

Abstract
The transferrin receptor (TfR1) is the principal means of iron importation for most mammalian cells, and regulation of mRNA stability is a major mechanism through which TfR1 expression is controlled in response to changing intracellular iron levels. An endonuclease activity degrades the TfR1 mRNA during iron-repletion, which reduces iron importation and contributes to the restoration of homeostasis. Correct identification of the TfR1 mRNA endonuclease activity is important as it has the potential to be a pharmacological target for the treatment of several pathologies in which iron homeostasis is perturbed. A recent RNA article identified both miR-7-5p and miR-141-3p as mediators of TfR1 mRNA degradation during iron-repletion. However, the proposed TfR1 microRNA binding sites are inconsistent with several earlier studies. To better understand the discrepancy, we tested the proposed sites within an assay developed to detect changes to TfR1 mRNA stability. The complete disruption of both proposed binding sites failed to impact the assay in all cell lines tested, which include cell lines derived from mouse connective tissue (L-M), a human colon adenocarcinoma (SW480), and a human ovarian carcinoma (A2780). The overexpression of a miR-7-5p mimic also failed to decrease expression of both the endogenous TfR1 mRNA and a luciferase-TfR1 reporter under conditions in which the expression of a previously identified mir-7-5p target is attenuated. As a result, it is unlikely that the microRNAs are directly mediating iron-responsive degradation of the TfR1 mRNA as recently proposed. Instead, three short hairpin loops within the TfR1 3′-UTR are shown to be more consistent as endonuclease recognition elements.
Keywords: transferrin receptor-1, endonuclease, iron homeostasis, miR-7-5p, miR-141-3p
INTRODUCTION
The coordination chemistry and redox potential of iron, in its ferrous and ferric forms, is integral to several proteins including those involved with DNA replication, energy generation, and oxygen transport. In addition to inadequate nutrition, iron deficiency anemia can also result from aging, chronic inflammation, chronic kidney disease, and several inborn genetic errors. It can cause congenital disorders, immune dysfunction and is associated with the progression of a large number of diseases (for review, see Weiss et al. 2019). Conversely, high concentrations of iron can generate reactive oxygen species resulting in tissue damage. Iron overload, typified by hemochromatosis, can be caused by several inborn genetic errors (for review, see Katsarou et al. 2019). If left untreated, iron overload can lead to cardiomyopathy, diabetes mellitus and liver cirrhosis. As a result, understanding iron homeostatic mechanisms at both the cellular and organismal levels is highly significant to both basic science and public health.
Transferrin is the principal carrier of iron in the plasma, and the transferrin receptor (TfR1) is the primary means of iron importation for most mammalian cells. The regulation of mRNA stability in response to changing intracellular iron is a major mechanism through which TfR1 expression is controlled. Iron regulatory proteins (IRPs) protect the TfR1 mRNA from an endonuclease activity during iron depletion through binding to iron-responsive elements (IREs), which are specialized stem–loop structures located within the 3′-UTR of the TfR1 mRNA. However, IRP binding is attenuated during iron-repletion resulting in unimpeded TfR1 mRNA degradation, which contributes to the restoration of homeostasis by reducing iron importation (for review, see Wang and Babitt 2019). The endonuclease catalyzing the mRNA degradation during iron-repletion has potential as a therapeutic target to correct for iron imbalances associated with multiple pathologies, making its correct identification important.
A recent RNA article identified the miR-7-5p and miR-141-3p microRNAs as mediators of TfR1 mRNA degradation during iron-repletion (Miyazawa et al. 2018). The putative binding sites for these microRNAs overlap with two IREs within the TfR1 3′-UTR, which suggested a compelling model where direct competition with the IRPs could account for iron-responsive regulation. However, the proposed binding site for miR-141-3p is not within a minimized region previously identified by two independent groups as sufficient to impart iron-responsive degradation at a level similar to that of the full-length TfR1 mRNA sequence (see, e.g., mutation pTR-90 in Müllner and Kühn 1988 and construct TRS1 in Casey et al. 1989). Although the proposed miR-7-5p binding site within the TfR1 3′-UTR falls within this essential region, the earlier studies are also difficult to reconcile with miR-7-5p having a direct role in TfR1 mRNA degradation (Müllner and Kühn 1988; Casey et al. 1989; Rupani and Connell 2016). In particular, the mRNA features within the minimized region that are essential for degradation include three non-IRE stem–loops that have no overlap with the proposed microRNA binding sites (loops I, III, and V in Fig. 1A). Because these three hairpin loops can cumulatively account for most of the iron-responsive degradation of the TfR1 mRNA (Rupani and Connell 2016), it is also unlikely that the proposed microRNA binding sites could have a significant redundant activity.
FIGURE 1.
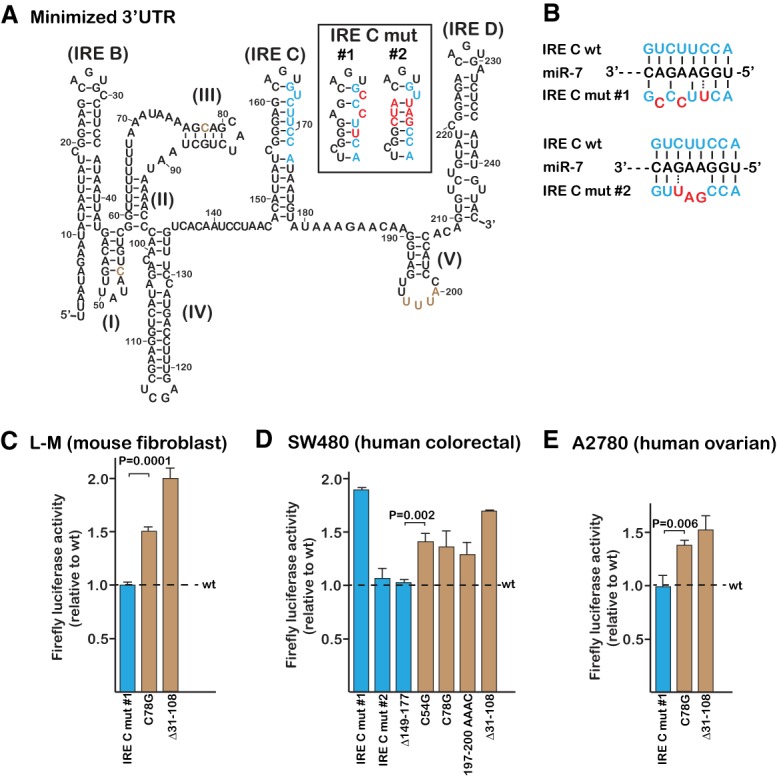
The proposed miR-7-5p binding site on the TfR1 mRNA is not functional. (A) Secondary structure of the minimized TfR1 mRNA previously determined to be required for efficient iron-responsiveness (Casey et al. 1989). Brown, sites of point mutations to the three 5 bp stem–loops (I, III, and V) that were previously identified as essential for iron-responsive degradation in mouse L-M cells (Rupani and Connell 2016); blue, the proposed miR-7-5p binding site. The RNA structure and labeling are based on earlier established convention (Horowitz and Harford 1992; Schlegl et al. 1997). Inset shows the predicted secondary structure of IRE C resulting from the mutations (red) used to disrupt the proposed miR-7-5p binding site; IRE C mut #1 is the Miyazawa et al. mutation. (B) Predicted base-pairing between the miR-7-5p seed sequence and the proposed TfR1 mRNA binding site should be disrupted by both IRE C mut #1 and #2. (C) Mutation of the proposed miR-7-5p binding site (blue) has no significant impact on mRNA stabilization when assayed in mouse L-M cells. Representative earlier mutations that do not overlap with the proposed binding site but impede TfR1 mRNA degradation were assayed in parallel (brown). (D) Although IRE C mut #1 appears to stabilize the reporter RNA in SW480 cells, neither IRE C mut #2 nor the complete deletion of the binding site (Δ149–177) has a significant effect. (E) IRE C mut #1 has no effect when assayed within A2780 cells. All error bars represent ±SEM of three biological replicates.
To explore the apparent discrepancies, we describe here the effect of mutations to the proposed miR-7-5p and miR-141-3p binding sites using an assay that was earlier developed to detect changes to TfR1 mRNA stability. If the proposed role for the microRNAs is correct, mutation of the postulated binding sites should result in increased stabilization of the TfR1 mRNA under iron-replete conditions when IRP protection is minimal. However, mutations to the proposed binding sites that should have inhibited miR-7-5p and miR-141-3p interactions had no significant impact in all cell lines tested. Furthermore, overexpression of a miR-7-5p mimic had no significant impact on the endogenous TfR1 mRNA or on a luciferase-TfR1 reporter even though the abundance of a previously defined target of miR-7-5p was attenuated. As a result, it is unlikely that the microRNAs could be directly mediating iron-responsive TfR1 mRNA degradation as proposed.
RESULTS
Mutation of the proposed miR-7-5p binding site has no impact on TfR1 mRNA stability within a mouse fibroblast cell line
The proposed miR-7-5p binding site within the TfR1 3′-UTR was tested within an assay that had been developed and validated for the identification of sequence elements impacting iron-responsive stability of the TfR1 mRNA (Rupani and Connell 2016). The 3′ TfR1 mRNA sequence that was earlier demonstrated to be sufficient and essential for efficient iron-responsive degradation was cloned behind a firefly luciferase reporter (Fig. 1A), which results in the luciferase activity becoming iron-responsive. Cells expressing the reporter are treated with iron-replete media to minimize potential complications from IRP binding and protection. The assay is a good proxy for monitoring mRNA levels, as mutations to the TfR1 sequence that increase mRNA abundance also result in a corresponding increased luciferase activity.
The mutation to the proposed miR-7-5p binding site that was used to support a functional role was initially retested in the luciferase assay (IRE C mut #1 in Fig. 1A,B; Miyazawa et al. 2018). If the proposed miR-7-5p binding site was functional, the mutation should have increased the mRNA abundance relative to the parental sequence, resulting in increased firefly luciferase activity. However, the mutation had no significant impact when assayed within L-M cells, a mouse fibroblast-like cell line (compare IRE C mut #1 with wt in Fig. 1C). This cell line was chosen because it had been exploited for much of the pioneering work on the identification of the TfR1 gene and the subsequent characterization of the iron-responsive mRNA instability (Kühn et al. 1984; Casey et al. 1988).
The lack of an effect from IRE C mut #1 is in contrast to several previously described mutations that significantly impact iron-responsive stability but are not part of the proposed miR-7-5p binding site (Müllner and Kühn 1988; Casey et al. 1989; Rupani and Connell 2016). Two representatives from these earlier mutations were reassayed in parallel with the miR-7-5p binding site mutation in Figure 1C. A single nucleotide change disrupting one of the non-IRE loops (loop III in Fig. 1A) increased the luciferase signal by ∼50% relative to the parental sequence (contrast C78G with wt in Fig. 1C). This is consistent with an earlier compensatory mutagenesis study that identified loops I, III, and V as being critical for iron-responsive degradation (Rupani and Connell 2016). A deletion that encompasses both loops I and III but is outside of the proposed miR-7-5p binding site was earlier shown to prevent iron-responsive TfR1 changes at both the protein and mRNA levels (see mutation pTR-82 in Müllner and Kühn 1988 and Δ31–108 in Rupani and Connell 2016). This deletion also increased luciferase activity when reassayed here (contrast Δ31–108 with wt in Fig. 1C). As a result, the proposed miR-7-5p binding site is neither consistent with the lack of an effect from mutations that should disrupt the miR-7-5p interaction nor with the effect of mutations that are well outside of the proposed miR-7-5p binding site.
The mRNA requirements for iron-responsive degradation are similar in mouse and human cell lines
It is expected that the major mechanism for iron-responsive TfR1 mRNA degradation should be similar among vertebrates because the minimized region that is essential and sufficient for degradation is highly conserved (Koeller et al. 1989). However, the possibility of redundant activities that could be of greater importance in some species and/or cell lineages cannot be excluded. To initially test this possibility, the assay of the miR-7-5p binding site mutation was repeated using SW480 cells, a cell line that was derived from a human colon adenocarcinoma and which was used for the Miyazawa et al. study. The assay in the SW480 cells is sensitive to mutations within the three non-IRE stem–loops (loops I, III, and V) as well as the Δ31–108 mutation (Fig. 1D), which is consistent with what was demonstrated earlier for the mouse L-M cells (Rupani and Connell 2016). These results suggest that the major mechanism mediating TfR1 mRNA stability is similar in the two species and cell lineages. Despite this underlying similarity, the mutation that was utilized by Miyazawa et al. to support a role for miR-7-5p (IRE C mut #1), has different effects in the two cell lines. Whereas the mutation has no significant impact in the mouse fibroblasts, it does appear to stabilize the TfR1 reporter mRNA within the SW480 cell line (contrast IRE C mut #1 to wt in Fig. 1C,D).
Additional mutations to the proposed miR-7-5p binding site were evaluated to assess the mechanism through which the Miyazawa et al. mutation was impacting TfR1 mRNA stability within the SW480 cells. A second mutation was made in a manner that should disrupt pairing with the microRNA seed sequence (Grimson et al. 2007; Kim et al. 2017), but it was designed to also minimize disruption of the IRE C structure (IRE C mut #2 in Fig. 1A). This was accomplished by making compensatory mutations to the opposite side of the helix to preserve the potential for base-pairing throughout the upper stem of the IRE, unlike with IRE C mut #1 (contrast mut #1 and #2 within inset of Fig. 1A). This second mutation to the proposed miR-7-5p binding site did not significantly differ from the parental sequence in the luciferase assay (compare IRE C mut #2 with wt in Fig. 1D). Moreover, deletion of the entire proposed miR-7-5p binding site also did not have a significant impact on the assay (compare Δ149–177 with wt in Fig. 1D), which argues strongly against the miR-7-5p binding site having a significant role in SW480 cells. As a result, it is likely that IRE C mut #1 is impacting the luciferase assay in the SW480 cells through a mechanism that is unrelated to the proposed miR-7-5p interaction.
To assess whether the different effect of IRE C mut #1 in the human SW480 and mouse L-M cells is a result of the species difference, the assay was repeated in the A2780 cell line, which is derived from a human ovarian carcinoma. As with the mouse L-M cells, IRE C mut #1 did not significantly impact the assay in the A2780 cells (Fig. 1E), indicating that the different behavior of the IRE C mut #1 in the SW480 cells is not an inherent characteristic of the species. In contrast, the assay in the A2780 cells is sensitive to mutation of the non-IRE stem–loops (Fig. 1E), which is consistent with these loops being part of a highly conserved regulatory mechanism.
Neither the proposed miR-7-5p nor miR-141-3p binding sites are functional within the context of an extended 3′-UTR
The proposed miR-141-3p binding site on IRE E is not within the minimized 3′-UTR region that two independent studies had earlier determined to be sufficient to impart iron-responsive degradation at a level similar to that of the full-length TfR1 mRNA sequence (Müllner and Kühn 1988; Casey et al. 1989). To investigate this discrepancy, a larger ∼700 nt sequence from the TfR1 3′-UTR which contains all five IREs was cloned behind the luciferase reporter. Repeating the assay with the extended 3′-UTR also provided the opportunity to test whether the proposed miR-7-5p binding site behaves differently in the context of the larger sequence.
Mutations to both proposed microRNA binding sites within the context of the extended 3′-UTR were designed to prevent interaction of the microRNAs in a manner that did not destabilize the hairpin loops of IRE C and E or the overall structure. This was accomplished both through the appropriate compensatory changes to maintain the potential for base-pairing and also through the substitution of the CAGUGU IRE loop sequence with a UAAC tetraloop (IRE C mut #3 and IRE E mut #1 within insets to Fig. 2A). In other systems, the UAAC tetraloop provides additional stability with minimal interference with tertiary structure (Zhao et al. 2012). The mutation of the IRE C loop has also previously been shown to increase degradation of the TfR1 mRNA (Casey et al. 1989; Rupani and Connell 2016), possibly through the disruption of residual IRP interactions that are maintained even during iron-replete conditions. In spite of the miR-7-5p binding site being disrupted by the mutation (IRE C mut #3 in Fig. 2B), the RNA appears less stable within both the L-M and SW480 cell lines (contrast IRE C mut #3 with wt in Fig. 2C,D). The proposed miR-141-3p binding site is also not consistent with being functional as the substitution mutation at the site did not have a significant impact on the reporter in both the L-M and SW480 cells (IRE E mut #1 in Fig. 2C,D). The complete deletion of the site likewise failed to have an impact when assayed within the SW480 cells (ΔIRE E in Fig. 2D). In contrast, the disruption of non-IRE loop III in the extended context still resulted in increased luciferase activity in both tested cell lines (C78G in Fig. 2C,D), consistent with degradation of the mRNA being impeded.
FIGURE 2.

The proposed miR-7-5p and miR-141-3p binding sites are not functional within the context of an extended 3′-UTR from TfR1. (A) Approximately 700 nt of the TfR1 3′-UTR, containing all five IREs (nt 3445–4135 in NM_003234.3), was cloned behind a firefly luciferase reporter. Insets indicate the predicted secondary structures of IRE C and IRE E resulting from the mutations (red) used to disrupt the proposed miR-7-5p (blue) and miR-141-3p (green) binding sites. (B) The interactions of miR-7-5p and miR-141-3p with their proposed binding sites should be disrupted by the indicated mutations. (C) The expression of the reporter mRNA within L-M cells was not increased by disruption of either proposed microRNA binding site. A point change within stem–loop III is shown for comparison (brown); this mutation is within the context of the extended 3′-UTR but numbering corresponds to C78G in Figure 1A. (D) Mutation of the proposed microRNA binding sites failed to increase the luciferase within the SW480 cells. The ΔIRE E deletion (nt 4017–4045 in NM_003234.3), which includes the entire proposed microRNA binding site, also had no impact. Luciferase was measured after 14 h treatment with FAC. All error bars represent ±SEM of three biological replicates.
Evaluation of the proposed miR-7-5p binding site within an optimized substrate
The proposed miR-7-5p binding site was further evaluated in the context of a minimized TfR1 3′-UTR reporter that had been previously optimized for instability (Rupani and Connell 2016). IRP binding to the optimized reporter is inhibited through the deletion of the 5′ C within each of the IRE loops (Fig. 3A). This has the advantage of unambiguously eliminating potential complications resulting from differences in iron-responsive protection. Even though the larger dynamic range of the optimized reporter increased the assay sensitivity in the SW480 cells, there was no significant effect of the IRE C mut #3 mutation within the optimized reporter (compare IRE C mut #3 with optimized in Fig. 3B). In contrast, point mutations within each of the three non-IRE hairpin loops increased luciferase activity to the level of the Δ31–108 mutation (compare mut loops I, III, V with Δ31–108 in Fig. 3B). A similar result was reported earlier when the loop mutations were assayed within the mouse L-M cells (Rupani and Connell 2016). The result suggests that most, if not all, of the iron-responsiveness can be accounted for by the three non-IRE loops because the Δ31–108 mutation was earlier shown to prevent iron-responsive instability (see mutation pTR-82 in Müllner and Kühn 1988 and Δ31–108 in Rupani and Connell 2016). It was not possible to reevaluate the proposed miR-141-3p site in this context because it is not included within the optimal sequence that was earlier determined to be necessary and sufficient for efficient iron-responsiveness degradation (Müllner and Kühn 1988; Casey et al. 1989).
FIGURE 3.

The proposed miR-7-5p binding site has no significant effect in the context of the TfR1 3′-UTR that had been optimized for instability. (A) Deletion of the 5′ C within each IRE hairpin loop (green) eliminates potential complications arising from IRP interactions and variations in iron-responsive protection. Brown, sites of point mutations within the three non-IRE hairpin loops that were earlier demonstrated to impede degradation. (B) IRE C mut #3 (Fig. 2A) in the context of the optimized 3′-UTR (opt) has no significant impact on the assay (ns, P > 0.1). Combined mutations (C54G, 81–82 GU and 197–200 AAAC) to the three non-IRE hairpin loops (I, III, and V) negate iron-responsiveness in SW480 cells to the same extent as the Δ31–108 mutation, which was earlier demonstrated to prevent iron-responsive degradation in the mouse L-M cells mRNA (see mutation pTR-82 in Mullner and Kuhn 1988 and Δ31–108 in Rupani and Connell 2016). Luciferase was measured after 14 h treatment with FAC. All error bars represent ±SEM of three biological replicates.
Overexpression of a miR-7-5p mimic does not impact the endogenous TfR1 mRNA levels or a luciferase reporter
A miR-7-5p mimic was used to further assess the proposed role for the microRNA in the regulation of endogenous TfR1 mRNA stability. The miR-7-5p mimic was adjusted to a concentration at which there was specific attenuation of the EGFR mRNA (Fig. 4A), which is a previously identified miR-7-5p target in SW480 cells (Suto et al. 2015). While the relative abundance of the EGFR mRNA was decreased approximately twofold, the miR-7-5p treatment did not affect the abundance of the endogenous TfR1 mRNA (Fig. 4B), which is inconsistent with the proposed binding site on the TfR1 mRNA being critical for degradation. Overexpression of the miR-7-5p mimic should likewise have decreased expression of the luciferase-TfR1 reporter, but this was also not observed (Fig. 4C).
FIGURE 4.

Overexpression of a miR-7-5p mimic does not decrease the abundance of either the endogenous TfR1 mRNA or a luciferase-TfR1 reporter. (A) Overexpression of a miR-7-5p mimic (gray) attenuates the EGFR mRNA, which is a previously identified miR-7-5p target in SW480 cells. A negative control mimic (yellow) with no known microRNA function is also indicated. Cells were harvested after 5 h treatment with 65 µg/mL FAC, similar to the Miyazawa et al. study. The fold change in EGFR mRNA abundance after treatment with the mimic was calculated relative to the nontransfected (vehicle only) control using an HPRT amplicon as the reference (Table 1). (B) The endogenous TfR1 mRNA is not significantly impacted by the same miR-7-5p treatment that attenuates the EGFR mRNA (ns, P = 0.7) (C) Overexpression of the miR-7-5p mimic failed to decrease the activity of the luciferase-TfR1 reporter, as would have been expected if the proposed site were functional. The luciferase reporter with the extended TfR1 3′-UTR (Fig. 2A) was used for the assay. Mutation of the proposed miR-7-5p site (IRE C mut #3) has no impact on the assay, regardless of whether the miR-7-5p mimic is present. Luciferase was measured after 5 h treatment with 65 µg/mL FAC. The activity of the miR-7-5p and negative control mimics is relative to the corresponding minus mimic (vehicle only) transfection. All error bars represent ±SEM of three biological replicates.
DISCUSSION
The proposed miR-7-5p and miR-141-3p binding sites overlap with IRP-binding sites on the TfR1 mRNA (Koeller et al. 1989; Connell et al. 2018), which suggested a potential mechanism for the iron-responsive regulation of TfR1 mRNA stability if the sites were functional (Miyazawa et al. 2018). MicroRNA mimic and antagomir overexpression strategies that are often exploited to test potential binding sites are highly vulnerable to indirect effects and to the formation of low-affinity interactions that become favored at high concentrations (for review, see Elton and Yalowich 2015). A widely used strategy to address these limitations is to assess the impact of the target's 3′-UTR when cloned behind a reporter. We demonstrated here that disruption of both the proposed miR-7-5p and miR-141-3p sites with several different mutations failed to have a significant impact on the reporter's expression in all cell lines tested (Figs. 1–3). This makes it unlikely that the proposed microRNA binding sites could be functional. The proposed miR-141-3p site is also not included within the minimal TfR1 3′-UTR sequence previously determined to be necessary for efficient iron-responsive degradation (Müllner and Kühn 1988; Casey et al. 1989). While the proposed miR-7-5p site is within the minimized region, the site is also not consistent with the earlier mutagenesis nor with the failure of the miR-7-5p mimic to impact either the endogenous TfR1 mRNA or a reporter (Fig. 4).
Although miR-7-5p does not appear to be directly involved with the iron-responsive TfR1 mRNA degradation, the mutation to the proposed binding site that initially supported a possible role in SW480 cells is interesting (Miyazawa et al. 2018). IRE C mut #1 has no significant impact when assayed in the mouse L-M and human A2780 cells (Fig. 1C,E), indicating that the effect observed in SW480 cells is not an inherent characteristic of the mutation or the species. The mutation, unlike the other substitutions, does not preserve the potential for base-pairing within the upper stem of the IRE (contrast IRE C mut #1 with IRE C mut #2 and IRE C mut #3 within insets to Figs. 1A, 2A). This is relevant because the apparent complexity of the TfR1 mRNA structure required for iron-responsive degradation increases the likelihood that mutations designed to test potential microRNA binding sites could inadvertently impede degradation by favoring alternative conformations. Interference with correct folding would also be consistent with the iron-responsiveness of combined mutations to both proposed microRNA binding sites having only a small apparent difference from the singly mutated sites used in the Miyazawa et al. study (compare mutC and mutE with mut C/E in Fig. 3C of Miyazawa et al. 2018). This result from the Miyazawa et al. study is otherwise difficult to explain as the effects from the proposed microRNA interactions would have been expected to be additive. It is unclear why the SW480 cells would be more sensitive to misfolding than the other two tested cell lines. Whatever the cause, the effect of IRE C mut #1 is not directly related to miR-7-5p as three other mutations that should have disrupted its proposed interaction, including the complete deletion of the site, failed to have an impact in the SW480 cells (IRE C mut #2, IRE C mut #3 and Δ149–177 in Figs. 1–3).
Three non-IRE hairpin loops were previously identified within the 3′-UTR of the TfR1 mRNA that are essential for iron-responsive degradation in all cell lines tested and are also highly conserved among vertebrates (Rupani and Connell 2016). Their importance is supported by the earlier mutagenesis studies (Müllner and Kühn 1988; Casey et al. 1989), making it likely that one or more of the hairpins function as the major recognition element for the endonuclease activity. Stem–loops I and III fit the general consensus sequence for constitutive decay elements (CDEs) which are found within several inflammatory mRNAs (for review, see Kim and Maquat 2019). The canonical CDE consists of a stem capped with a YRY trinucleotide loop (Leppek et al. 2013; Mino et al. 2015; Braun et al. 2018; Wilamowski et al. 2018), and it is recognized by several proteins that include Regnase/MCPIP-1 (Zc3h12a), Roquin-1 (Zc3h1) and its close paralog Roquin-2 (Zc3h2). Although the U-rich sequence of stem–loop V does not fit the consensus for the canonical CDE, it has similarities to noncanonical stem–loops that interact with Roquin and destabilize mRNA (Murakawa et al. 2015; Janowski et al. 2016; Braun et al. 2018; Rehage et al. 2018).
Regnase and Roquin function through distinct mechanisms even though there is some overlap in the mRNA sequence elements that are recognized (Mino et al. 2015). Regnase is an endonuclease that degrades CDE-containing mRNAs that are translationally active, and it has recently been proposed to recognize stem–loop III and degrade the TfR1 mRNA during iron-repletion (Yoshinaga et al. 2017). There are also several closely related endonucleases within the same family as Regnase (Zc3h12) that potentially could interact with the TfR1 mRNA hairpin loops. In contrast to Regnase, Roquin has no intrinsic endonuclease activity but interacts with nontranslated mRNAs and facilitates recruitment of deadenylation and decapping complexes within processing-body/stress granules (Glasmacher et al. 2010; Leppek et al. 2013). Although Roquin has been implicated in the regulation of TfR1 mRNA stability (Braun et al. 2018), its role and relative importance to iron-responsive TfR1 mRNA degradation has not yet been defined.
MATERIALS AND METHODS
Luciferase assay
The TfR1 reporters and mutations were synthesized at GenScript and cloned into the pMIRGLO luciferase vector (Promega), as previously described (Rupani and Connell 2016). The mouse L-M cell line (ATCC, CCL-1.3) was grown in DMEM, the SW480 colon adenocarcinoma cell line (ATCC, CCL-228) in L-15 and the human A2780 ovarian carcinoma cell line (provided by S. Ramakrishnan, University of Minnesota) in RPMI. All media were supplemented with 10% fetal bovine serum (Life Technologies, 16000-044). L-M cells were plated at 2000 per well, SW480 cells at 10,000 per well and the A2780 cells at 5000 per well within 96 well plates. Cells were transfected 20 h later with 30 ng of plasmid using 0.3 µL Lipofectamine 2000 (Life Technologies) in a 75 µL volume. Fresh media containing ferric ammonium citrate (FAC) was added after 24 h and the plates incubated a further 17 h; the L-M cells were treated with 100 µg/mL FAC, the SW480 cells with 65 µg/mL FAC, and the A2780 cells with 45 µg/mL FAC. Variation in transfection efficiency was controlled for in the assay through normalization to a Renilla luciferase, encoded on the same plasmid as the firefly luciferase reporter. Dual-Glo (Promega) reagents were used to assay luciferase activity and measurements were made with a luminometer (Tecan). Each TfR1 construct was assayed with a minimum of three independent sets of transfections, and each set was assayed in at least triplicate.
Effect of microRNA mimics on endogenous mRNA
SW480 cells were plated within 3.5 cm dishes at a density of 150,000 cells per plate. The cells were transfected 24 h later with either the hsa-miR-7-5p Mirvana microRNA mimic (5′-UGGAAGACUAGUGAUUUUGUUGUU-3′, Life Technologies MC11755) or Mirvana microRNA mimic negative control #1 (Life Technologies, 4464058) using 2 µL of Lipofectamine RNAiMAX (Life Technologies) in a final volume of 1.2 mL. The mimic and negative control mimic were at a final concentration of 30 nM. Nontransfected cultures contained the Lipofectamine with no RNA but were otherwise treated identically. The media was changed after 20 h and the cells incubated for 24 h prior to adding fresh media containing 65 µg/mL FAC and incubating for a further 5 h, similar to the Miyazawa et al. treatment (Fig. 2 of Miyazawa et al. 2018). Cells were then briefly rinsed with cold PBS and total RNA was prepared after directly scraping the cells into 1 mL TRIzol reagent (Chomczynski and Sacchi 2006).
The RT-qPCR was performed as previously described (Rupani and Connell 2016). The ΔΔCq method was used to quantify changes in the EGFR and TfR1 mRNAs in the mimic-treated cells relative to the nontransfected (vehicle only) control. HPRT mRNA was used as the reference mRNA, which was chosen because it is the same mRNA used as a reference in the Miyazawa et al. study (Miyazawa et al. 2018). Primers were chosen so that the amplicons would be of similar size (Table 1), using either Primer Express software (Life Technologies) or NCBI Primer BLAST. The amplification efficiencies were all measured at >95%. Minus reverse transcriptase controls consistently indicated <1% genomic DNA contamination. All values were calculated from three independent sets of biological replicates, which were each assayed in triplicate.
TABLE 1.
Amplicons used for qPCR

Effect of microRNA mimics on the luciferase reporter
SW480 cells were plated at a density of 10,000 cells per well within a 96 well plate and were transfected 24 h later. Transfections were done with 0.13 µL RNAiMAX in a 75 µL volume using the mimic or control at a final concentration of 30 nM. This is the same concentration of the miR-7-5p mimic that effectively inhibited EGFR, a previously identified miR-7-5p target (Fig. 4A). Nontransfected wells contained the RNAiMAX with no RNA and were otherwise treated identically. Media was changed 24 h later and cells were transfected with 30 ng of plasmid using 0.3 µL Lipofectamine 2000 (Life Technologies) in a 75 µL volume. After 14 h incubation, the cells were treated with 65 µg/mL FAC for 5 h prior to the quantification of the luciferase activity.
Statistical analysis
Statistical significance was analyzed by a two-tailed Student's t-test.
ACKNOWLEDGMENTS
This work was supported in part by a University of Minnesota Multicultural Summer Research Opportunity Program award to V.M.C., a University of Minnesota Undergraduate Research Opportunities Program award to E.R.S., and a Grant-in-Aid of Research from the Office of the Vice President for Research (GIA 211645; University of Minnesota) to G.J.C. We also thank Rick Eisenstein (University of Wisconsin—Madison) for helpful discussions.
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.072371.119.
REFERENCES
- Braun J, Fischer S, Xu ZZ, Sun H, Ghoneim DH, Gimbel AT, Plessmann U, Urlaub H, Mathews DH, Weigand JE. 2018. Identification of new high affinity targets for Roquin based on structural conservation. Nucleic Acids Res 46: 12109–12125. 10.1093/nar/gky908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casey JL, Hentze MW, Koeller DM, Caughman SW, Rouault TA, Klausner RD, Harford JB. 1988. Iron-responsive elements: regulatory RNA sequences that control mRNA levels and translation. Science 240: 924–928. 10.1126/science.2452485 [DOI] [PubMed] [Google Scholar]
- Casey JL, Koeller DM, Ramin VC, Klausner RD, Harford JB. 1989. Iron regulation of transferrin receptor mRNA levels requires iron-responsive elements and a rapid turnover determinant in the 3′ untranslated region of the mRNA. EMBO J 8: 3693–3699. 10.1002/j.1460-2075.1989.tb08544.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chomczynski P, Sacchi N. 2006. The single-step method of RNA isolation by acid guanidinium thiocyanate–phenol–chloroform extraction: twenty-something years on. Nat Protoc 1: 581–585. 10.1038/nprot.2006.83 [DOI] [PubMed] [Google Scholar]
- Connell GJ, Danial JS, Haastruthers CX. 2018. Evaluation of the iron regulatory protein-1 interactome. Biometals 31: 139–146. 10.1007/s10534-018-0076-8 [DOI] [PubMed] [Google Scholar]
- Elton TS, Yalowich JC. 2015. Experimental procedures to identify and validate specific mRNA targets of miRNAs. EXCLI J 14: 758–790. 10.17179/excli2015-319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glasmacher E, Hoefig KP, Vogel KU, Rath N, Du L, Wolf C, Kremmer E, Wang X, Heissmeyer V. 2010. Roquin binds inducible costimulator mRNA and effectors of mRNA decay to induce microRNA-independent post-transcriptional repression. Nat Immunol 11: 725–733. 10.1038/ni.1902 [DOI] [PubMed] [Google Scholar]
- Grimson A, Farh KK-H, Johnston WK, Garrett-Engele P, Lim LP, Bartel DP. 2007. MicroRNA targeting specificity in mammals: determinants beyond seed pairing. Mol Cell 27: 91–105. 10.1016/j.molcel.2007.06.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horowitz JA, Harford JB. 1992. The secondary structure of the regulatory region of the transferrin receptor mRNA deduced by enzymatic cleavage. New Biol 4: 330–338. [PubMed] [Google Scholar]
- Janowski R, Heinz GA, Schlundt A, Wommelsdorf N, Brenner S, Gruber AR, Blank M, Buch T, Buhmann R, Zavolan M, et al. 2016. Roquin recognizes a non-canonical hexaloop structure in the 3′-UTR of Ox40. Nat Commun 7: 11032 10.1038/ncomms11032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsarou M-S, Papasavva M, Latsi R, Drakoulis N. 2019. Hemochromatosis: hereditary hemochromatosis and HFE gene. Vitam Horm 110: 201–222. 10.1016/bs.vh.2019.01.010 [DOI] [PubMed] [Google Scholar]
- Kim YK, Maquat LE. 2019. UPFront and center in RNA decay: UPF1 in nonsense-mediated mRNA decay and beyond. RNA 25: 407–422. 10.1261/rna.070136.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Chang HR, Baek D. 2017. Rules for functional microRNA targeting. BMB Rep 50: 554–559. 10.5483/BMBRep.2017.50.11.179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koeller DM, Casey JL, Hentze MW, Gerhardt EM, Chan LN, Klausner RD, Harford JB. 1989. A cytosolic protein binds to structural elements within the iron regulatory region of the transferrin receptor mRNA. Proc Natl Acad Sci 86: 3574–3578. 10.1073/pnas.86.10.3574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kühn LC, McClelland A, Ruddle FH. 1984. Gene transfer, expression, and molecular cloning of the human transferrin receptor gene. Cell 37: 95–103. 10.1016/0092-8674(84)90304-0 [DOI] [PubMed] [Google Scholar]
- Leppek K, Schott J, Reitter S, Poetz F, Hammond MC, Stoecklin G. 2013. Roquin promotes constitutive mRNA decay via a conserved class of stem–loop recognition motifs. Cell 153: 869–881. 10.1016/j.cell.2013.04.016 [DOI] [PubMed] [Google Scholar]
- Mino T, Murakawa Y, Fukao A, Vandenbon A, Wessels H-H, Ori D, Uehata T, Tartey S, Akira S, Suzuki Y, et al. 2015. Regnase-1 and Roquin regulate a common element in inflammatory mRNAs by spatiotemporally distinct mechanisms. Cell 161: 1058–1073. 10.1016/j.cell.2015.04.029 [DOI] [PubMed] [Google Scholar]
- Miyazawa M, Bogdan AR, Hashimoto K, Tsuji Y. 2018. Regulation of transferrin receptor-1 mRNA by the interplay between IRE-binding proteins and miR-7/miR-141 in the 3′-IRE stem–loops. RNA 24: 468–479. 10.1261/rna.063941.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müllner EW, Kühn LC. 1988. A stem–loop in the 3′ untranslated region mediates iron-dependent regulation of transferrin receptor mRNA stability in the cytoplasm. Cell 53: 815–825. 10.1016/0092-8674(88)90098-0 [DOI] [PubMed] [Google Scholar]
- Murakawa Y, Hinz M, Mothes J, Schuetz A, Uhl M, Wyler E, Yasuda T, Mastrobuoni G, Friedel CC, Dölken L, et al. 2015. RC3H1 post-transcriptionally regulates A20 mRNA and modulates the activity of the IKK/NF-κB pathway. Nat Commun 6: 7367 10.1038/ncomms8367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehage N, Davydova E, Conrad C, Behrens G, Maiser A, Stehklein JE, Brenner S, Klein J, Jeridi A, Hoffmann A, et al. 2018. Binding of NUFIP2 to Roquin promotes recognition and regulation of ICOS mRNA. Nat Commun 9: 299 10.1038/s41467-017-02582-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rupani DN, Connell GJ. 2016. Transferrin receptor mRNA interactions contributing to iron homeostasis. RNA 22: 1271–1282. 10.1261/rna.056184.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlegl J, Gegout V, Schlager B, Hentze MW, Westhof E, Ehresmann C, Ehresmann B, Romby P. 1997. Probing the structure of the regulatory region of human transferrin receptor messenger RNA and its interaction with iron regulatory protein-1. RNA 3: 1159–1172. [PMC free article] [PubMed] [Google Scholar]
- Suto T, Yokobori T, Yajima R, Morita H, Fujii T, Yamaguchi S, Altan B, Tsutsumi S, Asao T, Kuwano H. 2015. MicroRNA-7 expression in colorectal cancer is associated with poor prognosis and regulates cetuximab sensitivity via EGFR regulation. Carcinogenesis 36: 338–345. 10.1093/carcin/bgu242 [DOI] [PubMed] [Google Scholar]
- Wang C-Y, Babitt JL. 2019. Liver iron sensing and body iron homeostasis. Blood 133: 18–29. 10.1182/blood-2018-06-815894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss G, Ganz T, Goodnough LT. 2019. Anemia of inflammation. Blood 133: 40–50. 10.1182/blood-2018-06-856500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilamowski M, Gorecki A, Dziedzicka-Wasylewska M, Jura J. 2018. Substrate specificity of human MCPIP1 endoribonuclease. Sci Rep 8: 7381 10.1038/s41598-018-25765-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshinaga M, Nakatsuka Y, Vandenbon A, Ori D, Uehata T, Tsujimura T, Suzuki Y, Mino T, Takeuchi O. 2017. Regnase-1 maintains iron homeostasis via the degradation of transferrin receptor 1 and prolyl-hydroxylase-domain-containing protein 3 mRNAs. Cell Rep 19: 1614–1630. 10.1016/j.celrep.2017.05.009 [DOI] [PubMed] [Google Scholar]
- Zhao Q, Huang HC, Nagaswamy U, Xia Y, Gao X, Fox GE. 2012. UNAC tetraloops: to what extent do they mimic GNRA tetraloops? Biopolymers 97: 617–628. 10.1002/bip.22049 [DOI] [PubMed] [Google Scholar]