

Abstract
The pistol ribozyme (Psr) is among the most recently discovered RNA enzymes and has been the subject of experiments aimed at elucidating the mechanism. Recent biochemical studies have revealed exciting clues about catalytic interactions in the active site not apparent from available crystallographic data. The present work unifies the interpretation of the existing body of structural and functional data on Psr by providing a dynamical model for the catalytically active state in solution from molecular simulation. Our results suggest that a catalytic Mg2+ ion makes inner-sphere contact with G33:N7 and outer-sphere coordination to the pro-RP of the scissile phosphate, promoting electrostatic stabilization of the dianionic transition state and neutralization of the developing charge of the leaving group through a metal-coordinated water molecule that is made more acidic by a hydrogen bond donated from the 2′OH of P32. This model is consistent with experimental activity-pH and mutagenesis data, including sensitivity to G33(7cG) and phosphorothioate substitution/metal ion rescue. The model suggests several experimentally testable predictions, including the response of cleavage activity to mutations at G42 and P32 positions in the ribozyme, and thio substitutions of the substrate in the presence of different divalent metal ions. Further, the model identifies striking similarities of Psr to the hammerhead ribozyme (HHr), including similar global fold, organization of secondary structure around an active site three-way junction, catalytic metal ion binding mode, and guanine general base. However, the specific binding mode and role of the Mg2+ ion, as well as a conserved 2′-OH in the active site, are interrelated but subtly different between the ribozymes.
Keywords: pistol ribozyme, RNA catalysis, molecular dynamics, metal binding, catalytic strategies
INTRODUCTION
Ribozymes are molecules of RNA that act as catalysts, and their discovery in 1982 had profound implications into theories of evolution (Gilbert 1986; DeRose 2002; Chen et al. 2007). In biology, ribozymes are known to play important roles in gene regulation, RNA processing and peptide synthesis (Steitz and Moore 2003; Winkler et al. 2004; Scott 2007). However, the function of many ribozymes has yet to be unveiled, and new ribozymes continue to be discovered, for example through comparative genomics (Weinberg et al. 2015). Further, synthetic ribozymes and even DNAzymes (Gu et al. 2013; Ponce-Salvatierra et al. 2016; Liu et al. 2017) have been engineered that extend the range of known reactions nucleic acids are able to catalyze (Sengle et al. 2001; Fusz et al. 2005; Serganov et al. 2005), and have great promise in the design of new biomedical technology (Sullenger and Gilboa 2002; Liu et al. 2009; Saragliadis et al. 2013) and therapeutics (Famulok et al. 2007; Krug et al. 2015).
Tremendous insight has been gained about mechanisms of RNA catalysis through the study of small nucleolytic ribozymes that catalyze site-specific cleavage (and in some cases ligation) of the RNA phosphodiester backbone (Bevilacqua et al. 2004; Bevilacqua and Yajima 2006; Wilson and Lilley 2013; Ward et al. 2014; Wilson et al. 2016b) and their nuanced dependence on divalent cations (DeRose 2003). In this reaction (formally a 2′O-transphosphorylation), a 2′-OH nucleophile becomes activated and makes an in-line attack to the adjacent scissile phosphate, proceeding through a pentavalent phosphorane transition state (or metastable intermediate) to form 2′,3′-cyclic phosphate and 5′OH cleavage products. These ribozymes use a number of different catalytic strategies to facilitate RNA cleavage that can be described using the framework suggested by Breaker and coworkers (Emilsson et al. 2003) illustrated in Figure 1. These general strategies are designated alpha (α) for the in-line fitness of the 2-OH′ nucleophile, beta (β) for the electrostatic stabilization/protonation of the nonbridging phosphoryl oxygens (NPOs) of the dianionic phosphorane transition state, gamma (γ) for the deprotonation (activation) of the 2′-OH nucleophile (e.g., by a general base), and delta (δ) for the electrostatic stabilization (neutralization/protonation) of the 5′-OH leaving group (e.g., by a general acid). Recently, we stratified Breaker's framework into a more precise ontology for discussion of RNA-cleaving enzymes by decomposing the β, γ, and δ strategies into primary (1°), secondary (2°), and tertiary (3°) contributions (Bevilacqua et al. 2019). Pivotal to these delineations is the concept of primary atomic positions, that is, atoms directly involved with the chemical space of covalent or ionic bonds associated with a particular catalytic strategy (Bevilacqua et al. 2019). A primary catalytic effect results from changes in the identity of one or more primary (β, γ, δ) atoms (e.g., general acid or base knockout), whereas a secondary catalytic effect is caused by a perturbation of the electronic environment of the primary atom(s) resulting from changes in the identity of nonprimary atoms (e.g., pKa tuning). A tertiary catalytic effect is caused by alteration of the position of the primary atoms resulting from modification of the structural scaffold or hydrogen bond network that organizes the enzyme active site.
FIGURE 1.
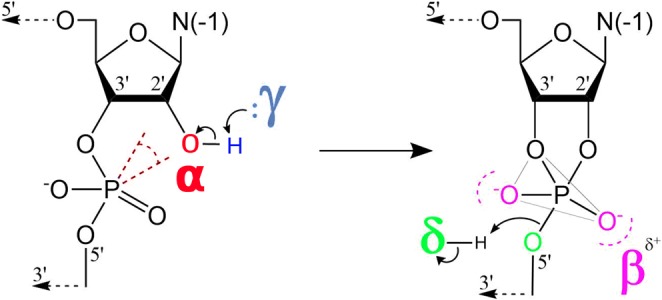
The four strategies that shape discussions of catalysis by small nucleolytic ribozymes (Emilsson et al. 2003): (α) in-line fitness of the 2-OH′ nucleophile, (β) electrostatic stabilization/protonation of the NPOs in the transition state, (γ) deprotonation (activation) of the 2′-OH nucleophile (e.g., by a general base), and (δ) stabilization/protonation of the 5′-O leaving group (e.g., by a general acid). Recently, an ontology has been introduced that extends this framework for more precise discussion of mechanisms of RNA-cleaving enzymes by decomposing the β, γ, and δ strategies into primary (1°), secondary (2°), and tertiary (3°) contributions (Bevilacqua et al. 2019).
As a field, we are now for the first time at the point where there are a sufficient number of nucleolytic ribozyme classes that have been discovered and characterized structurally that we can begin to unveil common themes, possible evolutionary connections and general principles about catalysis to guide the design of new biomedical technology and therapeutics. The Psr has been recently discovered through comparative genomics (Weinberg et al. 2015), and subsequently characterized by biochemical studies (Harris et al. 2015; Ren et al. 2016; Neuner et al. 2017; Wilson et al. 2019) and X-ray crystallography (Ren et al. 2016; Nguyen et al. 2017; Wilson et al. 2019). Initial interpretations of crystallographic data (Ren et al. 2016; Nguyen et al. 2017) differed to some degree, and in some cases were inconsistent with interpretations of biochemical data (Harris et al. 2015), particularly with respect to the identification of the putative general acid, and the role of metal ions in catalysis. Subsequent functional studies helped to clarify these roles, and provided strong evidence against initial interpretations that general acid catalysis was affected through the N3 position of an active site adenine residue (Neuner et al. 2017), as had at the time been suggested for the twister ribozyme (Liu et al. 2014). The role of A1:N3 in twister as the general acid has subsequently been convincingly demonstrated by molecular simulations that provide a dynamical ensemble of the active state in solution (Gaines and York 2016), and a rigorous atomic-level model from which to interpret concurrently reported functional studies (Wilson et al. 2016a). While this work was under review, a very comprehensive biochemical and structural study was reported by Wilson et al. (2019) that provided deep insight into the mechanism of Psr and its relation to the hammerhead ribozyme (HHr).
Recently Breaker has discussed several caveats in the elucidation of ribozyme mechanisms from theoretical, structural and biochemical data that potentially lead to conflicting interpretations (Breaker 2017). These include (i) inconsistent terminology used to discuss the same catalytic effects, (ii) diverse biophysical data sets and divergent structural models, and (iii) conflicting/nonunique interpretations of essentially the same biochemical functional data. Ultimately, the path forward involves recognizing these caveats and developing physics-based models that provide a structural and dynamical picture of catalysis that is consistent with available data, and that can be validated with further experiments. Molecular simulations in principle provide such a tool, and when carefully applied, can provide a unified interpretation of a wide range of structural and biochemical data (Panteva et al. 2015a).
The goal of the present work is to establish a molecular simulation model (i.e., a dynamical ensemble of structures) that connects experimental crystallographic and biochemical data and provides greater clarity into the mechanism of the pistol ribozyme (Psr) class. We will frame our discussion of catalytic effects in Psr using the four catalytic strategies for RNA cleavage (Fig. 1; Emilsson et al. 2003), using a recent ontology that further decomposes these strategies into primary, secondary, and tertiary contributions for more precise discussion (Bevilacqua et al. 2019). We perform simulations at several points along the catalytic pathway (including a transition state mimic model), departing separately from both of the currently available X-ray crystal structures (Ren et al. 2016; Nguyen et al. 2017) such that a unified set of conclusions can be drawn. Our results are compared with biochemical data (Harris et al. 2015; Neuner et al. 2017), including very recent data which have appeared during the review of this work (Wilson et al. 2019), in order to provide a consistent structural and dynamical interpretation of the active state of Psr in solution. Finally, we describe several experimentally testable predictions, some of which have been validated by the very recent experiments that have now appeared.
Thus, while the current body of experimental work on the Psr is extremely important and enlightening, the only structural model of the catalytically active state that has been proposed is based on modeling a divalent ion in a position indicated by the anomalous scatter of a Mn2+ ion in the most recent crystal structure (which at the time of this writing is not yet publicly available) and coordinated water molecules placed to be most consistent with mutagenesis data from the same study (Wilson et al. 2019). While such a model is quite reasonable to speculate, it remains to test whether the model can be supported energetically, and to ascertain its probability of being observed in the solution given realistic dynamical freedom. Further, as pointed out by the authors, the model does not explain all the available functional data, and there is likely a need for models of the transition state to complete the interpretations.
The present theoretical/computational modeling study is of considerable value as a complement to this recent experimental work, as it provides the first detailed structural ensemble of the Psr, derived from rigorous physical simulation models, for both the active state and transition state in solution. These results enable a more precise, quantitative and complete atomic-level interpretation of existing experimental data, and motivate new experiments. It is also of interest that the present results, departing from two crystallographic structures reported by different groups, converge to a common model for catalysis that also explains the most recent experimental results (Wilson et al. 2019) that were not considered in the simulations. Finally, the present work enables a detailed comparison of functional links between the pistol and HHrs, the latter for which an active state has been proposed from molecular simulation (Lee and York 2010; Lee et al. 2008, 2013) involving a metal-activated G8:O2′H acting as the general acid, the feasibility for which was later confirmed by experimental studies (Thomas and Perrin 2009) and further supported by very recent work (Wilson et al. 2019). While there is experimental support for this HHr catalysis model, it should be pointed out that alternative mechanisms have been suggested based on experimental evidence (Ward and DeRose 2012; Ward et al. 2014; Mir and Golden 2016; Mir et al. 2015) and it is possible that under different conditions multiple reactive channels are available (Frankel et al. 2017).
RESULTS
Current body of experimental structural and functional data on Psr
The Psr has been characterized structurally through X-ray crystallography, and at the time this work was submitted, two laboratories have published crystallographic structures: PDB ID 5K7C (Ren et al. 2016) and 5KTJ (Nguyen et al. 2017). During the initial review of this manuscript, a report of a third crystal structure, as well as related biochemical experiments, has appeared online (Wilson et al. 2019). Hence, the initial focus will concentrate on the 5K7C and 5KTJ structures, and later in the discussion, a section is devoted to bringing the present work into the context of the most recent report (Wilson et al. 2019). The sequences and tertiary structures for each of the crystal structures are compared in Figure 2. Both constructs (5K7C and 5KTJ) are trans-acting ribozymes consisting of enzyme and substrate strands and have very similar sequence conservation, tertiary contacts, and architecture of the active site. The Psr structures contain three stems (P1–P3) and one pseudoknot (PK) that is flanked by stems P2 and P3, and coaxially stacked with stem P1. The active site is located at the three-way junction formed by stems P2, P3, and the C41-G33 base pair near the PK.
FIGURE 2.

Tertiary structure schematics of the 5K7C (Ren et al. 2016) and 5KTJ (Nguyen et al. 2017) crystal structures, with boxes marking the ten residues of >97% sequence conservation.
The Psr cleavage site is located between a GU base step of the substrate strand (G53/U54 in 5K7C and G10/U11 in 5KTJ) with the scissile phosphate belonging to the U(+1) residue. The G and U nucleobases flanking the scissile phosphate are held in a splayed-out conformation that facilitates an active in-line attack geometry by interactions with conserved nucleobases U29 and G42 of the enzyme strand. Specifically, a U–U wobble base pair is formed between the cleavage site U(+1) and U29, and a bifurcated hydrogen bond interaction occurs between the exocyclic amine of cleavage site G and the O6 atom of G42.
The active site hydrogen bonding patterns in 5K7C and 5KTJ share common features for which mechanistic interpretations agree; however, they also contain differences that have led to some discrepancies. While both 5K7C and 5KTJ point to G40 as the likely general base candidate for γ catalysis, the respective candidates for stabilization of the NPO positions (β catalysis) are somewhat inconsistent, and explanations of general acid (δ catalysis) are still unclear. The neutralization of charge buildup on the NPOs has been attributed to a hydrated Mg2+ ion in the 5K7C structure (Ren et al. 2016), whereas analysis of the 5KTJ structure produced a hypothesis where only contributions to β catalysis were on the part of G40's exocyclic amine and transient binding of divalent cations from the solvent (Nguyen et al. 2017). Similar issues arose with attempts to clarify Psr's δ catalytic strategies. The original δ interpretation, based only on the crystallographic and biochemical data available at the time (Harris et al. 2015; Weinberg et al. 2015; Ren et al. 2016), concluded that both the N3 and 2′-OH of A32 were likely proton donors for activation of the 5′-OH leaving group. This seemed reasonable, following the discovery of an adenine N3 acting as the general acid of the recently discovered twister ribozyme (Liu et al. 2014), and a more recently reported crystal structure that provided further structural evidence consistent with this δ hypothesis (Nguyen et al. 2017).
In an insightful follow-up functional study (Neuner et al. 2017), both the N1 and N3 positions of purine 32 (A32 in 5K7C, G32 in 5KTJ) were convincingly eliminated as candidates for the general acid, modifying previous speculations based on crystallographic data, but also leaving open new questions about the specific mechanism for δ catalysis. The 2′-OH position of purine 32 was also considered as a candidate as a general acid, but chemical probing studies (Neuner et al. 2017) suggest the more likely role of this functional group was structural. A new δ hypothesis has been proposed (Neuner et al. 2017), attributing the role of general acid to water directly coordinated by a Mg2+ ion that was crystallographically resolved within outer-sphere contact distance (5 Å) of the 5′-OH in both 5K7C and 5KTJ. In this hypothesis, water bound to the same Mg2+ is expected to play a catalytic role, contributing to β and δ catalysis. In support of this model, the substitution of the N7 position of G33, 7-deazaguanosine-33 or simply G33(7cG), which would disrupt inner-sphere coordination of a Mg2+ bound at this position, has a drastic effect on cleavage (Neuner et al. 2017). The most recent functional studies have further clarified the importance of the G33(7cG) mutation, which is reported to have a 1.3 × 104-fold decrease in rate (Wilson et al. 2019). Hence, the recent atom-specific mutagenesis data called into question some key mechanistic interpretations of the original crystallographic data, and provided new insight into the importance of key functional groups and metal ion cofactors in the active site.
The purpose of the current work is to develop a simulation model of the Psr active state and transition state mimic in the solution that provides a detailed, atomistic structural and dynamical picture of catalysis that aids in the refinement and interpretation of the current body of structural and functional data. Breaker has recently warned that vastly different predictions about ribozyme mechanisms can arise from computer simulations that are built upon disparate structural models (Breaker 2017). It has been suggested that as starting points, computer simulation campaigns should consider all available structural and biophysical data sets and work to unify conclusions, and avoid embracing only a single incomplete or potentially flawed model. Toward this end, we perform simulations departing from both currently available crystallographic structures (Ren et al. 2016; Nguyen et al. 2017), and considering active site interactions and metal ion binding scenarios implicated by functional studies (Harris et al. 2015; Weinberg et al. 2015; Ren et al. 2016; Neuner et al. 2017), in order to provide a structural and dynamical model that unifies the current body of experimental data, and makes experimentally testable predictions. A summary of the evolution of catalytic hypotheses (within Breaker's suggested framework [Emilsson et al. 2003]) developed by interpretation of the original crystallographic and recent functional data, and refined in the current work, are illustrated in Figure 3. Subsequent sections delve into quantitative details of the simulation results that establish the identity and role of key residues and functional groups that participate in each catalytic strategy, and form connections with catalytic strategies of other known ribozymes, particularly the HHr.
FIGURE 3.

Comparison of catalytic hypotheses derived from interpretation of pistol X-ray crystal structures, 5K7C (Ren et al. 2016) and 5KTJ (Nguyen et al. 2017), and recent functional data (Neuner et al. 2017), as well as refined in the current work with MD simulation that considers all available structural and biophysical data sets. Discussion of the most recent structural and functional data (Wilson et al. 2019) that appeared during an initial review of this manuscript is deferred to a special section of the Discussion.
Promiscuous Mg2+ binding in the Psr active site supports catalysis
The 5K7C (Ren et al. 2016) and 5KTJ (Nguyen et al. 2017) crystal structures both contained an Mg2+ ion in the binding pocket at the active site (herein designated as the C-site), directly coordinating the G33:N7 position (2.1 and 2.3 Å, respectively). In the most recent structure, a divalent ion was modeled in a similar position indicated by the anomalous scatter of a Mn2+ ion (Wilson et al. 2019). However, inner-sphere binding of Mg2+ to the N7 position of guanine is exceptionally rare (and sometimes misassigned from the electron density) (Leonarski et al. 2017), and it is not clear whether the divalent metal ion binding mode observed crystallographically is the one required to support catalysis. Nonetheless, while the binding mode of Mg2+ to N7 of guanine is expected to be very infrequently observed as the most stable binding mode, it should be pointed out that such coordination does exist in the RNA literature and appears in structural databases, albeit sparsely.
It should be emphasized that the active state of Psr in solution is likely an improbable (rarely visited) state, different from that of the crystals, with requirements for G40 to be activated (deprotonated at N1), in an in-line attack conformation, and with a Mg2+ binding mode positioned to support the catalytic chemical steps of the reaction. MD simulations and molecular solvation theory/electrostatic (3D-RISM) calculations indicate the Psr active site, like the HHr (Lee et al. 2009), is highly electronegative, and able to recruit and bind divalent metal ions. In addition to G33:N7, there are a number of negatively charged phosphates positioned in close proximity to the Mg2+ ion in both 5K7C and 5KTJ crystals so as to possibly make indirect (or direct) coordination in the active state, including the pro-RP of G33, U35 and U(+1), as well as A/G32:O2′ and U(+1):O5′.
In order to ascertain the Mg2+ binding mode(s) capable to support catalysis, several possible binding modes were considered and are listed in Table 1. Long-time MD simulations were performed departing from each of the Mg2+ binding modes, but during the course of the simulations, it was observed that there was interconversion between different inner/outer sphere binding modes in the C-site binding pocket. In this way, the specific coordination environment of the Mg2+ at the C-site (although overall quite similar) was observed to be somewhat promiscuous, with several specific binding modes able to support the key catalytic strategies departing from both crystallographic structures.
TABLE 1.
Simulated binding modes of the hydrated C-site active site Mg2+ ion, defined using the contacts listed as columns

Of key relevance was the observation that direct coordination was often observed at G33:N7, in agreement with the crystallographic data, and also functional data for G33(7cG) mutation that has a significant impact on activity. In addition, direct (inner-sphere) coordination was also observed with partial occupancy at the pro-RP positions of G33 and occasionally U35, but not at the scissile phosphate U(+1). Rather outer-sphere Mg2+ binding was observed at the pro-RP of the scissile phosphate. This is consistent with phosphorothioate substitution experiments that indicate that nonstereospecific thio effects at U(+1) are not rescuable by titration with thiophilic metal ions (Neuner et al. 2017). More recent functional studies have identified the pro-RP of U(+1) as exhibiting a 4.5 × 104-fold thio effect, that is, only partially (150-fold) rescuable by titration with Mn2+ ions (Wilson et al. 2019). Taken together, this suggests that direct metal coordination at the scissile phosphate is not strictly required for catalysis.
In the next two sections, clustering will be used to identify which metal ion binding mode(s) are catalytically important. We first examine metal ion binding modes that support α and γ catalysis based on an “active state” model with the guanine general base deprotonated. Next, we examine metal ion binding modes that support β and δ catalysis based on a dianionic pentavalent phosphorane “transition state mimic” model. In the final section, these results are used to interpret functional data and suggest new experiments.
Multiple Mg2+ binding modes are able to promote α and γ catalysis
In this section, we present results of the Psr “active state” having G40 deprotonated at the N1 position, and a Mg2+ ion bound in the active site (C-site) in order to make inferences about α and γ catalysis. Table 2 provides a summary of active site interactions that support α and γ catalysis for the Mg2+ binding modes listed in Table 1.
TABLE 2.
Percentage of MD frames where the nucleophile is inline (α), and/or where it is hydrogen bonding with the N1 atom of G40 (γ)

α Catalysis
The concept of “near-attack conformations” (Torres and Bruice 1998) and “in-line fitness” (Soukup and Breaker 1999) as a prerequisite for catalysis in 2′O-transphosphorylation reactions has been well established, and herein is framed under the category of α catalysis (Emilsson et al. 2003). Selectively stabilizing the 2′-OH nucleophile in its most active in-line geometry is partially accomplished by the global fold of the typical self-cleaving ribozyme. Base-pairing, in addition to tertiary interactions, leads to the splaying of the two bases that flank the scissile phosphate, which in turn places the nucleophilic 2′-OH group in a reactive conformation. The Mg2+ binding mode that best supports α catalysis in both Psr constructs is the simultaneous direct coordination of G33:N7 and G33:pro-RP, producing nucleophile inline occupancies of 68.8% in 5K7C and 95.4% in 5KTJ simulations (Comparison with other binding modes is available in Table 2).
γ Catalysis
Crystallographic data indicate that G40 is in a position to act as a general base through the N1 position to deprotonate the nucleophile in all crystallized constructs (Ren et al. 2016; Nguyen et al. 2017; Wilson et al. 2019). These results are supported indirectly by biochemical analysis (Harris et al. 2015; Neuner et al. 2017), and convincingly demonstrated by recent activity-pH data on the wild type and a G40I mutant (Wilson et al. 2019). Our MD simulation results suggest, as with α catalysis, that Mg2+ binding at G33:N7/pro-RP supports γ catalysis, in addition to binding at the U35:pro-RP position in the 5KTJ construct. The Watson–Crick edge of G40 in its N1-deprotonated form (active state) has two adjacent hydrogen bond acceptors (N1 and O6) that increase the likelihood of the nucleophile adopting a conformation suitable for proton abstraction. This structural stabilization of the active site so as to position the deprotonated G40:N1 to abstract the proton from the 2′-OH nucleophile is designated tertiary γ catalysis (Bevilacqua et al. 2019).
Further, the Hoogsteen edge of G40 is exposed to solvent in an electronegative pocket, making the O6 position available to bind (perhaps transiently) monovalent or even divalent metal ions that can tune the pKa at the N1 position. Figure 4 illustrates that G40:O6 of Psr interacts with Na+ ions in solution considerably more than other guanine residues, and this interaction increases markedly when the general base is activated (deprotonated at the N1 position, causing delocalized charge to distribute to the O6 position). This lowers the pKa of the general base making the active state more probable near-neutral pH (Ward et al. 2014). This feature has been observed crystallographically (Mir and Golden 2016; Mir et al. 2015) and quantified computationally (Chen et al. 2017) for a Mg2+ ion in the HHr. MD simulations indicate placement of a Mg2+ ion in this position in Psr, either directly or indirectly coordinating G40:O6, is at least transiently stable, lending support to the hypothesis that Psr, like HHr, has engineered a cation recruiting site (e.g., Na+ or Mg2+) able to tune the general base pKa and facilitate γ catalysis. This tuning of the general base G40:N1 pKa by remote metal ion binding at G40:O6 is designated as a form of secondary γ catalysis (Bevilacqua et al. 2019).
FIGURE 4.
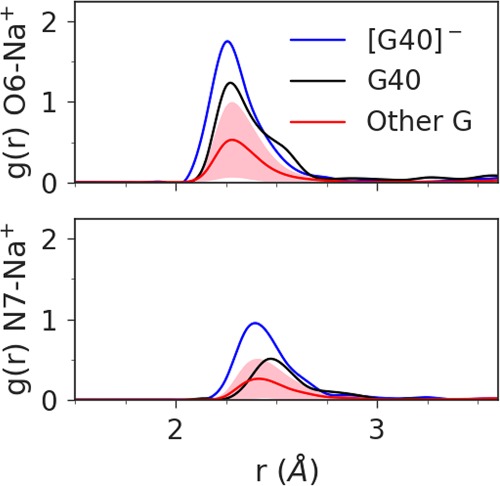
Radial distribution function of Na+ ions about the O6 (top plot) and N7 (bottom plot) atoms of G40 in its active state (blue) and native protonation state (black), compared to the average radial distribution function for the other 15 guanines in the 5K7C ribozyme (red), with the standard deviation shaded in pink. The average coordination number of the first Na+ ion shell about the O6 is 1.0 for G40−, 0.8 for G40, and 0.3 for Other G. The coordination number of the first sodium shell about the N7 is 0.7 for G40−, 0.4 for G40, and 0.2 for Other G.
C-site Mg2+ binding enables β and δ catalysis through metal-bound water molecules
In this section, we extend results of the Psr “active state” to also include analysis of the “transition state mimic” (similar to a vanadate TS mimic, but with structure and charge distribution of a true dianionic phosphorane TS), assuming an SN2-like, associative reaction mechanism. Figure 6 examines simulation results with respect to contact occupancies relevant to α and γ (active state) and β and δ (TS mimic) catalytic strategies as a function of Mg2+ binding mode (Table 1). Here, the pentavalent TS mimic corresponds to an assumed associative mechanism, as is characteristic of nucleolytic ribozymes that presumably utilize a guanine nucleobase to activate the nucleophile (Wilson et al. 2016b). It should be pointed out that under certain circumstances, metal-aided catalysis in RNA might proceed through a dissociative pathway that does not require nucleophile activation by a catalytic nucleobase, and has been predicted for group II introns (Casalino et al. 2016) and may also occur for the Hepatitis Delta Virus ribozyme (Ganguly et al. 2014; Thaplyal et al. 2015; Lee et al. 2016).
FIGURE 6.
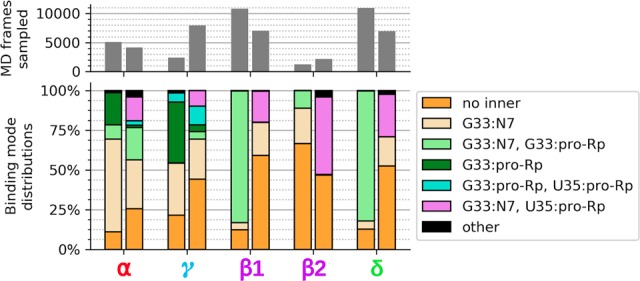
Data on α and γ contact occupancy came from active state simulations of the 5K7C and 5KTJ constructs, while data on β1, β2, and δ contact occupancies came from transition state mimic simulations of the 5K7C and 5KTJ constructs. Each pair of bars in the upper and lower plots correspond to the 5K7C construct as the left bar and the 5KTJ construct as the right bar. The gray bars in the upper plot indicate the portion of simulation data (out of 500,000 frames, equal to 1.0 µsec for each bar) that was found to satisfy catalytic occupancy regardless of the C-site binding mode. The β1 contact is U(+1):pro-RP–MgC:HOH, and the β2 contact is U(+1):pro-RP–G40:N2.
β Catalysis
It is not immediately clear from the 5K7C or the 5KTJ crystal structures which chemical groups in the active site are responsible for β catalysis. The pro-RP NPO of the scissile phosphate appears to interact with the hydration shell of the C-site Mg2+ ion in 5K7C, but in 5KTJ this NPO is instead accepting a hydrogen bond from the exocyclic amine of the general base (G40:N2). TS mimic simulations (of each crystal structure construct) suggest that the pro-RP NPO of the scissile phosphate is simultaneously stabilized by the exocyclic amine of the general base (G40:N2), and water (outer-sphere contact) of the C-site Mg2+. Both of these contacts help to explain the detrimental effects of the phosphorothioate substitution Psr's activity (Neuner et al. 2017), specifically at the pro-RP position (Wilson et al. 2019), since a larger sulfur would either sterically clash, or bind less favorably to each of the two hydrogen bond donors. The lack of a strong thiophilic metal rescue effect suggests the participation of a nucleobase functional group (e.g., G40:N2) and/or indirect coordination with a metal ion plays a role. The indirect coordination of a metal ion or hydrogen bonding of a nucleobase functional group to the scissile phosphate NPOs to provide electrostatic stabilization is designated as a form of secondary β catalysis (Bevilacqua et al. 2019).
Especially enlightening is the eightfold decrease in activity of a G40I mutant (which removed the N2 exocyclic amine of G), despite that the expected pKa shift of inosine (∼0.4 pKa units lower than guanine) alone might be expected to lead to an ∼2.5-fold increase in rate (γ catalysis) in the log-linear pH regime expected at pH 7. This suggests that the effect of removing the N2 exocyclic amine on secondary β catalysis may be as high as ∼20-fold (although this could also affect positioning of the general base, which is a form of tertiary γ catalysis). In the case of the RP thio substrate, G40I has a ∼140-fold mutational rescue effect, (kO/kS)WT/(kO/kS)G40I ∼ 140, reflecting the sensitivity to steric interactions and disruption of hydrogen bonding between the G40:N2 and pro-RP positions (Wilson et al. 2019).
δ Catalysis
Previously, based on the crystal structures, it was proposed that the N3 atom of the purine residue located at position 32 may act as the general acid, similar to the twister ribozyme (Liu et al. 2014; Wilson et al. 2016a; Gaines et al. 2019). Our MD simulation results suggest that the N3 of purine 32 is not favorably positioned to hydrogen bond to the O5′ leaving group and act as a general acid (Fig. 5B,D). This is consistent with recent experimental data that shows the deaza modification at the N3 position in purine 32, P32(3cP), does not substantially impact catalysis, definitively eliminating it as the general acid (Neuner et al. 2017). Simulations indicate the active site of Psr becomes compact and rigid upon binding of a hydrated metal ion in the C-site pocket defined by the scissile phosphate (outer-sphere and possibly inner-sphere to the pro-RP), the Hoogsteen edge of G33 (inner-sphere to N7), and the phosphate of G33 or U35 (possibly inner-sphere to the pro-RP NPO). A tight and stable hydrogen-bonding network arises from these conserved contacts. When the solvated metal ion is in this position, it is ideally suited to mediate δ catalysis through a stable, high-occupancy outer-sphere contact with the 5′-OH leaving group (Fig. 6), suggesting a pivotal role of a metal-bound water molecule acting as a general acid. These results are consistent with very recent cleavage rate measurements in the presence of different divalent metal ions (Co2+, Mn2+, Mg2+, and Ca2+) that yield an almost perfect log-linear relationship with pKa of the metal ion (Wilson et al. 2019). Under a model whereby a metal-bound water molecule acts as the general acid in a manner consistent with pKa trends, the variation of the identity of the metal ion itself (all other factors unchanged) is designated as a form of secondary δ catalysis (Bevilacqua et al. 2019). It would be of great interest to reexamine this divalent metal ion dependence in the presence of an enhanced leaving group such as a 5′ thio-substituted substrate that presumably does not require a general acid for activity. If the divalent metal ions are acting as a general acid through a coordinated water molecule, such a substrate should produce a much-reduced slope with respect to pKa of the metal ion.
FIGURE 5.

The four MD models studied in this work (top and bottom show different views). The active state model (A,C) contains a general base guanine deprotonated at the N1 position and is useful for investigating the α and γ catalytic strategies. The transition state mimic model (B,D) contains a pentacoordinate phosphorane in place of the scissile phosphate and is useful for investigating the β and δ catalytic strategies. A structurally important hydrogen bond was observed between the O2′ position of purine 32 and the exocyclic N2 amine of G42.
DISCUSSION
Critical comparison of similarities and differences between Psr and HHr
With the recent discovery of four new classes of nucleolytic ribozymes from comparative genomics (Breaker 2011; Weinberg et al. 2015), it is of great interest to understand the structural and mechanistic relationships between ribozymes. This would not only help unveil the necessary and sufficient requirements for the activity to guide design, but also provide hints as to possible evolutionary connections between them. In the present context, of the known nucleolytic ribozymes, the Psr bears the greatest resemblance to the HHr (Martick and Scott 2006; Martick et al. 2008; Ward and DeRose 2012; Lee et al. 2013; Mir and Golden 2016; Mir et al. 2015; Chen et al. 2017).
The purpose of this section is to make a critical comparison of some striking similarities and important distinctions between the Psr and HHr classes. At this point, molecular simulations have been performed previously on the HHr constructs 2OEU (Martick et al. 2008; Lee and York 2010; Wong et al. 2011; Lee et al. 2013) and 5DI2 (Mir et al. 2015; Chen et al. 2017), and in the present work simulations have been reported for the Psr based on the 5K7C and 5KTJ constructs, enabling a comprehensive comparison of structural and dynamical features. Analysis of the Psr simulations has led to the reexamination of original secondary structural assignments (Fig. 7D), and their reevaluation reveals striking similarities to those of the HHr as shown in Figure 7. As we establish below, the Psr and HHr have both structural and functional links, and yet exhibit some distinguishing features in their catalytic mechanisms, particularly in the acid step.
FIGURE 7.

Comparison of the structural features of Psr and HHr discussed in this work. The common stem–loops are color-coded and the common metal binding sites are identified in both the three-dimensional structures and secondary structures of HHr (A: 2OEU [Martick et al. 2008] and B: 5DI2 [Mir et al. 2015]) and Psr (C: 5K7C [Ren et al. 2016] and E: 5KTJ [Nguyen et al. 2017]) crystal structures. The secondary structure depiction published in all works on Psr to date (Harris et al. 2015; Weinberg et al. 2015; Müller et al. 2016; Ren et al. 2016, 2017; Kobori et al. 2017; Lee and Lee 2017; Neuner et al. 2017; Nguyen et al. 2017; Nomura et al. 2017; Seith et al. 2018) is color-coded (D) to show how it relates to the new HHr-style depiction of the Psr secondary structure (A–E). The pink spheres denote crystallographic manganese ions in the HHr structures and magnesium ions in the Psr structures. The three binding sites common to both HHr and Psr are the C, G, and E sites. The C site ion is positioned ideally to act as a general acid to the O5′ leaving group, while the G site ion is likely to influence the general base guanine through its Hoogsteen edge (Chen et al. 2017). Cobalt hexammine is resolved in place of hydrated metal ions for some binding sites of the 5KTJ Psr structure.
Psr and HHr have similar global folds, metal ion binding sites, and active site architectures
Both Psr crystal structures, 5K7C (Ren et al. 2016) and 5KTJ (Nguyen et al. 2017), show remarkable similarity in global fold to the HHr, 2OEU (Martick et al. 2008) and 5DI2 (Mir et al. 2015), with active sites located at the three-way junction, and similar Mg2+ ion binding near the active sites (Fig. 7). Previously, no secondary structure diagram of Psr has depicted the conserved G33-C41 base pair that is crucial for recognition of the three-way junction as we have in Figure 7. Upon inclusion of this base pair, the secondary structures of Psr and HHr crystal structures become remarkably similar (Fig. 7).
Comparing the HHr to Psr, the HHr L1 stem–loop is likened to the Psr P2 stem–loop, the HHr L3 stem–loop is likened to the Psr P3 stem–loop, and the small, yet-unnamed stem–loop formed by residues G33 through C41 in Psr is likened to the HHr L2 stem–loop, especially in its formation of a sequence-conserved pseudoknot with the P1(Psr)/L1(HHr) stem–loop. In support of this striking structural link between the two ribozymes, an unusual HHr variant was discovered by the Breaker group (Weinberg et al. 2015) with an L2 stem–loop nearly identical in nature to the single base pair stem–loop that makes up the active site side of the pseudoknot in Psr, and an L1 stem–loop comprised of two helices separated by a short single-stranded stretch of nucleotides.
The active site of Psr is located at the three-way junction formed by stems P2, P3, and the C41-G33 base pair near the PK, similar to the conserved three-way junction of the HHr (Martick and Scott 2006; Martick et al. 2008). For Psr, there were only two Mg2+ ions observed to be bound crystallographically in common sites between 5K7C and 5KTJ, and these are remarkably similar to the two most physiologically relevant and most commonly occupied metal-binding sites in other HHr crystal structures: the “C site” catalytic cation and the “E site” structural cation (Fig. 7A; Martick et al. 2008). Further, Psr crystal structures 5K7C and 5KTJ have a Mg2+ and cobalt hexammine, respectively, in a position similar to the to “G site” in HHr at the Hoogsteen edge of the general base. To facilitate comparison, we propose a naming scheme for Mg2+ binding sites in Psr to follow that of the HHr as described above and illustrated in Figure 7B.
Molecular simulations reveal that Psr and HHr have remarkably similar active site architectures (Fig. 8). Both Psr and HHr contain a conserved L-platform (Suslov et al. 2015) that enables positioning of the general base guanine via an L-anchoring residue, and recruitment of a divalent metal ion in the active site (C-site) using the Hoogsteen edge of a flanking guanine. The C-site binding in Psr and HHr is overall highly comparable, although specifics of the Mg2+ binding mode differ slightly and give rise to subtle differences in the mechanism of general acid catalysis.
FIGURE 8.
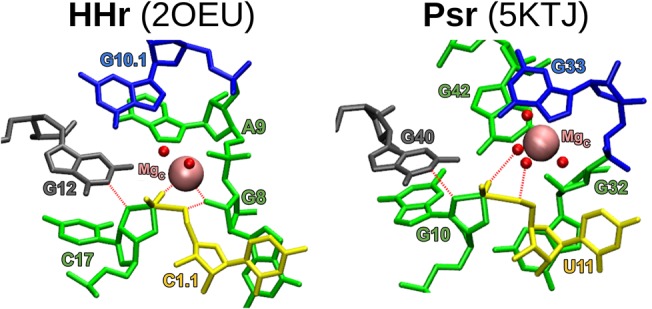
MD models of each crystal equilibrated in a water box as a transition-state-mimic with Mg2+ bound at each ribozyme's C-site. Color-coding corresponds to Figure 7, where the general base guanine is gray. The γ, β2, and δ interactions are labeled with red dashed lines.
The γ catalysis strategy is identical in Psr and HHr
Both Psr and HHr utilize an active site guanine (G40 in Psr and G12 in HHr) to promote γ catalysis by activating the nucleophile. In order to be functional as a general base, this guanine needs to be deprotonated at the N1 position. The unshifted pKa of guanine in aqueous solution is around 9.2. Naively, one might expect the electrostatics in the active site to shift the pKa considerably upward, away from neutrality, reducing the probability of observing the active deprotonated state. Rather, the apparent pKa is estimated for different variants to be between 7.2 and 9.9 in both Psr and HHr (Harris et al. 2015; Frankel et al. 2017), with most recent measurements in the presence of 1 mM Mg2+ and 2 M Na+ is 9.2 (HHr) and 9.8 (Psr) (Wilson et al. 2019).
The HHr utilizes a divalent metal ion binding at the Hoogsteen edge of the general base, involving either direct or indirect coordination with the O6 position, to induce a pKa shift toward neutrality. This has been suggested by Golden and co-workers (Mir and Golden 2016; Mir et al. 2015) in a study that demonstrated the appearance of a divalent metal ion at the G-site in HHr at an elevated pH of 8.5, where presumably the general base guanine would be deprotonated at the N1 position. Subsequent molecular dynamics and free energy simulations, together with quantum mechanical calculations, provide quantitative support for the crystallographic results and their interpretation (Chen et al. 2017). Examination of the resonance structures of deprotonated guanine indicates a formal −1 charge is distributed between the N1 and O6 positions, making the divalent metal binding at O6 a strong stabilizing factor on the ionized residue.
In the crystal structures, a Mg2+ ion is positioned 3.9 Å (5K7C) and a cobalt hexammine ion is positioned 4.3 Å (5KTJ) from the G40:O6. These outer-sphere contacts were stably maintained by Mg2+ on the time scale of our MD simulations. Further, our simulations illustrate that the G-site pocket is electronegative, and in the absence of Mg2+ attracts monovalent ions (e.g., Na+) in the solution that have elevated binding occupations with the G40:N7/O6 positions (Fig. 4). It is known that the inner-sphere binding of Mg2+ ions to the N7 position of guanines in RNA is quite rare (Leonarski et al. 2017), and in the case of Psr, it is likely that divalent metal ion binding is outer-sphere with G40:N7/O6 and overall weaker than in HHr. Nonetheless, it is clear from both crystallographic data (Ren et al. 2016; Nguyen et al. 2017; Wilson et al. 2019) and simulations that Psr and HHr utilize a similar strategy of creating an electronegative binding pocket at the G-site that is able to recruit cations from solution to tune the pKa and activate the guanine general base, and thereby promote γ catalysis.
The role of Mg2+ ions in β and δ catalysis subtly differs in Psr and HHr
In HHr, a Mn2+ ion (PDB ID: 2OEU [Martick et al. 2008]), and a Mg2+ ion (PDB ID: 5EAO [Mir and Golden 2016]), has been observed crystallographically directly coordinated to the N7 of G10.1, as well as the pro-RP of A9. Phosphorothioate-thiophilic metal ion rescue experiments on a truncated HHr (Wang et al. 1999; Osborne et al. 2005), supported by molecular simulations (Lee et al. 2007, 2008), suggest that in the active state (but not necessarily the ground state) the scissile phosphate acquires a functionally important inner-sphere coordination with a divalent metal ion that has yet to be observed crystallographically. The A9:pro-RP position also showed a significant thio effect on both the rate of cleavage and the Kd of binding in the ground state (and additionally was sensitive to the G10.1:N7 position), suggesting this pro-RP position acquires direct coordination from a divalent ion in the ground state that is important also for catalytic activity. Titration experiments involving a doubly substituted truncated HHr were consistent with a single metal ion model (Wang et al. 1999), and subsequent reexamination of an extended HHr construct that had enhanced activity in lower (e.g., 10 µM) concentrations of Mg2+ ions revealed similar putative metal-binding sites as the less-active truncated HHr (Osborne et al. 2005). In the first “full-length” (tertiary stabilized) HHr structure where the solvent was resolved (Martick et al. 2008), a divalent metal ion (Mn2+ in the crystal) was observed bound in the C-site position where it coordinates the A9 at pro-RP and the N7 of G10.1. In this structure, the pro-RP positions of the A9 and scissile phosphates were 4.3 Å apart, and subsequent computational studies strongly suggest that, upon deprotonation of the nucleophile or formation of a dianionic transition state, a Mg2+ ion bound at the C-site will migrate into a bridging position (B-site), consistent with catalytic metal-rescue measurements of the truncated HHr (Wang et al. 1999).
However, more recent metal-rescue effect data suggests that in the extended HHr, a Mg2+ ion occupies a B-site binding mode even in the ribozyme's ground state (Ward and DeRose 2012). To date, a divalent metal ion has not been observed crystallographically bound in the B-site binding mode in HHr, despite the wealth of experimental evidence for its importance for catalytic activity (Vogt et al. 2006). It should be noted that statistically, Mg2+ only rare binds at the N7 position of guanine in RNA (Leonarski et al. 2017), whereas Mn2+ binding in this position is stronger (Sigel and Sigel 2010). More recent theoretical work (Panteva et al. 2015c) using specially developed models for Mg2+ binding to sites in RNA examined the free energy for the Mg2+ ion migration in the extended HHr, and found that, while there was stable binding at the C-site (G10.1:N7 position), the slightly favored binding mode in the ground state was at the B-site, and this preference drastically increased upon deprotonation of the nucleophile. Hence, there seems compelling evidence that Mg2+ binds in the B-site in the ground state of the extended HHr, despite the fact that it has not yet been observed crystallographically in this position.
In any case, there is a general consensus that B-site Mg2+ binding occurs in the HHr active state, and is the mode required for catalysis. Once positioned in the B-site, the divalent metal interacts with the 2′-OH of G8, which has been proposed to act as a metal-activated acid to promote δ catalysis in HHr (Martick et al. 2008; Lee et al. 2013), although other studies have suggested that a metal-bound water molecule may act as the general acid (Ward and DeRose 2012; Mir and Golden 2016; Mir et al. 2015) and it is possible that under different conditions multiple pathways are available (Frankel et al. 2017).
Our previous extensive simulation work (Lee et al. 2013; Chen et al. 2017) are collectively consistent with the interpretation that the primary catalytic mode available to HHr under near-physiological conditions of monovalent and Mg2+ ion concentrations is one whereby a metal-activated 2-OH of G8 acts as the acid. This supposition is supported by recent activity-pH profiles of an extended HHr with 2′NH2 modification at G8 (Wilson et al. 2019) that illustrate convincingly that the shape of the profile is predictably influenced by the pKa of the functional group at the 2′ position in G8, suggesting its role as a general acid. Further, comparison of activity-pH profiles in both the presence and absence of Mg2+ ions support the supposition that the divalent metal ion helps to activate the 2′OH group (Wilson et al. 2019). In the presence of 2 M Na+ ions and the absence of Mg2+, the apparent pKa corresponding to the general acid was 8.7. Upon addition of 1 mM Mg2+, the apparent pKa shifts downward by 1.5 units to 7.2, consistent with our theoretical predictions that the catalytic divalent ion activates (increases the acidity of) the 2′OH of G8. For these reasons, theory from our group predicts, and experiments support the hypothesis that the primary mode of catalysis in HHr under near-physiological conditions is a metal-activated 2′OH of G8 that acts as the general acid.
Nonetheless, it is likely that there is a competing pathway that involves a metal-bound water molecule that can act as the acid. Close examination of recent activity-pH profiles of an extended HHr (Wilson et al. 2019) is consistent with this interpretation. Specifically, it is observed that in the absence of Mg2+ ions, the activity of a G8(dG) mutant (eliminating the 2′OH of G8) at high pH (∼9.5, where the rate reaches its plateau), leads to a dramatic reduction (>103-fold) in cleavage rate relative to the wild type. In the presence of Mg2+, on the other hand, where presumably the alternative metal-bound water pathway is available, the cleavage rate is reduced only by ∼10-fold with respect to the wild type. Further, the G8(dG) mutant causes a considerable shift in the activity-pH profile toward higher pH, consistent with the substitution of a general acid with a higher pKa. One might expect this from a Mg2+ ion that has a pKa considerably upshifted by inner-sphere coordination to two phosphates, as opposed to a guanine 2′OH that has a pKa considerably downshifted by interacting with a Mg2+ ion.
In Psr, stereospecific phosphorothioate substitution experiments indicate thio substitution at the pro-RP NPO of the scissile phosphate disrupts cleavage activity by 4.5 × 104-fold in the presence of Mg2+, and can only be partially (150-fold) rescued by Mn2+ ions (Wilson et al. 2019). This lack of activity rescue differs substantially from the effect in HHr, in which a roughly 4000-fold RP thio effect is almost completely rescued by the addition of 2 mM thiophilic Cd2+ ions (Wang et al. 1999). Thio substitution in Psr at the pro-SP NPO, on the other hand, has less than a twofold effect. An interpretation of this data is that sulfur substitution disrupts contact with a functional group from RNA (Harris et al. 2015; Wilson et al. 2019) rather than a directly coordinated divalent metal ion. We point out that Psr has a highly compact active site relative to HHr, and it is possible that thio substitution could also disrupt binding a divalent metal ion indirectly bound through a water molecule to the pro-RP of the scissile phosphate in a way that was not rescuable due to steric occlusion of the bulky sulfur atom. It is noteworthy to mention that methylation at the 2′ position of A32 leads to a 1.8 × 103-fold rate reduction, whereas the A32(dA) deoxy mutation produces only a 65-fold effect (Wilson et al. 2019). Our simulation results indicate that the 2′OH donates a hydrogen bond to inner-sphere water coordinating the C-site Mg2+ ion that is poised for δ catalysis (Fig. 9), and in this fashion, may serve to increase the acidity of the water. This is discussed in more detail below.
FIGURE 9.
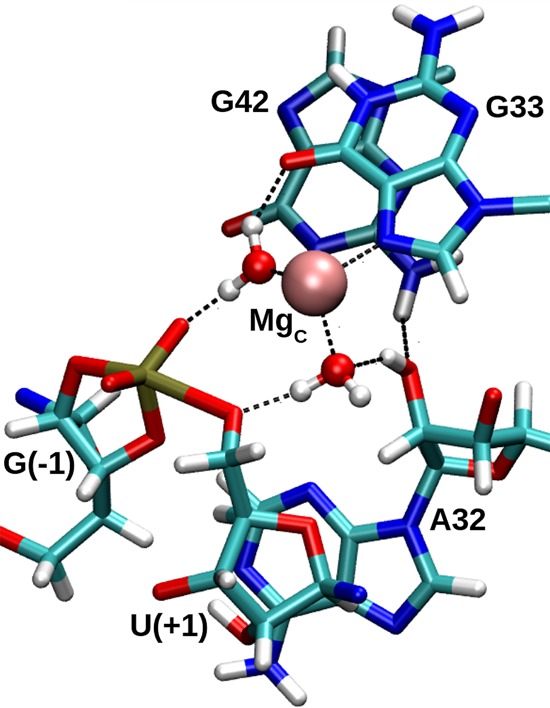
A hydrogen bonding network discovered in the course of a transition state mimic MD simulation, whereby the 2′-OH of P32 acidifies metal-bound water that is poised to protonate the leaving group. Another metal-bound water donates hydrogen bonds to both the pro-RP NPO of the scissile phosphate and the O6 of G33. The MgC metal coordinates three additional waters that are hidden for clarity.
In addition, the N7 position of G33 appears to directly coordinate a divalent metal ion in the Psr crystal (Ren et al. 2016) and has been identified as functionally important after the G33(7cG) mutant showed hardly any activity in the presence of Mg2+ (Neuner et al. 2017). The most recent structural and functional studies also identify a Mn2+ ion with the same G33:N7 direct coordination, and report that G33(7cG) mutation leads to a 1.3 × 104-fold decrease in rate (Wilson et al. 2019). This differs considerably from the 30-fold effect of G10.1(7cG) mutation in HHr (Nakamatsu et al. 2000). While this binding mode is likely important for Mg2+, it is noteworthy that Psr retains modest activity in the presence of the exchange-inert cobalt hexammine as the sole multivalent cation (Harris et al. 2015). Conditions of 1 mM cobalt hexammine in the absence of Mg2+ lead to a 390-fold rate reduction (relative to 1 mM Mg2+), which is consistent with the participation of a Mg2+-bound water molecule acting as acid in Psr (Neuner et al. 2017; Wilson et al. 2019), but perhaps suggesting the recruitment of the less suitable 2′-OH of purine 32 as a general acid (similar to the 2′OH of G8 proposed as an acid in HHr) in the absence of Mg2+.
The roles of a conserved active site 2′-OH are distinctly different in Psr and HHr
In the HHr, the 2′-OH of G8 has been implicated as functionally important (Blount and Uhlenbeck 2005; Nelson and Uhlenbeck 2008), and in the full-length (extended) HHr crystal (Martick and Scott 2006; Martick et al. 2008), is also in close proximity to the O5′ leaving group. This led to the suggestion that the 2′-OH of G8 may act as a general acid in HHr, but objections were raised by the argument that the pKa of this position (estimated to be >12) was likely too high to act as an acid. As described above, computational studies first predicted that in HHr, a Mg2+ ion occupies a bridging (B-site) position between the pro-RP NPOs of A9 and the scissile phosphate (Lee et al. 2007, 2008), both of which have been implicated as functionally important from phosphorothioate-thiophilic metal rescue experiments. In this position, the Mg2+ ion interacts with the 2′-OH of G8, increasing its acidity. Subsequent combined quantum mechanical/molecular mechanical simulations of the chemical steps of the reaction indicated that the Mg2+ ion stabilizes the accumulating charge of the leaving group and facilitates the 2′-OH of G8 to ultimately act as a general acid to promote δ catalysis (Wong et al. 2011).
In the case of the Psr, the 2′-OH of purine 32 exhibits different behavior. In initial crystal structure interpretations, the hydroxyl group was identified as a possible general acid because of its proximity to the leaving group (5.1 Å). Rate data of a deoxy-32 mutant seemed to support this hypothesis when its activity was found to be significantly reduced (Neuner et al. 2017). Further probing of this functional group, including 2′-H, 2′-OMe, and 2′-NH2 substitution motivated the hypothesis that the likely role of this functional group is in forming a hydrogen bonding scaffold that ultimately positions the general base G40. However, more recent experiments that examine mutations at position 42 provide a more refined interpretation (Wilson et al. 2019). In particular, the G42I mutation (removal of N2 exocyclic amine) has almost negligible (1.2-fold) effect on cleavage rate, demonstrating that the hydrogen bond between G42:N2 and the P32:O2′ is not critical. On the other hand, G42(AP) mutation (removal of guanine O6) has a 700-fold rate reduction. Chemical modifications at A32 provide more detailed insight into the role of P32:O2′. Specifically, whereas 2′NH2 modification of G8 in HHr produces an expected apparent pKa shift in accord with a role as general acid, the 2′NH2 modification of A32 in Psr leads to deviation from linearity at low pH, that is, wavy activity-pH profile (Frankel and Bevilacqua 2018), with apparent pKa 5.2 that is consistent with the interpretation that this group has an influence on cleavage rate but that does not participate directly in proton transfer in the acid step (Wilson et al. 2019). Taken together, these results provide strong evidence that the roles of the conserved 2′OH groups in the active sites of HHr and Psr play different mechanistic roles. The simulation results reported here establish detailed atomic-level models from which to interpret these mechanistic distinctions.
Our Psr simulations indicate that the 2′OH of P32, in addition to receiving a hydrogen bond from G42, can form an outer-sphere contact with the Mg2+ ion (Fig. 9). In the TS mimic simulation, the 2′OH of P32 can donate a hydrogen bond to a metal-coordinated water molecule that is in a position to act as the general acid. In this way, the 2′OH of P32 may play a structural role in forming part of the Mg2+ ion binding pocket, but also in increasing the acidity of a metal-bound water molecule to facilitate δ catalysis. This is likely important for an Mg2+ ion in order to downshift its pKa value from solution (11.4) to the apparent pKa value of 9.2 derived from the experimental activity-pH profile (Wilson et al. 2019). This model is consistent with the very large (>104-fold) decrease in cleavage rate of 2′O methylation of A32 (Wilson et al. 2019), as our model suggests this would disrupt the inner-sphere water poised to act as the general acid (Fig. 9), and might also disrupt Mg2+ binding altogether in the compact Psr active site through steric occlusion. It is also possible that 2′O methylation of P32 alters the sugar pucker to favor a C3′-endo conformer (which in the simulations is sometimes held in a C3′-endo conformation due to H-bonding interaction with G42 seen in Fig. 9), and hence it would be valuable to experimentally examine the effect of 2′F and locked nucleic acid residues at P32 in order to gain further insight into the importance of the sugar pucker and hydrogen bond donor/acceptor ability at this key position.
In this scenario suggested by our simulations, the roles of the divalent metal ion and conserved 2′OH in the active sites of Psr and HHr are closely related, but subtly reversed. In HHr, a plausible mechanistic hypothesis is that a Mg2+ ion directly coordinated to the pro-RP acts to increase the acidity of the 2′OH of G8 that then can act as the general acid in δ catalysis. In Psr, on the other hand, we predict that a Mg2+ ion indirectly coordinated to the pro-RP acts as the general acid through a coordinated water molecule, and the 2′OH of P32 acts to increase the acidity of the water making it more effective in δ catalysis. Experimental support for this hypothesis can be gleaned from a comparison of the activity-pH profiles for the HHr G8(dG) mutant and the wild-type Psr in the presence of 1 mM Mg2+ (Wilson et al. 2019). In the present model, the G8(dG) HHr mutant must revert to a catalytic pathway that uses a metal-bound water molecule as acid, which has a measured apparent pKa of 8.9. This value is considerably shifted from the apparent pKa value (7.8) of the wild-type Psr that also uses metal-bound water as the general acid. One can reconcile the 1.1 pKa unit shift in the apparent pKa values to the presence of the 2′OH of A32 in Psr that can donate a hydrogen bond to the general acid water as indicated in Figure 9, whereas the G8(dG) mutant in HHr lacks this capability. This prediction establishes the atomic-level origin of a functional link between HHr and Psr, and hints at the possibility of an evolutionary connection between the ribozymes.
Open questions and future work
The present simulations make several experimentally testable predictions. First, simulations suggest that the catalytic divalent metal ion may make inner-sphere contacts with the pro-RP NPO of G33 or possibly U35. The G33:pro-RP contact would be analogous to the A9 contact in HHr, which exhibits a strong stereospecific thio effect that can be rescued by thiophilic metals. Hence, probing these remote NPO positions stereospecifically may lead to more detailed insight into the catalytic Mg2+ ion binding mode. Further, given the hypothesis that it is the 2′OH of P32 that donates a hydrogen bond to the metal-bound water molecule to increase its acidity, it would be interesting to explore the metal-ion dependence of A32(dA) and/or G32(dG) mutants to see if the mutational effect was correlated with the pKa of the metal ion. Comparison of the activity-pH profile of the wild type and G32(dG) mutant with a series of metal ions with varying pKa values (e.g., Mg2+, Mn2+, Co2+, and Zn2+) would provide insight into the role of the 2′OH of P32 in tuning the pKa of the general acid.
Second, the intriguing results of 2′NH2 chemical modification to A32 could be further explored. The interpretation of functional results led to the supposition that the 2′ position of A32 required the ability to receive a hydrogen bond from G42:N2 in order to maintain the hydrogen bond network that positions G40, leading to high activity (Neuner et al. 2017). However, subsequent mutational studies that examined the G42I mutant, showed that cleavage rate decreased negligibly (1.2-fold decrease), indicating this is not the origin of the observed reduced activity (Wilson et al. 2019). Nonetheless, at pH below 6.2, the 2′-NH2 group is likely protonated (2′-NH3+) and can no longer receive a hydrogen bond from the N2 exocyclic amine of G42. As shown in Figure 5, this network is observed in simulations and does appear to provide organization to the active site, including the positioning of G40 to act in γ catalysis.
There remain several open questions that require additional study to answer. In particular, details surrounding the very large, partially rescuable stereospecific thio effect still need to be clarified. Further, the origin of the 1.8 × 104-fold effect of methylation of the O2′ position of A32 remains unclear. An understanding of how these, and other chemical modifications already performed in the active site such as thio substitutions, affect metal ion occupancy through electrostatic perturbation or steric occlusion is of paramount importance. Finally, rigorous examination of the chemical steps of the reaction using quantum mechanical methods will provide needed clarity on the interpretation of structural and functional data, and deep insight into the Psr catalytic mechanism.
Conclusions
We present a model for the active state of the Psr in solution from molecular dynamics simulations at different stages along the reaction pathway. The resulting structural ensemble represents a model that unifies the existing body of structural and functional data, provides new insight into the catalytic chemical mechanism, and establishes important functional connections with the HHr. Results indicate that multiple Mg2+ binding modes in the active site, including outer-sphere coordination to the scissile phosphate (C-site binding) consistent with experimental thio effects, can promote in-line fitness (α catalysis), electrostatic stabilization of the dianionic transition state (β catalysis) and neutralization of the developing charge of the leaving group (δ catalysis) through a metal-coordinated water molecule. Further, metal ion binding at the G-site (G40:O6) tunes the pKa of the general base in order to promote γ catalysis. The molecular simulation models explain the current body of functional data, including mutagenesis data involving P32(3cP) and G33(7cG), chemical modification of the 2′-OH of P32, and phosphorothioate substitution/metal ion rescue experiments at the scissile phosphate. Simulation results suggest a number of experimentally testable predictions, including metal ion titration experiments to probe the pH-dependence of the general acid, stereospecific thio substitution to test sensitivity at the pro-RP position of scissile phosphate, and A32(2′nA)/G42X double mutation to examine the pH-dependence and test the functional importance of a proposed hydrogen bond network that helps to position the general base and scissile phosphate for catalysis. Finally, analysis of the Psr simulations has led to the reexamination of original secondary structural assignments, and their reevaluation reveals striking similarities to those of the HHr. The present study helps to establish new functional connections between the Psr and HHr, including their remarkably similar global folds, metal ion binding sites, active site architectures, and γ (general base) catalysis strategies. The Psr and HHr have subtle differences in their δ (general acid) strategies, and have distinctly different roles for a functionally important active site 2′-OH group.
MATERIALS AND METHODS
Setup for molecular dynamics simulations
All molecular dynamics simulations were performed using the AMBER 16 packages (Case et al. 2016) and GPU-accelerated enhancements in AMBER 18 (Lee et al. 2018) using the AMBER FF14 force field (Maier et al. 2015), which includes corrections to α/γ conformers (Pérez et al. 2007) and glycosidic torsions (Zgarbova et al. 2011) in nucleic acids.
Both crystal structures, 5K7C (Ren et al. 2016) and chains C and D of 5KTJ (Nguyen et al. 2017), were stripped of all crystallographic waters and ions except the C- and E-site Mg2+ ions, then solvated in a truncated octahedral TIP4P-Ew water box (Horn et al. 2004) with a minimum of 15 Å between the ribozyme and box edge. Monovalent (Joung and Cheatham III 2008) and divalent (Li et al. 2013) ion parameters were selected for compatibility with the water model. This model for ions enables implicit first-order pairwise polarization effects that provide a better description of structural, thermodynamic, kinetic and mass transport properties (Panteva et al. 2015b). Next, Na+ ions were added to balance the total system charge (50 in the case of 5K7C, and 57 in the case of 5KTJ), and a number of NaCl ion pairs was added (33 for the 5K7C box, and 40 for the slightly larger 5KTJ box) to mimic a physiological salt concentration of ∼140 mM.
Both systems underwent 2000 cycles of solvent minimization before heating and annealing over 12 nsec to bring the system temperature to 300 K and density to roughly 1.0 g/cm3 (which required alternating between the NVT and NPT ensembles), all while keeping the ribozymes and Mg2+ ions constrained to their respective crystallographic coordinates. These harmonic force coordinate constraints were slowly reduced to a value of zero over ∼0.5 nsec, after which the ribozyme was allowed to undergo equilibration for 20 nsec in the NVT ensemble. In each of the two systems, the resulting structure represented the native protonation state, and served as a starting point for all following simulations. All subsequent simulations were run with a 12 Å cutoff for nonbonded interactions and particle mesh Ewald (Darden et al. 1993) for electrostatics outside the cutoff.
Probing binding mode effects on α and γ catalysis
Each of the two equilibrated ribozymes was transformed into the “active state,” which entails an anionic general base G40 deprotonated at its N1 position, and a neutral, protonated nucleophile. After deletion of the G40:N1 hydrogen, the G40 was assigned a new charge-set previously developed in our laboratory (Chen et al. 2017), prioritizing consistency with the AMBER FF14 force field. Distance restraints were applied to enforce a particular C-site Mg2+ binding mode at the outset of each simulation, with restraints turned off at 20 nsec before running a subsequent 100 nsec of unrestrained dynamics. Each binding mode was simulated in this way ten times, with independent regions of phase space accessed by enforcing different metal-RNA contacts and hydrogen bonding patterns during each restrained 20 nsec.
Probing binding mode effects on β and δ catalysis
Each of the two equilibrated ribozymes was transformed into the “transition state mimic,” which entails a dianionic pentacoordinate phosphorane at the scissile phosphate, where a new bond has been made between the nucleophile and the adjacent phosphorus. After deletion of the G(−1):O2′ hydrogen, both G(−1) and U(+1) were assigned new charge-sets previously developed in our laboratory (Heldenbrand et al. 2014), prioritizing consistency with the AMBER FF14 force field. Distance restraints were applied to enforce a particular C-site Mg2+ binding mode at the outset of each simulation, with restraints turned off at 20 nsec before running a subsequent 100 nsec of unrestrained dynamics. Each binding mode was simulated in this way 10 times, with independent regions of phase space accessed by enforcing different metal-RNA contacts and hydrogen bonding patterns during each restrained 20 nsec.
Radial distribution functions
The radial distribution functions plotted in Figure 4 were obtained by binning G40:O6-Na+ (top plot) and G40:N7-Na+ (bottom plot) distances over a 100 nsec simulation of 5K7C with G40 in the active state (blue and red curves), and a 100 nsec simulation of 5K7C with G40 in its standard protonation state (black curves). The average coordination numbers were calculated as a running sum of bins up to and including the edge of the first Na+ ion shell.
ACKNOWLEDGMENTS
We are grateful for the financial support provided by the National Institutes of Health (grant no. GM62248 to D.M.Y.). Computational resources were provided by the Office of Advanced Research Computing (OARC) at Rutgers University, supported by the National Institutes of Health under grant no. S10OD012346, the Rutgers Discovery Informatics Institute at Rutgers University, supported by the State of New Jersey, and by the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by the National Science Foundation (grant nos. ACI-1548562 and OCI-1053575). Additionally, this research is part of the Blue Waters sustained-petascale computing project, which is supported by the National Science Foundation (awards OCI-0725070 and ACI-1238993) and the state of Illinois. Blue Waters is a joint effort of the University of Illinois at Urbana-Champaign and its National Center for Supercomputing Applications.
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.071944.119.
REFERENCES
- Bevilacqua PC, Yajima R. 2006. Nucleobase catalysis in ribozyme mechanism. Curr Opin Chem Biol 10: 455–464. 10.1016/j.cbpa.2006.08.014 [DOI] [PubMed] [Google Scholar]
- Bevilacqua PC, Brown TS, Nakano S, Yajima R. 2004. Catalytic roles for proton transfer and protonation in ribozymes. Biopolymers 73: 90–109. 10.1002/bip.10519 [DOI] [PubMed] [Google Scholar]
- Bevilacqua PC, Harris ME, Piccirilli JA, Gaines C, Ganguly A, Kostenbader K, Ekesan Ş, York DM. 2019. An ontology for facilitating discussion of catalytic strategies of RNA-cleaving enzymes. ACS Chem Biol 14: 1068–1076. 10.1021/acschembio.9b00202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blount KF, Uhlenbeck OC. 2005. The structure-function dilemma of the hammerhead ribozyme. Annu Rev Biophys Biomol Struct 34: 415–440. 10.1146/annurev.biophys.34.122004.184428 [DOI] [PubMed] [Google Scholar]
- Breaker RR. 2011. Prospects for riboswitch discovery and analysis. Mol Cell 43: 867–879. 10.1016/j.molcel.2011.08.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breaker RR. 2017. Mechanistic debris generated by twister ribozymes. ACS Chem Biol 12: 886–891. 10.1021/acschembio.7b00010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casalino L, Palermo G, Rothlisberger U, Magistrato A. 2016. Who activates the nucleophile in ribozyme catalysis? An answer from the splicing mechanism of group II introns. J Am Chem Soc 138: 10374–10377. 10.1021/jacs.6b01363 [DOI] [PubMed] [Google Scholar]
- Case DA, Betz RM, Cerutti DS, Cheatham TE III, Darden TA, Duke RE, Giese TJ, Gohlke H, Goetz AW, Homeyer N, et al. 2016. AMBER 16. University of California, San Francisco, CA. [Google Scholar]
- Chen X, Li N, Ellington AD. 2007. Ribozyme catalysis of metabolism in the RNA world. Chem Biodivers 4: 633–655. 10.1002/cbdv.200790055 [DOI] [PubMed] [Google Scholar]
- Chen H, Giese TJ, Golden BL, York DM. 2017. Divalent metal ion activation of a guanine general base in the hammerhead ribozyme: insights from molecular simulations. Biochemistry 56: 2985–2994. 10.1021/acs.biochem.6b01192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darden T, York D, Pedersen L. 1993. Particle mesh Ewald: an N log(N) method for Ewald sums in large systems. J Chem Phys 98: 10089–10092. 10.1063/1.464397 [DOI] [Google Scholar]
- DeRose VJ. 2002. Two decades of RNA catalysis. Chem Biol 9: 961–969. 10.1016/S1074-5521(02)00217-X [DOI] [PubMed] [Google Scholar]
- DeRose VJ. 2003. Metal ion binding to catalytic RNA molecules. Curr Opin Struct Biol 13: 317–324. 10.1016/S0959-440X(03)00077-0 [DOI] [PubMed] [Google Scholar]
- Emilsson GM, Nakamura S, Roth A, Breaker RR. 2003. Ribozyme speed limits. RNA 9: 907–918. 10.1261/rna.5680603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Famulok M, Hartig JS, Mayer G. 2007. Functional aptamers and aptazymes in biotechnology, diagnostics, and therapy. Chem Rev 107: 3715–3743. 10.1021/cr0306743 [DOI] [PubMed] [Google Scholar]
- Frankel EA, Bevilacqua PC. 2018. Complexity in pH-dependent ribozyme kinetics: dark pKa shifts and wavy rate-pH profiles. Biochemistry 57: 483–488. 10.1021/acs.biochem.7b00784 [DOI] [PubMed] [Google Scholar]
- Frankel EA, Strulson CA, Keating CD, Bevilacqua PC. 2017. Cooperative interactions in the hammerhead ribozyme drive pKa shifting of G12 and its stacked base C17. Biochemistry 56: 2537–2548. 10.1021/acs.biochem.7b00174 [DOI] [PubMed] [Google Scholar]
- Fusz S, Eisenführ A, Srivatsan SG, Heckel A, Famulok M. 2005. A ribozyme for the aldol reaction. Chem Biol 12: 941–950. 10.1016/j.chembiol.2005.06.008 [DOI] [PubMed] [Google Scholar]
- Gaines CS, York DM. 2016. Ribozyme catalysis with a twist: active state of the twister ribozyme in solution predicted from molecular simulation. J Am Chem Soc 138: 3058–3065. 10.1021/jacs.5b12061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaines CS, Giese TJ, York DM. 2019. Cleaning up mechanistic debris generated by twister ribozymes using computational RNA enzymology. ACS Catal 9: 5803–5815. 10.1021/acscatal.9b01155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganguly A, Thaplyal P, Rosta E, Bevilacqua PC, Hammes-Schiffer S. 2014. Quantum mechanical/molecular mechanical free energy simulations of the self-cleavage reaction in the hepatitis delta virus ribozyme. J Am Chem Soc 136: 1483–1496. 10.1021/ja4104217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert W. 1986. The RNA world. Nature 319: 618 10.1038/319618a0 [DOI] [Google Scholar]
- Gu H, Furukawa K, Weinberg Z, Berenson DF, Breaker RR. 2013. Small, highly active DNAs that hydrolyze DNA. J Am Chem Soc 135: 9121–9129. 10.1021/ja403585e [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris KA, Lünse CE, Li S, Brewer KI, Breaker RR. 2015. Biochemical analysis of pistol self-cleaving ribozymes. RNA 21: 1852–1858. 10.1261/rna.052514.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heldenbrand H, Janowski PA, Giambasşu G, Giese TJ, Wedekind JE, York DM. 2014. Evidence for the role of active site residues in the hairpin ribozyme from molecular simulations along the reaction path. J Am Chem Soc 136: 7789–7792. 10.1021/ja500180q [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horn HW, Swope WC, Pitera JW, Madura JD, Dick TJ, Hura GL, Head-Gordon T. 2004. Development of an improved four-site water model for biomolecular simulations: TIP4P-Ew. J Chem Phys 120: 9665–9678. 10.1063/1.1683075 [DOI] [PubMed] [Google Scholar]
- Joung IS, Cheatham TE III. 2008. Determination of alkali and halide monovalent ion parameters for use in explicitly solvated biomolecular simulations. J Phys Chem B 112: 9020–9041. 10.1021/jp8001614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobori S, Takahashi K, Yokobayashi Y. 2017. Deep sequencing analysis of aptazyme variants based on a pistol ribozyme. ACS Synth Biol 6: 1283–1288. 10.1021/acssynbio.7b00057 [DOI] [PubMed] [Google Scholar]
- Krug N, Hohlfeld JM, Kirsten AM, Kornmann O, Beeh KM, Kappeler D, Korn S, Ignatenko S, Timmer W, Rogon C, et al. 2015. Allergen-induced asthmatic responses modified by a GATA3-specific DNAzyme. New Engl J Med 372: 1987–1995. 10.1056/NEJMoa1411776 [DOI] [PubMed] [Google Scholar]
- Lee KY, Lee BJ. 2017. Structural and biochemical properties of novel self-cleaving ribozymes. Molecules 22: 678 10.3390/molecules22040678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee TS, York DM. 2010. Computational mutagenesis studies of hammerhead ribozyme catalysis. J Am Chem Soc 132: 13505–13518. 10.1021/ja105956u [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee TS, Silva-López C, Martick M, Scott WG, York DM. 2007. Insight into the role of Mg2+ in hammerhead ribozyme catalysis from x-ray crystallography and molecular dynamics simulation. J Chem Theory Comput 3: 325–327. 10.1021/ct6003142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee TS, Silva López C, Giambasşu GM, Martick M, Scott WG, York DM. 2008. Role of Mg2+ in hammerhead ribozyme catalysis from molecular simulation. J Am Chem Soc 130: 3053–3064. 10.1021/ja076529e [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee TS, Giambaşu GM, Sosa CP, Martick M, Scott WG, York DM. 2009. Threshold occupancy and specific cation binding modes in the hammerhead ribozyme active site are required for active conformation. J Mol Biol 388:195–206. 10.1016/j.jmb.2009.02.054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee TS, Wong KY, Giambasu GM, York DM. 2013. Bridging the gap between theory and experiment to derive a detailed understanding of hammerhead ribozyme catalysis. Prog Mol Biol Transl Sci 120: 25–91. 10.1016/B978-0-12-381286-5.00002-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee TS, Radak BK, Harris ME, York DM. 2016. A two-metal-ion-mediated conformational switching pathway for HDV ribozyme activation. ACS Catal 6: 1853–1869. 10.1021/acscatal.5b02158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee TS, Cerutti DS, Mermelstein D, Lin C, LeGrand S, Giese TJ, Roitberg A, Case DA, Walker RC, York DM. 2018. GPU-accelerated molecular dynamics and free energy methods in Amber18: performance enhancements and new features. J Chem Inf Model 58: 2043–2050. 10.1021/acs.jcim.8b00462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonarski F, D'Ascenzo L, Auffinger P. 2017. Mg2+ ions: do they bind to nucleobase nitrogens? Nucleic Acids Res 45: 987–1004. 10.1093/nar/gkw1175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P, Roberts BP, Chakravorty DK, Merz KM Jr. 2013. Rational design of particle Mesh Ewald compatible Lennard-Jones parameters for +2 metal cations in explicit solvent. J Chem Theory Comput 9: 2733–2748. 10.1021/ct400146w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Cao Z, Lu Y. 2009. Functional nucleic acid sensors. Chem Rev 109: 1948–1998. 10.1021/cr030183i [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Wilson TJ, McPhee SA, Lilley DMJ. 2014. Crystal structure and mechanistic investigation of the twister ribozyme. Nat Chem Biol 10: 739–744. 10.1038/nchembio.1587 [DOI] [PubMed] [Google Scholar]
- Liu H, Yu X, Chen Y, Zhang J, Wu B, Zheng L, Haruehanroengra P, Wang R, Li S, Lin J, et al. 2017. Crystal structure of an RNA-cleaving DNAzyme. Nat Commun 8: 2006–2015. 10.1038/s41467-017-02203-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier JA, Martinez C, Kasavajhala K, Wickstrom L, Hauser KE, Simmerling C. 2015. ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB. J Chem Theory Comput 11: 3696–3713. 10.1021/acs.jctc.5b00255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martick M, Scott WG. 2006. Tertiary contacts distant from the active site prime a ribozyme for catalysis. Cell 126: 309–320. 10.1016/j.cell.2006.06.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martick M, Lee TS, York DM, Scott WG. 2008. Solvent structure and hammerhead ribozyme catalysis. Chem Biol 15: 332–342. 10.1016/j.chembiol.2008.03.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mir A, Golden BL. 2016. Two active site divalent ions in the crystal structure of the hammerhead ribozyme bound to a transition state analogue. Biochemistry 55: 633–636. 10.1021/acs.biochem.5b01139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mir A, Chen J, Robinson K, Lendy E, Goodman J, Neau D, Golden BL. 2015. Two divalent metal ions and conformational changes play roles in the hammerhead ribozyme cleavage reaction. Biochemistry 54: 6369–6381. 10.1021/acs.biochem.5b00824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller S, Appel B, Balke D, Hieronymus R, Nübel C. 2016. Thirty-five years of research into ribozymes and nucleic acid catalysis: where do we stand today? F1000Res 5: 1511 10.12688/f1000research.8601.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamatsu Y, Warashina M, Kuwabara T, Tanaka Y, Yoshinari K, Taira K. 2000. Significant activity of a modified ribozyme with N7-deazaguanine at G10.1: the double-metal-ion mechanism of catalysis in reactions catalysed by hammerhead ribozymes. Genes Cells 5: 603–612. 10.1046/j.1365-2443.2000.00352.x [DOI] [PubMed] [Google Scholar]
- Nelson JA, Uhlenbeck OC. 2008. Minimal and extended hammerheads utilize a similar dynamic reaction mechanism for catalysis. RNA 14: 43–54. 10.1261/rna.717908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuner S, Falschlunger C, Fuchs E, Himmelstoss M, Ren A, Patel DJ, Micura R. 2017. Atom-specific mutagenesis reveals structural and catalytic roles for an active-site adenosine and hydrated Mg2+ in pistol ribozymes. Angew Chem Int Ed 56: 15954–15958. 10.1002/anie.201708679 [DOI] [PubMed] [Google Scholar]
- Nguyen LA, Wang J, Steitz TA. 2017. Crystal structure of Pistol, a class of self-cleaving ribozyme. Proc Natl Acad Sci 114: 1021–1026. 10.1073/pnas.1611191114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomura Y, Chien HC, Yokobayashi Y. 2017. Direct screening for ribozyme activity in mammalian cells. Chem Commun 53: 12540–12543. 10.1039/C7CC07815C [DOI] [PubMed] [Google Scholar]
- Osborne EM, Schaak JE, Derose VJ. 2005. Characterization of a native hammerhead ribozyme derived from schistosomes. RNA 11: 187–196. 10.1261/rna.7950605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panteva MT, Dissanayake T, Chen H, Radak BK, Kuechler ER, Giambaşu GM, Lee TS, York DM. 2015a. Multiscale methods for computational RNA enzymology. Methods Enzymol 553: 335–374. 10.1016/bs.mie.2014.10.064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panteva MT, Giambaşu GM, York DM. 2015b. Comparison of structural, thermodynamic, kinetic and mass transport properties of Mg2+ ion models commonly used in biomolecular simulations. J Comput Chem 36: 970–982. 10.1002/jcc.23881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panteva MT, Giambaşu GM, York DM. 2015c. Force field for Mg2+, Mn2+, Zn2+, and Cd2+ ions that have balanced interactions with nucleic acids. J Phys Chem B 119: 15460–15470. 10.1021/acs.jpcb.5b10423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez A, Marchán I, Svozil D, Sponer J, Cheatham TE III, Laughton CA, Orozco M. 2007. Refinement of the AMBER force field for nucleic acids: improving the description of α/γ conformers. Biophys J 92: 3817–3829. 10.1529/biophysj.106.097782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponce-Salvatierra A, Wawrzyniak-Turek K, Steuerwald U, Höbartner C, Pena V. 2016. Crystal structure of a DNA catalyst. Nature 529: 231–234. 10.1038/nature16471 [DOI] [PubMed] [Google Scholar]
- Ren A, Vušurovic N, Gebetsberger J, Gao P, Juen M, Kreutz C, Micura R, Patel D. 2016. Pistol ribozyme adopts a pseudoknot fold facilitating site-specific in-line cleavage. Nat Chem Biol 12: 702–708. 10.1038/nchembio.2125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren A, Micura R, Patel DJ. 2017. Structure-based mechanistic insights into catalysis by small self-cleaving ribozymes. Curr Opin Chem Biol 41: 71–83. 10.1016/j.cbpa.2017.09.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saragliadis A, Krajewski SS, Rehm C, Narberhaus F, Hartig JS. 2013. Thermozymes synthetic RNA thermometers based on ribozyme activity. RNA Biol 10: 1009–1016. 10.4161/rna.24482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott WG. 2007. Ribozymes. Curr Opin Struct Biol 17: 280–286. 10.1016/j.sbi.2007.05.003 [DOI] [PubMed] [Google Scholar]
- Seith DD, Bingaman JL, Veenis AJ, Button AC, Bevilacqua PC. 2018. Elucidation of catalytic strategies of small nucleolytic ribozymes from comparative analysis of active sites. ACS Catal 8: 314–327. 10.1021/acscatal.7b02976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengle G, Eisenführ A, Arora PS, Nowick JS, Famulok M. 2001. Novel RNA catalysts for the Michael reaction. Chem Biol 8: 459–473. 10.1016/S1074-5521(01)00026-6 [DOI] [PubMed] [Google Scholar]
- Serganov A, Keiper S, Malinina L, Tereshko V, Skripkin E, Höbartner C, Polonskaia A, Phan AT, Wombacher R, Micura R, et al. 2005. Structural basis for Diels–Alder ribozyme-catalyzed carbon–carbon bond formation. Nat Struct Biol 12: 218–224. 10.1038/nsmb906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigel RKO, Sigel H. 2010. A stability concept for metal ion coordination to single-stranded nucleic acids and affinities of individual sites. Acc Chem Res 43: 974–984. 10.1021/ar900197y [DOI] [PubMed] [Google Scholar]
- Soukup GA, Breaker RR. 1999. Relationship between internucleotide linkage geometry and the stability of RNA. RNA 5: 1308–1325. 10.1017/S1355838299990891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steitz TA, Moore PB. 2003. RNA, the first macromolecular catalyst: the ribosome is a ribozyme. Trends Biochem Sci 28: 411–418. 10.1016/S0968-0004(03)00169-5 [DOI] [PubMed] [Google Scholar]
- Sullenger BA, Gilboa E. 2002. Emerging clinical applications of RNA. Nature 418: 252–258. 10.1038/418252a [DOI] [PubMed] [Google Scholar]
- Suslov NB, DasGupta S, Huang H, Fuller JR, Lilley DMJ, Rice PA, Piccirilli JA. 2015. Crystal structure of the Varkud satellite ribozyme. Nat Chem Biol 11: 840–846. 10.1038/nchembio.1929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thaplyal P, Ganguly A, Hammes-Schiffer S, Bevilacqua PC. 2015. Inverse thio effects in the hepatitis delta virus ribozyme reveal that the reaction pathway is controlled by metal ion charge density. Biochemistry 54: 2160–2175. 10.1021/acs.biochem.5b00190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas JM, Perrin DM. 2009. Probing general acid catalysis in the hammerhead ribozyme. J Am Chem Soc 131: 1135–1143. 10.1021/ja807790e [DOI] [PubMed] [Google Scholar]
- Torres RA, Bruice TC. 1998. Molecular dynamics study displays near in-line attack conformations in the hammerhead ribozyme self-cleavage reaction. Proc Natl Acad Sci 95: 11077–11082. 10.1073/pnas.95.19.11077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogt M, Lahiri S, Hoogstraten CG, Britt DR, DeRose VJ. 2006. Coordination environment of a site-bound metal ion in the hammerhead ribozyme determined by 15N and 2H ESEEM spectroscopy. J Am Chem Soc 128: 16764–16770. 10.1021/ja057035p [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Karbstein K, Peracchi A, Beigelman L, Herschlag D. 1999. Identification of the hammerhead ribozyme metal ion binding site responsible for rescue of the deleterious effect of a cleavage site phosphorothioate. Biochemistry 38: 14363–14378. 10.1021/bi9913202 [DOI] [PubMed] [Google Scholar]
- Ward WL, Derose VJ. 2012. Ground-state coordination of a catalytic metal to the scissile phosphate of a tertiary-stabilized Hammerhead ribozyme. RNA 18: 16–23. 10.1261/rna.030239.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward WL, Plakos K, DeRose VJ. 2014. Nucleic acid catalysis: metals, nucleobases, and other cofactors. Chem Rev 114: 4318–4342. 10.1021/cr400476k [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg Z, Kim PB, Chen TH, Li S, Harris KA, Lünse CE, Breaker RR. 2015. New classes of self-cleaving ribozymes revealed by comparative genomics analysis. Nat Chem Biol 11: 606–610. 10.1038/nchembio.1846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson TJ, Lilley DM. 2013. A mechanistic comparison of the Varkud satellite and hairpin ribozymes. Prog Mol Biol Transl Sci 120: 93–121. 10.1016/B978-0-12-381286-5.00003-2 [DOI] [PubMed] [Google Scholar]
- Wilson TJ, Liu Y, Domnick C, Kath-Schorr S, Lilley DMJ. 2016a. The novel chemical mechanism of the twister ribozyme. J Am Chem Soc 138: 6151–6162. 10.1021/jacs.5b11791 [DOI] [PubMed] [Google Scholar]
- Wilson TJ, Liu Y, Lilley DMJ. 2016b. Ribozymes and the mechanisms that underlie RNA catalysis. Front Chem Sci Eng 10: 178–185. 10.1007/s11705-016-1558-2 [DOI] [Google Scholar]
- Wilson TJ, Liu Y, Li NS, Dai Q, Piccirilli JA, Lilley DM. 2019. Comparison of the structures and mechanisms of the pistol and hammerhead ribozymes. J Am Chem Soc 141: 7865–7875. 10.1021/jacs.9b02141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler WC, Nahvi A, Roth A, Collins JA, Breaker RR. 2004. Control of gene expression by a natural metabolite-responsive ribozyme. Nature 428: 281–286. 10.1038/nature02362 [DOI] [PubMed] [Google Scholar]
- Wong KY, Lee TS, York DM. 2011. Active participation of the Mg2+ ion in the reaction coordinate of RNA self-cleavage catalyzed by the hammerhead ribozyme. J Chem Theory Comput 7: 1–3. 10.1021/ct100467t [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zgarbová M, Otyepka M, Šponer J, Mládek A, Banáš P, Cheatham TE III, Jurečka P. 2011. Refinement of the Cornell et al. nucleic acids force field based on reference quantum chemical calculations of glycosidic torsion profiles. J Chem Theory Comput 7: 2886–2902. 10.1021/ct200162x [DOI] [PMC free article] [PubMed] [Google Scholar]