorthogonal reactions. These reactions can be utilized, for example, for conjugation of biomolecules like antibodies or nanoparticles to radiolabels.25,32,33 In the past decade, many different bioorthogonal reactions have been developed like the Staudinger and traceless Staudinger ligation34,35 and the strain-promoted alkyne–azide cycoaddition (SPAAC, a subclass of “click reactions”).36,37 In addition, the copper-catalyzed alkyne–azide cycloaddition (CuAAC)38 has been proposed as a bioorthogonal ligation reaction.39 While these reactions are suited for conjugation in vitro, they have limitations for in vivo applications due to their low reactivity in the case of Staudinger ligations and SPAACs or to the use of cytotoxic copper as a catalyst. In 2008, Fox and co-workers and Weissleder and co-workers independently introduced the tetrazine (Tz) trans-cyclooctene (TCO) ligation.40,41 This reaction is an inverse electron demand Diels–Alder [4 + 2] cycloaddition (iEDDA) between an electron-rich dienophile and an electron-poor diene, followed by a cycloreversion under the loss of nitrogen leading to a dihydropyridazine (Figure 1). This type of reaction does not need a catalyst and is up to 10000-fold faster than CuAAC.37,42 Unsubstituted tetrazines (Tz; R1 = H) possess the highest reactivity but degrade in blood serum. However, the less-reactive, methyl-substituted Tzs (R1 = CH3) have been shown to be stable under these conditions.43 Therefore, this reaction with methylated Tzs is well suited for bioorthogonal click reactions in vivo.
Figure 1.

Inverse electron demand Diels–Alder-initiated ligation between 1,2,4,5-tetrazines and trans-cyclooctenes. The reaction between Tz-functionalized polymeric nanoparticles and trans-cyclooctene functionalized antibodies is given as an example.
Here we present the route to incorporate methylated Tz moieties as highly reactive click reagents into the hydrophilic block of amphiphilic block copolymers either at the chain end or randomly distributed. In addition, we prepare and characterize (e.g., core-cross-linked) polymeric micelles and test their reactivity for the ligation of functionalized antibodies (Figure 1).
2. Materials and Methods
2.1. Materials
All reagents and solvents were purchased from Acros Organics (Nidderau, Germany), Sigma-Aldrich (Munich, Germany), Roth (Karlsruhe, Germany), or Fluka (Munich, Germany) and used without further purification if not marked otherwise. Solvents with technical grade were distilled before use and solvents p.a. (pro analysis) were utilized as received. Dichloromethane and chloroform were distilled from CaH2. Tetrahydrofuran (THF), hexane, diethyl ether, and 1,4-dioxane were distilled from Na/K with benzophenone as an indicator. Anhydrous dimethyl sulfoxide (DMSO) was stored in a septum-sealed bottle over an activated molecular sieve (3 Å). Dimethylformamide (DMF) was purchased from VWR (Darmstadt, Germany), dried over BaO, and subsequently distilled in vacuo onto predried molecular sieves (3 Å). 2,2′-Azobis(4-methoxy-2,4-dimethylvaleronitrile) (AMDVN, V-70) was purchased from Wako Chemicals (Neuss, Germany). Lauryl methacrylate (LMA) was purchased from Sigma-Aldrich and distilled prior to use. Poly(d,l-lactide) (PDLLA) was purchased as Resomer 203 S with a molecular weight of 18000–28000 g/mol from Sigma-Aldrich. Dialysis was performed with Spectra/Por membranes (Roth) with a nominal cutoff of 3500 g/mol. Deuterated solvents were obtained from Deutero GmbH dried and stored over molecular sieves. Oregon Green 488, 5-isomer was purchased from ThermoFisher Scientific. 6-Methyl-tetrazine-amine (HCl-salt) was obtained from Jena Bioscience (Jena, Germany). Human blood plasma was provided from the Transfusionszentrale of the Medical Department of the Johannes Gutenberg-University Mainz. It was pooled from six healthy donors and stabilized with EDTA.
2.2. Characterization
1H, 13C, and 19F NMR spectra were recorded on either a Bruker 300 or 400 MHz spectrometer. Chemical shifts (δ) are reported in parts per million (ppm) relative to tetramethylsilane and referenced; the following abbreviations are used in the experimental section for the description of 1H NMR spectra: singlet (s), doublet (d), triplet (t), multiplet (m), and broad (br). The chemical shifts of complex multiplets are given as the range of their occurrence. Reactions were monitored by thin layer chromatography (TLC, performed on Merck silica gel 60 F254, not modified, precoated silica gel on aluminum-supported plates). The polymers were dried overnight at 40 °C under vacuum and afterward submitted to gel permeation chromatography (GPC). GPC was performed in tetrahydrofuran (THF) or 1,1,1,3,3,3-hexafluoroisopropanol (HFIP) as solvents and with the following parts: pump PU 1580, auto sampler AS 1555, UV-detector UV 1575, RI-detector RI 1530 from Jasco, and miniDAWN Tristar light scattering detector from Wyatt. Columns were used from MZ-Analysentechnik: MZ-Gel SDplus 102 Å, MZ-Gel SDplus 104 Å, and MZ-Gel SDplus 106 Å. HPMA polymers were analyzed by HFIP as solvent containing 3 g/L potassium trifluoroacetate. For HFIP GPC, a pump PU 2080+, an autosampler AS1555, and an RI detector RI2080+ from Jasco were used. The elution diagrams were analyzed using the WinGPC Uni Chrom. Calibration was done using polystyrene (THF) or PMMA (HFIP) standards. The flow rate was 1 mL/min at a temperature of 25 °C (THF) and 40 °C (HFIP). For size analysis, a Malvern Zetasizer NanoZS was used. Samples were prepared at 1 mg/mL in Milli-Q water. Each sample was independently measured five times and analyzed by its mean average and standard deviation.
2.3. Synthesis
Synthesis of Pentafluoromethacrylate (PFPMA)
PFPMA was synthesized according to the literature.44
Synthesis of Hymecromonemethacrylate (HCMA)
HCMA was synthesized according to the literature.45
Synthesis of Pentafluorophenol 4-Cyan-4-(phenylcarobonothioylthio)pentanoate (PFP-CTA)
Synthesis of PFP-CTA was performed as recently described in the literature.46
Synthesis of 2-Cyano-5-((4-(6-methyl-1,2,4,5-tetrazine-3-yl)benzyl)amino)-5-oxopentan-2-yl (Tz-CTA)
In a Schlenk tube equipped with a stir bar, PFP-CTA (1 g, 2.25 mmol) was dissolved in 50 mL of anhydrous dioxane under an argon atmosphere. First, triethylamine (227 mg, 2.25 mmol) was added dropwise, and then (4-(6-methyl-1,2,4,5-tetrazine-3-yl)phenyl)methanamine (m-Tz; 347 mg, 1.17 mmol) dissolved in dioxane was added. The red solution was stirred for 19 h at RT under an argon atmosphere. It was concentrated in vacuo and then purified by column chromatography (eluent petrol ether/ethyl acetate = 1:1), obtaining 2-cyano-5-((4-(6-methyl-1,2,4,5-tetrazine-3-yl)benzyl)amino)-5-oxopentan-2-yl benzodithioate as a red solid (497 mg). 1H NMR (CDCl3, 400 MHz): δ [ppm] = 8.54 (d, 2H, Ar–H), 7.90 (dd, 2H, Ar–H), 7.56 (m, 1H, Ar–H), 7.49 (d, 2H, Ar–H), 7.39 (m, 2H, Ar–H), 6.12 (br, 1H, O=C–N–H), 4.57 (m, 2H, NH–CH2), 3.09 (s, 3H, Tz-CH3), 2.71–2.42 (m, 4H, C=O–CH2–CH2), 1.95 (s, 3H, CH3).
Synthesis of p(PFPMA) Macro-CTA
The RAFT polymerization of PFPMA was performed using Tz-CTA for end group functionalized polymers and 4-cyano-4-(phenylcarbonothioylthio)pentanoic acid (acid-CTA) as chain transfer agent and AMDVN as initiator. The polymerization was achieved in different ratios (molar ratios monomer/CTA 40–150/1). The ratio of AMDVN was always 10% of the CTA. The reagents were dissolved in absolute dioxane. After three freeze–vacuum–thaw cycles, the Schlenk tube was immersed in an oil bath at 40 °C for 18 h. The pink polymer solution was then precipitated three times in hexane and dried overnight in a vacuum oven at 40 °C. The product p(PFPMA) was obtained as a pink powder and could be used without further purification as macro-CTA for further polymerization. Yield: 46–57%. 1H NMR (400 MHz, CDCl3): δ [ppm] 2.1–2.5 (br, 2H), 1.3–1.6 (m, 3H). 19F NMR (400 MHz, CDCl3): δ [ppm] −151.5 to −153.1 (br, 2F), −157.9 to −158.2 (br, 1F), −162.9 to −163.4 (br, 2F).
Synthesis of Precursor Block Copolymer
The p(PFPMA) macro-CTA was dissolved in absolute dioxane in a Schlenk tube. LMA, HCMA, and AMDVN were also dissolved in absolute dioxane and added to the polymer solution. After three freeze–vacuum–thaw cycles, the solution was immersed in an oil bath at 40 °C for 4 days. The polymer solution was then precipitated three times in cold methanol and dried in a vacuum oven at 40 °C overnight. The block copolymer p(PFPMA)-b-p(LMA)-ran-p(HCMA) was obtained as a slightly pink powder. Yield: 88%. 1H NMR (400 MHz, CDCl3): δ [ppm] 7.6 (br, 1H, Ar–H), 7.1 (br, 2H, Ar–H), 6.3 (br, 1 H, Ar–H), 3.9 (br, 2H, −CH2), 2.4–0.9 (m, 5H, polymer backbone). 19F NMR (400 MHz, CDCl3): δ [ppm] −151.5 to −153.1 (br, 2F), −157.9 to −158.2 (br, 1F), −162.9 to −163.4 (br, 2F)
p(PFPMA)-b-p(LMA) precursor polymers without cross-linkable HCMA units were synthesized as indicated above without adding HCMA monomer.
Removal of the Dithiobenzoate End Group
The precursor polymer was dissolved in absolute dioxane, and a 20-fold excess of AMDVN in relation to the polymer end group was added. The solution was heated at 40 °C for 16 h. Afterward, the copolymer was precipitated in cold methanol and collected by centrifugation. The copolymer was dried under vacuum at 40 °C overnight, and a pink powder was obtained. Yield: 95–98%.
Synthesis of Amphiphilic Block Copolymers
In a typical reaction, 200 mg of the precursor polymers without the dithiobenzoate end group were dissolved in 2 mL of absolute dioxane. Triethylamine (4 equiv) was then added dropwise to the solution. Anhydrous 2-hydroxypropylamine (HPA; 2 equiv) was dissolved in 2 mL of anhydrous DMSO and added to the solution under an argon atmosphere. In the case of the Tz side chain functionalized polymers, first the desired Tz was added in the designated equivalents and stirred for 6 h at 40 °C before adding HPA. After 24 h, 2 equiv of HPA were added, and the solution was stirred for another 6 h before the transfer of the reaction mixture to a 3500 Da MWCO dialysis bag. The final polymer was dialyzed against water for 3 days, changing the water twice a day. Subsequently, the polymer was lyophilized, obtaining a pink powder. Yield: 84–96%. 1H NMR (400 MHz, DMSO-d6): δ [ppm] = 7.6 (br, 1H), 7.4–7.3 (br, 1H, −NH), 7.1 (br, 2H, Ar–H), 6.3 (br, 1 H, Ar–H), 4.7 (br, 1H, −C–OH), 4.4 (br, 1H, −COOCH2−), 3.7 (br, 1H, CH-HPA), 3.1–2.6 (br, 2H, −NH–CH2–HPA), 1.44–0.76 (br, polymer backbone).
2.4. Nanoparticle Preparation
Micelle Preparation
A total of 3 mg of HPMA-LMA/HCMA polymer with the desired Tz functionality were dissolved in 1 mL of anhydrous DMSO, HFIP, or a mixture of methanol and THF and dialyzed against water using 3500 Da MWCO dialysis bags for 3 days, changing the water three times a day. The micelles were extracted and core cross-linked by UV light for 10 min. Following this, the particles were lyophilized and can be redissolved for further application.
Colloid Preparation
For the miniemulsion process, in a typical reaction, 10 mg of poly(d,l-lactide) were dissolved in 2 g of chloroform. The macroemulsion was prepared by adding the aqueous phase consisting of 10 mg of dissolved block copolymer in 4.5 g of water to the organic phase and subsequent magnetic stirring of the mixture at 1200 rpm for 60 min. Afterward, the macroemulsion was subjected to ultrasonication under ice cooling for 180 s at 70% amplitude in a pulse regime (10 s sonication, 10 s pause) using a Branson ultrasonic device W450 digital, 1/400 tip. The obtained miniemulsion was stirred overnight at room temperature for complete evaporation of the organic solvent. To avoid a reduction of the aqueous phase, lost water was added.
2.5. Reaction Kinetics of Tetrazines
Reaction kinetics were determined using pseudo-first-order measurements with an excess of TCO in PBS (pH = 7.4) at 37.0 ± 0.1 °C.47 Measurements were performed in sextuplicates using a SX20 stopped flow photometer (Applied Photophysics, U.K.). Data analysis was performed by nonlinear fit using Prism 6 (Graphpad) to determine the observed rate constants, which were converted into second order rate constants by division through TCO concentration.
Measurements of reference compounds HELIOS 347Me and HELIOS 388Me48 with PEG4-TCO (see SI, Figure S1) were performed in the fluorescence mode by following the increase in fluorescence above 400 nm. Therefore, the SX20 was equipped with a 360 nm LED light source and a photomultiplier type R374 in combination with a 400 nm long-pass filter as detector. Measurements of Tz containing polymers were performed in the absorbance mode using a 530 nm LED light source and following the decrease of absorbance at this wavelength.
2.6. Structure of the TCO-Functionalized Antibodies
The anti-TAG72 mAb CC49 was conjugated with IRDye 800 CW (LI-COR) and TCO following established procedures.49 Briefly, the a mAb solution in PBS was mixed with 3 molar equiv of NHS-functionalized dye (10 mg/mL solution in dry DMSO), and the pH was adjusted to 8.5 with 1 M sodium carbonate (5 mg/mL CC49 final concentration). The reaction mixture was incubated for 2 h at room temperature in the dark, and then the unreacted dye was removed using a PD-10 desalting cartridge (GE Healthcare Life Sciences). The mAb solution was then diluted to a 1 mg/mL concentration with PBS, added with 10 molar equiv of NHS-functionalized TCO, and the pH was adjusted to 9 with 1 M sodium carbonate. After 2 h incubation at room temperature in the dark, the mixture was 2-fold concentrated using an Amicon centrifugal filter (50 kDa MW cutoff, Merck), and subsequently, the TCO-CC49-IRDye-800CW was purified via PD-10. After purification, the antibody construct was analyzed by SDS-PAGE (see SI, Figure S2). The mAb concentration and dye functionalization grade (0.6 dyes/mAb) were measured by UV (Infinite M200 PRO, TECAN) using extinction coefficients ε280 = 210000 M–1 cm–1 and ε774 = 168000 M–1 cm–1 for the mAb and the dye, respectively. The TCO functionalization grade was measured with a tetrazine titration, as previously reported.49 Briefly, an aliquot of TCO-CC49-IRDye-800CW was reacted with a known excess of an 111In-labeled TZ. After 1 h incubation at 37 °C, the reaction mixture was analyzed by SDS-PAGE and phosphorimager (Typhoon FLA 7000, GE Healthcare Life Scences) in triplicate. The amount of CC49-bound TCO (8 TCOs/mAb) was then calculated from the fraction of mAb-bound radioactivity.
2.7. Protocol for Linking Antibodies and Polymeric Micelles
Click Reaction in PBS Buffer
For the bioorthogonal reaction in PBS buffer, 72 μL of 1 mg/mL solution of the Tz-micelles were added to 2 μL of TCO-antibody solution, with a concentration of 3.69 mg/mL, and incubated for 1 h at 37 °C under continuous agitation. In this case, 61 Tz units would be combined with 1 TCO unit.
Click Reaction in Human Blood Plasma
For the bioorthogonal reaction in human blood plasma, 2 μL of TCO-antibody solution, with a concentration of 3.69 mg/mL, were added to 222 μL of pure plasma, and subsequently, 72 μL of 1 mg/mL solution of the Tz-micelles were added to the mixture. The plasma solution was incubated for 1 h at 37 °C under continuous agitation. In this case, 61 Tz units would be combined with 1 TCO unit, diluted 4-fold with plasma.
2.8. Fluorescence Correlation Spectroscopy Measurements
FCS experiments were performed on a commercial confocal microscope (LSM 880, Carl Zeiss, Jena, Germany) customized for near-infrared (NIR) measurements, as described in detail elsewhere.50 Briefly, the excitation of the IR-Dye CW-800 labels was done by a Ti:Sa laser (Mai Tai, Newport, U.S.A.) operating at 780 nm and focused into the sample by a high numerical aperture water immersion objective (C-Apochromat 40x/1.2 W, Carl Zeiss, Jena, Germany). The fluorescence was collected with the same objective and, after passing through a dichroic mirror, a confocal pinhole (54 μm), and 835/70 emission filter, was delivered to an avalanche photodiode detector, integrated in a FLIM and FCS upgrade kit (PicoQuant, Berlin, Germany) fiber coupled to the microscope.
After the respective incubation times, the micelle–antibody solutions were further diluted 10-fold with blood plasma and poured into eight-well polystyrene-chambered coverglass (Laboratory-Tek, Nalge Nunc International, Penfield, NY, U.S.A.) mounted in a microscope incubator (PM 2000 RBT, Pecon, Erbach, Germany) that kept the temperature at 37 °C during the FCS measurements.
For each sample, a series of 10 measurements (10 s each) were performed. The time-dependent fluctuations of the fluorescence intensity δI(t), caused by the diffusion of the fluorescent species through the confocal observation volume, were recorded and analyzed by an autocorrelation function:
| 1 |
As it has been shown theoretically for an ensemble of m different types of freely diffusing fluorescent species, G(τ) has the following analytical form:51
| 2 |
Here, N is the average number
of diffusing fluorescent species in the observation volume, τD,i is the diffusion time of the i-th species, fi is the
fraction of component i, and S is
the so-called structure parameter 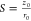 , where z0 and r0 represent
the axial and radial dimension of
the confocal volume, respectively. Furthermore, the diffusion time
τD,i is related to the respective
diffusion coefficient Di, through
, where z0 and r0 represent
the axial and radial dimension of
the confocal volume, respectively. Furthermore, the diffusion time
τD,i is related to the respective
diffusion coefficient Di, through 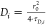 . The experimental autocorrelation
curves
were fitted with eq 2, yielding the fractions, the corresponding diffusion times, and
subsequently, the diffusion coefficients of the fluorescent species.
Finally, the hydrodynamic radii Rh were
calculated using the Stokes–Einstein relation as
. The experimental autocorrelation
curves
were fitted with eq 2, yielding the fractions, the corresponding diffusion times, and
subsequently, the diffusion coefficients of the fluorescent species.
Finally, the hydrodynamic radii Rh were
calculated using the Stokes–Einstein relation as 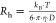 , where T is the absolute
temperature, kB is the Boltzmann constant,
and η is the viscosity of the solvent.
, where T is the absolute
temperature, kB is the Boltzmann constant,
and η is the viscosity of the solvent.
3. Results and Discussion
3.1. Polymer Synthesis
The goal of this study was to prepare different biocompatible amphiphilic block copolymers, from which non-cross-linked or core-cross-linked polymer micelles can be prepared. Non-cross-linked or core-cross-linked polymer micelles behave rather similar in water or aqueous buffer. Core-cross-linked micelles have, however, advantages in in vivo applications (they are more stable) because they cannot dissociate into unimers.52 The polymers shall be functionalized with Tzs and tested in bioorthogonal ligation reactions. For this purpose, we chose a HPMA-based polymer system. These polymers are well-known in literature for their biocompatibility and already used in medical applications.53,54
Due to the fact that this study focuses on the reactivity toward iEDDA click reaction, we chose two different ways to incorporate the Tz units into the amphiphilic HPMA-based block copolymers. The first is to include the Tz into the end group, the second is to attach the Tz into the hydrophilic block by a polymer analogues reaction. The first approach has the advantage that the reactive group is at the end of the hydrophilic chain of the polymer, which should lead to good accessibility of the Tz. The second strategy has the benefit that multiple reactive Tzs can be incorporated into the hydrophilic block. However, their accessibility might be reduced. For the first strategy we synthesized a new chain transfer agent (CTA) for reversible addition–fragmentation polymerization (RAFT). The synthesis of this novel structure is shown in Figure 2. The methyl tetrazine-CTA (Tz-CTA) was synthesized by aminolysis of the pentafluorophenyl 4-cyano-4-((phenylcarbonothioyl)thio)-pentanoate PFP-CTA with (4-(6-methyl-1,2,4,5-tetrazine-3-yl)phenyl)methanamine and characterized by 1H NMR (see Figure 3a).
Figure 2.

Synthesis of the methyl tetrazine-CTA.
Figure 3.

(a) 1H NMR (400 Hz, CDCl3) spectrum of Tz-CTA. (b) SEC elugram from p(PFPMA) and p(PFPMA)-b-p(LMA) polymers, synthesized via Tz-CTA (solvent: THF, calibration: PS).
This Tz-functionalized CTA was applied in a RAFT polymerization using the reaction conditions established for other functionalized CTAs.55 The successful polymerization with the Tz-CTA is pictured in Figure 3b. For polymerization of the p(PFPMA) homopolymer (macro-CTA), different chain lengths were prepared as depicted in Table S1 in the SI. To prevent degradation of the end group the initiator 2,2-azobis(4-methoxy)-2,4-dimethylvaleronitrile (AMDVN, V-70) was used, which initiates at 40 °C. The resulting macro-CTA was subsequently used for block copolymerization with either only lauryl methacrylate (LMA), yielding p(PFPMA)-b-p(LMA) precursor polymers or applied in a statistical copolymerization with LMA and hymecromonemethacrylate (HCMA) to produce the cross-linkable p(PFPMA)-b-p(LMA)-ran-p(HCMA) precursor polymer, as displayed in Figure 4. The synthesis of both types of block copolymers worked well and, especially for the non-cross-linkable polymers, a narrow dispersity could be achieved (Table 1). To avoid side reactions in the polymeranalogous reaction later on, the dithiobenzoate group was removed after polymerization by adding an excess of the initiator. In the last step, the precursor polymers were converted into the amphiphilic block copolymers. This was done with hydroxyl propyl amine (HPA) and triethyl amine (NEt3) in an aminolysis reaction, yielding the final amphiphilic p(HPMA)-b-p(LMA) and the cross-linkable p(HPMA)-b-p(LMA)-ran-p(HCMA), respectively (Figure 4).45
Figure 4.

Synthesis of end-group-functionalized polymers P1–P6.
Table 1. Characterization of End-Group Functionalized Polymers P1a–P6a and the Final Amphiphilic Block Copolymers P1–P6.
| label | polymer | Mn in kg/mol | amt of hydrophobic units mol % | PDI |
|---|---|---|---|---|
| P1a | Tz-PFPMA-LMA | 15.9 | 26 | 1.22 |
| P2a | 19.6 | 25 | 1.27 | |
| P3a | 21.7 | 25 | 1.24 | |
| P4a | Tz-PFPMA-LMA/HCMA | 11.4 | 25 | 1.43 |
| P5a | 16.7 | 23 | 1.48 | |
| P6a | 19.6 | 21 | 1.52 | |
| P1 | Tz-HPMA-LMA | 9.5 | 26 | |
| P2 | 11.8 | 25 | ||
| P3 | 13.0 | 25 | ||
| P4 | Tz-HPMA-LMA/HCMA | 6.8 | 25 | |
| P5 | 10.0 | 23 | ||
| P6 | 11.8 | 21 |
For side chain functionalization we used the same RAFT technique as described above. But instead of the new Tz-CTA, we deployed the acid-CTA as CTA (see Figures 2 and 5), as recently described.45 In the last step, we transformed the precursor polymers in the respective amphiphilic HPMA-based block copolymer (Figure 5). Hereby, we could incorporate Tz in various quantities during the polymer analogous reaction into the hydrophilic part and characterize them by 1H NMR technique (see SI, Figure S9). The non-cross-linkable polymers are displayed in Table 2, and the HCMA containing cross-linkable polymers are depicted in Table 3. During this process it is, of course, also possible to add additional Tz to a polymer already functionalized at the chain end (see polymer P13.1 in Table 3).
Figure 5.

Synthesis of side chain functionalized polymers P7 to P12.
Table 2. Side-Chain-Functionalized, Non-Cross-Linkable Polymers P7–P9.
| label | polymer | tetrazine units | Mn in kg/mol | amt of hydrophobic units mol % | PDI |
|---|---|---|---|---|---|
| P7 | PFPMA-LMA | 16.8 | 23 | 1.24 | |
| P7.1 | Tz/HPMA-LMA | 1 | 10.1 | ||
| P7.5 | 5 | 10.1 | |||
| P7.8 | 8 | 10.1 | |||
| P8 | PFPMA-LMA | 24 | 22 | 1.39 | |
| P8.1 | Tz/HPMA-LMA | 1 | 14.4 | ||
| P8.5 | 5 | 14.4 | |||
| P8.8 | 8 | 14.4 | |||
| P9 | PFPMA-LMA | 30.2 | 24 | 1.4 | |
| P9.1 | Tz/HPMA-LMA | 1 | 18.1 | ||
| P9.5 | 5 | 18.1 | |||
| P9.8 | 8 | 18.1 |
Table 3. Side-Chain-Functionalized Cross-Linkable Polymers P10–P12 and End and Side-Chain-Functionalized Polymer P13.
| label | polymer | tetrazine units | Mn in kg/mol | amt of hydrophobic units mol % | PDI |
|---|---|---|---|---|---|
| P10 | PFPMA-LMA/HCMA | 15.0 | 19 | 1.43 | |
| P10.1 | Tz/HPMA-LMA/HCMA | 1 | 9.0 | ||
| P10.5 | 5 | 9.0 | |||
| P10.8 | 8 | 9.0 | |||
| P11 | PFPMA-LMA/HCMA | 23.2 | 21 | 1.47 | |
| P11.1 | Tz/HPMA-LMA/HCMA | 1 | 13.9 | ||
| P11.5 | 5 | 13.9 | |||
| P11.8 | 8 | 13.9 | |||
| P12 | PFPMA-LMA/HCMA | 29.0 | 20 | 1.44 | |
| P12.1 | Tz/HPMA-LMA/HCMA | 1 | 17.4 | ||
| P12.5 | 5 | 17.4 | |||
| P12.8 | 8 | 17.4 | |||
| P13 | Tz-PFPMA-LMA/HCMA | 19.0 | 19 | 1.44 | |
| P13.1 | Tz-Tz/HPMA-LMA/HCMA | 4 | 12.8 |
The block copolymers described above form spontaneously micellar structures (nanoparticles) in water or PBS buffer. This is important for possible applications, because the biodistribution of nanoparticles can be influenced by their size. And the size of the micellar structures from the HPMA-LMA/HCMA block copolymers can be varied by the process during their preparation from 20 to 160 nm in diameter. Nanoparticles prepared from the tetrazine containing polymers are collected in Table S2. They can be stabilized by cross-linking after micellar formation if they contain the HCMA unit.45 Even larger nanoparticles (170–400 nm diameter) can be manufactured in a miniemulsion process as recently described.56 Thus, particles, which can diffuse into a tumor (below 100 nm) or particles, which will end up in the liver (>100 nm) are rapidly accessible.1
If large nanoparticles are desired, colloids filled with a hydrophobic polymer can be prepared in a miniemulsion process as shown in the Supporting Information (Figure S14 and Nanoparticle Preparation).56 Here poly(d,l-lactide) (PDLLA) was used as hydrophobic polymer due to its biocompatible and biodegradable properties.57 The size of the colloids can be characterized by dynamic light scattering (DLS). In this way colloids with a diameter ranging from 170 to 370 nm are accessible (see Table S2).
The preparation of much smaller core cross-linked micelles is realized by a solvent switch process. As published recently the size of these nanoparticles can be adjusted by the choice of method and solvent.45 Their size can be determined by DLS (Zetasizer). The sizes of all resulting particles, independent of the preparation protocol, are compiled in the SI (Table S2).
3.2. Reaction Kinetics
In order to determine the reaction kinetics of the differently functionalized polymers the rate constants were measured by stopped-flow spectroscopy in aqueous PBS buffer using PEG4-TCO as a water-soluble small molecule reactant (see Supporting Information, Figure S1). Under these conditions the polymers have formed micellar structures with the Tz units distributed between the hydrophilic corona and the hydrophobic core. So, in fact, the reactivity of the hydrophilic structure is determined directly.
As a reference, the rate constants of the reaction between PEG4-TCO and fluorogenic phenylene-aryl tetrazines HELIOS 347Me and HELIOS 388Me were measured under the same conditions and revealed second order rate constants of 620 and 500 M–1 s–1, respectively.58 Measured rate constants for end-group-functionalized cross-linkable and non-cross-linkable polymers (P1–P6, Figure 6a) were determined to be around 600 M–1 s–1, independent of the molecular weight ranging from 7 to 13 kDa. The rate constants for side chain functionalized polymers P7.1–P7.8 and P10.1–P10.8 exhibit similar rates to the end group bearing polymers (P1–P6) with rates around 600 M–1 s–1 when normalized to Tz amount. Second order rate constants for polymers prepared from the precursor polymers P8, P9, P11, and P12 (Tables 2 and 3) show similar values of approximately 600 M–1 s–1. The high reactivity of the methyl tetrazine is thus observed for all tested polymers. It can therefore be concluded, that in these polymers the reactivity of the Tz-units with small molecule TCOs is not influenced by the local environment and especially by the fact, if they are chemically linked to the chain end or are statistically distributed in the hydrophilic block. Also, the binding of up to eight tetrazines per polymer chain, which might lead to stronger hydrophobic effects and a hiding of the tetrazines in the hydrophobic core, does not lead to a significant reduction in reactivity. Thus, it is unlikely that much of the tetrazines “hide permanently” in the hydrophobic core. As a result, polymers P7.1–P7.8 and P10.1–P10.8 offer the possibility to increase reactivity by incorporation of several units of Tz. Normalizing the reactivity to the polymer concentration increases the observed second order rate constants reaching rates of several thousand M–1 s–1, for example, in the case of P7.8 (Figure 6b).
Figure 6.

(a) Second order rate constants per Tz unit for the reaction between polymers and TCO-PEG4 for non-cross-linkable polymers (red) and cross-linkable polymers (blue) in PBS at 25 °C. (b) Second order rate constants of P7.1–P7.8 and P10.1–P10.8, bearing 1, 5, and 8 units of Tz normalized to the polymer concentration.
Experiments to verify if all tetrazines are accessible for the reaction with TCO-units were performed by UV–vis measurements and are presented in the SI (Figure S17). They could, however, not finally solve the question due to a strong increase of scattering of the micellar solution upon approaching full conversion with a TCO Amine (see SI, Figure S20 for the structure). This makes it impossible to determine, if small amounts of tetrazines are left unreacted.
3.3. Bioconjugation to Functionalized Antibodies
Next, we tested the ability of the core cross-linked micelles (nanoparticles) to undergo a click reaction with large biomolecules like antibodies (see Figure 7). Previous studies (click reactions performed between azide and cyclooctyne) had shown that this reaction gets very slow due to the slow diffusion of the large reactants.25 Thus, it could only be performed in extremely concentrated systems, which is, of course, incompatible with in vivo conditions. To increase the chance for a reaction, it is therefore desirable to increase the reactivity of the click reaction and to increase the number of reactive groups per particle. In this context, it is not necessary to look for a stochiometric reaction. The quick formation of some stable bonds is sufficient. Fluorescence correlation spectroscopy (FCS) had proven to be a perfect measurement technique to follow the ligation between the polymer nanoparticle and the modified antibody.25 Moreover, it can be applied not only in aqueous solutions, but also in blood plasma or full blood.50
Figure 7.

Schematic illustration of particle preparation followed by cross-linking to prepare the core-cross-linked micelles and the subsequent antibody click reaction (pink: Tz, green: dye, yellow: TCO, red: ligation product).
Here we choose a more reactive system, which consists of a fluorescently labeled CC49 antibody (IR-Dye CW-800 as fluorescent label), which is functionalized with eight trans-cyclooctene units49 (see SI, Figure S2, for characterization) and let it react with nanoparticles functionalized with the more reactive tetrazines. In this context it is important that the less reactive methyl substituted Tzs (R1 = CH3) have been shown to be stable in blood plasma.43 Concerning the block copolymers, we selected P13.1, which contains altogether four Tz units per polymer (one at the chain end and three in the hydrophilic block). From those we prepared two types of core-cross-linked micelles to prevent the dissociation dynamics (P13.1A and P13.1B) by variation of the preparation procedure. They are compiled in Table 4 and characterized by dynamic light scattering. They differ in diameter by about 15 nm. As both are not fluorescently labeled, FCS can then detect the increase of the size of the fluorescently labeled antibody, while it gets ligated to the polymer nanoparticle functionalized with Tz.
Table 4. Hydrodynamic Diameters (Dh) of the Particles P13.1A and P13.1Ba.
| particle type | preparation | polymer label | size (Dh) in nm | PdI |
|---|---|---|---|---|
| core-cross-linked micelle | dialysis from MeOH/THF P13.1A | P13.1 | 17.2 | 0.21 |
| dialysis from HFIP P13.1B | P13.1 | 32.3 | 0.36 |
The micelles are obtained by the dialysis approach using MeOH/THF or HFIP as solvent.
As the click reaction with Tz is discussed for in vivo click reactions, we verified in the next step that this reaction can also be performed in human blood plasma. For this purpose, we tested the ligation of trans-cyclooctene functionalized antibodies with the core-cross-linked micelles P13.1A and P13.1B and followed their size increase by FCS measurements (see Figure 8a,b). For the click reaction, we choose a concentration of 0.243 mg of core-cross-linked micelles and 0.025 mg of antibody per mL of human plasma. This concentration represents a possible in vivo scenario, because concentrations of 1 mg (or above) of polymeric micelles per mL of aqueous PBS buffer can be prepared easily. From such a solution, 200 μL can be given to mice, where it would lead to a concentration of 0.133 mg/mL in the blood pool (assuming about 1.5 mL of blood per 20 g mouse). Therefore, the concentrations tested here in vitro are in a similar range.
Figure 8.

(a) Antibody click with particle P13.1A in plasma. Normalized FCS autocorrelation curves of the antibody in plasma (black) and the antibody-particle-click after 1 h incubation in plasma (blue). For comparison the curve of the antibody-particle-click obtained after 1 h incubation in PBS and then diluted in plasma is also shown (green). (b) Antibody click with particle P13.1B in plasma. Normalized FCS autocorrelation curves of the antibody in plasma (black) and of the antibody-particle-click after 1 h incubation in plasma (blue). For comparison, the curve of the antibody-particle-click obtained after 1 h incubation in PBS and then diluted in plasma is also shown (green). (c) Fractions of the click-product (red) and the free antibody (black) vs the incubation time for the antibody-particle-mixture in plasma. (d) Antibody click with particle P13.1A in PBS. Size measurement of the particle (black) and the antibody-particle-click (red) after 1 h incubation in PBS.
As FCS measurements require a highly diluted solution, we incubated both reactants for 1 h at 37 °C in human plasma at the concentration given above and diluted them afterward with more plasma. The FCS measurements (Figure 8, note that the figures give the hydrodynamic radius, not the diameter) show that the size of the fluorescent antibodies (14 nm in diameter in plasma) increases as they got ligated to the nonfluorescent core-cross-linked micelles, for which the diameter had been determined independently by dynamic light scattering (Table 4). This increase in size was observed for the two batches of core-cross-linked micelles (increase to 26 and 50 nm in diameter) prepared by variation of their preparation conditions.42 For the system with smaller core-cross-linked micelles we studied also the kinetic of the process by performing FCS measurements at defined time intervals after mixing. The recorded FCS autocorrelation curves were fitted with a two-component fit (m = 2 in eq 2 in the Materials and Methods) to account for the small freely diffusing antibodies and for the larger antibody-particle-clicks, respectively. The fractions (fi in eq 2 in Materials and Methods) of these components were obtained from the fits and plotted versus incubation time in Figure 8c. It shows that after about 10 min most of the ligation did happen and 90% of the antibodies got linked to the micellar structures, resulting in a click efficiency of approximately 90%. Thus, this reaction should also be applicable in a living body. Finally, we repeated the experiment in aqueous PBS buffer for the core cross-linked particle P13.1A (see Table 4) and determined the size by DLS (Zetasizer). Again, a comparable size increase from 17.2 nm for the micelle to 22.2 nm for the micelle-antibody-click-product could be determined (Figure 8d).
To exclude that the size increase is a consequence of an unspecific interaction (protein corona formation) and to demonstrate that it does result from the click-reaction, we performed a control experiment. Therefore, we added the TCO functionalized (fluorescent) antibodies to core-cross-linked micelles without Tz units in the corona and performed the FCS-measurement again (see SI, Figure S18). In this case, no size increase can be detected, which proves that the tetrazines are required for the ligation.
In this context, we looked also at the disappearance of the tetrazine band at 520 nm in an UV–vis experiment (see SI, Figure S19). In UV, a strong reduction of the tetrazine band is clearly visible at the beginning of the reaction. The end point of the reaction can, however, not be determined as the overall adsorption is increasing due to the adsorption of the antibodies. However, it should be stressed here again that the click reaction between nanoparticle and antibody is not supposed to be stochiometric, and it is not the intention to let all tetrazines react with all TCO units.
This shows that these Tz-covered nanoparticles have the ability to react fast and effectively with large biomacromolecules, such as antibodies, and that this reaction is not changed in a biologically relevant medium like human blood plasma.
4. Conclusion
The aim of this study was to develop a fast reacting, bioorthogonal HPMA-based amphiphilic block copolymer system for further functionalization with antibodies and radiolabels. Different effective pathways to introduce reactive tetrazine groups into the hydrophilic part of the polymer could be established. These amphiphilic block copolymers showed very fast reaction kinetics. It can be concluded that the attachment of tetrazines to the polymer does not influence the second order rate constant of the click reaction compared to small molecules. In addition, the inclusion of several tetrazine units per polymer can further increase the reactivity toward TCO. The polymers could be successfully employed to produce nanoparticles of various sizes. These nanocarriers can be employed in different biomedical applications, due to the size variation between 20 and 400 nm in diameter. Additionally, we showed that these nanoparticles have a high efficacy and fast kinetics in conjugations with TCO bearing antibodies, even in human blood plasma. This approves one of the major applications for these particles, the rapid and bioorthogonal reaction with biomacromolecules.
Acknowledgments
This project has received funding from the European Union's Horizon 2020 research and innovation programme under Grant Agreement no. 668532 (Click-It).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.biomac.9b00868.
More details concerning (1) the new polymers and their (2) aggregate formation and the (3) kinetics of their click reaction, as well as (4) the TCO-modified antibodies and (5) control experiments to verify the click reaction in plasma (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Sun T.; Zhang Y. S.; Pang B.; Hyun D. C.; Yang M.; Xia Y. Engineered Nanoparticles for Drug Delivery in Cancer Therapy. Angew. Chem., Int. Ed. 2014, 53, 12320–64. 10.1002/anie.201403036. [DOI] [PubMed] [Google Scholar]
- Padmanabhan P.; Kumar A.; Kumar S.; Chaudhary R. K.; Gulyás B. Nanoparticles in Practice for Molecular-Imaging Applications: An Overview. Acta Biomater. 2016, 41, 1–16. 10.1016/j.actbio.2016.06.003. [DOI] [PubMed] [Google Scholar]
- Grabbe S.; Landfester K.; Schuppan D.; Barz M.; Zentel R. Nanoparticles and the Immune System. Nanomedicine (London, U. K.) 2016, 11 (20), 2621–2624. 10.2217/nnm-2016-0281. [DOI] [PubMed] [Google Scholar]
- Duncan R. The Dawning Era of Polymer Therapeutics. Nat. Rev. Drug Discovery 2003, 2 (5), 347–360. 10.1038/nrd1088. [DOI] [PubMed] [Google Scholar]
- Kunjachan S.; Ehling J.; Storm G.; Kiessling F.; Lammers T. Noninvasive Imaging of Nanomedicines and Nanotheranostics: Principles, Progress, and Prospects. Chem. Rev. 2015, 115, 10907–37. 10.1021/cr500314d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelaz B.; Alexiou C.; Alvarez-Puebla R. A.; Alves F.; Andrews A. M.; Ashraf S.; Balogh L. P.; Ballerini L.; Bestetti A.; Brendel C.; et al. Diverse Applications of Nanomedicine. ACS Nano 2017, 11, 2313–2381. 10.1021/acsnano.6b06040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J.; Kantoff P. W.; Wooster R.; Farokhzad O. C. Cancer Nanomedicine: Progress, Challenges and Opportunities. Nat. Rev. Cancer 2017, 17 (1), 20–37. 10.1038/nrc.2016.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dube A.; Lemmer Y.; Hayeshi R.; Balogun M.; Labuschagne P.; Swai H.; Kalombo L. State of the Art and Future Directions in Nanomedicine for Tuberculosis. Expert Opin. Drug Delivery 2013, 10 (12), 1725–1734. 10.1517/17425247.2014.846905. [DOI] [PubMed] [Google Scholar]
- Karthivashan G.; Ganesan P.; Park S.-Y.; Kim J.-S.; Choi D.-K. Therapeutic Strategies and Nano-Drug Delivery Applications in Management of Ageing Alzheimer’s Disease. Drug Delivery 2018, 25 (1), 307–320. 10.1080/10717544.2018.1428243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etheridge M. L.; Campbell S. A.; Erdman A. G.; Haynes C. L.; Wolf S. M.; McCullough J. The Big Picture on Nanomedicine: The State of Investigational and Approved Nanomedicine Products. Nanomedicine 2013, 9 (1), 1–14. 10.1016/j.nano.2012.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Z.; Yan X.; Sun X.; Shen B.; Gambhir S. S. Tumor Molecular Imaging with Nanoparticles. Engineering 2016, 2 (1), 132–140. 10.1016/J.ENG.2016.01.027. [DOI] [Google Scholar]
- Matsumura Y.; Maeda H. A New Concept for Macromolecular Therapeutics in Cancer Chemotherapy: Mechanism of Tumoritropic Accumulation of Proteins and the Antitumor Agent Smancs. Cancer Res. 1986, 46, 6387–6392. [PubMed] [Google Scholar]
- Maeda H.; Nakamura H.; Fang J. The EPR Effect for Macromolecular Drug Delivery to Solid Tumors: Improvement of Tumor Uptake, Lowering of Systemic Toxicity, and Distinct Tumor Imaging in Vivo. Adv. Drug Delivery Rev. 2013, 65 (1), 71–79. 10.1016/j.addr.2012.10.002. [DOI] [PubMed] [Google Scholar]
- Maeda H.; Tsukigawa K.; Fang J. A Retrospective 30 Years After Discovery of the Enhanced Permeability and Retention Effect of Solid Tumors: Next-Generation Chemotherapeutics and Photodynamic Therapy—Problems, Solutions, and Prospects. Microcirculation 2016, 23 (3), 173–182. 10.1111/micc.12228. [DOI] [PubMed] [Google Scholar]
- Youn Y. S.; Bae Y. H. Perspectives on the Past, Present, and Future of Cancer Nanomedicine. Adv. Drug Delivery Rev. 2018, 130, 3–11. 10.1016/j.addr.2018.05.008. [DOI] [PubMed] [Google Scholar]
- Kiessling F.; Mertens M. E.; Grimm J.; Lammers T. Nanoparticles for Imaging: Top or Flop ?. Radiology 2014, 273, 10–28. 10.1148/radiol.14131520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saleem A.; Charnley N.; Price P. Clinical Molecular Imaging with Positron Emission Tomography. Eur. J. Cancer 2006, 42 (12), 1720–1727. 10.1016/j.ejca.2006.02.021. [DOI] [PubMed] [Google Scholar]
- Chapman S.; Dobrovolskaia M.; Farahani K.; Goodwin A.; Joshi A.; Lee H.; Meade T.; Pomper M.; Ptak K.; Rao J.; et al. Nanoparticles for Cancer Imaging: The Good, the Bad, and the Promise. Nano Today 2013, 8, 454–460. 10.1016/j.nantod.2013.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissleder R.; Pittet M. J. Imaging in the Era of Molecular Oncology. Nature 2008, 452, 580–589. 10.1038/nature06917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Key J.; Leary J. F. Nanoparticles for Multimodal in Vivo Imaging in Nanomedicine. Int. J. Nanomed. 2014, 9, 711–726. 10.2147/IJN.S53717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kluetz P. G.; Meltzer C. C.; Villemagne V. L.; Kinahan P. E.; Chander S.; Martinelli M. A.; Townsend D. W. Combined PET/CT Imaging in Oncology: Impact on Patient Management. Clin. Positron Imaging 2000, 3 (6), 223–230. 10.1016/S1095-0397(01)00055-3. [DOI] [PubMed] [Google Scholar]
- Pichler B. J.; Kolb A.; Nagele T.; Schlemmer H.-P. PET/MRI: Paving the Way for the Next Generation of Clinical Multimodality Imaging Applications. J. Nucl. Med. 2010, 51, 333–336. 10.2967/jnumed.109.061853. [DOI] [PubMed] [Google Scholar]
- Meyer J.; Adumeau P.; Lewis J. S.; Zeglis B. M. Click Chemistry and Radiochemistry: The First 10 Years. Bioconjugate Chem. 2016, 27, 2791–2807. 10.1021/acs.bioconjchem.6b00561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozma E.; Demeter O.; Kele P. Bio-Orthogonal Fluorescent Labelling of Biopolymers through Inverse-Electron-Demand Diels – Alder Reactions. ChemBioChem 2017, 18 (6), 486–501. 10.1002/cbic.201600607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck S.; Schultze J.; Räder H. J.; Holm R.; Schinnerer M.; Barz M.; Koynov K.; Zentel R. Site-Specific DBCO Modification of DEC205 Antibody for Polymer Conjugation. Polymers 2018, 10 (2), 1–25. 10.3390/polym10020141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deri M. A.; Zeglis B. M.; Francesconi L. C.; Lewis J. S. PET Imaging with Zr: From Radiochemistry to the Clinic. Nucl. Med. Biol. 2013, 40 (1), 3–14. 10.1016/j.nucmedbio.2012.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dziel T.; Listkowska A.; Tyminski Z. Standardisation and Half-Life Measurements of 111In. Appl. Radiat. Isot. 2016, 109, 345–348. 10.1016/j.apradiso.2015.11.105. [DOI] [PubMed] [Google Scholar]
- Rossin R.; Renart Verkerk P.; van den Bosch S. M.; Vulders R. C. M.; Verel I.; Lub J.; Robillard M. S. In Vivo Chemistry for Pretargeted Tumor Imaging in Live Mice. Angew. Chem., Int. Ed. 2010, 49 (19), 3375–3378. 10.1002/anie.200906294. [DOI] [PubMed] [Google Scholar]
- Zeglis B. M.; Sevak K. K.; Reiner T.; Mohindra P.; Carlin S. D.; Zanzonico P.; Weissleder R.; Lewis J. S. A Pretargeted PET Imaging Strategy Based on Bioorthogonal Diels-Alder Click Chemistry. J. Nucl. Med. 2013, 54 (8), 1389–1396. 10.2967/jnumed.112.115840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altai M.; Membreno R.; Cook B.; Tolmachev V.; Zeglis B. M. Pretargeted Imaging and Therapy. J. Nucl. Med. 2017, 58 (10), 1553–1559. 10.2967/jnumed.117.189944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossin R.; Robillard M. S. Pretargeted Imaging Using Bioorthogonal Chemistry in Mice. Curr. Opin. Chem. Biol. 2014, 21, 161–169. 10.1016/j.cbpa.2014.07.023. [DOI] [PubMed] [Google Scholar]
- Devaraj N. K.; Weissleder R. Biomedical Applications of Tetrazine Cycloadditions. Acc. Chem. Res. 2011, 44, 816–827. 10.1021/ar200037t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svatunek D.; Denk C.; Rosecker V.; Sohr B.; Hametner C.; Allmaier G.; Fröhlich J.; Mikula H. Efficient Low-Cost Preparation of Trans -Cyclooctenes Using a Simplified Flow Setup for Photoisomerization. Monatsh. Chem. 2016, 147, 579–585. 10.1007/s00706-016-1668-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxon E.; Bertozzi C. R. Cell Surface Engineering by a Modified Staudinger Reaction. Science 2000, 287 (5460), 2007–2010. 10.1126/science.287.5460.2007. [DOI] [PubMed] [Google Scholar]
- Saxon E.; Armstrong J. I.; Bertozzi C. R. A “ Traceless ” Staudinger Ligation for the Chemoselective Synthesis of Amide Bonds. Org. Lett. 2000, 2 (14), 2141–2143. 10.1021/ol006054v. [DOI] [PubMed] [Google Scholar]
- Agard N. J.; Prescher J. A.; Bertozzi C. R. A Strain-Promoted [3 + 2] Azide-Alkyne Cycloaddition for Covalent Modification of Biomolecules in Living Systems. J. Am. Chem. Soc. 2004, 126 (46), 15046–15047. 10.1021/ja044996f. [DOI] [PubMed] [Google Scholar]
- Oliveira B. L.; Guo Z.; Bernardes G. J. L. Inverse Electron Demand Diels-Alder Reactions in Chemical Biology. Chem. Soc. Rev. 2017, 46, 4895–4950. 10.1039/C7CS00184C. [DOI] [PubMed] [Google Scholar]
- Rostovtsev V. V.; Green L. G.; Fokin V. V.; Sharpless K. B. A Stepwise Huisgen Cycloaddition Process: Copper (I) -Catalyzed Regioselective “Ligation” of Azides and Terminal Alkynes. Angew. Chem., Int. Ed. 2002, 41 (14), 2596–2599. . [DOI] [PubMed] [Google Scholar]
- Li L.; Zhang Z. Development and Applications of the Copper-Catalyzed Azide-Alkyne Cycloaddition (CuAAC) as a Bioorthogonal Reaction. Molecules 2016, 21, 1393. 10.3390/molecules21101393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devaraj N. K.; Weissleder R.; Hilderbrand S. A. Bioconjugate Chem. 2008, 19, 2297–2299. 10.1021/bc8004446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackman M. L.; Royzen M.; Fox J. M. J. Am. Chem. Soc. 2008, 130, 13518–13519. 10.1021/ja8053805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darko A.; Wallace S.; Dmitrenko O.; Machovina M. M.; Mehl R. A.; Chin W.; Fox J. M. Conformationally Strained Trans-Cyclooctene with Improved Stability and Excellent Reactivity in Tetrazine Ligation. Chem. Sci. 2014, 5, 3770–3776. 10.1039/C4SC01348D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karver M. R.; Weissleder R.; Hilderbrand S. A. Synthesis and Evaluation of a Series of 1,2,4,5-Tetrazines for Bioorthogonal Conjugation. Bioconjugate Chem. 2011, 22, 2263–2270. 10.1021/bc200295y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eberhardt M.; Mruk R.; Zentel R.; Théato P. Synthesis of Pentafluorophenyl(Meth)Acrylate Polymers: New Precursor Polymers for the Synthesis of Multifunctional Materials. Eur. Polym. J. 2005, 41 (7), 1569–1575. 10.1016/j.eurpolymj.2005.01.025. [DOI] [Google Scholar]
- Kramer S.; Kim K. O.; Zentel R. Size Tunable Core Crosslinked Micelles from HPMA-Based Amphiphilic Block Copolymers. Macromol. Chem. Phys. 2017, 218 (19), 1700113. 10.1002/macp.201700113. [DOI] [Google Scholar]
- Roth P. J.; Wiss K. T.; Zentel R.; Theato P. Synthesis of Reactive Telechelic Polymers Based on Pentafluorophenyl Esters. Macromolecules 2008, 41, 8513–8519. 10.1021/ma801681b. [DOI] [Google Scholar]
- Svatunek D.; Denk C.; Mikula H. A Computational Model to Predict the Diels – Alder Reactivity of Aryl/Alkyl-Substituted Tetrazines. Monatsh. Chem. 2018, 149 (4), 833–837. 10.1007/s00706-017-2110-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meimetis L. G.; Carlson J. C. T.; Giedt R. J.; Kohler R. H.; Weissleder R. Ultrafluorogenic Coumarin-Tetrazine Probes for Real-Time Biological Imaging. Angew. Chem., Int. Ed. 2014, 53 (29), 7531–7534. 10.1002/anie.201403890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossin R.; van Duijnhoven S. M. J.; Läppchen T.; Van Den Bosch S. M.; Robillard M. S. Trans -Cyclooctene Tag with Improved Properties for Tumor Pretargeting with the Diels - Alder Reaction. Mol. Pharmaceutics 2014, 11, 3090–3096. 10.1021/mp500275a. [DOI] [PubMed] [Google Scholar]
- Negwer I.; Best A.; Schinnerer M.; Schafer O.; Capeloa L.; Wagner M.; Schmidt M.; Mailander V.; Helm M.; Barz M.; Butt H.-J.; Koynov K. Monitoring Drug Nanocarriers in Human Blood by Near-Infrared Fluorescence Correlation Spectroscopy. Nat. Commun. 2018, 9 (1), na. 10.1038/s41467-018-07755-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigler R.; Wennmalm S.; Edman L. FCS in Single Molecule Analysis. Springer Ser. Chem. Phys. 2001, 65, 459–476. 10.1007/978-3-642-59542-4_22. [DOI] [Google Scholar]
- Talelli M.; Barz M.; Rijcken C. J. F.; Kiessling F.; Hennink W. E.; Lammers T. Core-Crosslinked Polymeric Micelles: Principles, Preparation, Biomedical Applications and Clinical Translation. Nano Today 2015, 10 (1), 93–117. 10.1016/j.nantod.2015.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan R.; Gac-Breton S.; Keane R.; Musila R.; Sat Y.N.; Satchi R.; Searle F. Polymer – Drug Conjugates, PDEPT and PELT: Basic Principles for Design and Transfer from the Laboratory to Clinic. J. Controlled Release 2001, 74, 135–146. 10.1016/S0168-3659(01)00328-5. [DOI] [PubMed] [Google Scholar]
- Duncan R.; Vicent M. J. Do HPMA Copolymer Conjugates Have a Future as Clinically Useful Nanomedicines? A Critical Overview of Current Status and Future Opportunities. Adv. Drug Delivery Rev. 2010, 62 (2), 272–282. 10.1016/j.addr.2009.12.005. [DOI] [PubMed] [Google Scholar]
- Nuhn L.; Kaps L.; Diken M.; Schuppan D.; Zentel R. Reductive Decationizable Block Copolymers for Stimuli-Responsive MRNA Delivery. Macromol. Rapid Commun. 2016, 37, 924–933. 10.1002/marc.201600046. [DOI] [PubMed] [Google Scholar]
- Kelsch A.; Tomcin S.; Rausch K.; Barz M.; Mailänder V.; Schmidt M.; Landfester K.; Zentel R. HPMA Copolymers as Surfactants in the Preparation of Biocompatible Nanoparticles for Biomedical Application. Biomacromolecules 2012, 13 (12), 4179–4187. 10.1021/bm301453g. [DOI] [PubMed] [Google Scholar]
- Soppimath K. S; Aminabhavi T. M; Kulkarni A. R; Rudzinski W. E Biodegradable Polymeric Nanoparticles as Drug Delivery Devices. J. Controlled Release 2001, 70, 1–20. 10.1016/S0168-3659(00)00339-4. [DOI] [PubMed] [Google Scholar]
- Meimetis L. G.; Carlson J. C. T.; Giedt R. J.; Kohler R. H.; Weissleder R. Ultrafluorogenic Coumarin – Tetrazine Probes for Real-Time Biological Imaging **. Angew. Chem., Int. Ed. 2014, 53, 7531–7534. 10.1002/anie.201403890. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.