

Abstract
HIV-1 infection of the nervous system causes various neurological diseases, and synaptic degeneration is likely a critical step in the neuropathogenesis. Our prior studies revealed a significant decrease of synaptic protein, specifically in the spinal dorsal horn of patients with HIV-1 in whom pain developed, suggesting a potential contribution of synaptic degeneration to the pathogenesis of HIV-associated pain. However, the mechanism by which HIV-1 causes the spinal synaptic degeneration is unclear. Here, we identified a critical role of microglia in the synaptic degeneration. In primary cortical cultures (day in vitro 14) and spinal cords of 3- to 5-month-old mice (both sexes), microglial ablation inhibited gp120-induced synapse decrease. Fractalkine (FKN), a microglia activation chemokine specifically expressed in neurons, was upregulated by gp120, and knockout of the FKN receptor CX3CR1, which is predominantly expressed in microglia, protected synapses from gp120-induced toxicity. These results indicate that the neuron-to-microglia intercellular FKN/CX3CR1 signaling plays a role in gp120-induced synaptic degeneration. To elucidate the mechanism controlling this intercellular signaling, we tested the role of the Wnt/β–catenin pathway in regulating FKN expression. Inhibition of Wnt/β-catenin signaling blocked both gp120-induced FKN upregulation and synaptic degeneration, and gp120 stimulated Wnt/β-catenin-regulated FKN expression via NMDA receptors (NMDARs). Furthermore, NMDAR antagonist APV, Wnt/β-catenin signaling suppressor DKK1, or knockout of CX3CR1 alleviated gp120-induced mechanical allodynia in mice, suggesting a critical contribution of the Wnt/β–catenin/FKN/CX3R1 pathway to gp120-induced pain. These findings collectively suggest that HIV-1 gp120 induces synaptic degeneration in the spinal pain neural circuit by activating microglia via Wnt3a/β-catenin-regulated FKN expression in neurons.
SIGNIFICANCE STATEMENT Synaptic degeneration develops in the spinal cord dorsal horn of HIV patients with chronic pain, but the patients without the pain disorder do not show this neuropathology, indicating a pathogenic contribution of the synaptic degeneration to the development of HIV-associated pain. However, the mechanism underlying the synaptic degeneration is unclear. We report here that HIV-1 gp120, a neurotoxic protein that is specifically associated with the manifestation of pain in HIV patients, induces synapse loss via microglia. Further studies elucidate that gp120 activates microglia by stimulating Wnt/β-catenin-regulated fractalkine in neuron. The results demonstrate a critical role of microglia in the pathogenesis of HIV-associated synaptic degeneration in the spinal pain neural circuit.
Keywords: HIV-1, microglia, pain, spinal cord, synaptic degeneration, Wnt
Introduction
HIV-1 enters the CNS soon after patients are infected (Resnick et al., 1988) and causes a range of neurological and cognitive complications (NeuroAIDS; Gendelman, 2012). Synaptic degeneration correlates closely with the presence and severity of HIV-associated cognitive impairment (Everall et al., 1999; Sá et al., 2004; Letendre et al., 2011). In support of this notion, HIV-associated pain is associated with a significant decrease of synaptic markers such as synapsin I (Syn I), postsynaptic density-95 (PSD-95), and NMDA receptor subunit 1 (NR1) in the spinal dorsal horn (SDH) of “pain-positive” HIV-1 patients but not in “pain-negative” patients (Yuan et al., 2014). Similarly, synaptic degeneration is also observed in the mouse HIV pain model generated by intrathecal injection of gp120 (Yuan et al., 2014). These prior observations suggest that synaptic degeneration in the spinal cord contributes to the development of HIV-associated pain. However, the mechanism by which HIV-1 causes the synaptic degeneration is unclear.
Microglia are the primary target for HIV infection in the CNS (Lee et al., 1993; Cenker et al., 2017). Microglia can remove excess and dysfunctional synapses by phagocytosis during neural circuit formation and in specific disease conditions (Stevens et al., 2007; Kettenmann et al., 2013; Brown and Neher, 2014; Xavier et al., 2014; Hong et al., 2016b; Vasek et al., 2016). However, the potential role of microglia in synapse degeneration that is induced by HIV-1 has not been determined. During HIV infection, neuronal damage is likely a bystander effect of infected cells since neurons are not infected. Reactive microglia may also release neurotoxic factors, including proinflammatory cytokines (Tyor et al., 1992; Saha and Pahan, 2003; Viviani et al., 2006; Shah et al., 2011), chemokines (e.g., CXCL12, CXCL10, and CCL2; Kelder et al., 1998; Langford et al., 2002; Sui et al., 2004; Peng et al., 2006; Sui et al., 2006), excitatory amino acids, platelet-activating factor, and free radicals (Giulian et al., 1990; Perry et al., 1998; Turchan et al., 2003; Sacktor et al., 2004; Tian et al., 2012) to induce synaptic degeneration (Ru and Tang, 2017).
Microglial phagocytosis of synapses is elicited by signals that regulate microglia–neuron interactions. These intercellular signals include the complement and fractalkine (FKN) pathways. To remove the “unwanted” synapses, complement components C1q and C3 sequentially translocate to the synapses to act as “eat-me” signals (Schafer et al., 2012), which bind to the microglial receptor CR3 to stimulate synapse elimination (Stevens et al., 2007; Schafer et al., 2012; Hong et al., 2016b; Vasek et al., 2016). FKN is primarily expressed by neurons and regulates synapse phagocytosis by stimulating its sole receptor CX3CR1 on microglia (Harrison et al., 1998; Paolicelli et al., 2011; Zhan et al., 2014). FKN is upregulated in HIV patients, and this upregulation is thought to relate to the development of HIV-associated dementia (Pereira et al., 2001; Cotter et al., 2002; Erichsen et al., 2003).
Because HIV infection is associated with FKN upregulation (Tong et al., 2000; Foussat et al., 2001; Pereira et al., 2001; Sporer et al., 2003; Letendre et al., 2011), we hypothesize that FKN signaling-mediated neuron–microglia cross talk plays a critical role in HIV infection-induced synaptic degeneration. We tested this hypothesis using both in vitro (primary cortical cultures) and in vivo (spinal cord) models of HIV-1 gp120-induced synaptic degeneration. Our results revealed an important role of microglia in gp120-induced synaptic degeneration. We also showed that microglia-mediated synapse degeneration depended on CX3CR1 signaling. We further elucidated that activity-dependent Wnt3a/β-catenin signaling controls gp120-induced FKN upregulation. These findings collectively suggest a critical role for the Wnt3a-FKN-CX3CR1 intercellular signaling from neurons to microglia in regulating gp120-induced synaptic degeneration.
Materials and Methods
Animals.
CX3CR1 knock-out mice (00582. B6.129P-Cx3cr1tm1Litt/J) were obtained from The Jackson Laboratory. In these mice, the CX3CR1 gene was replaced by a green fluorescent protein (GFP) reporter gene (Jung et al., 2000). CD11b-DTR mice were from The Jackson Laboratory [006000; B6. FVB-Tg (ITGAM-DTR/EGFP)34Lan/J]. The diphtheria toxin receptor is expressed under the control of the human ITGAM (integrin α M) promoter (CD11b), and the administration of diphtheria toxin leads to transient depletion of microglia/macrophages in the transgenic mice (Duffield et al., 2005). gp120 transgenic (Tg) mice (from Dr. Marcus Kaul, Sanford-Burnham Medical Research Institute, La Jolla, CA) express HIV-1 LAV gp120 under the control of the glial fibrillary acidic protein (GFAP) promoter (Toggas et al., 1994). The β-cateninflox mouse (004152. B6.129-Ctnnb1tm2Kem/KnwJ) and the nestin-Cre mouse (003771. B6. Cg-Tg(Nes-cre)1Kln/J) were from The Jackson Laboratory. For electrophysiological recording, 6- to 8-week-old male GAD67/GFP mice [003718. FVB-Tg(GadGFP)45704Swn/J, The Jackson Laboratory] were used. These mice express GFP exclusively in GABAergic neurons (Yowtak et al., 2011; Cadwell et al., 2016).
All animal procedures followed protocols that were approved by the Institutional Animal Care and Use Committee at the University of Texas Medical Branch. Mice were housed in a constant temperature environment (23 ± 3°C) with standard bedding, free access to food and water, and under a 12 h light/dark cycle. Mice (3–5 months old) were used. Both male and female mice were used in cellular and molecular analysis. Only males were used for behavioral testing.
Materials.
Maraviroc (catalog #11580) and HIV-1 gp120Bal recombinant protein (catalog #4961) were obtained from the National Institutes of Health (NIH) AIDS Reagent Program. APV (catalog #165304) was purchased from Calbiochem, and minocycline hydrochloride (catalog #M9511) and diphtheria toxin (DT; catalog #D0564) were from Sigma-Aldrich. The use of DT was approved by the Institutional Chemical Safety Committee. To deplete microglia in CD11b-DTR mice, DT was intrathecally injected daily for 3 d (20 ng/injection). HIV-1 gp120Bal in PBS was aliquoted and stored at −80°C until use.
The following antibodies were used for immunoblotting: anti-PSD-95 (1:2000; catalog #04–1066, EMD Millipore; RRID:AB_11212892); anti-Syn I (1:2000; catalog #AB1543, EMD Millipore; RRID:AB_2200400); anti-synaptotagmin-1 (Syt-1; 1:1000; D33B7, catalog #14558, Cell Signaling Technology; RRID:AB_2798510); anti-cleaved caspase-3 (Asp175; 1:1000; 5A1E; catalog #9661, Cell Signaling Technology; RRID:AB_2341188); anti-IBa1 (ionized calcium binding adaptor molecule 1; 1:1000; catalog #016–20001, Wako; RRID:AB_839506); anti-CD11b (1:2000; catalog #52478, Abcam; RRID:AB_868788); anti-CCR5 (1:1000; catalog #4088, NIH AIDS Reagent Program); anti-CXCR4 (1:1000; catalog #11236, NIH AIDS Reagent Program); anti-total-β-catenin (1:5000; catalog #610153, BD Biosciences; RRID:AB_397554); anti-Wnt3a (1:1000; catalog #MAB1324, R&D Systems; RRID:AB_2215416); anti-FKN (1:1000; catalog #ab25088, Abcam; RRID:AB_448600); and anti-β-actin (1:1000; catalog #sc-1616-R, Santa Cruz Biotechnology; RRID:AB_630836).
The following antibodies were used for immunofluorescent staining: anti-PSD-95 (1:200; UCT80; Dr. Randall Walikonis, University of Connecticut, Storrs, CT); anti-Syn I (1:500; catalog #106011, Synaptic Systems; RRID:AB_2619772); and anti-IBa1 (1:200; catalog #016–20001, Wako; RRID:AB_839506).
Primary cortical cultures.
Primary cortical cultures were prepared from postnatal mice as described previously (Ru et al., 2012). The newborn C57BL/6J pups (both male and female) were killed at postnatal days 0–1. Cortices were dissected and placed in iced HBSS (Invitrogen). Cells were dissociated with trypsin in HBSS with gentle trituration and resuspended in DMEM (Invitrogen) containing 10% Gibco FBS (Thermo Fisher Scientific). Dissociated cells were then plated on poly-d-lysine (PDL)-coated dishes (30,000–70,000; Sigma-Aldrich) at a density of 1.5 × 105 cells/cm2 for Western blotting and at 4 × 104 cells/cm2 onto PDL-coated glass coverslips (Biocoat, catalog #354085, BD) for immunofluorescence staining. Cells were grown in a humidified atmosphere of 5% CO2 and 95% air at 37°C. Three hours after plating, the media were replaced with Invitrogen Neurobasal Medium (Thermo Fisher Scientific) supplemented with 2% Invitrogen B27 (Thermo Fisher Scientific) and 0.5 mm Invitrogen l-glutamine (Thermo Fisher Scientific). Half of the media was replenished every 5 d until 14 d in vitro (DIV). Only morphologically healthy cultures were used for the further experiments.
Transcutaneous intrathecal injection.
Mice were anesthetized under 2% isoflurane. A 30.5 gauge stainless steel needle attached to a 10 μl syringe (Hamilton) was used for intrathecal injections. gp120Bal (500 ng/5 μl) in PBS or PBS (5 μl) was injected into the subarachnoid space between the L5 and L6 vertebrae. A correct intrathecal placement of the needle tip was judged by a tail twitch. Mice injected with vehicle were used as controls. Other drugs administered intrathecally, as follows: DT (20 ng/5 μl; daily), DKK1 (1 μg/5 μl, 30 min before gp120 injection), or APV (5 μg/5 μl 30 min before gp120 injection).
Western blotting analysis.
Mice were killed with excess anesthesia (isoflurane), and the L4–L5 lumbar spinal cord segments of mice were collected and homogenized in RIPA lysis buffer (50 mm Tris-HCl, pH 7.4, 150 mm NaCl; 0.1% SDS, 1% Nonidet P-40; 10% glycerol, 1 mm EDTA, pH 8.0, containing PMSF and protease inhibitor mixture; catalog #P8340; Sigma-Aldrich). Protein concentration in the homogenates was determined using a Pierce BCA Protein Assay Kit (product 23227, Thermo Fisher Scientific). Protein concentration was titrated to 2 μg/μl for immunoblotting analysis. For cortical cultures, the cells in 12-well plates (14 DIV) were rinsed with cold PBS and lysed immediately with 150 μl of 2× SDS-PAGE sample buffer (125 mm Tris, pH 6.8, 4% SDS, 10% 2-mercaptoethanol, 20% glycerol, and 0.0025% bromophenol blue) for 30 min. Equal amounts of protein (30 μg/lane) were loaded and separated by SDS-PAGE, followed by transferring to polyvinylidene difluoride membranes (Millipore). Immunoblots were blocked by 5% nonfat milk in Tris-buffered saline–Tween-20 (20 mm Tris-HCl, 150 mm NaCl, pH 7.5, 0.1% Tween-20) for 2 h at room temperature. The blots were then sequentially incubated with primary and secondary antibodies. Protein bands were visualized using the Pierce Enhanced Chemiluminescence Kit from Thermo Fisher Scientific. β-Actin was blotted as a loading control. The intensity of bands was quantified by densitometry analysis with NIH ImageJ.
Immunofluorescent staining and quantification.
For cultured cells, immunofluorescent staining was conducted as previously described (Ru and Tang, 2016). Briefly, the cultured cells were fixed in 4% paraformaldehyde (PFA) for 20 min and then incubated in blocking buffer (5% BSA and 0.3% Triton X-100 in 0.1 m PBS) for 1 h at room temperature. After blocking, the cells were incubated with anti-PSD-95 and anti-Syn I at 4°C overnight, followed by incubating with FITC and cy3-conjungated secondary antibodies (1:200; Jackson ImmunoResearch) for 1 h at room temperature. Images were captured with a confocal microscope (model A1, Nikon). NIH ImageJ software and NeuronJ plugin were used to trace the neuronal processes and determine the regions of interest (ROIs). For each channel, “Subtract Background” was applied to remove the background, and threshold (50–225) was set to remove outliers. A puncta analyzer then was used to count the number of PSD-95+, SynI+, and colocalized puncta within ROIs. At least three cultured coverslips were included for each condition, and four neurons from each coverslip were randomly picked for synaptic puncta analysis.
For tissue immunofluorescent staining, mice were anesthetized with isoflurane and perfused transcardially with cold PBS. The lumbar spinal region was removed and immediately placed in 4% PFA in PBS at 4°C overnight and then cryoprotected in 30% sucrose solution in PBS for at least 24 h at 4°C. Tissues were embedded in optimal cutting temperature compound (Sakura Finetek). Transverse sections (35 μm) were prepared on a cryostat (CM 1900, Leica). The sections were kept in Hito floating section storage solution (Hitobiotec) at −20°C until they were stained for immunocytochemistry. For immunostaining, sections were rinsed with PBS two times to remove storage solution and blocked with 5% BSA and 0.3% Triton X-100 in PBS for 2 h at room temperature. This was followed by 48 h incubation with anti-Syn I antibody (SYSY), anti-PSD-95 (UCT80), or anti-IBa1 (Wako) antibody. After five washes with PBS, the sections were incubated with FITC-conjugated donkey anti-rabbit IgG (1:200; Jackson ImmunoResearch) and/or 594-conjugated donkey anti-mouse lgG (1:200; Jackson ImmunoResearch) before mounting. Images were captured using a confocal microscope (model A1, Nikon). Confocal z-stack images were captured with a confocal microscope (model A1, Nikon) within layer I–II of spinal cord dorsal horn. For each mouse, at least three fixed-frozen sections were included for each experiment, and at least three z-stacks images at 20× or 60× magnification were captured. Thirty to 50 consecutive optical sections with 0.5 μm interval thickness at 60× magnification or 15–25 consecutive optical sections with 1 μm interval thickness at 20× magnification were captured for each z-stack image. For image analysis, Subtract Background (50 pixels) was applied to remove the background, and threshold (50–225) was set to remove outliers. The colocalization analysis was perform with Fiji software. The Syn I+ PSD-95+ puncta were counted as intact synapse. Microglia (IBa1+ or IBa1+TMEM119+) were also quantified.
Measurement of mechanical sensitivity.
Paw withdrawal thresholds (PWTs) of hindpaws were measured by von Frey testing as described previously (Callahan et al., 2008). Briefly, the testing on male adult animals (3–4 months) was performed 6 h before treatment and 24 h after each gp120Bal or vehicle intrathecal injection. After mice habituated to the surrounding environment, a set of calibrated von Frey filaments (Stoelting) was applied to the plantar surface of each hindpaw. The PWTs were determined by using the Dixon up/down method. Testing was conducted by an experimenter who did not have information about the treatments.
Spinal cord slice preparation and whole-cell patch recording.
After gp120Bal or vehicle intrathecal administration, 6- to 8-week-old male mice were deeply anesthetized with 3% isoflurane. Following decapitation, the lumbar spinal cord was removed and placed into a preoxygenated, cold (0–4°C) NMDG (N-methyl-d-glucamine) solution containing the following: 93 mm NMDG, 2.5 mm KCl, 1.2 mm NaH2PO4, 30 mm NaHCO3, 20 mm HEPES, 25 mm glucose, 5 mm sodium ascorbate, 2 mm thiourea, 3 mm sodium pyruvate, 10 mm MgSO4, and 0.5 mm CaCl2(pH 7.4) saturated with 95% O2 and 5% CO2. The spinal cord was then sliced transversely at a thickness of 350 μm using a Vibratome (VT1200S, Leica). Following incubation for 30 min in the NMDG solution at 33°C, the spinal cord slices were transferred to artificial CSF (ACSF) containing the following: 124 mm NaCl, 2.5 mm KCl, 1.2 mm NaH2PO4, 24 mm NaHCO3, 5 mm HEPES, 12.5 mm glucose, 2 mm MgSO4, and 2 mm CaCl2, pH 7.4, for 30 min at 33°C. Then, the slices were placed in a recording chamber perfused with ACSF saturated with 95% O2 and 5% CO2 at a rate of 2 ml/min. Patch-clamp recordings were performed 1 d after the last gp120 injection. The patch pipettes (4–8 MΩ) were filled with internal solution containing the following: 120 mm K-gluconate, 10 mm KCl, 2 mm Mg-ATP, 0.5 mm Na-GTP, 0.5 mm EGTA, 20 mm HEPES, and 10 mm phosphocreatine, pH 7.3. Whole-cell recordings were made using a Multiclamp 700B Amplifier (Molecular Devices), and DigiDATA (Molecular Devices) and pClamp software (version 10.6; Molecular Devices) with a 10 kHz sampling rate and 2 kHz filtering rate (Sigma-Aldrich).
Experimental design and statistical analysis.
In this study, we used primary cortical cultures as an in vitro model system and the L4–L5 lumbar spinal cord segments of mice as an in vivo model system to study HIV gp120-induced synaptic degeneration. To demonstrate synaptic degeneration after gp120 stimulation, we accessed the synaptic protein levels by Western blotting and calculated the synaptic terminal after immunofluorescent staining in both primary cortical cultures and mouse spinal cords. We also examined the gp120-induced functional changes of synapse in the pain circuit by patch-clamp recording and pain behavioral analysis. We then identified the mechanisms of gp120-induced synapse loss. Finally, we verified the proposed mechanism by manipulating microglial activation and interrupting the neuron–microglia intercellular signaling pathway. All data analysis was performed using Prism software (GraphPad Software) statistical analysis. Quantitative summary data were expressed as the mean ± SEM. Analyses were performed with Student's t test for comparing two samples with 95% confidence. One-way ANOVA with a Tukey post hoc test was used for comparisons among multiple groups, and two-way ANOVA with a Bonferroni post hoc test was used to analyze two categorical explanatory variables.
Results
Gp120 induces synaptic degeneration in vitro and in vivo
To test the effect of gp120 on synapses, we treated primary cortical neuron–glia cocultures with gp120 and then measured the levels of synaptic markers, including the presynaptic markers Syn I and Syt-1 and postsynaptic marker PSD-95. We found that both presynaptic and postsynaptic proteins decreased in a time-dependent manner after exposure to gp120 (200 pm). A significant decrease of PSD-95 was observed from 6 to 24 h after gp120 treatment (Fig. 1A). The decrease of Syn I occurred between 12 and 24 h (Fig. 1B), and similarly decreased profiles of Syt-1 were observed (Fig. 1C). Heat-inactivated gp120 control could not lead to the reduction of PSD-95 and Syn I (Fig. 1D,E). To exclude the possibility of a decrease of the synaptic markers that was caused by cell death, we tested whether the experiment induced apoptosis in the cultures. We found that treatment with gp120 did not alter the protein level of cleaved caspase-3 (Fig. 1F). Thus, gp120-induced synaptic protein decreases were not due to apoptosis. To further confirm that the decrease of synaptic markers was associated with a decrease of synapse density, we performed immunostaining of synapses in cultured neurons and quantified the intact synapses with costaining of Syn I and PSD-95 as described previously (Micheva et al., 2010; Okerlund et al., 2010; Galli et al., 2014). The results showed that gp120 exposure caused a significant loss of intact synapses (Fig. 1G–K).
Figure 1.

HIV-1 gp120 induces synapse loss in a time-dependent manner in primary cortical cultures. A–C, Time course of gp120-induced decreases of synaptic proteins. Cortical neuron–glia cocultures (14 DIV) were incubated with 200 pm gp120 for the indicated times. Cell lysates were analyzed by immunoblotting using antibodies against PSD-95 (A), Syn I (B), or Syt-1 (C). For A and B, N = 4; for C, N = 3 cultures/condition. D, E, Heat-inactivated gp120 did not affect PSD-95 (D) and Syn I (E) in cultures; N = 3. F, Western blot of cleaved caspase-3 from gp120-treated cell lysates; N = 4. G–J, Representative immunofluorescence images showing PSD-95 and Syn I after 12 h vehicle (Veh) or gp120 treatment (PSD-95 is red, Syn I is green, and colocalized region is yellow). I, J, Higher magnifications of boxed region. Scale bars: G, H, 20 μm; I, J, 5 μm. K, Bar graph summarizes the effects of gp120 on synapse density. N = 12 neurons from three cultured coverslips/condition (mean ± SEM). n.s., no significant difference.
To determine the effects of gp120 on synapses in vivo, we injected gp120 (500 ng/injection) intrathecally into adult mice and measured the synaptic proteins PSD-95 and Syn I in the L4–L5 spinal segments. Our results showed that both PSD-95 and Syn I levels were lower at day 7 after gp120 injection and returned to the baseline at day 14 (Fig. 2B). These observations indicate that transient gp120 first induces synapse degeneration, which is followed by a process of regeneration. It is important to note that the synapse regeneration is not necessarily able to restore the specificity of synaptic connection and thus the functionality of the previously disrupted neural circuits. We also performed double staining of Syn I and PSD-95, and quantified intact synapses with colocalized PSD-95 and Syn I on confocal images. We observed that the synapse number was significantly decreased in the SDH at day 7 post injection (Fig. 2D,E). It is interesting to note that gp120 treatment on cultures induces much quicker synapse degeneration (Fig. 1) than that in spinal cords (Fig. 2), although the underlying mechanism remains unclear.
Figure 2.

gp120 causes a synapse decrease in the spinal cord dorsal horn. A, The paradigm of intrathecal gp120 treatment. B, PSD-95 and Syn I protein levels in the L4–L5 spinal cord after intrathecal gp120 injection (N = 6 mice/time point). C, PSD-95 and Syn I protein levels in the L4–L5 spinal cord of the gp120 Tg and WT mice (N = 3 mice/condition). D, Confocal images of SDH sections showing anti-Syn I and anti-PSD-95 antibodies staining colocalized areas of Syn I and PSD-95 were calculated (white arrows). Scale bars: top, 100 μm; bottom, 5 μm. E, Quantification: images of layers 1–2 of the SDH from three to five mice were included for each group. For each mouse, three to four spinal slices were included, and two to three images were obtained from each section. F, G, Representative whole-cell recording of mEPSC from the SDH GABAergic interneurons (GFP labeled) from mice treated via intrathecal injection with vehicle (Sham; F) or gp120 (G). Five microliters of gp120 (500 ng) or PBS was intrathecally injected into GAD67-GFP transgenic mice once every other day for 7 d. Cells were held at Vm (membrane potential) = −70 mV. H, I, Summary graphs showing the differences of frequency (H) and amplitude (I) of mEPSCs between the sham- and gp120-treated mice. N = 15 (sham) and N = 31 (gp120) neurons were recorded from three sham and five intrathecally injected gp120 mice for mEPSCs. Error bars represent SEM. ns, no significant difference, ***P < 0.001.
To better model the clinically relevant effect of long-term gp120 exposure in vivo, we also analyzed potential synaptic degeneration in the gp120 Tg mice (Toggas et al., 1994). We observed that both PSD-95 and Syn I levels were lower in the spinal cords (Fig. 2C) of the transgenic mice. We further confirmed the gp120-induced synapse loss by electrophysiology. We recorded miniature EPSCs (mEPSCs) of GABAergic neurons in laminar 1 and 2 of SDH. The GABAergic neurons labeled with GFP (Yowtak et al., 2011; Cadwell et al., 2016) were identified for patching. The results showed that gp120 administration significantly decreased the frequency of mEPSCs (Fig. 2F–I), indicating the reduction of functional synapses.
Microglia are critical for gp120-induced synapse loss
Recent studies reveal a critical role of microglia in synapse pruning (Kettenmann et al., 2013; Brown and Neher, 2014; Chung et al., 2015). Hence, we wanted to test the potential involvement of microglia in gp120-induced synapse loss. Immunofluorescent staining of primary cortical cultures demonstrated the existent of microglia, identified as an IBa1+ cell, in our in vitro system (Fig. 3-1). Moreover, immunoblotting analysis showed IBa1, which is upregulated in reactive microglia, was significantly increased after gp120 treatment (Fig. 3A). IBa1 was also upregulated in the spinal cords of gp120 transgenic mice (Fig. 3B) and intrathecally injected gp120 mice (Fig. 3C). Another microglial marker, CD11b, was also elevated after gp120 injection (Fig. 3C). Immunofluorescent staining of the spinal sections revealed significant increases in the number of microglia in gp120 Tg (Fig. 3D,E) and intrathecally injected gp120 mice (Fig. 3G,H). High-magnification images showed an increase of processes and enlarged soma (Fig. 3F). These findings indicated that gp120 induced microglial activation. Since IBa1 is also expressed by macrophage, to rule out the possibility that the increase of IBa1+ cells is due to macrophage infiltration, we performed double staining of IBa1 and TMEM119. Because TMEM119 is microglia specific, IBa1+/TMEM119+ cells were counted as microglia while IBa1+/TMEM119− cells were counted as macrophages. The results showed that ∼95% IBa1+ cells were microglia (i.e., TMEM119+), under the conditions with or without gp120 treatment (Fig. 3I,J), with no significant increase of IBa1+/TMEM119− cells, indicating that gp120 did not induce significant macrophages infiltration.
Figure 3.

Gp120 causes microglial activation. A, Levels of IBa1 in 200 pm gp120-treated primary cortical cultures (N = 4 cultures in each condition). The cell composition of the cocultures is shown in Figure 3-1. B, IBa1 in L4–L5 spinal cords from gp120 transgenic and WT mice. WT, N = 3 mice; gp120 Tg, N = 4 mice. C, IBa1 and CD11b in L4–L5 spinal cords from mice after intrathecal gp120 injection (500 ng/injection). N = 4 mice/time point. D, Confocal images of IBa1+ cells in the SDH from WT and gp120 Tg mice. Scale bars, 50 μm. E, Quantitative summary of D (mean ± SEM); N = 6 mice/group. F, High-magnification images showing morphological characteristics of reactive microglia. Scale bars, 10 μm. G, Confocal images of IBa1+ cells in the SDH of mice intrathecally injected with PBS or gp120. Scale bars, 50 μm. H, Quantitative summary of G (mean ± SEM); PBS, N = 4 mice; gp120/3 d, N = 5 mice; gp120/7 d, N = 6 mice. I, Confocal images of IBa1 and TMEM119 double-positive cells in the SDH of mice intrathecally injected with PBS or gp120. Scale bars, 30 μm. J, Quantitative summary of I (mean ± SEM); PBS, N = 4 mice; gp120/3 d; N = 5 mice; gp120/7 d, N = 6 mice. n.s., no significant difference.
The cortical primary cultures contains neuron, microglia and astrocytes. Immnofluoresecent staining was perform at DIV 12 to identify neuron (MAP2+, blue; 50%), microglia (IBa1+, red; 20%) and astrocytes (GFAP+, green; 30%). Scale bar: 50μm. This extended data is included to support Figure 3. Download Figure 3-1, TIF file (3.8MB, tif)
Next, we set out to determine the role of microglia in gp120-induced synapse loss. As seen in previous experiments (Fig. 1A,B), gp120 caused transient decreases of PSD-95 and Syn I in cortical cultures (Fig. 4A,B). The gp120-induced decreases of synaptic proteins diminished when we cotreated the cultures with minocycline (Fig. 4A,B) to inhibit microglial activation (Kobayashi et al., 2013). To determine the role of microglia in vivo, we generated the gp120/CD11b-DTR double-transgenic mouse and ablated microglia in the mice by administration of DT (Cailhier et al., 2005; Duffield et al., 2005). Similar to the gp120 Tg mice, the gp120/CD11b-DTR mice also displayed decreases of PSD-95 and Syn I in spinal cord (Fig. 4D). Intrathecal administration of DT (20 ng/injection, once a day for 3 d) led to microglial atrophy (Fig. 4-1). Importantly, DT administration reversed the reduction of the synaptic markers in the double-transgenic mice (Fig. 4D). These complementary data from in vitro pharmacological and in vivo genetic approaches indicate that microglia play a crucial role in gp120-induced synapse loss.
Figure 4.

Microglial activation is crucial for gp120-induced synapse loss. A, B, Cotreatment of primary cortical cultures with minocycline prevented gp120-induced decreases in PSD-95 (A) and Syn I (B). Cortical cultures (14 DIV) were treated with gp120 (200 pm), minocycline (30 μm), or gp120+minocycline for various time periods. Time 0 is the baseline without any drug treatment. N = 4 cultures/condition. C, Diagram showing the generation of gp120/CD11b-DTR mice for microglial ablation by DT. D, Effect of DT administration on PSD-95 and Syn I levels in the spinal cord of the gp120/CD11b-DTR mice. WT, gp120Tg, and gp120Tg/CD11b-DTR mice were administrated 20 ng DT (in 5 μl of PBS) or 5 μl of PBS intrathecally only once/day for 3 d. L4–L5 spinal cords were used for immunoblotting analysis of PSD-95 and Syn I. N, WT/ PBS, 3; gp120Tg/DT, 4; gp120gp/CD11b-DTR/PBS, 4; gp120gp/CD11b-DTR/DT, 3. Quantitative graphs are shown as mean ± SEM. DT-induced microglial atrophy in CD11b-DTR mice is shown in Figure 4-1. n.s., no significant difference.
DT treatment caused microglial atrophy. 20ng DT (in 5μl PBS) or Veh (5μl PBS) was i.t. injected into CD11b-DTR mice, once a day for 3 days. Twenty-four hours after the last treatment, spinal cords were harvested for immunofluorescent staining with IBa1. DT treatment showed significant loss of microglial processes and shrunken cell body (* indicates microglia). Scale bar: 20μm. This extended data is included to support Figure 4. Download Figure 4-1, TIF file (5.2MB, tif)
The gp120-induced synapse loss is mediated by FKN/CX3CR1 signaling
FKN/CX3CR1 signaling is critical for neuron–microglia interaction. CX3CR1 (a receptor of FKN) is exclusively expressed on microglia in the CNS, whereas FKN is specifically addressed in neurons (Harrison et al., 1998; Cardona et al., 2006; Ransohoff, 2009). To determine the effect of gp120 on fractalkine expression, we measured the FKN protein level in gp120-treated primary cortical cultures. Western blotting results revealed that FKN was gradually upregulated over time (Fig. 5A). Time-dependent FKN upregulation was also observed in the spinal cords following intrathecal gp120 administration (Fig. 5B). To determine the role of FKN signaling in gp120-induced synapse loss, we used a mouse strain in which the CX3CR1 gene was replaced by a GFP reporter gene (Jung et al., 2000). Immunoblotting analysis showed that gp120 intrathecal administration for 7 d caused significant reduction of PSD-95 and Syn I in the wild-type (WT) mice (Fig. 5C). However, knocking out CX3CR1 blocked the effects of gp120 on PSD-95 and Syn I (Fig. 5C). Because synaptic degeneration is associated with the development of HIV-related pain in patients (Yuan et al., 2014), we sought to test whether the blockage of synaptic degeneration by the CX3XR1 knockout is associated with an inhibition of the gp120-induced mechanical pain in animal models. Von Frey tests showed that intrathecal gp120 administration increased the mechanical sensitivity in WT mice, as indicated by the decreased threshold of responses to mechanical stimulation. However, it failed to cause a similar effect in the CX3CR1 knock-out mice (Fig. 5D). These results suggest that the disruption of FKN/CX3CR1 signaling blocks gp120-induced synaptic degeneration and mechanical pain.
Figure 5.

FKN/CX3CR1 signaling is required for gp120-induced synaptic degeneration. A, gp120-stimulated FKN expression in cortical cultures (N = 3 cultures/treatment). B, gp120-stimulated (by intrathecal administration) FKN expression in the mouse spinal cords (N = 6 mice/time point). C, Protein levels of PSD 95 and Syn I in the spinal cord of CX3CR1−/− or WT mice at day 7 after intrathecal injection with vehicle (Veh) or gp120 (N = 5 mice/group). D, Behavioral tests of gp120-induced mechanical allodynia in WT and CX3CR1−/− mice. Threshold of mechanical sensitivity in the hindpaw was measured by von Frey tests (N = 6 mice/group). Error bars indicate the mean ± SEM.
Gp120 upregulated FKN via the Wnt canonical pathway
Wnt signaling regulates the FKN–CX3CR1 signal pathway in remifentanil-induced hyperalgesia (Gong et al., 2016). We sought to test the role of this pathway in gp120-induced FKN elevation. We observed that gp120 upregulated the expression of the prototypic Wnt ligand for the canonical pathway Wnt3a in both cortical cultures (Fig. 6A) and spinal cords (Fig. 6B). To determine the effects of disruption of the Wnt canonical pathway on gp120-induced FKN expression, we used the conditional knock-out approach to delete β-catenin in neural cells by crossing the floxed β-catenin mouse with the nestin-Cre mouse line (Tronche et al., 1999). Only heterozygotes were obtained (Cat+/−), indicating that the homozygotes died during the embryonic stages. The spinal β-catenin level in the heterozygotes was ∼60% of that in the WT (Fig. 6C). We found that, unlike the gp120-induced FKN upregulation observed in the WT mice, intrathecal gp120 administration failed to induce FKN elevation in the Cat+/− mice (Fig. 6D). This result suggests that gp120 upregulates FKN expression via the Wnt/β-catenin pathway. Because FKN signaling is critical for gp120-induced synapse loss (Fig. 5), we hypothesized that the Wnt/β-catenin pathway regulates gp120-induced synapse degeneration. To test this idea, we neutralized Wnt3a using a specific antibody and observed that the neutralization blocked the gp120-induced reduction of PSD-95 and Syn I (Fig. 6E). We also tested whether gp120 induced mechanical pain through the Wnt canonical pathway and found that intrathecal injection of purified DKK1 protein, a specific endogenous inhibitor of the Wnt canonical pathway, blocked gp120-caused mechanical pain (Fig. 6G). These results indicated that the Wnt/β-catenin signaling pathway is crucial for gp120-induced FKN upregulation and synaptic degeneration associated with pain pathogenesis.
Figure 6.

Wnt3a/β-catenin pathway mediates gp120-induced FKN upregulation and synaptic degeneration. A, Temporal profiles of Wnt3a expression in gp120-treated primary cortical cultures (N = 6 cultures/group). B, Temporal profiles of Wnt3a expression in the spinal cords (L4–L5) from mice after intrathecal injection with gp120 (N = 3 mice/group). C, β-catenin protein level in spinal cords (L4–L5) from WT and Cat+/− mice (N = 6 mice for WT; and N = 7 mice for Cat+/−). D, Comparison of the effect of gp120 on FKN expression in WT and Cat+/− mice, with intrathecal injection with gp120 every other day for 7 d. Spinal (L4–L5) FKN protein was analyzed by Western blotting. N = 3 mice/group for Veh/WT, Veh/Cat+/−, and gp120/Cat+/−; N = 4 mice for gp120/WT. E, Effect of Wnt3a neutralizing antibody on gp120-induced synaptic degeneration in primary cortical cultures. Anti-Wnt3a antibody (2 μg/ml) was added to the cultures for 30 min, and then gp120 (200pΜ) was added for an additional 12 h (N = 3 independent cultures/group). F, Drug administration paradigm. DKK1 (1 μg/5 μl) was injected 30 min before gp120 (500 ng/5 μl, i.t., in PBS) injection. G, Behavioral tests of the effect of DKK1 on gp120-induced mechanical allodynia. The threshold of mechanical sensitivity in the hindpaw was measured by von Frey tests (N = 6 mice/group). Error bars indicated the mean ± SEM. n.s., no significant difference, *P < 0.05, **P < 0.01, ***P < 0.001.
Gp120-induced FKN expression and synapse loss via NMDA receptor activation
Previous studies show that synaptic activity regulates Wnt3a/β-catenin signaling activation via NMDA receptor (NMDAR; Chen et al., 2006). It is known that gp120 can directly activate NMDARs by binding to their glycine binding sites (Fontana et al., 1997; Pattarini et al., 1998). Gp120 also can indirectly affect the activity of NMDARs after binding to its coreceptor, which triggers a series of signaling cascades as well as cytokine/chemokine release, thus facilitating NMDAR activation (Catani et al., 2000; Kaul et al., 2001; Nicolai et al., 2010; Marchionni et al., 2012; Maung et al., 2014; Ru and Tang, 2016). We observed that coreceptors CCR5 and CXCR4 were mainly located on the postsynapse, and blockage of CCR5 abolished gp120-induced synaptic degeneration (Fig. 7-1). Then, we wanted to determine whether gp120-induced Wnt3a and FKN upregulation required NMDARs in the spinal cord (Fig. 7A). We found that intrathecal injection of the NMDAR antagonist APV prevented gp120-induced increases of Wnt3a and FKN (Fig. 7B,C). We then tested whether APV could also block gp120-induced mechanical pain. The results showed a progressive inhibitory effect of APV on gp120-induced hyperalgesia. APV had no evident effect initially (days 0–1) but showed significant inhibition at later stages (after day 3; Fig. 7D).
Figure 7.

The NMDAR is crucial for gp120 to induce Wnt3a and FKN expression. A, Mice were intrathecally injected with either APV (5 μg/5 μl in PBS) or vehicle (Veh; 5 μl of PBS) 30 min before gp120 injection (500 ng/5 μl/2 d). B, C, Spinal cords (L4–L5) were collected at day 7 for immunoblotting analysis of Wnt3a (B) and FKN (C). N = 6 mice for the groups of Veh and APV; N = 7 for the groups of gp120 and gp120+APV. D, Von Frey tests of the effect of APV on gp120-induced mechanical pain (N = 5 mice/group). *p < 0.05; **p < 0.01 (gp120 vs gp120/APV at the same time point); #p < 0.05; ###p < 0.001 (between different time point within the gp120/APV group). The involvement of gp120 coreceptor is suggested by Figure 7-1. n.s., no significant difference.
gp120 co-receptors mediated synapse loss. A. Immunoblots showed the expression level of CXCR4 and CCR5 in forebrain, synaptosome and postsynaptic density homogenate. B. Effects of maraviroc pre-treatment on gp120-induced decrease of PSD95 and Syn I. Cortical cultures were pre-treated with the maraviroc (50μM) for 30 min and then co-treated with gp120 (200pM) for another 12 hours. Cell lysates were analyzed by immunoblotting. N=3 cultures per condition. B. Data are expressed as mean ± SEM. This extended data is included to support Figure 7. Download Figure 7-1, TIF file (2.7MB, tif)
We next determined the potential role of NMDARs in gp120-induced synapse degeneration both in primary cortical cultures and in the spinal cords. We found that APV application to the cultures completely blocked the gp120-induced decreases of PSD-95 (Fig. 8A) and Syn I (Fig. 8B). In addition, intrathecal injection of APV also blocked gp120-induced decrease of PSD-95 (Fig. 8C) and Syn I (Fig. 8D). Thus, NMDAR activity is critical for gp120-induced synaptic degeneration both in vitro and in vivo.
Figure 8.

gp120-induced synapse loss depends on NMDAR activation. A, B, Effects of APV on gp120-induced decreases of PSD-95 (A) and Syn I (B) in primary cortical cultures. APV (100 nm) was added to the cultures (14 DIV) 30 min before gp120 (200 pΜ) application. The cultures were exposed to gp120 for 12 h before harvesting for immunoblotting (N = 4 independent cultures/group). C, D, Effects of APV on gp120-induced decreases of PSD-95 (C) and Syn I (D) in the spinal cords (L4–L5). Mice were intrathecally injected with APV (5 μg/5 μl PBS) or vehicle (Veh; 5 μl of PBS) 30 min before gp120 injection (500 ng/5 μl/2 d). L4–L5 spinal cords were collected for immunoblotting analysis of PSD-95 (C) and Syt-1 (D). N = 3 mice/group (mean ± SEM). n.s., no significant difference.
Discussion
The synaptic alteration in the spinal cord contributes to central sensitization and pain hypersensitivity (Latremoliere and Woolf, 2009). Recent in vivo two-photon imaging revealed synaptic structural changes in cortical pain circuits following peripheral nerve injury, implicating structural change of cortical synapses in the development of neuropathic pain (Kim et al., 2012). Balance between excitatory and inhibitory neurons plays a critical role in pain processing (Kuner and Flor, 2017). For instance, an increase of spine density (Tan et al., 2012; Simonetti et al., 2013; Lu et al., 2015), NMDAR activation, and AMPA receptor insertion (Luo et al., 2014; Bliss et al., 2016), or presynaptic neurotransmitter release (Luo et al., 2012) is implicated in the pathogenesis of chronic pain. On the other hand, reduced inhibitory control in the spinal cord may also contribute to neuropathic pain development (Moore et al., 2002). We observed significant reduction of synaptic markers in the SDH in HIV patients with chronic pain but not in HIV patients without pain (Yuan et al., 2014). This synaptic degeneration also developed in the mouse HIV-1 gp120 pain model (Yuan et al., 2014). It indicated that synaptic degeneration might be related to HIV-induced neuropathic pain, although the mechanism by which synaptic degeneration contributes to pain manifestation is unclear. One possibility is that gp120 induces loss of inhibitory synapses on excitatory pain transmission neurons, which then become disinhibited. Alternatively, HIV-gp120 may cause loss of excitatory synapses on inhibitory neurons, which then become less active and consequently lead to hyperactivation of pain transmission neurons. Indeed, our electrophysiology study showed that gp120 decreased the frequency of mEPSCs of GABAergic interneurons (Fig. 2E–H). The findings from this study identify the first potential molecular and cellular mechanisms that underlie synaptic degeneration during the pathogenesis of HIV-associated pain.
Our results show that microglial blockage by two different approaches protected synapses from gp120 toxicity (Fig. 4). These data suggest that microglia play an important role in gp120-induced synaptic degeneration. Prior studies revealed a contribution of neurotoxic immunomodulatory factors from reactive and/or infected microglia (Garden, 2002). More recent work identified a key role of microglial phagocytosis in synaptic remodeling and maturation (Paolicelli et al., 2011; Schafer et al., 2012; Kettenmann et al., 2013; Hong et al., 2016a). Consistent with a role of microglia in synapse maintenance, our data showed that long-term treatment with minocycline alone seemed to decrease synaptic proteins (e.g., PSD-95 at 24 h; Fig. 4A). The findings from this work significantly expand the understanding of the role of microglia by identifying their involvement in synapse remodeling in a specific disease condition caused by the HIV-1 coat toxic protein. These results also potentially connect the microglia-mediated synaptic degeneration with the development of HIV-associated pathological pain.
Previous studies suggested certain signaling pathways, including TNF-α signaling (De et al., 2002; Olmos and Lladó, 2014) and cannabinoid signaling (Avraham et al., 2014; Zhang and Thayer, 2018), in mediating HIV-induced synaptic degeneration. In this study, we identify a FKN/CX3XR1 signaling pathway that may regulate neuron–microglia interaction during gp120-induced synaptic degeneration. FKN/CX3XR1 was reported to regulate microglia-mediated synapse pruning (Zabel et al., 2016). We observed that FKN was significantly upregulated by gp120 both in vitro and in vivo (Fig. 5), and deletion of the FKN receptor CX3CR1 blocked the synapse loss induced by gp120 (Fig. 5). The data suggest that FKN/CX3CR1 signaling is essential for gp120-induced synaptic degeneration. FKN acts as both a membrane-tethered and a secreted chemokine that binds to its receptor, CX3CR1, to regulate microglial phagocytosis. Previous studies indicate that the FKN/CX3CR1 signaling modulates synapse pruning during development (Paolicelli et al., 2011; Hoshiko et al., 2012). It is thus possible that the FKN/CX3CR1 signaling stimulates microglial phagocytosis of damaged synapses caused by gp120 exposure, although the direct evidence for this is still lacking. In addition to its potential role in microglial phagocytosis of synapses, FKN/CX3XR1 signaling may also regulate the microglial clearance of pathogens and extracellular protein aggregates (Lee et al., 2010; Liu et al., 2010). Therefore, another possibility is that FKN/CX3CR1 suppresses neurotoxin clearance and then promotes gp120-induced synaptic degeneration. FKN/CX3XR1 signaling also regulates microgliosis and the local neuroinflammatory milieu around the pathogenic insults (Lee et al., 2010; Tang et al., 2014). Thus, a third possibility is that FKN/CX3CR1 signaling may facilitate gp120-induced synaptic degeneration by enhancing neuroinflammation that is induced by gp120. Because CX3CR1 is also expressed in macrophages, we cannot rule out the possibility that the effect of CX3CR1 knockout on inhibiting gp120-induced synaptic degeneration is also contributed by macrophages. However, since gp120 did not evoke obvious macrophage infiltration (Fig. 3I,J), such a contribution of macrophages, if indeed it existed, should be minimal.
We further identified the Wnt/β-catenin signaling as a critical upstream regulator of FKN expression induced by gp120. Our data show that β-catenin is required for gp120 to upregulate FKN (Fig. 6). Consistent with our findings, recent studies indicate that the spinal Wnt/β-catenin pathway modulates FKN/CX3CR1 signaling during the development of opioid-induced hyperalgesia (Gong et al., 2016). In addition, we also show that gp120-induced Wnt3a and FKN expression depend on activation of NMDAR and the blockage of NMDAR activation by APV abolished the effects of gp120 on Wnt/β-catenin-FKN/CX3CR1 pathway and synapse loss (Figs. 7, 8). In support of NMDAR/Wnt/FKN signaling axis, we previously showed that NMDAR-regulated Wnt//β-catenin signaling upregulates ADAM10 (a disintegrin and metalloproteinase 10; Wan et al., 2012), which is implicated in FNK shedding (Hundhausen et al., 2003) and sensory lesion-induced microglial synapse elimination (Gunner et al., 2019).
APV did not completely inhibit gp120-induced mechanical allodynia, at least during the early phase (Fig. 7D, 1–3 d). This result indicates a contribution of other pathways induced by gp120 in the pain pathogenesis. For instance, gp120 coreceptors on the neurons are likely involved. Indeed, we observed that blocking CCR5 abolished gp120-induced synaptic degeneration (Fig. 7-1). By binding to its coreceptor, gp120 may modulate pain circuit function by regulating synaptic activity (Catani et al., 2000; Kaul et al., 2001; Nicolai et al., 2010; Marchionni et al., 2012; Maung et al., 2014; Ru and Tang, 2016, 2017) and/or cause neuron damages (Lazarini et al., 2000; Garden et al., 2004; Medders et al., 2010).
The synapse loss initiated by gp120-inudced NMDAR activation would be associated with a decrease of synaptic proteins, including Syn I, PSD-95, and others such as NR1 as reported previously (Yuan et al., 2014). Another possibility is that gp120 treatment causes a specific decease of specific synaptic proteins such as NR1 as a potential feedback mechanism for synapse protection. However, the observations of simultaneous decrease of multiple synaptic markers, the decrease of mEPSC frequency (Fig. 2E–H), and synapse density (Figs. 1, 2) provide strong evidence for synapse loss.
A single dose of gp120 administration caused transient decrease of synaptic proteins (Figs. 1, 2), and the levels of synaptic proteins recovered afterward. These observations indicate that gp120-induced synaptic degeneration is followed by compensatory processes of synapse formation. Interestingly, under the same condition, the upregulation of FKN and Wnt3a persisted even after the recovery of synaptic proteins (Figs. 5, 6). This finding indicates that FKN and Wnt3A, although essential for gp120 to induce synapse degeneration, are not sufficient to cause synapse degeneration by themselves. It is possible that continuous the presence of active gp120 is essential for FNK and Wnt3a to execute their activity in stimulating detectable synapse degeneration. Without active gp120, for example, synapse formation may overcome the synapse degeneration processes, although the newly formed synapse may not be recruited into the same neural circuit as the degenerated synapses.
In summary, we have uncovered a critical role of microglia in HIV-1 gp120-induced synaptic degeneration, especially in the spinal neural circuits. We also elucidate an important mechanism that regulates the microglia-mediated synaptic degeneration. Specifically, our results indicate that gp120 stimulates the NMDAR-dependent Wnt/β-catenin pathway to upregulate the FKN/CX3CR1 neuron-to-microglia signaling to activate microglia-mediated elimination of synapses (Fig. 9). Although our findings suggest important roles of microglia in gp120-induced synaptic degeneration, these results do not preclude the potential contribution of other glia (e.g., astrocytes) to this process. In addition, because CX3CR1 was suggested as an HIV coreceptor (Garin et al., 2003), it would be interesting for future studies to test whether gp120 may directly stimulate microglial CX3CR1 to elicit synapse degeneration.
Figure 9.
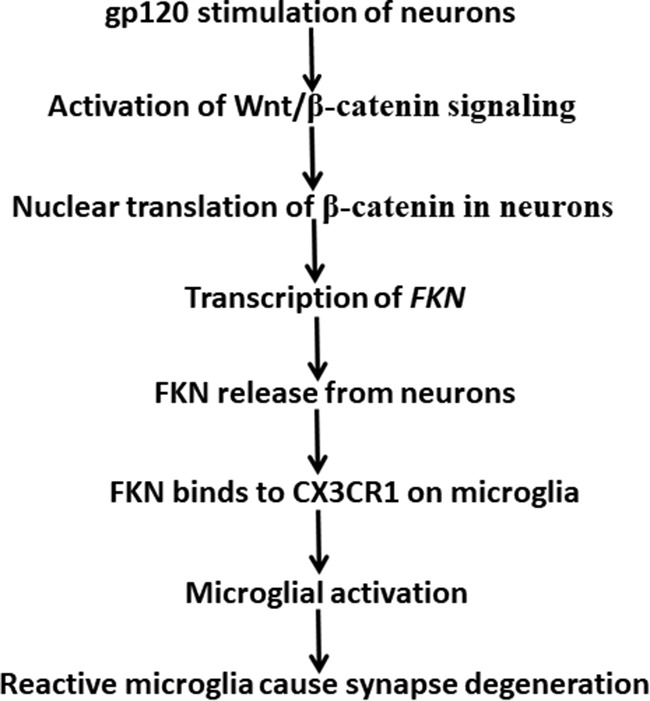
A model of gp120-induced synaptic degeneration in the spinal pain neural circuit. gp120 stimulates neurons and activates Wnt/β-catenin signaling that leads to β-catenin-mediated FKN transcription. FKN protein released from neurons binds to CX3CR1 to activate microglia to induce synaptic degeneration.
Footnotes
This work was supported by grants from the National Institute of Neurological Disorders and Stroke/National Institutes of Health (NIH; R01-NS-079166 and R01-NS-095747 to S.-J.T.) and the National Institute on Drug Abuse/NIH (R01-DA-036165 to S.-J.T.), and the William Willis Jr. MD PHD Professorship in Neuroscience (to S.-J.T.). We thank Dr. Marcus Kaul for providing the gp120 transgenic mouse.
The authors declare no competing financial interests.
References
- Avraham HK, Jiang S, Fu Y, Rockenstein E, Makriyannis A, Zvonok A, Masliah E, Avraham S (2014) The cannabinoid CB2 receptor agonist AM1241 enhances neurogenesis in GFAP/Gp120 transgenic mice displaying deficits in neurogenesis. Br J Pharmacol 171:468–479. 10.1111/bph.12478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bliss TV, Collingridge GL, Kaang BK, Zhuo M (2016) Synaptic plasticity in the anterior cingulate cortex in acute and chronic pain. Nat Rev Neurosci 17:485–496. 10.1038/nrn.2016.68 [DOI] [PubMed] [Google Scholar]
- Brown GC, Neher JJ (2014) Microglial phagocytosis of live neurons. Nat Rev Neurosci 15:209–216. 10.1038/nrn3710 [DOI] [PubMed] [Google Scholar]
- Cadwell CR, Palasantza A, Jiang X, Berens P, Deng Q, Yilmaz M, Reimer J, Shen S, Bethge M, Tolias KF, Sandberg R, Tolias AS (2016) Electrophysiological, transcriptomic and morphologic profiling of single neurons using patch-seq. Nat Biotechnol 34:199–203. 10.1038/nbt.3445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cailhier JF, Partolina M, Vuthoori S, Wu S, Ko K, Watson S, Savill J, Hughes J, Lang RA (2005) Conditional macrophage ablation demonstrates that resident macrophages initiate acute peritoneal inflammation. J Immunol 174:2336–2342. 10.4049/jimmunol.174.4.2336 [DOI] [PubMed] [Google Scholar]
- Callahan BL, Gil AS, Levesque A, Mogil JS (2008) Modulation of mechanical and thermal nociceptive sensitivity in the laboratory mouse by behavioral state. J Pain 9:174–184. 10.1016/j.jpain.2007.10.011 [DOI] [PubMed] [Google Scholar]
- Cardona AE, Pioro EP, Sasse ME, Kostenko V, Cardona SM, Dijkstra IM, Huang D, Kidd G, Dombrowski S, Dutta R, Lee JC, Cook DN, Jung S, Lira SA, Littman DR, Ransohoff RM (2006) Control of microglial neurotoxicity by the fractalkine receptor. Nat Neurosci 9:917–924. 10.1038/nn1715 [DOI] [PubMed] [Google Scholar]
- Catani MV, Corasaniti MT, Navarra M, Nisticò G, Finazzi-Agrò A, Melino G (2000) gp120 induces cell death in human neuroblastoma cells through the CXCR4 and CCR5 chemokine receptors. J Neurochem 74:2373–2379. 10.1046/j.1471-4159.2000.0742373.x [DOI] [PubMed] [Google Scholar]
- Cenker JJ, Stultz RD, McDonald D (2017) Brain microglial cells are highly susceptible to HIV-1 infection and spread. AIDS Res Hum Retroviruses 33:1155–1165. 10.1089/aid.2017.0004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Park CS, Tang SJ (2006) Activity-dependent synaptic wnt release regulates hippocampal long term potentiation. J Biol Chem 281:11910–11916. 10.1074/jbc.M511920200 [DOI] [PubMed] [Google Scholar]
- Chung WS, Welsh CA, Barres BA, Stevens B (2015) Do glia drive synaptic and cognitive impairment in disease? Nat Neurosci 18:1539–1545. 10.1038/nn.4142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotter R, Williams C, Ryan L, Erichsen D, Lopez A, Peng H, Zheng J (2002) Fractalkine (CX3CL1) and brain inflammation: implications for HIV-1-associated dementia. J Neurovirol 8:585–598. 10.1080/13550280290100950 [DOI] [PubMed] [Google Scholar]
- De SK, Devadas K, Notkins AL (2002) Elevated levels of tumor necrosis factor alpha (TNF-alpha) in human immunodeficiency virus type 1-transgenic mice: prevention of death by antibody to TNF-alpha. J Virol 76:11710–11714. 10.1128/JVI.76.22.11710-11714.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffield JS, Forbes SJ, Constandinou CM, Clay S, Partolina M, Vuthoori S, Wu S, Lang R, Iredale JP (2005) Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J Clin Invest 115:56–65. 10.1172/JCI200522675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erichsen D, Lopez AL, Peng H, Niemann D, Williams C, Bauer M, Morgello S, Cotter RL, Ryan LA, Ghorpade A, Gendelman HE, Zheng J (2003) Neuronal injury regulates fractalkine: relevance for HIV-1 associated dementia. J Neuroimmunol 138:144–155. 10.1016/S0165-5728(03)00117-6 [DOI] [PubMed] [Google Scholar]
- Everall IP, Heaton RK, Marcotte TD, Ellis RJ, McCutchan JA, Atkinson JH, Grant I, Mallory M, Masliah E (1999) Cortical synaptic density is reduced in mild to moderate human immunodeficiency virus neurocognitive disorder. HNRC group. HIV Neurobehavioral Research Center. Brain Pathol 9:209–217. 10.1111/j.1750-3639.1999.tb00219.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontana G, Valenti L, Raiteri M (1997) Gp120 can revert antagonism at the glycine site of NMDA receptors mediating GABA release from cultured hippocampal neurons. J Neurosci Res 49:732–738. [DOI] [PubMed] [Google Scholar]
- Foussat A, Bouchet-Delbos L, Berrebi D, Durand-Gasselin I, Coulomb-L'Hermine A, Krzysiek R, Galanaud P, Levy Y, Emilie D (2001) Deregulation of the expression of the fractalkine/fractalkine receptor complex in HIV-1-infected patients. Blood 98:1678–1686. 10.1182/blood.V98.6.1678 [DOI] [PubMed] [Google Scholar]
- Galli S, Lopes DM, Ammari R, Kopra J, Millar SE, Gibb A, Salinas PC (2014) Deficient wnt signalling triggers striatal synaptic degeneration and impaired motor behaviour in adult mice. Nat Commun 5:4992. 10.1038/ncomms5992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garden GA. (2002) Microglia in human immunodeficiency virus-associated neurodegeneration. Glia 40:240–251. 10.1002/glia.10155 [DOI] [PubMed] [Google Scholar]
- Garden GA, Guo W, Jayadev S, Tun C, Balcaitis S, Choi J, Montine TJ, Möller T, Morrison RS (2004) HIV associated neurodegeneration requires p53 in neurons and microglia. FASEB J 18:1141–1143. 10.1096/fj.04-1676fje [DOI] [PubMed] [Google Scholar]
- Gendelman HE. (2012) The neurology of AIDS, Ed 3 Oxford: Oxford UP. [Google Scholar]
- Giulian D, Vaca K, Noonan CA (1990) Secretion of neurotoxins by mononuclear phagocytes infected with HIV-1. Science 250:1593–1596. 10.1126/science.2148832 [DOI] [PubMed] [Google Scholar]
- Gong G, Hu L, Qin F, Yin L, Yi X, Yuan L, Wu W (2016) Spinal WNT pathway contributes to remifentanil induced hyperalgesia through regulating fractalkine and CX3CR1 in rats. Neurosci Lett 633:21–27. 10.1016/j.neulet.2016.09.006 [DOI] [PubMed] [Google Scholar]
- Gunner G, Cheadle L, Johnson KM, Ayata P, Badimon A, Mondo E, Nagy MA, Liu L, Bemiller SM, Kim KW, Lira SA, Lamb BT, Tapper AR, Ransohoff RM, Greenberg ME, Schaefer A, Schafer DP (2019) Sensory lesioning induces microglial synapse elimination via ADAM10 and fractalkine signaling. Nat Neurosci 22:1075–1088. 10.1038/s41593-019-0419-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison JK, Jiang Y, Chen S, Xia Y, Maciejewski D, McNamara RK, Streit WJ, Salafranca MN, Adhikari S, Thompson DA, Botti P, Bacon KB, Feng L (1998) Role for neuronally derived fractalkine in mediating interactions between neurons and CX3CR1-expressing microglia. Proc Natl Acad Sci U S A 95:10896–10901. 10.1073/pnas.95.18.10896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong S, Dissing-Olesen L, Stevens B (2016a) New insights on the role of microglia in synaptic pruning in health and disease. Curr Opin Neurobiol 36:128–134. 10.1016/j.conb.2015.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong S, Beja-Glasser VF, Nfonoyim BM, Frouin A, Li S, Ramakrishnan S, Merry KM, Shi Q, Rosenthal A, Barres BA, Lemere CA, Selkoe DJ, Stevens B (2016b) Complement and microglia mediate early synapse loss in alzheimer mouse models. Science 352:712–716. 10.1126/science.aad8373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshiko M, Arnoux I, Avignone E, Yamamoto N, Audinat E (2012) Deficiency of the microglial receptor CX3CR1 impairs postnatal functional development of thalamocortical synapses in the barrel cortex. J Neurosci 32:15106–15111. 10.1523/JNEUROSCI.1167-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hundhausen C, Misztela D, Berkhout TA, Broadway N, Saftig P, Reiss K, Hartmann D, Fahrenholz F, Postina R, Matthews V, Kallen KJ, Rose-John S, Ludwig A (2003) The disintegrin-like metalloproteinase ADAM10 is involved in constitutive cleavage of CX3CL1 (fractalkine) and regulates CX3CL1-mediated cell-cell adhesion. Blood 102:1186–1195. 10.1182/blood-2002-12-3775 [DOI] [PubMed] [Google Scholar]
- Jung S, Aliberti J, Graemmel P, Sunshine MJ, Kreutzberg GW, Sher A, Littman DR (2000) Analysis of fractalkine receptor CX(3)CR1 function by targeted deletion and green fluorescent protein reporter gene insertion. Mol Cell Biol 20:4106–4114. 10.1128/MCB.20.11.4106-4114.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaul M, Garden GA, Lipton SA (2001) Pathways to neuronal injury and apoptosis in HIV-associated dementia. Nature 410:988–994. 10.1038/35073667 [DOI] [PubMed] [Google Scholar]
- Kelder W, McArthur JC, Nance-Sproson T, McClernon D, Griffin DE (1998) Beta-chemokines MCP-1 and RANTES are selectively increased in cerebrospinal fluid of patients with human immunodeficiency virus-associated dementia. Ann Neurol 44:831–835. 10.1002/ana.410440521 [DOI] [PubMed] [Google Scholar]
- Kettenmann H, Kirchhoff F, Verkhratsky A (2013) Microglia: new roles for the synaptic stripper. Neuron 77:10–18. 10.1016/j.neuron.2012.12.023 [DOI] [PubMed] [Google Scholar]
- Kim SK, Eto K, Nabekura J (2012) Synaptic structure and function in the mouse somatosensory cortex during chronic pain: in vivo two-photon imaging. Neural Plast 2012:640259. 10.1155/2012/640259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi K, Imagama S, Ohgomori T, Hirano K, Uchimura K, Sakamoto K, Hirakawa A, Takeuchi H, Suzumura A, Ishiguro N, Kadomatsu K (2013) Minocycline selectively inhibits M1 polarization of microglia. Cell Death Dis 4:e525. 10.1038/cddis.2013.54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuner R, Flor H (2017) Structural plasticity and reorganisation in chronic pain. Nat Rev Neurosci 18:113. 10.1038/nrn.2017.5 [DOI] [PubMed] [Google Scholar]
- Langford D, Sanders VJ, Mallory M, Kaul M, Masliah E (2002) Expression of stromal cell-derived factor 1alpha protein in HIV encephalitis. J Neuroimmunol 127:115–126. 10.1016/S0165-5728(02)00068-1 [DOI] [PubMed] [Google Scholar]
- Latremoliere A, Woolf CJ (2009) Central sensitization: a generator of pain hypersensitivity by central neural plasticity. J Pain 10:895–926. 10.1016/j.jpain.2009.06.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarini F, Casanova P, Tham TN, De Clercq E, Arenzana-Seisdedos F, Baleux F, Dubois-Dalcq M (2000) Differential signalling of the chemokine receptor CXCR4 by stromal cell-derived factor 1 and the HIV glycoprotein in rat neurons and astrocytes. Eur J Neurosci 12:117–125. 10.1046/j.1460-9568.2000.00894.x [DOI] [PubMed] [Google Scholar]
- Lee SC, Hatch WC, Liu W, Kress Y, Lyman WD, Dickson DW (1993) Productive infection of human fetal microglia by HIV-1. Am J Pathol 143:1032–1039. [PMC free article] [PubMed] [Google Scholar]
- Lee S, Varvel NH, Konerth ME, Xu G, Cardona AE, Ransohoff RM, Lamb BT (2010) CX3CR1 deficiency alters microglial activation and reduces beta-amyloid deposition in two Alzheimer's disease mouse models. Am J Pathol 177:2549–2562. 10.2353/ajpath.2010.100265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letendre SL, Zheng JC, Kaul M, Yiannoutsos CT, Ellis RJ, Taylor MJ, Marquie-Beck J, Navia B (2011) Chemokines in cerebrospinal fluid correlate with cerebral metabolite patterns in HIV-infected individuals. J Neurovirol 17:63–69. 10.1007/s13365-010-0013-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Condello C, Schain A, Harb R, Grutzendler J (2010) CX3CR1 in microglia regulates brain amyloid deposition through selective protofibrillar amyloid-beta phagocytosis. J Neurosci 30:17091–17101. 10.1523/JNEUROSCI.4403-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Luo C, Bali KK, Xie RG, Mains RE, Eipper BA, Kuner R (2015) A role for kalirin-7 in nociceptive sensitization via activity-dependent modulation of spinal synapses. Nat Commun 6:6820. 10.1038/ncomms7820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo C, Gangadharan V, Bali KK, Xie RG, Agarwal N, Kurejova M, Tappe-Theodor A, Tegeder I, Feil S, Lewin G, Polgar E, Todd AJ, Schlossmann J, Hofmann F, Liu DL, Hu SJ, Feil R, Kuner T, Kuner R (2012) Presynaptically localized cyclic GMP-dependent protein kinase 1 is a key determinant of spinal synaptic potentiation and pain hypersensitivity. PLoS Biol 10:e1001283. 10.1371/journal.pbio.1001283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo C, Kuner T, Kuner R (2014) Synaptic plasticity in pathological pain. Trends Neurosci 37:343–355. 10.1016/j.tins.2014.04.002 [DOI] [PubMed] [Google Scholar]
- Marchionni I, Beaumont M, Maccaferri G (2012) The chemokine CXCL12 and the HIV-1 envelope protein gp120 regulate spontaneous activity of cajal-retzius cells in opposite directions. J Physiol 590:3185–3202. 10.1113/jphysiol.2011.224873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maung R, Hoefer MM, Sanchez AB, Sejbuk NE, Medders KE, Desai MK, Catalan IC, Dowling CC, de Rozieres CM, Garden GA, Russo R, Roberts AJ, Williams R, Kaul M (2014) CCR5 knockout prevents neuronal injury and behavioral impairment induced in a transgenic mouse model by a CXCR4-using HIV-1 glycoprotein 120. J Immunol 193:1895–1910. 10.4049/jimmunol.1302915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medders KE, Sejbuk NE, Maung R, Desai MK, Kaul M (2010) Activation of p38 MAPK is required in monocytic and neuronal cells for HIV glycoprotein 120-induced neurotoxicity. J Immunol 185:4883–4895. 10.4049/jimmunol.0902535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micheva KD, Busse B, Weiler NC, O'Rourke N, Smith SJ (2010) Single-synapse analysis of a diverse synapse population: proteomic imaging methods and markers. Neuron 68:639–653. 10.1016/j.neuron.2010.09.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore KA, Kohno T, Karchewski LA, Scholz J, Baba H, Woolf CJ (2002) Partial peripheral nerve injury promotes a selective loss of GABAergic inhibition in the superficial dorsal horn of the spinal cord. J Neurosci 22:6724–6731. 10.1523/JNEUROSCI.22-15-06724.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolai J, Burbassi S, Rubin J, Meucci O (2010) CXCL12 inhibits expression of the NMDA receptor's NR2B subunit through a histone deacetylase-dependent pathway contributing to neuronal survival. Cell Death Dis 1:e33. 10.1038/cddis.2010.10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okerlund ND, Kivimäe S, Tong CK, Peng IF, Ullian EM, Cheyette BN (2010) Dact1 is a postsynaptic protein required for dendrite, spine, and excitatory synapse development in the mouse forebrain. J Neurosci 30:4362–4368. 10.1523/JNEUROSCI.0354-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olmos G, Lladó J (2014) Tumor necrosis factor alpha: a link between neuroinflammation and excitotoxicity. Mediators Inflamm 2014:861231. 10.1155/2014/861231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paolicelli RC, Bolasco G, Pagani F, Maggi L, Scianni M, Panzanelli P, Giustetto M, Ferreira TA, Guiducci E, Dumas L, Ragozzino D, Gross CT (2011) Synaptic pruning by microglia is necessary for normal brain development. Science 333:1456–1458. 10.1126/science.1202529 [DOI] [PubMed] [Google Scholar]
- Pattarini R, Pittaluga A, Raiteri M (1998) The human immunodeficiency virus-1 envelope protein gp120 binds through its V3 sequence to the glycine site of N-methyl-D-aspartate receptors mediating noradrenaline release in the hippocampus. Neuroscience 87:147–157. 10.1016/S0306-4522(98)00125-0 [DOI] [PubMed] [Google Scholar]
- Peng H, Erdmann N, Whitney N, Dou H, Gorantla S, Gendelman HE, Ghorpade A, Zheng J (2006) HIV-1-infected and/or immune activated macrophages regulate astrocyte SDF-1 production through IL-1beta. Glia 54:619–629. 10.1002/glia.20409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira CF, Middel J, Jansen G, Verhoef J, Nottet HS (2001) Enhanced expression of fractalkine in HIV-1 associated dementia. J Neuroimmunol 115:168–175. 10.1016/S0165-5728(01)00262-4 [DOI] [PubMed] [Google Scholar]
- Perry SW, Hamilton JA, Tjoelker LW, Dbaibo G, Dzenko KA, Epstein LG, Hannun Y, Whittaker JS, Dewhurst S, Gelbard HA (1998) Platelet-activating factor receptor activation. an initiator step in HIV-1 neuropathogenesis. J Biol Chem 273:17660–17664. 10.1074/jbc.273.28.17660 [DOI] [PubMed] [Google Scholar]
- Ransohoff RM. (2009) Chemokines and chemokine receptors: standing at the crossroads of immunobiology and neurobiology. Immunity 31:711–721. 10.1016/j.immuni.2009.09.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Resnick L, Berger JR, Shapshak P, Tourtellotte WW (1988) Early penetration of the blood-brain-barrier by HIV. Neurology 38:9–14. 10.1212/WNL.38.1.9 [DOI] [PubMed] [Google Scholar]
- Ru W, Tang SJ (2016) HIV-1 gp120Bal down-regulates phosphorylated NMDA receptor subunit 1 in cortical neurons via activation of glutamate and chemokine receptors. J Neuroimmune Pharmacol 11:182–191. 10.1007/s11481-015-9644-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ru W, Tang SJ (2017) HIV-associated synaptic degeneration. Mol Brain 10:40. 10.1186/s13041-017-0321-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ru W, Peng Y, Zhong L, Tang SJ (2012) A role of the mammalian target of rapamycin (mTOR) in glutamate-induced down-regulation of tuberous sclerosis complex proteins 2 (TSC2). J Mol Neurosci 47:340–345. 10.1007/s12031-012-9753-1 [DOI] [PubMed] [Google Scholar]
- Sá MJ, Madeira MD, Ruela C, Volk B, Mota-Miranda A, Paula-Barbosa MM (2004) Dendritic changes in the hippocampal formation of AIDS patients: a quantitative Golgi study. Acta Neuropathol 107:97–110. 10.1007/s00401-003-0781-3 [DOI] [PubMed] [Google Scholar]
- Sacktor N, Haughey N, Cutler R, Tamara A, Turchan J, Pardo C, Vargas D, Nath A (2004) Novel markers of oxidative stress in actively progressive HIV dementia. J Neuroimmunol 157:176–184. 10.1016/j.jneuroim.2004.08.037 [DOI] [PubMed] [Google Scholar]
- Saha RN, Pahan K (2003) Tumor necrosis factor-alpha at the crossroads of neuronal life and death during HIV-associated dementia. J Neurochem 86:1057–1071. 10.1046/j.1471-4159.2003.01942.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer DP, Lehrman EK, Kautzman AG, Koyama R, Mardinly AR, Yamasaki R, Ransohoff RM, Greenberg ME, Barres BA, Stevens B (2012) Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 74:691–705. 10.1016/j.neuron.2012.03.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah A, Verma AS, Patel KH, Noel R, Rivera-Amill V, Silverstein PS, Chaudhary S, Bhat HK, Stamatatos L, Singh DP, Buch S, Kumar A (2011) HIV-1 gp120 induces expression of IL-6 through a nuclear factor-kappa B-dependent mechanism: suppression by gp120 specific small interfering RNA. PLoS One 6:e21261. 10.1371/journal.pone.0021261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonetti M, Hagenston AM, Vardeh D, Freitag HE, Mauceri D, Lu J, Satagopam VP, Schneider R, Costigan M, Bading H, Kuner R (2013) Nuclear calcium signaling in spinal neurons drives a genomic program required for persistent inflammatory pain. Neuron 77:43–57. 10.1016/j.neuron.2012.10.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sporer B, Kastenbauer S, Koedel U, Arendt G, Pfister HW (2003) Increased intrathecal release of soluble fractalkine in HIV-infected patients. AIDS Res Hum Retroviruses 19:111–116. 10.1089/088922203762688612 [DOI] [PubMed] [Google Scholar]
- Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, Micheva KD, Mehalow AK, Huberman AD, Stafford B, Sher A, Litke AM, Lambris JD, Smith SJ, John SW, Barres BA (2007) The classical complement cascade mediates CNS synapse elimination. Cell 131:1164–1178. 10.1016/j.cell.2007.10.036 [DOI] [PubMed] [Google Scholar]
- Sui Y, Potula R, Dhillon N, Pinson D, Li S, Nath A, Anderson C, Turchan J, Kolson D, Narayan O, Buch S (2004) Neuronal apoptosis is mediated by CXCL10 overexpression in simian human immunodeficiency virus encephalitis. Am J Pathol 164:1557–1566. 10.1016/S0002-9440(10)63714-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sui Y, Stehno-Bittel L, Li S, Loganathan R, Dhillon NK, Pinson D, Nath A, Kolson D, Narayan O, Buch S (2006) CXCL10-induced cell death in neurons: role of calcium dysregulation. Eur J Neurosci 23:957–964. 10.1111/j.1460-9568.2006.04631.x [DOI] [PubMed] [Google Scholar]
- Tan AM, Samad OA, Fischer TZ, Zhao P, Persson AK, Waxman SG (2012) Maladaptive dendritic spine remodeling contributes to diabetic neuropathic pain. J Neurosci 32:6795–6807. 10.1523/JNEUROSCI.1017-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Z, Gan Y, Liu Q, Yin JX, Liu Q, Shi J, Shi FD (2014) CX3CR1 deficiency suppresses activation and neurotoxicity of microglia/macrophage in experimental ischemic stroke. J Neuroinflammation 11:26. 10.1186/1742-2094-11-26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian C, Sun L, Jia B, Ma K, Curthoys N, Ding J, Zheng J (2012) Mitochondrial glutaminase release contributes to glutamate-mediated neurotoxicity during human immunodeficiency virus-1 infection. J Neuroimmune Pharmacol 7:619–628. 10.1007/s11481-012-9364-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toggas SM, Masliah E, Rockenstein EM, Rall GF, Abraham CR, Mucke L (1994) Central nervous system damage produced by expression of the HIV-1 coat protein gp120 in transgenic mice. Nature 367:188–193. 10.1038/367188a0 [DOI] [PubMed] [Google Scholar]
- Tong N, Perry SW, Zhang Q, James HJ, Guo H, Brooks A, Bal H, Kinnear SA, Fine S, Epstein LG, Dairaghi D, Schall TJ, Gendelman HE, Dewhurst S, Sharer LR, Gelbard HA (2000) Neuronal fractalkine expression in HIV-1 encephalitis: roles for macrophage recruitment and neuroprotection in the central nervous system. J Immunol 164:1333–1339. 10.4049/jimmunol.164.3.1333 [DOI] [PubMed] [Google Scholar]
- Tronche F, Kellendonk C, Kretz O, Gass P, Anlag K, Orban PC, Bock R, Klein R, Schütz G (1999) Disruption of the glucocorticoid receptor gene in the nervous system results in reduced anxiety. Nat Genet 23:99–103. 10.1038/12703 [DOI] [PubMed] [Google Scholar]
- Turchan J, Pocernich CB, Gairola C, Chauhan A, Schifitto G, Butterfield DA, Buch S, Narayan O, Sinai A, Geiger J, Berger JR, Elford H, Nath A (2003) Oxidative stress in HIV demented patients and protection ex vivo with novel antioxidants. Neurology 60:307–314. 10.1212/01.WNL.0000042048.85204.3D [DOI] [PubMed] [Google Scholar]
- Tyor WR, Glass JD, Griffin JW, Becker PS, McArthur JC, Bezman L, Griffin DE (1992) Cytokine expression in the brain during the acquired immunodeficiency syndrome. Ann Neurol 31:349–360. 10.1002/ana.410310402 [DOI] [PubMed] [Google Scholar]
- Vasek MJ, Garber C, Dorsey D, Durrant DM, Bollman B, Soung A, Yu J, Perez-Torres C, Frouin A, Wilton DK, Funk K, DeMasters BK, Jiang X, Bowen JR, Mennerick S, Robinson JK, Garbow JR, Tyler KL, Suthar MS, Schmidt RE, et al. (2016) A complement-microglial axis drives synapse loss during virus-induced memory impairment. Nature 534:538–543. 10.1038/nature18283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viviani B, Gardoni F, Bartesaghi S, Corsini E, Facchi A, Galli CL, Di Luca M, Marinovich M (2006) Interleukin-1 beta released by gp120 drives neural death through tyrosine phosphorylation and trafficking of NMDA receptors. J Biol Chem 281:30212–30222. 10.1074/jbc.M602156200 [DOI] [PubMed] [Google Scholar]
- Wan XZ, Li B, Li YC, Yang XL, Zhang W, Zhong L, Tang SJ (2012) Activation of NMDA receptors upregulates a disintegrin and metalloproteinase 10 via a Wnt/MAPK signaling pathway. J Neurosci 32:3910–3916. 10.1523/JNEUROSCI.3916-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xavier AL, Menezes JR, Goldman SA, Nedergaard M (2014) Fine-tuning the central nervous system: microglial modelling of cells and synapses. Philos Trans R Soc Lond B Biol Sci 369:20130593. 10.1098/rstb.2013.0593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yowtak J, Lee KY, Kim HY, Wang J, Kim HK, Chung K, Chung JM (2011) Reactive oxygen species contribute to neuropathic pain by reducing spinal GABA release. Pain 152:844–852. 10.1016/j.pain.2010.12.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan SB, Shi Y, Chen J, Zhou X, Li G, Gelman BB, Lisinicchia JG, Carlton SM, Ferguson MR, Tan A, Sarna SK, Tang SJ (2014) Gp120 in the pathogenesis of human immunodeficiency virus-associated pain. Ann Neurol 75:837–850. 10.1002/ana.24139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zabel MK, Zhao L, Zhang Y, Gonzalez SR, Ma W, Wang X, Fariss RN, Wong WT (2016) Microglial phagocytosis and activation underlying photoreceptor degeneration is regulated by CX3CL1-CX3CR1 signaling in a mouse model of retinitis pigmentosa. Glia 64:1479–1491. 10.1002/glia.23016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan Y, Paolicelli RC, Sforazzini F, Weinhard L, Bolasco G, Pagani F, Vyssotski AL, Bifone A, Gozzi A, Ragozzino D, Gross CT (2014) Deficient neuron-microglia signaling results in impaired functional brain connectivity and social behavior. Nat Neurosci 17:400–406. 10.1038/nn.3641 [DOI] [PubMed] [Google Scholar]
- Zhang X, Thayer SA (2018) Monoacylglycerol lipase inhibitor JZL184 prevents HIV-1 gp120-induced synapse loss by altering endocannabinoid signaling. Neuropharmacology 128:269–281. 10.1016/j.neuropharm.2017.10.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The cortical primary cultures contains neuron, microglia and astrocytes. Immnofluoresecent staining was perform at DIV 12 to identify neuron (MAP2+, blue; 50%), microglia (IBa1+, red; 20%) and astrocytes (GFAP+, green; 30%). Scale bar: 50μm. This extended data is included to support Figure 3. Download Figure 3-1, TIF file (3.8MB, tif)
DT treatment caused microglial atrophy. 20ng DT (in 5μl PBS) or Veh (5μl PBS) was i.t. injected into CD11b-DTR mice, once a day for 3 days. Twenty-four hours after the last treatment, spinal cords were harvested for immunofluorescent staining with IBa1. DT treatment showed significant loss of microglial processes and shrunken cell body (* indicates microglia). Scale bar: 20μm. This extended data is included to support Figure 4. Download Figure 4-1, TIF file (5.2MB, tif)
gp120 co-receptors mediated synapse loss. A. Immunoblots showed the expression level of CXCR4 and CCR5 in forebrain, synaptosome and postsynaptic density homogenate. B. Effects of maraviroc pre-treatment on gp120-induced decrease of PSD95 and Syn I. Cortical cultures were pre-treated with the maraviroc (50μM) for 30 min and then co-treated with gp120 (200pM) for another 12 hours. Cell lysates were analyzed by immunoblotting. N=3 cultures per condition. B. Data are expressed as mean ± SEM. This extended data is included to support Figure 7. Download Figure 7-1, TIF file (2.7MB, tif)