

Abstract
A fundamental question regarding the etiology of amyotrophic lateral sclerosis (ALS) is whether the various gene mutations associated with the disease converge on a single molecular pathway or act through multiple pathways to trigger neurodegeneration. Notably, several of the genes and cellular processes implicated in ALS have also been linked to frontotemporal dementia (FTD), suggesting these two diseases share common origins with varied clinical presentations. Scientists are rapidly identifying ALS/FTD suppressors that act on conserved pathways from invertebrates to vertebrates to alleviate degeneration. The elucidation of such genetic modifiers provides insight into the molecular pathways underlying this rapidly progressing neurodegenerative disease, while also revealing new targets for therapeutic development.
Keywords: ALS, FTD, genetic modifiers, RBPs, stress granules, disease models
Introduction
Amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD) have long been considered distinct neurodegenerative diseases whose precise pathogenesis remains elusive. Each is associated with a loss in specific neuronal populations. The loss of neurons in the frontal and/or temporal lobe of the brain in FTD produces changes in personality, language, and memory; whereas in ALS, functional disruptions in both upper and lower motor neurons first manifest as muscle weakness, with a rapid, progressive, and complete loss of voluntary movement in the arms and legs, followed by reduced respiratory muscle function and failure, usually 2–5 years after diagnosis. Despite these differences in clinical presentation, there is much genetic, clinical, and pathological overlap between ALS and FTD, suggesting that they may be a single disease with a range of phenotypic manifestations. For example, ∼50% of people with ALS exhibit cognitive and/or behavioral deficiencies, and 15%–20% reach a diagnosis of FTD (Marin et al., 2017; Nguyen et al., 2018). To date, no treatments for FTD exist, and the few ALS therapies provide only a minimal extension in lifespan of a few months. There is an urgent need to develop new therapeutics that slow the loss of motor function and prevent further neurodegeneration. To achieve such goals, we need a better understanding of what causes ALS and what cellular pathways are compromised in diseased individuals. Treatments could then target the causative factor and/or the resulting dysfunctional cellular pathway(s) that precede motor neuron death.
ALS does not arise from a single insult or one specific genetic mutation. Approximately 5%–10% of patients suffer from familial ALS (fALS), which is caused by a mutation in one of >25 different genes that act in a variety of cellular processes. A handful of these genes (C9ORF72, SOD1, TARDBP, FUS, PFN1) harbor the majority of fALS mutations. Genetic perturbations, such as the expansion of a hexanucleotide repeat in the C9Orf72 locus, result in abnormal RNA production and/or the accumulation of dipeptide repeat protein products (DPRs) resulting from RAN translation. ALS mutations can also disrupt superoxide dismutase (SOD1) activity, the normal function of RNA splicing factors (TARDBP and FUS) or of a cytoskeletal regulator (PFN1). The challenge before us is to determine how mutations in these genes all result in the same disease phenotype. Do they converge on a single downstream molecular pathway whose disruption triggers the neurodegeneration typical of ALS and FTD? Or do they affect distinct pathways, any one of which independently leads to the common disease phenotype?
Animal models provide a means to study aspects of complex diseases using tractable, well-characterized systems. In particular, invertebrates, such as Caenorhabditis elegans and Drosophila melanogaster, allow rapid, large-scale genetic and pharmacological studies (Sin et al., 2014; Fernández-Hernández et al., 2016; Deal and Yamamoto, 2018; Ma et al., 2018). Not only do >40% of the C. elegans genome (7569 genes) and 75% of the Drosophila genome (15,500 genes) have human orthologs (Pandey and Nichols, 2011; Kim et al., 2018), these model organisms share the same fundamental cellular processes governing neuronal function and survival. The functional conservation of the motor circuit between Drosophila and mammals further emphasizes their value in the rapid identification of disease-relevant pathways and genetic modifiers, which can then be studied in rodent models and in human-derived-induced pluripotent stem cell (iPSC) models of ALS/FTD. The identification of gene variants that suppress neurodegeneration in animal models of ALS can provide critical molecular insights into the mechanistic basis of ALS/FTD. Are there phenotypic suppressors that alleviate degeneration across all cases of ALS and FTD, regardless of cause? If so, such suppressors are valuable in the identification of targets for developing new, more effective therapeutics. This review addresses efforts to identify suppressors using different experimental approaches and different genetic models of ALS/FTD, through the examination of diverse cellular mechanisms ranging from stress response to local protein synthesis, uniting around the concept of identifying factors that modify the disease phenotype. In each case, we explore specific issues related to our overarching questions: does ALS/FTD arise from one common defect, or from a defect in any one of many cellular processes, each of which has the ability to orchestrate degeneration of motor neurons, and/or cortical neurons?
Diverse cellular pathways contribute to TDP-43 proteinopathy in C. elegans models of ALS/FTD
Mutations in TARDBP, encoding the DNA/RNA binding TDP-43 protein, cause <1% of ALS cases, but ∼95% of ALS and ∼50% of FTD (FTLD-TDP) cases exhibit cytoplasmic or nuclear aggregates containing the protein TDP-43 (Mackenzie and Neumann, 2016; Prasad et al., 2019). Because TDP-43 dysfunction can cause disease, understanding the biology driving this process is of critical importance. TDP-43 proteinopathies, such as ALS and FTLD-TDP, have been modeled by expressing human TDP-43 in neurons of C. elegans. Expressing WT hTDP-43 causes relatively mild behavioral or functional phenotypes, including uncoordinated locomotion, modest progressive neurodegeneration, shortened lifespan, and accumulation of pathologically truncated, ubiquitinated, phosphorylated, and insoluble TDP-43; expression of mutant hTDP-43 causes more severe presentation of these phenotypes (Ash et al., 2010; Liachko et al., 2010; Zhang et al., 2011; Vaccaro et al., 2012). The increased toxicity of mutant TDP-43 is also observed in vertebrate models, indicating that C. elegans can be used to investigate conserved mechanisms of TDP-43 biology (Tan et al., 2017).
TDP-43 is a DNA- and RNA-binding protein and has diverse roles regulating transcription, pre-mRNA splicing, micro-RNA processing, mRNA stability and transport, and stress granule (SG) assembly (Prasad et al., 2019). In human disease, aggregated TDP-43 exhibits post-translational modifications, including ubiquitination, SUMO-ylation, phosphorylation, and truncated protein species. These modifications are consistently observed across ALS/FTLD spanning multiple genetic causes and are typical of the majority of sporadic cases of disease. Phosphorylation of TDP-43 decreases TDP-43 protein turnover, promotes mislocalization from the nucleus to the cytoplasm, and increases protein aggregation. C. elegans models accumulate prominent phosphorylated TDP-43, which increases with age and correlates with neuronal loss and functional impairment (Liachko et al., 2010). Studies in both C. elegans and in vitro systems have identified conserved TDP-43 kinases and phosphatases responsible for this post-translational modification of TDP-43 (Hasegawa et al., 2008; Liachko et al., 2013, 2014, 2016; Li et al., 2017; Gu et al., 2018). These include the kinases CK1, CDC7, TTBK1, and TTBK2, which directly phosphorylate TDP-43 at serines 409 and 410 and promote its relocalization from the nucleus to cytoplasm, where aggregates are most commonly found. Notably, these kinases colocalize with TDP-43-positive inclusions in cortical and spinal cord tissue from ALS and FTLD-TDP patients, whereas the expression of TTBK1/2 protein is elevated in FTLD-TDP patients (Liachko et al., 2013, 2014; Taylor et al., 2018). TDP-43 is also phosphorylated at threonines 153 and 155 by MEK, a kinase in the MAPK/ERK signaling family, although it is unclear whether these epitopes are involved in disease (Li et al., 2017). The importance of tightly regulating TDP-43 phosphorylation has also been suggested by studies showing that genetic loss of calcineurin, a phosphatase that removes the pathological C-terminal phosphates on TDP-43, increases motor dysfunction, and accelerates neurodegeneration in C. elegans-expressing hTDP-43, and pharmacological inhibition or siRNA-mediated reduction in calcineurin expression promotes accumulation of phosphorylated endogenous TDP-43 in human cultured cells (Liachko et al., 2016). Phosphorylation of TDP-43 may be a consequence of disruptions in cellular homeostasis; therefore, intervening to decrease or eliminate TDP-43 phosphorylation, either by inhibiting TDP-43-targeted kinases or activating TDP-43 phosphatases, is an attractive strategy for treating TDP-43 proteinopathies, such as ALS and FTLD-TDP.
A recent genomewide RNA interference (RNAi) screen has been conducted using C. elegans-expressing human fALS-linked mutant TDP-43 pan-neuronally. The screen surveyed 16,767 genes, covering >86% of the C. elegans genome (Kamath et al., 2003). TDP-43-induced uncoordinated locomotion was suppressed by 46 RNAi targets, 24 of which have human homologs. These genes have known functions in diverse areas of cell biology, including energy production, cellular metabolism, extracellular matrix, cytoskeleton, ion transport, proteostasis, and signaling, highlighting the complexity of pathways contributing to TDP-43 proteinopathy. One suppressor identified hse-5, a heparan sulfate epimerase that catalyzes the inversion stereochemistry of d-glucuronic acid to l-iduronic acid. Elucidating the mechanism underlying hse-5-mediated suppression, as well as that of other identified suppressors, is future important work.
Disruption of RNA processing: a common denominator in ALS and FTD
The groundbreaking discovery of TDP-43 as a component of ubiquitinated inclusions in neurons of patients with sporadic ALS and FTD (Arai et al., 2006; Neumann et al., 2006) was the first evidence pointing to a role for altered RNA processing in these disorders (Lagier-Tourenne et al., 2010). Indeed, TDP-43 is mislocalized in the vast majority of ALS patients and ∼45% of patients with FTD. This finding was rapidly followed by the identification of mutations and/or the mislocalization of several RNA-binding proteins (RBPs) in ALS and FTD, including Fused in sarcoma (FUS) (Kwiatkowski et al., 2009; Vance et al., 2009), Ewing's sarcoma (EWSR1) (Couthouis et al., 2012), TATA-binding protein-associated factor 15 (TAF-15) (Couthouis et al., 2011), heterogeneous ribonucleoparticle proteins A2B1 and A1 (hnRNPA2B1 and hnRNPA1) (Kim et al., 2013), Matrin 3 (MATR3) (Johnson et al., 2014), and the T cell-restricted intracellular antigen-1 (TIA1) (Mackenzie et al., 2017). These RBPs have been implicated in several steps of RNA metabolism, including RNA splicing, transport, stability, or translation, and their disruption leads to widespread defects in RNA processing (Nussbacher et al., 2019).
Genomewide approaches have provided major insights into the multiple ways these RBPs influence the processing of their RNA targets, with hundreds of expression or splicing alterations identified in ALS/FTD models and patient tissues (Nussbacher et al., 2015). Importantly, several splicing defects consistent with a loss of TDP-43 function were identified in brain tissues of ALS/FTD patients (Polymenidou et al., 2011; Prudencio et al., 2012; Shiga et al., 2012; Ling et al., 2015), likely as a result of TDP-43 relocating from the nucleus to the cytoplasm. Indeed, a striking nuclear clearance of TDP-43 is widely observed in both sporadic and familial cases, including in patients with expansions in the C9orf72 gene. Notably, TDP-43 pathology, rather that DPR aggregates, correlates with neurodegeneration in postmortem tissues from C9orf72 patients (Chew et al., 2015, 2019; Vatsavayai et al., 2016; Solomon et al., 2018). Considering the widespread disruption of RNA processing associated with TDP-43 pathology, a major challenge is to determine the relative contribution to disease of TDP-43 versus DPR aggregates.
The human RNA encoding Stathmin-2, a neuronal microtubule-associated protein also known as SCG10, was recently shown to be the most affected transcript upon TDP-43 loss (Klim et al., 2019; Melamed et al., 2019). TDP-43 disruption induces aberrant splicing and premature polyadenylation within the first intron of STMN2, producing a nonfunctional truncated mRNA. Altered STMN2 expression was found in TDP-43-mutant or -depleted neurons directly converted from fibroblasts and iPSC-derived motor neurons, as well as in motor cortex and spinal motor neurons from sporadic and C9orf72 ALS/FTD patients. Stathmin-2 is one of four mammalian stathmins known to directly bind dimers of α/β Tubulin and to affect microtubule dynamics (Belmont and Mitchison, 1996; Chauvin and Sobel, 2015). Stathmin-2 has been suggested as an axonal-maintenance factor (Shiga et al., 2012) and as an essential component for axonal regeneration (Mason et al., 2002). Upon axonal injury, Stathmin-2 is upregulated and recruited to growth cones of regenerating axons (Shin et al., 2014) with a demonstrated requirement for neuromuscular junction stability in Drosophila (Graf et al., 2011). Antisense oligonucleotides targeting either STMN2 or TARDBP transcripts were recently shown to preclude regeneration following axotomy of iPSC-derived human motor axons (Melamed et al., 2019). Remarkably, although reduction in TDP-43 is associated with hundreds of RNA alterations, restoration of Stathmin-2 levels was sufficient to rescue axonal regenerative capacity in motor neurons lacking TDP-43.
The potential consequences of other TDP-43-mediated RNA defects and their contribution to disease remain to be demonstrated, and further investigations are required to better understand the role of Stathmin-2 in axonal biology and renervation/denervation of synapses, especially neuromuscular junctions. Nevertheless, the weight of evidence supports rescue of Stathmin-2 levels as a potential therapeutic approach in neurodegenerative diseases affected by TDP-43 proteinopathy.
ALS/FTD mutations in the RNA-binding protein FUS suppress local axonal protein synthesis
The gene encoding another RBP, FUS, like TARDBP (TDP-43), is the site of ALS/FTD-causing mutations (Kwiatkowski et al., 2009; Vance et al., 2009). RBPs have been shown to play key roles in highly polarized neurons, including the selective transport of mRNAs along axons and their localization at synapses allowing for local synthesis of proteins (Donlin-Asp et al., 2017). A wealth of data has led to a consensus that protein synthesis occurs in mature dendrites and growth cones and that this synthesis is critical for memory, learning, and synaptic plasticity (Biever et al., 2019). Whereas there is ample evidence that invertebrate axons can synthesize proteins (Piper and Holt, 2004; Twiss and Fainzilber, 2009) and that local translation is required for axonal growth during development and repair following injury, there is no consensus local translation contributes to neuronal homeostasis and degeneration in mature mammalian axons. In mammals, some axons, including those of motor neurons, can extend >1 m with an estimated volume of up to 160 million μm3 (>5000 times the volume of typical cell). This poses a challenge for protein homeostasis in distal synaptic structures, such as at neuromuscular junctions, whose loss is one of the earliest events observed in ALS patients.
It has recently been shown that ALS/FTD FUS mutations suppress local axonal protein synthesis through activation of an integrated stress response in mouse hippocampi, which in turn leads to impaired neuronal activity and synaptic deficits (López-Erauskin et al., 2018). Using humanized mice in which ALS/FTD-causing FUS mutations provoke age-dependent progressive motor and cognitive deficits, the inhibition of local translation in sciatic nerves was observed before development of disease and was accompanied by increased axonal accumulation of mutant FUS. This work provided in vivo evidence for local intra-axonal translation in adult mice and uncovered a role of FUS in local protein synthesis, extending its cellular functions beyond its known roles in RNA processing, microRNA biogenesis, and mRNA transport (Butti and Patten, 2018). It also raises the possibility that, like deficits in translation observed in C9orf72 and TDP-43-associated ALS/FTD (Kanekura et al., 2016; Cestra et al., 2017; Balendra and Isaacs, 2018; Lehmkuhl and Zarnescu, 2018), impaired local translation may contribute to, or even cause, the age-dependent neurodegeneration associated with mutations in FUS. Future efforts will build on these findings to define the local “translatome” (mRNAs that are actively translated) and proteome (newly synthesized proteins) in healthy and diseased motor axons. Together, these efforts have the potential to further our understanding of how mature axons and their synapses with muscles are lost in ALS.
Targeting stress response pathways and interactions within RNA granules to mitigate persistent protein aggregation
A common feature of ALS and FTD, as well as many other neurodegenerative diseases, is the presence of abnormal protein aggregates in neurons in the brain and spinal cord. Environmental stresses can contribute to the formation of aggregates, and multiple forms of stress have been implicated in neurodegenerative disease pathogenesis, including oxidative stress, ER stress, and inflammation (Sprenkle et al., 2017). Most of these stresses activate the integrated stress response pathway leading to phosphorylation of the α subunit of the Eukaryotic initiation factor 2 and subsequent translational repression (Bosco, 2018). In cultured cells, stress-induced translational repression often coincides with the formation dynamic cytoplasmic foci called stress granules (SGs) (Kedersha and Anderson, 2002). SGs are membraneless organelles that contain hundreds of RBPs, components of the translation machinery, and repressed mRNA transcripts (Souquere et al., 2009; Sidrauski et al., 2015; Jain et al., 2016; Khong et al., 2017). The biological role of SGs under conditions of stress has not been fully elucidated (Protter and Parker, 2016); but within the last 10 years, the hypothesis that SGs are pathogenic in the context of neurodegenerative disease has emerged. This hypothesis is supported by observations that disease-linked variants associate with SGs in cell culture and frequently appear to alter their physical properties, including size, abundance, and/or dynamics. These effects have been demonstrated for the ALS/FTD-associated RBPs TDP-43, FUS, and hnRNPA2B1 (Liu-Yesucevitz et al., 2010; McDonald et al., 2011; Baron et al., 2013; Kim et al., 2013; Sama et al., 2014; Gomes and Shorter, 2019), and other ALS/FTD-associated proteins, including SOD1 (Gal et al., 2016), UBQLN2 (Alexander et al., 2018), MAPT (Vanderweyde et al., 2016), and DPRs encoded by the C9orf72 nucleotide repeat expansion (Boeynaems et al., 2017). Moreover, SG-related proteins colocalize within pathological aggregates from postmortem CNS tissues from human ALS and FTD patients (Dormann et al., 2010; Liu-Yesucevitz et al., 2010; Alexander et al., 2018), supporting the idea that SGs could serve as precursors of inclusions found in neurodegenerative disease (Aulas and Vande Velde, 2015).
In recent years, aberrant SG behavior has been proposed to integrate genetic and environmental factors to facilitate the formation of permanent insoluble inclusions. Both SG formation and permanent aggregation depend on liquid–liquid phase separation, a cellular process by which some proteins form highly concentrated liquid-like droplets that are demixed from the aqueous cellular milieu. Of particular relevance to ALS/FTD, mutations in TARDBP, FUS, and HNRNPA2/B1, as well as expansion of the hexanucleotide repeat in the C9orf72 locus, interfere with liquid–liquid phase separation behavior and impact either the formation or disassembly of SGs (Murakami et al., 2015; Patel et al., 2015; Boeynaems et al., 2016; Lee et al., 2016; Martinez et al., 2016; Mackenzie et al., 2017; Markmiller et al., 2018). Importantly, therapeutic reduction of the SG-associated proteins Ataxin-2 and TIA1 components was recently shown to provide neuroprotective effects in animal models of ALS (Becker et al., 2017; Apicco et al., 2018).
Two important considerations remain. First, the molecular interactions governing the behavior of these granules is still incompletely understood, and a more complete understanding of how SG composition and behavior are affected by genetic, environmental, and aging-related factors is needed. Second, in vivo mammalian models of stress and disease are needed to rigorously test the hypothesis that SGs play a role in human neurodegenerative disease pathogenesis. To address the first of these questions, robust cellular models and APEX-mediated proximity labeling with high-content imaging was used to analyze the protein composition of SGs under different conditions to identify candidate targets that regulate SG biology and stress-induced protein aggregation (Markmiller et al., 2018). Reducing the levels of some candidate targets rescued degenerative phenotypes in three different Drosophila models of neurodegeneration (Markmiller et al., 2018). Additional work in ALS patient-derived iPSC-derived motor neurons showed that transient exposure to stress leads to persistent accumulation of insoluble TDP-43 and other ALS-associated RBPs, as well as long-lasting perturbations of global mRNA localization and increased neuronal cell death. A small-molecule screen by the same group identified a class of compounds that can prevent the RNA-dependent recruitment of TDP-43 to persistent aggregates and increases survival in transiently stressed ALS mutant iPSC-derived motor neurons (Fang et al., 2019). Last, experiments designed to systematically dissect the role of post-translational protein ubiquitylation in regulating RNA granules and protein aggregation came to the surprising conclusion that unconjugated ubiquitin, but not polyubiquitin, accumulates in SGs and that active polyubiquitylation is not necessary for SG dynamics (Markmiller et al., 2019).
To date, the majority of research on SGs has been performed in cell culture, and there is clear evidence that forced oligomerization of SG proteins is toxic to cells (Zhang et al., 2019). There are multiple factors to consider as we translate our knowledge of SGs from cell culture to in vivo models. One factor is the type of stress that can induce SG formation in vivo. Generally, nonphysiological stressors, such as sodium arsenite, sorbitol, and protein overexpression, are used to induce SG assembly in cultured cells (Sama et al., 2014). Whether granules formed by protein overexpression are bona fide SGs is unclear, as these granules exhibit different dynamic properties compared with SGs formed in response to acute stress. Furthermore, disease-relevant stressors, such as glutamate-induced excitotoxicity, did not induce SG assembly in neurons; thus, not all types of stress necessarily result in SG formation (Tischbein et al., 2019). A recent study in Drosophila provided novel proof of concept that physical trauma can serve as an acute form of stress that triggers SG assembly in vivo (Anderson et al., 2018). Unpublished work from the D.A.B. laboratory supports the hypothesis that head trauma can also induce SG formation in mammals. Whether trauma-induced granules in vivo can precipitate larger pathological inclusions remains to be determined.
If SGs are found to promote disease pathology and/or worsen neurodegenerative disease outcomes in vivo, SGs may be appropriate therapeutic targets for these disorders. Small molecules, such as ISRIB, which prevent SG formation in culture, could be considered for suppressing SG-related phenotypes in vivo (Sidrauski et al., 2015). However, some cell-culture studies suggest that SGs protect cells from the toxic behaviors of proteins, such as FUS (Shelkovnikova et al., 2013) and TDP-43 (McGurk et al., 2018), and that these proteins exert toxicity independently of their association with SGs (Gasset-Rosa et al., 2019; Mann et al., 2019). Therefore, it will be important to carefully examine SGs in vivo under various conditions of acute and chronic stress, as well as disease models, to elucidate whether these species play a role in neurodegenerative disease pathogenesis.
Genetic suppressors of multiple models of ALS identify common dysregulated pathways
A preponderance of data shows that a wide range of molecular and cellular processes are affected by ALS-associated mutations. Studies on both patient populations and animal models have revealed that specific second-site gene perturbations can modify ALS phenotypes (Yanagi et al., 2019). Despite the fact that 40–50 such gene perturbations have been identified that modify more than one ALS gene, few have been further tested systematically across additional ALS/FTD models. A recent genomewide screen in Drosophila by the Artavanis–Tsakonas group identified specific genetic lesions that could modify both TDP-43 and FUS-associated degeneration, with many tested against a C9orf72 G4C2 expansion model. The K.A.W. laboratory tested a subset of these genetic modifiers in a fourth ALS model, a knock-in of the SOD1-G85R lesion into the endogenous locus (dSod1G85R) (Sahin et al., 2017), for suppression of degenerative phenotypes. More than 30 genes were identified that could modify all four ALS models. The affected genes fall into different functional categories, including energy metabolism, stress response, cell polarity, cytoskeletal regulators, and chromatin regulation. It has been shown that motor circuit dysfunction associated with dSod1G85R can be suppressed through the activation of BMP signaling in nonmotor neurons or in motor neurons (Held et al., 2019). ALS models where cell type-specific suppression can be assessed will provide important information about the cells in which modulation of ALS-associated phenotypes is possible by particular genetic modifiers.
The majority of genetic models of ALS/FTD involve the overexpression of a mutant human gene or the homologous host gene harboring a patient allele. More recently, the potential impact of gene dosage or levels of mutant protein is being considered as it may alter stoichiometries and molecular interactions in affected pathways. The use of a knock-in model has the advantage of providing a platform to identify and study suppressors of neurodegeneration in a genetic and homeostatic context that is closer to that seen in patients, as opposed to that in most overexpression models. While an exhaustive comparison of suppressors of knock-in versus overexpression models of ALS has not been done, it is of considerable interest that a significant number of gene modifiers were identified that suppress degeneration associated with both types of models. Interestingly, further genetic analysis of two modifiers whose gene products are known to physically interact (one affecting stress response and the other metabolism) indicates that they exhibit a genetic interaction that modifies their ability to rescue neurodegeneration phenotypes caused by some, but not all, ALS models tested. Moreover, the ability of the two modifier genes to rescue neurodegeneration-related phenotypes is inhibited by their functional interdependence in the case of one ALS model, whereas in the other these modifiers appear to act independently. Together, these results indicate that not all ALS models respond in the same way to modifiers.
In conclusion, the advances presented here provide valuable insights into biological processes involved in onset and/or progression of ALS/FTD (Fig. 1). They show that ALS/FTD is accompanied by a variety of different cellular dysfunctions and that the repair of just one dysfunction may be sufficient to restore function. Such studies will continue to shed light on molecules that underlie disease mechanism and provide targets for therapeutic intervention in ALS/FTD. As we move forward, considering the interactions between modifiers and components of identified pathways will inform us of the cellular basis of disease, as well as the effectiveness and mode of action of newly developed therapeutics.
Figure 1.
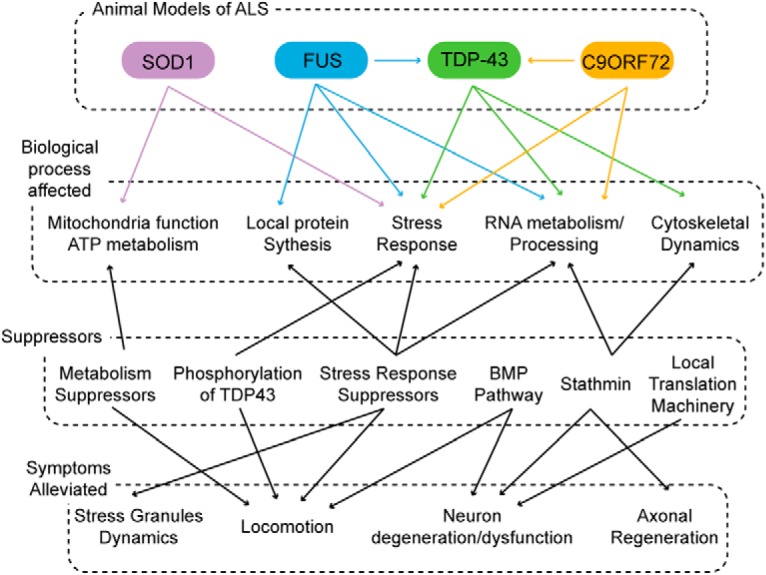
Suppressors of dysfunctional cellular pathways in ALS/FTD identify putative targets for therapeutic development. The etiology of ALS/FTD is complex, involving a number of independent cellular processes that lead to varied degenerative symptoms. Nevertheless, a single suppressor can reduce multiple symptoms, an encouraging outlook for therapeutic target identification by genetic modifier screens.
Footnotes
M.B and K.A.W. were funded by The Judith and Jean Pape Adams Charitable Foundation, ALS: Finding a Cure Foundation, and Biogen. D.A.B. was funded by NIH/NINDS R01NS108769 and the Healy Endowment. S.D.C. was funded by The Wellcome Trust and Medical Research Council, Target ALS, the Muscular Dystrophy Association (grant MDA352600), a Milton Safenowitz postdoctoral fellowship, NIH/NIGMS T32 GM008666, the Ludwig Institute for Cancer Research and Medical Research Council, Alzheimer Research UK, Alzheimer Society (United Kingdom Dementia Research Institute), the National Institute for Health Research (NIHR) Dementia Biomedical Research Unit at South London, Maudsley NHS Foundation Trust, and King's College London. C.L.-T. is supported by the Sean M. Healey & AMG Center for ALS at Mass General, NIH (NS087227 and NS108769), the Department of Defense (USAMRAA 18-1-0092), Target ALS, the ALS Association, the Association Française contre les myopathies and the Binational Science Foundation. N.L. thanks the Department of Veterans Affairs (Merit Review Grant I01BX004044 to N.L.) for funding.
The authors declare no competing financial interests.
References
- Alexander EJ, Ghanbari Niaki A, Zhang T, Sarkar J, Liu Y, Nirujogi RS, Pandey A, Myong S, Wang J (2018) Ubiquilin 2 modulates ALS/FTD-linked FUS-RNA complex dynamics and stress granule formation. Proc Natl Acad Sci U S A 115:E11485–E11494. 10.1073/pnas.1811997115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson EN, Gochenaur L, Singh A, Grant R, Patel K, Watkins S, Wu JY, Pandey UB (2018) Traumatic injury induces stress granule formation and enhances motor dysfunctions in ALS/FTD models. Hum Mol Genet 27:1366–1381. 10.1093/hmg/ddy047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apicco DJ, Ash PE, Maziuk B, LeBlang C, Medalla M, Al Abdullatif A, Ferragud A, Botelho E, Ballance HI, Dhawan U, Boudeau S, Cruz AL, Kashy D, Wong A, Goldberg LR, Yazdani N, Zhang C, Ung CY, Tripodis Y, Kanaan NM, et al. (2018) Reducing the RNA-binding protein TIA1 protects against tau-mediated neurodegeneration in vivo. Nat Neurosci 21:72–80. 10.1038/s41593-017-0022-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arai T, Hasegawa M, Akiyama H, Ikeda K, Nonaka T, Mori H, Mann D, Tsuchiya K, Yoshida M, Hashizume Y, Oda T (2006) TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun 351:602–611. [DOI] [PubMed] [Google Scholar]
- Ash PE, Zhang YJ, Roberts CM, Saldi T, Hutter H, Buratti E, Petrucelli L, Link CD (2010) Neurotoxic effects of TDP-43 overexpression in C. elegans. Hum Mol Genet 19:3206–3218. 10.1093/hmg/ddq230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aulas A, Vande Velde C (2015) Alterations in stress granule dynamics driven by TDP-43 and FUS: a link to pathological inclusions in ALS? Front Cell Neurosci 9:423. 10.3389/fncel.2015.00423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balendra R, Isaacs AM (2018) C9orf72-mediated ALS and FTD: multiple pathways to disease. Nat Rev Neurol 14:544–558. 10.1038/s41582-018-0047-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baron DM, Kaushansky LJ, Ward CL, Sama RR, Chian RJ, Boggio KJ, Quaresma AJC, Nickerson JA, Bosco DA (2013) Amyotrophic lateral sclerosis-linked FUS/TLS alters stress granule assembly and dynamics. Mol Neurodegener 8:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker LA, Huang B, Bieri G, Ma R, Knowles DA, Jafar-Nejad P, Messing J, Kim HJ, Soriano A, Auburger G, Pulst SM, Taylor JP, Rigo F, Gitler AD (2017) Therapeutic reduction of ataxin-2 extends lifespan and reduces pathology in TDP-43 mice. Nature 544:367–371. 10.1038/nature22038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belmont LD, Mitchison TJ (1996) Identification of a protein that interacts with tubulin dimers and increases the catastrophe rate of microtubules. Cell 84:623–631. 10.1016/S0092-8674(00)81037-5 [DOI] [PubMed] [Google Scholar]
- Biever A, Donlin-Asp PG, Schuman EM (2019) Local translation in neuronal processes. Curr Opin Neurobiol 57:141–148. 10.1016/j.conb.2019.02.008 [DOI] [PubMed] [Google Scholar]
- Boeynaems S, Bogaert E, Kovacs D, Konijnenberg A, Timmerman E, Volkov A, Guharoy M, De Decker M, Jaspers T, Ryan VH, Janke AM, Baatsen P, Vercruysse T, Kolaitis RM, Daelemans D, Taylor JP, Kedersha N, Anderson P, Impens F, Sobott F, et al. (2017) Phase separation of C9orf72 dipeptide repeats perturbs stress granule dynamics. Mol Cell 65:1044–1055.e5. 10.1016/j.molcel.2017.02.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeynaems S, Bogaert E, Michiels E, Gijselinck I, Sieben A, Jovicic A, De Baets G, Scheveneels W, Steyaert J, Cuijt I, Verstrepen KJ, Callaerts P, Rousseau F, Schymkowitz J, Cruts M, Van Broeckhoven C, Van Damme P, Gitler AD, Robberecht W, Van Den Bosch L (2016) Drosophila screen connects nuclear transport genes to DPR pathology in c9ALS/FTD. Sci Rep 6:20877. 10.1038/srep20877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosco DA. (2018) Translation dysregulation in neurodegenerative disorders. Proc Natl Acad Sci U S A 115:12842–12844. 10.1073/pnas.1818493115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butti Z, Patten SA (2018) RNA dysregulation in amyotrophic lateral sclerosis. Front Genet 9:712. 10.3389/fgene.2018.00712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cestra G, Rossi S, Di Salvio M, Cozzolino M (2017) Control of mRNA translation in ALS proteinopathy. Front Mol Neurosci 10:85. 10.3389/fnmol.2017.00085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chauvin S, Sobel A (2015) Neuronal stathmins: a family of phosphoproteins cooperating for neuronal development, plasticity and regeneration. Prog Neurobiol 126:1–18. 10.1016/j.pneurobio.2014.09.002 [DOI] [PubMed] [Google Scholar]
- Chew J, Gendron TF, Prudencio M, Sasaguri H, Zhang YJ, Castanedes-Casey M, Lee CW, Jansen-West K, Kurti A, Murray ME, Bieniek KF, Bauer PO, Whitelaw EC, Rousseau L, Stankowski JN, Stetler C, Daughrity LM, Perkerson EA, Desaro P, Johnston A, et al. (2015) Neurodegeneration: C9ORF72 repeat expansions in mice cause TDP-43 pathology, neuronal loss, and behavioral deficits. Science 348:1151–1154. 10.1126/science.aaa9344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chew J, Cook C, Gendron TF, Jansen-West K, Del Rosso G, Daughrity LM, Castanedes-Casey M, Kurti A, Stankowski JN, Disney MD, Rothstein JD, Dickson DW, Fryer JD, Zhang YJ, Petrucelli L (2019) Aberrant deposition of stress granule-resident proteins linked to C9orf72-associated TDP-43 proteinopathy. Mol Neurodegener 14:9. 10.1186/s13024-019-0310-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couthouis J, Hart MP, Shorter J, DeJesus-Hernandez M, Erion R, Oristano R, Liu AX, Ramos D, Jethava N, Hosangadi D, Epstein J, Chiang A, Diaz Z, Nakaya T, Ibrahim F, Kim HJ, Solski JA, Williams KL, Mojsilovic-Petrovic J, Ingre C, et al. (2011) A yeast functional screen predicts new candidate ALS disease genes. Proc Natl Acad Sci U S A 108:20881–20890. 10.1073/pnas.1109434108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couthouis J, Hart MP, Erion R, King OD, Diaz Z, Nakaya T, Ibrahim F, Kim HJ, Mojsilovic-Petrovic J, Panossian S, Kim CE, Frackelton EC, Solski JA, Williams KL, Clay-Falcone D, Elman L, McCluskey L, Greene R, Hakonarson H, Kalb RG, et al. (2012) Evaluating the role of the FUS/TLS-related gene EWSR1 in amyotrophic lateral sclerosis. Hum Mol Genet 21:2899–2911. 10.1093/hmg/dds116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deal SL, Yamamoto S (2018) Unraveling novel mechanisms of neurodegeneration through a large-scale forward genetic screen in Drosophila. Front Genet 9:700. 10.3389/fgene.2018.00700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donlin-Asp PG, Rossoll W, Bassell GJ (2017) Spatially and temporally regulating translation via mRNA-binding proteins in cellular and neuronal function. FEBS Lett 591:1508–1525. 10.1002/1873-3468.12621 [DOI] [PubMed] [Google Scholar]
- Dormann D, Rodde R, Edbauer D, Bentmann E, Fischer I, Hruscha A, Than ME, Mackenzie IR, Capell A, Schmid B, Neumann M, Haass C (2010) ALS-associated fused in sarcoma (FUS) mutations disrupt transportin-mediated nuclear import. EMBO J 29:2841–2857. 10.1038/emboj.2010.143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang MY, Markmiller S, Vu AQ, Javaherian A, Dowdle WE, Jolivet P, Bushway JP, Castello NA, Baral A, Chan M, Linsley JW, Linsley D, Mercola M, Finkbeiner S, Lecuyer E, Lewcock JW, Yeo GW (2019) Small-molecule modulation of TDP-43 recruitment to stress granules prevents persistent TDP-43 accumulation in ALS/FTD. Neuron 103:S0896-6273(19)30524-0. 10.1016/j.neuron.2019.05.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández-Hernández I, Scheenaard E, Pollarolo G, Gonzalez C (2016) The translational relevance of Drosophila in drug discovery. EMBO Rep 17:471–472. 10.15252/embr.201642080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gal J, Kuang L, Barnett KR, Zhu BZ, Shissler SC, Korotkov KV, Hayward LJ, Kasarskis EJ, Zhu H (2016) ALS mutant SOD1 interacts with G3BP1 and affects stress granule dynamics. Acta Neuropathol 132:563–576. 10.1007/s00401-016-1601-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasset-Rosa F, Lu S, Yu H, Chen C, Melamed Z, Guo L, Shorter J, Da Cruz S, Cleveland DW (2019) Cytoplasmic TDP-43 de-mixing independent of stress granules drives inhibition of nuclear import, loss of nuclear TDP-43, and cell death. Neuron 102:339–357.e7. 10.1016/j.neuron.2019.02.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes E, Shorter J (2019) The molecular language of membraneless organelles. J Biol Chem 294:7115–7127. 10.1074/jbc.TM118.001192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graf ER, Heerssen HM, Wright CM, Davis GW, DiAntonio A (2011) Stathmin is required for stability of the Drosophila neuromuscular junction. J Neurosci 31:15026–15034. 10.1523/JNEUROSCI.2024-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu J, Wang W, Miao S, Chen F, Wu F, Hu W, Iqbal K, Gong CX, Liu F (2018) Protein phosphatase 1 dephosphorylates TDP-43 and suppresses its function in tau exon 10 inclusion. FEBS Lett 592:402–410. 10.1002/1873-3468.12976 [DOI] [PubMed] [Google Scholar]
- Hasegawa M, Arai T, Nonaka T, Kametani F, Yoshida M, Hashizume Y, Beach TG, Buratti E, Baralle F, Morita M, Nakano I, Oda T, Tsuchiya K, Akiyama H (2008) Phosphorylated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Ann Neurol 64:60–70. 10.1002/ana.21425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Held A, Major P, Sahin A, Reenan RA, Lipscombe D, Wharton KA (2019) Circuit dysfunction in SOD1-ALS model first detected in sensory feedback prior to motor neuron degeneration is alleviated by BMP signaling. J Neurosci 39:2347–2364. 10.1523/JNEUROSCI.1771-18.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain S, Wheeler JR, Walters RW, Agrawal A, Barsic A, Parker R (2016) ATPase-modulated stress granules contain a diverse proteome and substructure. Cell 164:487–498. 10.1016/j.cell.2015.12.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson JO, Pioro EP, Boehringer A, Chia R, Feit H, Renton AE, Pliner HA, Abramzon Y, Marangi G, Winborn BJ, Gibbs JR, Nalls MA, Morgan S, Shoai M, Hardy J, Pittman A, Orrell RW, Malaspina A, Sidle KC, Fratta P, et al. (2014) Mutations in the matrin 3 gene cause familial amyotrophic lateral sclerosis. Nat Neurosci 17:664–666. 10.1038/nn.3688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamath RS, Fraser AG, Dong Y, Poulin G, Durbin R, Gotta M, Kanapin A, Le Bot N, Moreno S, Sohrmann M, Welchman DP, Zipperlen P, Ahringer J (2003) Systematic functional analysis of the Caenorhabditis elegans genome using RNAi. Nature 421:231–237. 10.1038/nature01278 [DOI] [PubMed] [Google Scholar]
- Kanekura K, Yagi T, Cammack AJ, Mahadevan J, Kuroda M, Harms MB, Miller TM, Urano F (2016) Poly-dipeptides encoded by the C9ORF72 repeats block global protein translation. Hum Mol Genet 25:1803–1813. 10.1093/hmg/ddw052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kedersha N, Anderson P (2002) Stress granules: sites of mRNA triage that regulate mRNA stability and translatability. Biochem Soc Trans 30:963–969. 10.1042/bst0300963 [DOI] [PubMed] [Google Scholar]
- Khong A, Matheny T, Jain S, Mitchell SF, Wheeler JR, Parker R (2017) The stress granule transcriptome reveals principles of mRNA accumulation in stress granules. Mol Cell 68:808–820.e5. 10.1016/j.molcel.2017.10.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HJ, Kim NC, Wang YD, Scarborough EA, Moore J, Diaz Z, MacLea KS, Freibaum B, Li S, Molliex A, Kanagaraj AP, Carter R, Boylan KB, Wojtas AM, Rademakers R, Pinkus JL, Greenberg SA, Trojanowski JQ, Traynor BJ, Smith BN, et al. (2013) Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature 495:467–473. 10.1038/nature11922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim W, Underwood RS, Greenwald I, Shaye DD (2018) OrthoList 2: a new comparative genomic analysis of human and Caenorhabditis elegans genes. Genetics 210:445–461. 10.1534/genetics.118.301307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klim JR, Williams LA, Limone F, Guerra San Juan I, Davis-Dusenbery BN, Mordes DA, Burberry A, Steinbaugh MJ, Gamage KK, Kirchner R, Moccia R, Cassel SH, Chen K, Wainger BJ, Woolf CJ, Eggan K (2019) ALS-implicated protein TDP-43 sustains levels of STMN2, a mediator of motor neuron growth and repair. Nat Neurosci 22:167–179. 10.1038/s41593-018-0300-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwiatkowski TJ Jr, Bosco DA, Leclerc AL, Tamrazian E, Vanderburg CR, Russ C, Davis A, Gilchrist J, Kasarskis EJ, Munsat T, Valdmanis P, Rouleau GA, Hosler BA, Cortelli P, de Jong PJ, Yoshinaga Y, Haines JL, Pericak-Vance MA, Yan J, Ticozzi N, et al. (2009) Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 323:1205–1208. 10.1126/science.1166066 [DOI] [PubMed] [Google Scholar]
- Lagier-Tourenne C, Polymenidou M, Cleveland DW (2010) TDP-43 and FUS/TLS: emerging roles in RNA processing and neurodegeneration. Hum Mol Genet 19:R46–R64. 10.1093/hmg/ddq137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KH, Zhang P, Kim HJ, Mitrea DM, Sarkar M, Freibaum BD, Cika J, Coughlin M, Messing J, Molliex A, Maxwell BA, Kim NC, Temirov J, Moore J, Kolaitis RM, Shaw TI, Bai B, Peng J, Kriwacki RW, Taylor JP (2016) C9orf72 dipeptide repeats impair the assembly, dynamics, and function of membrane-less organelles. Cell 167:774–788.e17. 10.1016/j.cell.2016.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmkuhl EM, Zarnescu DC (2018) Lost in translation: evidence for protein synthesis deficits in ALS/FTD and related neurodegenerative diseases. Adv Neurobiol 20:283–301. 10.1007/978-3-319-89689-2_11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liachko NF, Guthrie CR, Kraemer BC (2010) Phosphorylation promotes neurotoxicity in a Caenorhabditis elegans model of TDP-43 proteinopathy. J Neurosci 30:16208–16219. 10.1523/JNEUROSCI.2911-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liachko NF, McMillan PJ, Guthrie CR, Bird TD, Leverenz JB, Kraemer BC (2013) CDC7 inhibition blocks pathological TDP-43 phosphorylation and neurodegeneration. Ann Neurol 74:39–52. 10.1002/ana.23870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liachko NF, McMillan PJ, Strovas TJ, Loomis E, Greenup L, Murrell JR, Ghetti B, Raskind MA, Montine TJ, Bird TD, Leverenz JB, Kraemer BC (2014) The tau tubulin kinases TTBK1/2 promote accumulation of pathological TDP-43. PLoS Genet 10:e1004803. 10.1371/journal.pgen.1004803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liachko NF, Saxton AD, McMillan PJ, Strovas TJ, Currey HN, Taylor LM, Wheeler JM, Oblak AL, Ghetti B, Montine TJ, Keene CD, Raskind MA, Bird TD, Kraemer BC (2016) The phosphatase calcineurin regulates pathological TDP-43 phosphorylation. Acta Neuropathol 132:545–561. 10.1007/s00401-016-1600-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Reeb AN, Lin B, Subramanian P, Fey EE, Knoverek CR, French RL, Bigio EH, Ayala YM (2017) Heat shock-induced phosphorylation of TAR DNA-binding protein 43 (TDP-43) by MAPK/ERK kinase regulates TDP-43 function. J Biol Chem 292:5089–5100. 10.1074/jbc.M116.753913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling JP, Pletnikova O, Troncoso JC, Wong PC (2015) TDP-43 repression of nonconserved cryptic exons is compromised in ALS/FTD. Science 349:650–655. 10.1126/science.aab0983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu-Yesucevitz L, Bilgutay A, Zhang YJ, Vanderweyde T, Citro A, Mehta T, Zaarur N, McKee A, Bowser R, Sherman M, Petrucelli L, Wolozin B (2010) Tar DNA binding protein-43 (TDP-43) associates with stress granules: analysis of cultured cells and pathological brain tissue. PLoS One 5:e13250. 10.1371/journal.pone.0013250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Erauskin J, Tadokoro T, Baughn MW, Myers B, McAlonis-Downes M, Chillon-Marinas C, Asiaban JN, Artates J, Bui AT, Vetto AP, Lee SK, Le AV, Sun Y, Jambeau M, Boubaker J, Swing D, Qiu J, Hicks GG, Ouyang Z, Fu XD, et al. (2018) ALS/FTD-linked mutation in FUS suppresses intra-axonal protein synthesis and drives disease without nuclear loss-of-function of FUS. Neuron 100:816–830.e7. 10.1016/j.neuron.2018.09.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L, Zhao Y, Chen Y, Cheng B, Peng A, Huang K (2018) Caenorhabditis elegans as a model system for target identification and drug screening against neurodegenerative diseases. Eur J Pharmacol 819:169–180. 10.1016/j.ejphar.2017.11.051 [DOI] [PubMed] [Google Scholar]
- Mackenzie IR, Nicholson AM, Sarkar M, Messing J, Purice MD, Pottier C, Annu K, Baker M, Perkerson RB, Kurti A, Matchett BJ, Mittag T, Temirov J, Hsiung GR, Krieger C, Murray ME, Kato M, Fryer JD, Petrucelli L, Zinman L, et al. (2017) TIA1 mutations in amyotrophic lateral sclerosis and frontotemporal dementia promote phase separation and alter stress granule dynamics. Neuron 95:808–816.e9. 10.1016/j.neuron.2017.07.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie IR, Neumann M (2016) Molecular neuropathology of frontotemporal dementia: insights into disease mechanisms from postmortem studies. J Neurochem 138 [Suppl 1]:54–70. [DOI] [PubMed] [Google Scholar]
- Mann JR, Gleixner AM, Mauna JC, Gomes E, DeChellis-Marks MR, Needham PG, Copley KE, Hurtle B, Portz B, Pyles NJ, Guo L, Calder CB, Wills ZP, Pandey UB, Kofler JK, Brodsky JL, Thathiah A, Shorter J, Donnelly CJ (2019) RNA binding antagonizes neurotoxic phase transitions of TDP-43. Neuron 102:321–338.e8. 10.1016/j.neuron.2019.01.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marin B, Boumédiene F, Logroscino G, Couratier P, Babron MC, Leutenegger AL, Copetti M, Preux PM, Beghi E (2017) Variation in worldwide incidence of amyotrophic lateral sclerosis: a meta-analysis. Int J Epidemiol 46:57–74. 10.1093/ije/dyw061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markmiller S, Soltanieh S, Server KL, Mak R, Jin W, Fang MY, Luo EC, Krach F, Yang D, Sen A, Fulzele A, Wozniak JM, Gonzalez DJ, Kankel MW, Gao FB, Bennett EJ, Lécuyer E, Yeo GW (2018) Context-dependent and disease-specific diversity in protein interactions within stress granules. Cell 172:590–604.e13. 10.1016/j.cell.2017.12.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markmiller S, Fulzele A, Higgins R, Leonard M, Yeo GW, Bennett EJ (2019) Active protein neddylation or ubiquitylation is dispensable for stress granule dynamics. Cell Rep 27:1356–1363.e3. 10.1016/j.celrep.2019.04.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez FJ, Pratt GA, Van Nostrand EL, Batra R, Huelga SC, Kapeli K, Freese P, Chun SJ, Ling K, Gelboin-Burkhart C, Fijany L, Wang HC, Nussbacher JK, Broski SM, Kim HJ, Lardelli R, Sundararaman B, Donohue JP, Javaherian A, Lykke-Andersen J, et al. (2016) Protein-RNA networks regulated by normal and ALS-associated mutant HNRNPA2B1 in the nervous system. Neuron 92:780–795. 10.1016/j.neuron.2016.09.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason MR, Lieberman AR, Grenningloh G, Anderson PN (2002) Transcriptional upregulation of SCG10 and CAP-23 is correlated with regeneration of the axons of peripheral and central neurons in vivo. Mol Cell Neurosci 20:595–615. 10.1006/mcne.2002.1140 [DOI] [PubMed] [Google Scholar]
- McDonald KK, Aulas A, Destroismaisons L, Pickles S, Beleac E, Camu W, Rouleau GA, Vande Velde C (2011) TAR DNA-binding protein 43 (TDP-43) regulates stress granule dynamics via differential regulation of G3BP and TIA-1. Hum Mol Genet 20:1400–1410. 10.1093/hmg/ddr021 [DOI] [PubMed] [Google Scholar]
- McGurk L, Gomes E, Guo L, Mojsilovic-Petrovic J, Tran V, Kalb RG, Shorter J, Bonini NM (2018) Poly(ADP-ribose) prevents pathological phase separation of TDP-43 by promoting liquid demixing and stress granule localization. Mol Cell 71:703–717.e9. 10.1016/j.molcel.2018.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melamed Z, López-Erauskin J, Baughn MW, Zhang O, Drenner K, Sun Y, Freyermuth F, McMahon MA, Beccari MS, Artates JW, Ohkubo T, Rodriguez M, Lin N, Wu D, Bennett CF, Rigo F, Da Cruz S, Ravits J, Lagier-Tourenne C, Cleveland DW (2019) Premature polyadenylation-mediated loss of stathmin-2 is a hallmark of TDP-43-dependent neurodegeneration. Nat Neurosci 22:180–190. 10.1038/s41593-018-0293-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami T, Qamar S, Lin JQ, Schierle GS, Rees E, Miyashita A, Costa AR, Dodd RB, Chan FT, Michel CH, Kronenberg-Versteeg D, Li Y, Yang SP, Wakutani Y, Meadows W, Ferry RR, Dong L, Tartaglia GG, Favrin G, Lin WL, et al. (2015) ALS/FTD mutation-induced phase transition of FUS liquid droplets and reversible hydrogels into irreversible hydrogels impairs RNP granule function. Neuron 88:678–690. 10.1016/j.neuron.2015.10.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, McCluskey LF, Miller BL, Masliah E, Mackenzie IR, Feldman H, Feiden W, Kretzschmar HA, Trojanowski JQ, Lee VM (2006) Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314:130–133. [DOI] [PubMed] [Google Scholar]
- Nguyen HP, Van Broeckhoven C, van der Zee J (2018) ALS genes in the genomic era and their implications for FTD. Trends Genet 34:404–423. 10.1016/j.tig.2018.03.001 [DOI] [PubMed] [Google Scholar]
- Nussbacher JK, Batra R, Lagier-Tourenne C, Yeo GW (2015) RNA-binding proteins in neurodegeneration: Seq and you shall receive. Trends Neurosci 38:226–236. 10.1016/j.tins.2015.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nussbacher JK, Tabet R, Yeo GW, Lagier-Tourenne C (2019) Disruption of RNA metabolism in neurological diseases and emerging therapeutic interventions. Neuron 102:294–320. 10.1016/j.neuron.2019.03.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey UB, Nichols CD (2011) Human disease models in Drosophila melanogaster and the role of the fly in therapeutic drug discovery. Pharmacol Rev 63:411–436. 10.1124/pr.110.003293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel A, Lee HO, Jawerth L, Maharana S, Jahnel M, Hein MY, Stoynov S, Mahamid J, Saha S, Franzmann TM, Pozniakovski A, Poser I, Maghelli N, Royer LA, Weigert M, Myers EW, Grill S, Drechsel D, Hyman AA, Alberti S (2015) A liquid-to-solid phase transition of the ALS protein FUS accelerated by disease mutation. Cell 162:1066–1077. 10.1016/j.cell.2015.07.047 [DOI] [PubMed] [Google Scholar]
- Piper M, Holt C (2004) RNA translation in axons. Annu Rev Cell Dev Biol 20:505–523. 10.1146/annurev.cellbio.20.010403.111746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polymenidou M, Lagier-Tourenne C, Hutt KR, Huelga SC, Moran J, Liang TY, Ling SC, Sun E, Wancewicz E, Mazur C, Kordasiewicz H, Sedaghat Y, Donohue JP, Shiue L, Bennett CF, Yeo GW, Cleveland DW (2011) Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nat Neurosci 14:459–468. 10.1038/nn.2779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad A, Bharathi V, Sivalingam V, Girdhar A, Patel BK (2019) Molecular mechanisms of TDP-43 misfolding and pathology in amyotrophic lateral sclerosis. Front Mol Neurosci 12:25. 10.3389/fnmol.2019.00025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Protter DS, Parker R (2016) Principles and properties of stress granules. Trends Cell Biol 26:668–679. 10.1016/j.tcb.2016.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prudencio M, Jansen-West KR, Lee WC, Gendron TF, Zhang YJ, Xu YF, Gass J, Stuani C, Stetler C, Rademakers R, Dickson DW, Buratti E, Petrucelli L (2012) Misregulation of human sortilin splicing leads to the generation of a nonfunctional progranulin receptor. Proc Natl Acad Sci U S A 109:21510–21515. 10.1073/pnas.1211577110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahin A, Held A, Bredvik K, Major P, Achilli TM, Kerson AG, Wharton K, Stilwell G, Reenan R (2017) Human SOD1 ALS mutations in a Drosophila knock-in model cause severe phenotypes and reveal dosage-sensitive gain- and loss-of-function components. Genetics 205:707–723. 10.1534/genetics.116.190850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sama RR, Ward CL, Bosco DA (2014) Functions of FUS/TLS from DNA repair to stress response: implications for ALS. ASN Neuro 6:1759091414544472. 10.1177/1759091414544472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelkovnikova TA, Robinson HK, Connor-Robson N, Buchman VL (2013) Recruitment into stress granules prevents irreversible aggregation of FUS protein mislocalized to the cytoplasm. Cell Cycle 12:3194–3202. 10.4161/cc.26241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiga A, Ishihara T, Miyashita A, Kuwabara M, Kato T, Watanabe N, Yamahira A, Kondo C, Yokoseki A, Takahashi M, Kuwano R, Kakita A, Nishizawa M, Takahashi H, Onodera O (2012) Alteration of POLDIP3 splicing associated with loss of function of TDP-43 in tissues affected with ALS. PLoS One 7:e43120. 10.1371/journal.pone.0043120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin JE, Geisler S, DiAntonio A (2014) Dynamic regulation of SCG10 in regenerating axons after injury. Exp Neurol 252:1–11. 10.1016/j.expneurol.2013.11.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidrauski C, McGeachy AM, Ingolia NT, Walter P (2015) The small molecule ISRIB reverses the effects of eIF2alpha phosphorylation on translation and stress granule assembly. eLife 4: 10.7554/eLife.05033. 10.7554/eLife.05033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sin O, Michels H, Nollen EA (2014) Genetic screens in Caenorhabditis elegans models for neurodegenerative diseases. Biochim Biophys Acta 1842:1951–1959. 10.1016/j.bbadis.2014.01.015 [DOI] [PubMed] [Google Scholar]
- Solomon DA, Stepto A, Au WH, Adachi Y, Diaper DC, Hall R, Rekhi A, Boudi A, Tziortzouda P, Lee YB, Smith B, Bridi JC, Spinelli G, Dearlove J, Humphrey DM, Gallo JM, Troakes C, Fanto M, Soller M, Rogelj B, et al. (2018) A feedback loop between dipeptide-repeat protein, TDP-43 and karyopherin-alpha mediates C9orf72-related neurodegeneration. Brain 141:2908–2924. 10.1093/brain/awy241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souquere S, Mollet S, Kress M, Dautry F, Pierron G, Weil D (2009) Unravelling the ultrastructure of stress granules and associated P-bodies in human cells. J Cell Sci 122:3619–3626. 10.1242/jcs.054437 [DOI] [PubMed] [Google Scholar]
- Sprenkle NT, Sims SG, Sánchez CL, Meares GP (2017) Endoplasmic reticulum stress and inflammation in the central nervous system. Mol Neurodegener 12:42. 10.1186/s13024-017-0183-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan RH, Ke YD, Ittner LM, Halliday GM (2017) ALS/FTLD: experimental models and reality. Acta Neuropathol 133:177–196. 10.1007/s00401-016-1666-6 [DOI] [PubMed] [Google Scholar]
- Taylor LM, McMillan PJ, Liachko NF, Strovas TJ, Ghetti B, Bird TD, Keene CD, Kraemer BC (2018) Pathological phosphorylation of tau and TDP-43 by TTBK1 and TTBK2 drives neurodegeneration. Mol Neurodegener 13:7. 10.1186/s13024-018-0237-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tischbein M, Baron DM, Lin YC, Gall KV, Landers JE, Fallini C, Bosco DA (2019) The RNA-binding protein FUS/TLS undergoes calcium-mediated nuclear egress during excitotoxic stress and is required for GRIA2 mRNA processing. J Biol Chem 294:10194–10210. 10.1074/jbc.RA118.005933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twiss JL, Fainzilber M (2009) Ribosomes in axons: scrounging from the neighbors? Trends Cell Biol 19:236–243. 10.1016/j.tcb.2009.02.007 [DOI] [PubMed] [Google Scholar]
- Vaccaro A, Tauffenberger A, Aggad D, Rouleau G, Drapeau P, Parker JA (2012) Mutant TDP-43 and FUS cause age-dependent paralysis and neurodegeneration in C. elegans. PLoS One 7:e31321. 10.1371/journal.pone.0031321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vance C, Rogelj B, Hortobágyi T, De Vos KJ, Nishimura AL, Sreedharan J, Hu X, Smith B, Ruddy D, Wright P, Ganesalingam J, Williams KL, Tripathi V, Al-Saraj S, Al-Chalabi A, Leigh PN, Blair IP, Nicholson G, de Belleroche J, Gallo JM, et al. (2009) Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 323:1208–1211. 10.1126/science.1165942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanderweyde T, Apicco DJ, Youmans-Kidder K, Ash PE, Cook C, Lummertz da Rocha E, Jansen-West K, Frame AA, Citro A, Leszyk JD, Ivanov P, Abisambra JF, Steffen M, Li H, Petrucelli L, Wolozin B (2016) Interaction of tau with the RNA-binding protein TIA1 regulates tau pathophysiology and toxicity. Cell Rep 15:1455–1466. 10.1016/j.celrep.2016.04.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vatsavayai SC, Yoon SJ, Gardner RC, Gendron TF, Vargas JN, Trujillo A, Pribadi M, Phillips JJ, Gaus SE, Hixson JD, Garcia PA, Rabinovici GD, Coppola G, Geschwind DH, Petrucelli L, Miller BL, Seeley WW (2016) Timing and significance of pathological features in C9orf72 expansion-associated frontotemporal dementia. Brain 139:3202–3216. 10.1093/brain/aww250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanagi KS, Wu Z, Amaya J, Chapkis N, Duffy AM, Hajdarovic KH, Held A, Mathur AD, Russo K, Ryan VH, Steinert BL, Whitt JP, Fallon JR, Fawzi NL, Lipscombe D, Reenan RA, Wharton KA, Hart AC (2019) Meta-analysis of genetic modifiers reveals candidate dysregulated pathways in amyotrophic lateral sclerosis. Neuroscience 396:A3–A20. 10.1016/j.neuroscience.2018.10.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, Fan B, Yang P, Temirov J, Messing J, Kim HJ, Taylor JP (2019) Chronic optogenetic induction of stress granules is cytotoxic and reveals the evolution of ALS/FTD pathology. eLife 8:e39578. 10.7554/eLife.39578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T, Mullane PC, Periz G, Wang J (2011) TDP-43 neurotoxicity and protein aggregation modulated by heat shock factor and insulin/IGF-1 signaling. Hum Mol Genet 20:1952–1965. 10.1093/hmg/ddr076 [DOI] [PMC free article] [PubMed] [Google Scholar]