

Abstract
The ability to control transcription in a time-dependent manner in vitro promises numerous applications in molecular biology and nanotechnology. Here we demonstrate an approach that enables precise, independent control over the production of multiple RNA transcripts in vitro using single guide RNA (sgRNA)-directed transcription blockades by catalytically dead Streptococcus pyogenes CRISPR-Cas9 enzyme (dCas9). We show that when bound to a DNA template, the dCas9:sgRNA complex forms a robust blockade to transcription by RNA polymerases (RNAPs) from bacteriophages SP6, T3, and T7 (>99.5% efficiency), and a partial blockade to transcription by Escherichia coli RNAP (∼70% efficiency). We find that all three bacteriophage RNAPs dissociate from the DNA template upon encountering the dCas9 blockade, while E. coli RNAP stays bound for at least the 90-min duration of our experiments. The blockade maintains >95% efficiency when four mismatches are introduced into the 5′ end of the sgRNA target sequence. Notably, when using such a mismatched blockade, production of specific RNA species can be activated on demand by addition of a double-stranded competitor DNA perfectly matching the sgRNA. This strategy enables the independent production of multiple RNA species in a temporally controlled fashion from the same DNA template, demonstrating a new approach for transcription control.
Keywords: transcription, RNA polymerase, CRISPR, reversibility
INTRODUCTION
Across all organisms, the temporal modulation of transcription is one of the most prevalent mechanisms of gene regulation. In bacteria, certain riboswitches control the expression of downstream genes in real-time by fine-tuning the efficiency of transcription termination in response to fluctuations in metabolite and ion concentrations (Sherwood and Henkin 2016). Similarly, chromatin remodeling enzymes can activate or deactivate transcription by repositioning nucleosomes, and transcription factors can do so by binding to promoters and/or directly interacting with RNA polymerase (RNAP) (Yang and Lewis 2014; Ji and Sharrocks 2015). Furthermore, the benefits of temporal control over transcription are not limited to intracellular processes: Modulation of transcription in vitro has proven to be a valuable tool in the fields of molecular biology and nanotechnology, enabling the study of cotranscriptional folding of RNA (Watters et al. 2016) and the production of molecular motors and RNA nanostructures (Geary et al. 2014; Valero et al. 2018).
Engineered control over transcription in cells has been achieved through a number of different methods, including the use of a catalytically dead mutant of the type II CRISPR enzyme Cas9 (dCas9) from Streptococcus pyogenes (Spy) either as a passive blockade to transcription initiation or elongation (Bikard et al. 2013; Qi et al. 2013) or to position activating or repressing transcription factors near particular genes of interest (Bikard et al. 2013; Gilbert et al. 2013). Both wild-type (WT) Spy Cas9 and dCas9 bind very tightly to double-stranded DNA targets that are complementary to a 20-nt region at the 5′ end of a single guide RNA (sgRNA), utilizing a species-specific 3-nt protospacer adjacent motif (PAM) to select potential binding sites (Fig. 1A; Jinek et al. 2012). These features enable the down-regulation of transcription by physically blocking the progress of RNAP in a site-specific manner; however, rapid reversibility of this effect has not been demonstrated so far. In fact, blockage in vivo of E. coli (Eco) RNAP using dCas9:sgRNA complexes is effective, but—as a result of the extremely slow dissociation rate of the dCas9:sgRNA complex—requires cell division for reversal (Qi et al. 2013; Li et al. 2016). Therefore, a different strategy is needed to confer reversibility on dCas9-mediated transcription control in vitro.
FIGURE 1.

Transcription control in vitro using dCas9 blockades. (A) dCas9 is directed to a target site on a DNA template encoding U5 by an sgRNA harboring a target sequence complementary to that of the DNA, and a PAM sequence in the DNA adjacent to the target site. (B) DNA sequence encoding U5 snRNA, with colored lines indicating the target sites of the sgRNAs used in this study and with the corresponding PAMs highlighted in the same color. The dashed line above the sequence indicates sgRNA4, which binds to the template DNA (tDNA), while solid lines below the sequence indicate sgRNAs that bind to the nontemplate DNA (ntDNA). The two guanosine residues at the 5′ end were inserted in order to facilitate transcription initiation. (C) Denaturing urea-polyacrylamide gel showing that full-length U5 is produced in the absence of sgRNA or dCas9, whereas blockage products of approximately the expected lengths are produced in the presence of each sgRNA:dCas9 blockade. In this and all the following figures, the displayed gel images were subjected to a rolling ball background correction. The vertical line indicates the location where an unnecessary lane was spliced out.
Numerous proteins have been found to act as blockades to transcription elongation by various RNAPs in vitro, including lac repressor (Deuschle et al. 1986), catalytically dead restriction enzymes (Pavco and Steege 1990, 1991), and Eco RNAP initiation complexes (Kassavetis et al. 1978). However, each of these proteins exhibits specificity for a single binding sequence, limiting its versatility. Furthermore, while in some cases the blockade can be removed and transcription resumed (Pavco and Steege 1990), the conditions necessary for removal often interfere with transcription (Pavco and Steege 1991). As a result, there is a need for a transcription control method with high sequence flexibility and rapid reversibility under mild conditions. Such an approach would have numerous applications for studying how cotranscriptional folding is affected by temporary pausing by RNAP, for creating RNA nanostructures with domains that must fold sequentially, and for obtaining fine-tuned temporal control over transcription-based nanodevices.
In this work, we show that Spy dCas9 forms an effective blockade to in vitro transcription by RNAPs from bacteriophages SP6, T3, and T7, and a partial blockade to transcription by Eco RNAP. As a model system, we utilized the U5 small nuclear RNA (snRNA) from S. cerevisiae, which is involved in positioning the two exons for ligation during the second chemical reaction of pre-messenger RNA splicing (Newman 1997; O'Keefe and Newman 1998). We show that robust blockage is maintained even with four mismatches in the sgRNA:DNA hybrid, but that this mismatched complex can be removed on demand by addition of a competitor DNA duplex. After addition of the competitor, transcription of the full-length RNA is recovered within 5 min. We show that multiple blockades can be assembled on a single DNA template and removed independently of one another, allowing relative production of multiple RNA transcripts to be controlled in real-time. Our mechanistic foundation demonstrates a simple, yet versatile paradigm for both down- and up-regulating the relative populations of RNA species in vitro over time, a methodology that we anticipate to open up numerous opportunities for temporal systems control in molecular biology and nanotechnology.
RESULTS
dCas9 complexes binding the nontemplate strand act as efficient transcription blockades
In seeking to develop a modular platform for transcription control in vitro, we prepared sgRNAs with targets at a series of positions along a DNA template encoding U5 snRNA (Fig. 1B). We assembled sgRNA:dCas9 blockades at these positions and found that with each of the sgRNAs tested, the complex blocked transcription by T7 RNAP at the expected location (Fig. 1C). We found that the complex assembled with sgRNA4, which binds to the tDNA, did not block transcription as effectively as complexes assembled with sgRNAs 1, 2, and 3, which bind to the ntDNA (note the band labeled “Full-length U5 snRNA” in Fig. 1C), analogous to in vivo observations of transcription by Eco RNAP (Qi et al. 2013). Clarke et al. (2018) investigated the ability of transcribing T7 RNAP and Pol II to displace an sgRNA:WT Cas9 complex following DNA cleavage by Cas9. Testing a broad panel of sgRNAs in vitro, they found that Cas9 generally gets displaced by RNAP when the sgRNA targets the tDNA and does not get displaced when the sgRNA targets the ntDNA.
We next compared the ability of the dCas9 blockade to halt transcription by bacteriophage SP6, T3, and T7 RNAPs as well as Eco RNAP (Fig. 2). For these experiments, we utilized sgRNA5, which binds to the ntDNA near the promoter-distal end. This had the advantage of making the blockage product longer and therefore more readily detected, and of making the exonuclease digestion product of interest (discussed below) shorter, allowing changes in its length to be more easily resolved. SYBR gold staining was used to visualize the transcription products as well as all other nucleic acid species (Fig. 2A). We found that SP6, T3, and T7 RNAPs were each blocked with >99.5% efficiency by sgRNA5:dCas9 complexes (Fig. 2B,C). Previous studies utilizing other proteins as roadblocks found that bacteriophage RNAPs can be blocked with efficiencies ranging from 0% by nucleosomes (Lorch et al. 1987), to ∼40% by the catalytically dead Gln-111 variant of EcoRI (Pavco and Steege 1991), to ∼90% by the Eco replication protein Tus (Guajardo and Sousa 1999) to ∼100% by promoter-bound Eco RNAP (Kassavetis et al. 1978). It is reasonable to assume that sgRNA:dCas9 complexes may reside at the high end of that range, as they exhibit bound-state lifetimes on DNA of >45 min and a KD of <1 nM (Sternberg et al. 2014), well under the 80 nM DNA concentration used in these experiments. Unexpectedly, we found that Eco RNAP was blocked with only ∼70% efficiency (Fig. 2D,E). This stands in contrast to the nearly 100% blockage of Eco RNAP by dead EcoRI variant Gln-111 (Pavco and Steege 1990); however, less efficient Eco RNAP blockage was observed with lac repressor (Deuschle et al. 1986). Since our reactions were all carried out under identical conditions, it has to be assumed that the initial occupancy of the dCas9 binding site was identical for all four RNAPs. We hypothesize that the propensity of Eco RNAP to stay bound when it encounters dCas9 may give it more opportunities to proceed through the blockade than the bacteriophage RNAPs, which appear to dissociate rapidly (see next section). In addition, recent work has indicated that transcribing Eco RNAP has the ability to displace bound dCas9 blockades (Vigouroux et al. 2018). Since we used nearly equimolar amounts of DNA and dCas9, leaving little free dCas9, if a dCas9 blockade gets removed, it is likely that RNAP will be able to transcribe beyond the binding site before another blockade can bind.
FIGURE 2.

Blockage of various RNAPs by sgRNA5:dCas9 complexes. (A) Cy5-labeled DNA template encoding U5 was transcribed by SP6, T3, T7, or Eco RNAP, and reaction aliquots were removed and digested using Exonuclease III. The tDNA strand was visualized on a denaturing urea-polyacrylamide gel by detecting Cy5 (magenta star), and all nucleic acid species were visualized via staining with SYBR gold (green halos). Each species is given a letter that is used to identify its band. Band A: full-length DNA template; B: full-length U5; C: protected ntDNA fragment; D: blockage product; E: sgRNA5; and F: protected tDNA fragment. (B) Urea-polyacrylamide gels analyzing transcription reactions using SP6, T3, and T7 RNAPs, with Cy5 (magenta) and SYBR gold (green) scans overlaid. Reactions were performed with, from left to right, SP6 RNAP in the absence of a blockade, and SP6, T3, and T7 RNAPs in the presence of a blockade. Nucleic acid bands are labeled as indicated in panel A. The time points represented by the gray triangle are 5, 10, 15, 30, 60, and 90 min, left to right. (C) Quantification of the number of molecules of blockage product (band D) produced per molecule of RNAP for the reactions containing SP6, T7, and T3 RNAPs. (Inset) Quantification of molecules of full-length U5 (band B) produced per molecule of RNAP. (D) Urea-polyacrylamide gel analyzing a transcription reaction using Eco RNAP, with Cy5 (magenta) and SYBR gold (green) scans overlaid. Bands are labeled as in panel B. (Right) Band F displayed at higher contrast, showing new bands (F*) that are dependent on the presence of both Exo III and RNAP. (E) Quantification of the number of molecules of full-length U5 and blockage product produced per molecule of Eco RNAP.
Bacteriophage RNAPs dissociate rapidly upon encountering a dCas9 blockade, while Eco RNAP remains stably template-bound
For a transcription control platform based on blockage of RNAP, the scope of potential applications depends on whether RNAP stays bound upon encountering the blockade. Applications such as the production of RNA nanostructures benefit from multiple turnovers, which require that RNAP dissociates and re-initiates after blockage. Applications mimicking RNAP pausing require RNAP to remain bound and resume transcription at a later time. We therefore investigated whether RNAP stays bound to the DNA template when it encounters the sgRNA:dCas9 blockade. To avoid dissociation resulting from “bumping” of trailing RNAPs into blocked RNAPs (Zhou and Martin 2006), we utilized a low (1:4) RNAP:DNA ratio, which avoids the complication of having multiple RNAP molecules transcribing the same DNA template molecule. To determine whether RNAP stays bound, we analyzed both the relative stoichiometries of RNA products and RNAP and the protection pattern of the DNA template from digestion by Exonuclease III (Exo III). We placed a Cy5 fluorophore label at the 5′ end of the tDNA so that the labeled fragment protected from Exo III would increase in length if RNAP were present (Fig. 2A). If RNAP were to remain stably bound at the blockade, transcription of the blockage product would be limited to a single round, and the accumulation of RNAPs stuck at the blockade would cause the production of the full-length RNA to cease as well. In addition, longer products would be observed upon Exo III digestion, resulting from the protection of the DNA by RNAP as well as by dCas9.
Using the above strategy on the bacteriophage RNAPs, we can set an upper limit of the bound-state lifetime after encountering the sgRNA:dCas9 blockade to <5 min. This was concluded based on the facts that the Exo III digestion pattern was identical in the presence and absence of RNAP (Fig. 2B; for each image, compare band F in the −RNAP +Exo III lane to the transcription time course lanes), and that substantially more than one RNA molecule was produced per RNAP molecule (Fig. 2C). The dCas9:sgRNA blockade itself was resistant to Exo III, with the digestion of the Cy5-labeled tDNA producing multiple bands of lengths within 5–10 nt of each other. In contrast, T7 RNAP elongation complexes are stable for hours when stalled by the absence of NTPs (Gopal et al. 1999) and such complexes are resistant to Exo III digestion (Huang and Sousa 2000). Instead, consistent with our findings, it has previously been shown that when T7 RNAP is stalled by a blockading DNA-binding protein, its bound-state lifetime decreases to ∼9 min (Guajardo and Sousa 1999).
Strikingly, while Eco RNAP was blocked with lower efficiency than the bacteriophage RNAPs (compare Fig. 2E to the inset in Fig. 2C), it produced only ∼1 molecule of blockage product per molecule of RNAP, and the production of RNA ceased early in the time course. When Eco RNAP transcription reactions were digested with Exo III, two new bands appeared, representing DNA species ∼25 nt longer than those observed in the absence of RNAP (Fig. 2D, right-hand side). This is consistent with studies on Eco transcription complexes blocked by inactive restriction enzymes, which showed that 22–28 nt are protected by RNAP (Pavco and Steege 1990), as well as with in vivo studies that found that at dCas9 blockades the nascent RNA remains tightly bound to the transcription elongation complex, allowing it to be recovered via immunoprecipitation of RNAP (Qi et al. 2013). Together, our results suggest that SP6, T3, and T7 RNAPs dissociate when they encounter the dCas9 blockade and consequently undergo multiple turnovers, while Eco RNAP occasionally reads through but otherwise remains stably bound at the blockade.
Blockades formed from mismatched sgRNAs can be removed with matching trap duplexes
For many applications, the extremely tight binding of sgRNA:dCas9 complexes to DNA (Sternberg et al. 2014) is a significant benefit. These include applications in which dCas9 is used to position effector proteins (Gilbert et al. 2013) or to fluorescently label certain positions within the genome (Ma et al. 2016). However, this property can be a drawback for applications requiring temporal control over transcription, and previous work has found that after expression of dCas9 has ceased, transcription in cells is recovered only upon cell division (Qi et al. 2013; Li et al. 2016). Cas9 complexes can be “weakened” by mismatches between the target DNA and the sgRNA, but if these mismatches are located at the PAM-distal end of the target sequence, tight binding affinity and high cleavage activity are maintained (Jinek et al. 2012; Singh et al. 2016). Specifically, up to four consecutive mismatches at the PAM-distal end are tolerated before in vitro cleavage activity of WT Cas9 is diminished (Jinek et al. 2012). To generate reversible transcription blockades, we therefore compared fully matched, wild-type (“WT”) variants of sgRNA1 to variants containing four mismatches (“4MM”). We found that in the absence of active transcription, blockades containing 4MM sgRNA1 could be removed by the addition of a competitor DNA with a target sequence perfectly complementary to that of 4MM sgRNA1, whereas blockades containing WT sgRNA1 could not (Fig. 3A,B). This process may occur via spontaneous dissociation of the 4MM sgRNA1:dCas9 complex followed by rebinding to the trap, or by a strand invasion process akin to toehold displacement (Zhang and Seelig 2011; Li et al. 2018). To differentiate these two possibilities, we added competitor DNA containing the WT target sequence to 4MM sgRNA1:dCas9:DNA complexes. We found that the WT trap, which lacks complementarity to the 4-nt “toehold” presented by 4MM sgRNA1:dCas9 blockades, was as effective at removing the complex as the trap containing the 4MM target sequence (Fig. 3B). This observation supports a mechanism based on spontaneous dissociation followed by rebinding. Interestingly, WT sgRNA:dCas9 and 4MM sgRNA:dCas9 complexes migrate distinctly in nondenaturing gels, both in the presence and absence of DNA (Fig. 3C), suggesting a difference in structure between them.
FIGURE 3.
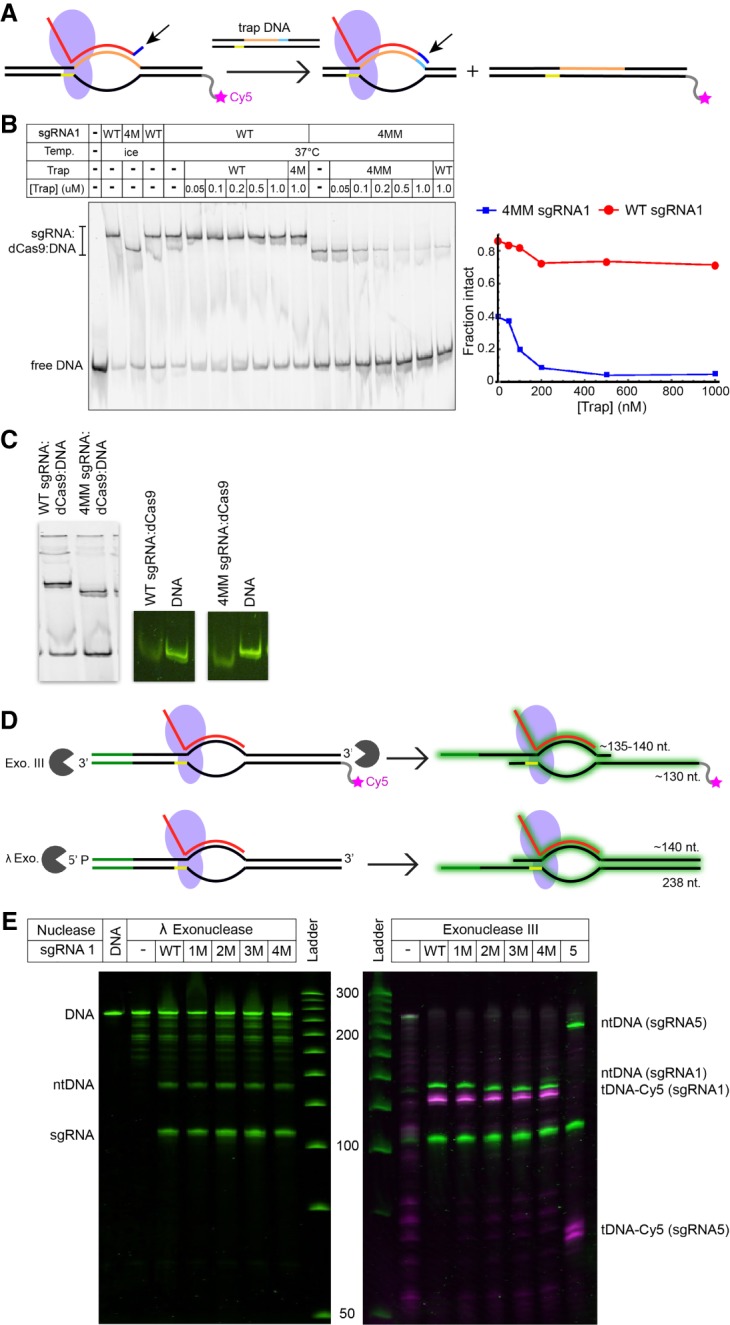
Binding and displacement of blockades containing mismatched sgRNAs. (A) Schematic of a dCas9 blockade with mismatches (blue, indicated by an arrow) between the DNA target sequence and the 5′ end of the sgRNA. A perfectly matched trap DNA is added as a competitor, displacing the blockade from the original target. (B) Cy5 scan of a native polyacrylamide gel showing WT or 4MM sgRNA1:dCas9:DNA complexes after the addition of different concentrations of trap DNA. Traps containing target sequences perfectly matched to the WT and 4MM sgRNAs are compared in their ability to displace blockades. The fraction intact is quantified on the right. (C) Close-up of the gel in B showing the difference in the migration position between WT and 4MM complexes (left). Close-up of the DNA region of an independent native polyacrylamide gel after SYBR gold staining reveals that the difference in migration occurs with the sgRNA:dCas9 complex alone (right). (D) Schematics of digestion of sgRNA:dCas9-bound DNA with Exo III and λ Exo. In Exo III digestions, the tDNA is detected via Cy5 labeling (magenta star), while in both digestions, all nucleic acid species are detected via SYBR gold staining (green halo). (E) Comparison of exonuclease digestions of DNA with WT sgRNA:dCas9 blockades and blockades containing one to four mismatches, with Cy5 (magenta) and SYBR gold (green) scans overlaid. In the Exo III gel, a digestion of DNA with an sgRNA5:dCas9 blockade is shown for comparison.
To investigate how an approaching RNAP might be affected by mismatches in the sgRNA:DNA hybrid, we performed exonuclease digestions to probe the accessibility of the DNA at the upstream and downstream edges of the blockade when different numbers of mismatches were present (Fig. 3D). We utilized λ Exonuclease (λ Exo), which digests 5′ phosphorylated strands within dsDNA, and Exo III. We placed the 5′ phosphate group such that λ Exo probes the promoter-proximal side of the ntDNA, while Exo III probes the promoter-proximal side of the tDNA and the promoter-distal side of the ntDNA. We found that Exo III is able to intrude into the mismatched region of the sgRNA:DNA hybrid when it approaches from the promoter-distal end of the ntDNA. However, no additional intrusion is observed from the promoter-proximal end of the tDNA or ntDNA, consistent with our observation that the length of the transcription blockage product is the same for both the WT and 4MM sgRNA blockades (Fig. 4A,B). This is also consistent with structural studies that found that Cas9 envelops the PAM-proximal end of the sgRNA:DNA hybrid, with the repeat:anti-repeat duplex of the sgRNA stacking on it (Nishimasu et al. 2014).
FIGURE 4.

Displacement of sgRNA1:dCas9 blockades during transcription by T7 RNAP. (A) SYBR gold-stained urea-polyacrylamide gel showing transcription time courses in the presence of 4MM sgRNA:dCas9 blockades. Lanes labeled “KCl” represent aliquots taken after the addition of 10 mM KCl, while lanes labeled “DNA” represent aliquots taken after the addition of 10 mM KCl containing a DNA trap duplex matched to the 4MM sgRNA1 target sequence. The bands that were quantified in panels C and D are labeled. (B) Transcription time courses in the presence of WT sgRNA:dCas9 blockades. Lanes labeled “DNA” represent aliquots taken after the addition of 10 mM KCl containing a DNA trap matched to the WT sgRNA1 target sequence. (C) Quantification of the U5, sgRNA, and blockage product bands in the reactions with 4MM sgRNA1. The intensity of the band of interest is reported relative to the intensity of the DNA band, which is used as a loading control. The time at which the KCl or the DNA trap was added is indicated in each plot. Error bars represent the standard deviation of three independent replicates. (D) Quantification of the U5, sgRNA, and blockage product bands in the reactions with WT sgRNA1.
We utilized these mismatched blockades to implement real-time control of the RNA products produced by T7 RNAP. We assembled WT sgRNA:dCas9 and 4MM sgRNA:dCas9 blockades and tested the ability of the competitor DNA duplexes introduced above to remove them from the DNA template during transcription. When 4MM blockades were present, within 5 min of adding the trap DNA, accumulation of blockage product slowed and production of full-length RNA drastically increased (Fig. 4C). When WT blockades were present, the addition of the trap duplex, even at 25-fold excess over the template, had little effect on the relative production of blockage and full-length products (Fig. 4D). In control reactions where no trap DNA was added, only a slight increase in the production of full-length RNA was observed over time. We note that the speed of unblocking could potentially differ in the context of transcription by the stably stalled Eco RNAP versus the rapidly dissociating bacteriophage RNAPs.
A single blockade can be selectively removed from a multiblockaded template
The RNA-guided sequence programmability of Cas9 should enable the selective removal of a specific dCas9 blockade while others remain bound to the same DNA. This selectivity should be obtainable by incorporating mismatches into only certain blockades and providing trap DNAs specific to those blockades. To test this idea, we investigated a variety of methods for assembling double-blockade complexes using the WT or 4MM variants of sgRNA1 and sgRNA5, the latter of which binds at a nonoverlapping site further downstream. Using native polyacrylamide gel electrophoresis, we assessed the effects of order of addition and of preincubating the sgRNAs with dCas9 before addition to the DNA on the efficiency of double-blockade assembly (Fig. 5). We found that for both WT and 4MM sgRNA1, the most efficient method involved incubating the DNA with a slight excess of dCas9 and a large excess of sgRNA5, then adding to that a preassembled sgRNA1:dCas9 complex (designated Method F in Fig. 5).
FIGURE 5.

Assembly of multiple sgRNA:dCas9 blockades on a single DNA template. (A) Schematic outlining six sequences of mixing and incubation that were tested for their efficiency in yielding double-blockade complexes. One blockade consists of either WT sgRNA1:dCas9 or 4MM sgRNA1:dCas9, and the second blockade consists of sgRNA5:dCas9. In cases where sgRNA and dCas9 are shown on top of one another, these DNA-free complexes were assembled by incubating the indicated sgRNA and dCas9 for 15 min at 37°C before adding them to the remaining components. After each “Add” stage, the mixture was incubated for 15 min at 37°C. Stoichiometries are indicated relative to a starting quantity of 0.7 equivalents of DNA. Methods A and B differ only in the sgRNA stoichiometry and are shown in a single schematic. (B) Nondenaturing polyacrylamide gel imaged via a Cy5 label on the DNA. The WT sgRNA-only, 4MM sgRNA-only, sgRNA 5-only, and double-blockade complexes all exhibit distinct migration rates, allowing the observed bands to be separated into zero-blockade, single-blockade, and double-blockade species. (C) Quantification of the above gel, indicating the fraction of DNA that migrates as part of a zero-, single- or double-blockade complex following each assembly sequence.
Analogous to the experiments shown in Figure 4, we then assembled these complexes and utilized them in transcription reactions with T7 RNAP, testing the ability of trap DNA to activate the production of longer RNA products. We found that when blockades containing 4MM sgRNA1 and sgRNA5 were assembled on the same DNA template, only the 4MM sgRNA1 blockage product was observed in significant quantity before the addition of the trap (Fig. 6). When trap DNA was added, within 5 min production of 4MM blockage product slowed drastically and sgRNA5 blockage product began to rapidly accumulate. In control reactions to which trap DNA was not added, no such activation occurred. These results demonstrate that by selectively weakening a single dCas9 blockade and not others, and by introducing a trap duplex that is specific to the weakened blockade, the production of multiple RNA species that are shorter than the full-length RNA can be controlled in real-time.
FIGURE 6.

Selective displacement of 4MM sgRNA1:dCas9 blockade from a complex in which both 4MM sgRNA1:dCas9 and sgRNA5:dCas9 blockades are present. Complexes were assembled via method F, with the change that the quantity of 4MM sgRNA1:dCas9 added for the second incubation was doubled. (A) SYBR gold-stained urea-polyacrylamide gel showing transcription time courses using T7 RNAP in which 10 mM KCl containing a trap DNA perfectly matched to the 4MM sgRNA1 target sequence was added after the 5-min time point (left), or in which 10 mM KCl alone was added (right). (B) Quantification of the U5, sgRNA5 blockage product, sgRNAs, and 4MM sgRNA1 blockage product bands, reported relative to the intensity of the DNA band. The time at which the KCl or DNA trap was added is indicated in each plot. Error bars represent the standard deviation of three independent replicates.
DISCUSSION
In this work, we have demonstrated a versatile approach that allows the relative abundances of different RNA species to be fine-tuned during active transcription in vitro. We found noteworthy similarities and differences in the behavior of RNAPs at dCas9 blockades as compared to previously studied protein blockades (Table 1). For bacteriophage RNAPs, dCas9 appears to be one of the most robust blockades yet reported, matched only by stalled Eco RNAP initiation complexes (Kassavetis et al. 1978; Lorch et al. 1987; Pavco and Steege 1991; Guajardo and Sousa 1999). The ∼70% blockage of Eco RNAP by dCas9 contrasts with the near-100% blockage previously seen with restriction enzyme blockades (Pavco and Steege 1990). However, it is known that in vivo, the blockage efficiency of Eco RNAP by dCas9 is limited by the propensity of RNAP to displace the blockade (Vigouroux et al. 2018). This process becomes significant under nonsaturating conditions, where the finite rebinding rate of removed blockades cannot be neglected. Our experiments were done at a dCas9:DNA molar ratio only slightly greater than 1:1, which is saturating when equilibrium binding is probed at 40–80 nM DNA. However, this ratio is unlikely to be saturating when physical displacement contributes to the unbinding rate, rather than only the spontaneous dissociation of the blockade. In the studies of restriction enzyme blockades, a molar ratio of at least 7.5:1 protein:DNA was required for both complete binding at equilibrium and complete blockage of transcription (Pavco and Steege 1990), becoming saturating even when displacement occurs during transcription. We further discovered that—in contrast to the bacteriophage RNAPs—Eco RNAP stays bound when it encounters dCas9 blockades, potentially enabling applications such as mimicking transcriptional pausing or installing site-specific modifications in the nascent RNA.
TABLE 1.
Summary of a selection of previous work on blockage of transcription elongation by DNA-binding proteins in vitro

We note that the blockage efficiency and the stability of RNAP at the blockade may be affected by factors such as the target sequence and target strand (which determines the orientation of the blockade relative to the approaching RNAP). Clarke et al. investigated the ability of transcribing T7 RNAP and Pol II to displace an sgRNA:WT Cas9 complex following DNA cleavage (Clarke et al. 2018). Testing a broad panel of sgRNAs in vitro, they found that Cas9 generally gets displaced by both RNAPs when the sgRNA targets the tDNA, but not when it targets the ntDNA. Analogous to these as well as in vivo results on Eco RNAP (Qi et al. 2013), the one tDNA-targeting sgRNA we investigated showed a lower blockage efficiency than any of the four ntDNA-targeting ones (Fig. 1C). For both T7 RNAP and Pol II, Clarke and coworkers found that the quantity of free, active Cas9 resulting from displacement varies significantly among different tDNA-targeting sgRNAs, but nearly no displacement occurs with any ntDNA-targeting sgRNA tested (Clarke et al. 2018). Again consistent with these results, we found that all four of the ntDNA-targeting sgRNAs we tested almost fully blocked transcription by T7 RNAP. It therefore appears that the robustness of ntDNA-targeted blockades is relatively insensitive to the sgRNA target sequence.
We found that blockage of T7 RNAP remains effective with four mismatches in the PAM-distal end of the sgRNA:DNA hybrid. However, these mismatches make the complex susceptible to disruption by a trap duplex, allowing transcription to be recovered on demand. It is noteworthy that the competitor DNA is able to displace 4MM sgRNA1:dCas9 both in the absence (Fig. 3B) and in the presence (Fig. 4B) of active transcription, while WT sgRNA1:dCas9 complexes could not be displaced under either condition. Previous work has shown that collisions with T3 and T7 RNAPs can physically displace an otherwise very stable DNA-binding protein, allowing it to be sequestered by a competitor oligonucleotide. However, in those studies the competitor had no effect in the absence of active transcription, suggesting that the protein blockade had a very slow inherent dissociation rate (Guajardo and Sousa 1999). In our system, the 4MM sgRNA1:dCas9 complex must have an inherent dissociation rate (in the absence of transcription) that is slower than the frequency of encounters with RNAP during active transcription, but faster than the 5 min over which transcription is resumed after trap addition. The bound-state lifetime of WT sgRNA1:dCas9, in contrast, must be longer than the 60 min for which it was incubated with trap DNA in the experiment reported in Figure 3B, consistent with previous reports (Sternberg et al. 2014). The notion that displacement of 4MM sgRNA1:dCas9 blockades is governed by the inherent dissociation rate of that complex (rather than a toehold displacement mechanism) is supported by the observation that a high concentration of trap DNA matched to the WT sgRNA1 target sequence is equally effective at displacing the blockade as trap DNA that is perfectly matched to 4MM sgRNA1 (Fig. 3B). It has been shown, however, that a toehold-like mechanism allows cleaved nontarget DNA to be displaced from sgRNA:WT Cas9:DNA complexes (Richardson et al. 2016).
Finally, we found that complexes can be assembled with multiple dCas9 blockades in place that can be removed individually, allowing the relative abundances of at least three RNA species to be controlled in real-time. This represents a very significant advantage of our approach in that it allows the selective removal of a particular blockade without any changes to the reaction conditions that would affect all blockades equally and/or interfere with transcription itself (such as an increase in salt concentration). Using our approach, specificity can be obtained through two complementary mechanisms—by introducing mismatches into some blockades and not others, and by proving trap DNA complementary only to a particular blockade. Incorporating mismatches into the dCas9 blockade circumvents the limited reversibility of previously existing approaches (Qi et al. 2013), allowing transcription to resume within minutes of addition of trap DNA. This general approach can also be applied to gene regulation approaches that do not involve direct blockage of transcription. For example, dCas9 complexes that position effector proteins (Gilbert et al. 2013) could be similarly weakened by the introduction of mismatches and then rendered inactive at the desired time by the addition of a trap duplex.
As with many approaches based on CRISPR, the selection of blockage sites is limited by the availability of PAM sequences. For Spy Cas9, however, the PAM is defined simply by two adjacent guanosine residues, a motif that will be found by chance far more frequently than, for example, the 6-nt EcoRI binding sequence (Pavco and Steege 1991) or the 21-nt lac repressor binding sequence (Deuschle et al. 1986) that have previously been used to position transcription blockades. Furthermore, other members of the Cas9 superfamily and their analogs in other organisms often recognize different PAM sequences (Nakade et al. 2017), and/or have significantly different mechanisms of PAM and sgRNA recognition (Jinek et al. 2014). Additional sequence flexibility may therefore be obtained through the use of CRISPR variants from different organisms or those that have been engineering for a rapidly expanding CRISPR toolset (Kleinstiver et al. 2015a,b; Hu et al. 2018; Nishimasu et al. 2018). Blockades assembled from other Cas9 variants may also have different “weak points” from Spy dCas9, allowing the use of distinct approaches to remove them.
In conclusion, we have shown that dCas9 can be used to block transcription in vitro in a site-specific manner and is effective at blocking transcription by several different RNAPs and that mismatches in the sgRNA:DNA hybrid confer reversibility on the blockade. This work lays the mechanistic foundation for future applications, including the production of RNA nanostructures and the study of cotranscriptional folding. Our approach should also be applicable to more complex designs such as in vitro coupled transcription–translation assays and cell-free gene circuits.
MATERIALS AND METHODS
Materials
All oligonucleotides were obtained from Integrated DNA Technologies and those containing fluorophores were purified via HPLC by the manufacturer. T7 RNA polymerase was prepared in-house, while T3, SP6, and Eco RNAPs, Exo III and λ Exo were purchased from New England BioLabs. dCas9 was a generous gift from the laboratory of Dr. Carol Fierke. Transcription templates encoding U5 snRNA were generated by PCR from plasmid m571 (O'Keefe et al. 1996) using a variety of primers according to the intended application (sequences given in Table 2). Promoters for SP6, T3, and T7 templates were inserted by including a 5′ overhang on the forward primer, and for certain experiments the forward primer was 5′ phosphorylated or the reverse primer was 5′ Cy5-labeled. To prepare Eco RNAP transcription templates, the sequence encoding U5 was cloned into the pUC18 plasmid between the KpnI and EcoRI restriction sites, and the Eco RecA promoter was inserted immediately upstream between the BamHI and KpnI sites. This region of the plasmid was then amplified via PCR using the primers indicated in Table 2.
TABLE 2.
PCR primers used for the preparation of U5 transcription templates

sgRNA preparation
Transcription templates for sgRNAs were prepared from the plasmid pX330 using a two-step PCR protocol, as previously described (Shao et al. 2014) (primer sequences given in Table 3). 200 nM template was then transcribed in a 100 µL reaction in the presence of 120 mM HEPES KOH pH 7.5, 30 mM MgCl2, 7.5 mM ATP, 7.5 mM CTP, 7.5 mM GTP, 7.5 mM UTP, 40 mM DTT, 0.01% Triton X-100, 2 mM spermidine, 0.1 U inorganic pyrophosphatase, and 10 µg/mL T7 RNAP. The RNA was purified by denaturing urea-polyacrylamide gel electrophoresis using UV shadowing to identify the product band. The RNA was extracted from the gel slice by soaking it overnight in 300 mM NaOAc pH 5.3, 1 mM EDTA, then collected by ethanol precipitation.
TABLE 3.
PCR primers used for the preparation of sgRNA transcription templates

In vitro transcription experiments with nuclease digestion
sgRNA:dCas9 blockades were assembled by incubating the DNA template at 400 nM with 567 nM dCas9 and 1133 nM sgRNA in Cas9 reaction buffer (20 mM Tris-HCl pH 7.5, 100 mM KCl, 5 mM MgCl2, 1 mM DTT, 5% glycerol) for 15 min at 37°C. Immediately after removal from the heating bath, this mixture was diluted to a final concentration of 80 nM DNA in New England Biolabs RNA polymerase reaction buffer (40 mM Tris-HCl pH 7.9, 6 mM MgCl2, 1 mM DTT, 2 mM spermidine) additionally containing each NTP at 400 µM, then the RNAP being tested was added to a concentration of 20 nM. Reactions with Eco RNAP additionally included initiating dinucleotide ApC (TriLink Biotechnologies) at a concentration of 100 µM. The transcription reaction was incubated at 37°C and at each time point, an 8 µL aliquot was removed, transferred to room temperature (RT) and treated with 17 units of Exo III. The reaction was quenched 5 min later with an equal volume of formamide loading buffer (95% formamide, 20 mM EDTA). A separate 8 µL mixture was prepared and treated with Exo III without the addition of RNAP, and a final aliquot was removed at the end of the reaction and quenched without being treated with Exo III. Aliquots were resolved by denaturing urea-polyacrylamide gel electrophoresis and the nucleic acid species were visualized by staining the gel with 1× SYBR gold (ThermoFisher S11494) in 1× TBE buffer for 30 min. The ladder shown on each relevant gel is the 25-bp DNA step ladder (Promega G4511). The gel was then imaged on a Typhoon variable mode imager using the “Cy3” setting to detect SYBR gold and the “Cy5” setting to detect Cy5. All gel images displayed in figures have been subjected to a rolling ball background correction. For digestions using λ Exo (not performed during transcription), 1.6 units of the enzyme were added.
To determine the relative stoichiometry of the various components of the reaction, the intensities of the blockage product and sgRNA bands were quantified using ImageQuant TL (GE Healthcare Life Sciences) with a rolling ball background correction applied. The sgRNA was used as both a loading control and calibration band. The ratio of blockage product intensity to sgRNA intensity was determined, then, assuming that the SYBR gold signal per nanogram of RNA was equal across the two RNA species, it was adjusted by the ratio of their molecular weights to obtain the molar ratio of blockage product to sgRNA. This was then adjusted by the known molar ratio of 10.725 mol of sgRNA per mole of RNAP in the reaction to obtain the ratio of blockage product to RNAP.
In vitro transcription experiments with DNA traps
For experiments with a single blockade, dCas9:sgRNA:DNA complexes were assembled as described above. Additionally, a mixture containing 10 µM of each trap DNA duplex strand (sequences given in Table 4) was annealed in 10 mM KCl by heating it to 90°C for 2 min and then allowing it to cool at RT for 10 min. For experiments with multiple blockades, a mixture containing 1.13 µM dCas9 and 2.27 µM 4MM sgRNA1 in Cas9 reaction buffer was incubated for 15 min at 37°C. Meanwhile, another mixture containing the DNA template at 400 nM, 567 nM dCas9, and 1133 nM sgRNA5 in Cas9 reaction buffer was incubated for 15 min at 37°C. The two mixtures were combined and the resulting sample was incubated for 15 min at 37°C. Immediately after being removed from the heating bath, the sgRNA:dCas9:DNA complex was diluted in transcription buffer (120 mM HEPES KOH pH 7.5, 15 mM MgCl2, 3.75 mM ATP, 3.75 mM CTP, 3.75 mM GTP, 3.75 mM UTP, 40 mM DTT, 0.0025 U/µL inorganic pyrophosphatase) to a final concentration of 40 nM DNA, then T7 RNAP was added to a concentration of 200 nM and the reaction was incubated at 37°C. Aliquots of size 0.5–1 μL were removed at the indicated times and quenched into an equal volume of formamide loading buffer (95% formamide, 20 mM EDTA). After the 5-min time point was removed, the annealed trap solution was added to a final concentration of 1 µM DNA. For “no trap” reactions, 10 mM KCl was added to a final concentration of 1 mM. Additional aliquots were removed and quenched at the indicated times. Reactions were resolved by denaturing urea-polyacrylamide gel electrophoresis, stained in SYBR gold and imaged on a Typhoon variable mode imager using the Cy3 setting. To analyze these experiments, the intensities of the blockage product(s), sgRNA, full-length U5, and DNA bands were determined using ImageQuant TL, and each was divided by the intensity of the DNA band.
TABLE 4.
Oligonucleotides used as traps for WT sgRNA1:dCas9 and 4MM sgRNA1:dCas9 complexes

dCas9 binding assays
dCas9 blockades were prepared as described above on a template containing a Cy5 label on the 5′ end of the tDNA. Trap duplexes (assembled as described above) were added in the indicated concentrations and the mixture was incubated for 60 min at 37°C, then mixed with an equal volume of loading buffer (1× TBE, 50% glycerol). Control samples were kept on ice throughout the 60 min. The samples were then resolved on 6% nondenaturing polyacrylamide gels prepared with 0.5× TBE, which were run at 4°C using 0.5× TBE as the running buffer. The gels were then imaged on a Typhoon variable mode imager using the Cy5 setting.
ACKNOWLEDGMENTS
We thank Andrea Stoddard from the laboratory of Dr. Carol Fierke for providing us with purified dCas9, and Dr. Paul Lund for useful discussions. This work was supported by the National Institutes of Health (GM122803 to N.G.W. and GM120457 to J.R.W.) and the Albion College Faculty Development Fund to C.E.R. V.R. acknowledges the Undergraduate Research Opportunity Program and Department of Biophysics for additional funding.
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.071613.119.
Freely available online through the RNA Open Access option.
REFERENCES
- Bikard D, Jiang W, Samai P, Hochschild A, Zhang F, Marraffini LA. 2013. Programmable repression and activation of bacterial gene expression using an engineered CRISPR-Cas system. Nucleic Acids Res 41: 7429–7437. 10.1093/nar/gkt520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke R, Heler R, MacDougall MS, Yeo NC, Chavez A, Regan M, Hanakahi L, Church GM, Marraffini LA, Merrill BJ. 2018. Enhanced bacterial immunity and mammalian genome editing via RNA-polymerase-mediated dislodging of Cas9 from double-strand DNA breaks. Mol Cell 71: 42–55. 10.1016/j.molcel.2018.06.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deuschle U, Gentz R, Bujard H. 1986. lac repressor blocks transcribing RNA polymerase and terminates transcription. Proc Natl Acad Sci 83: 4134–4137. 10.1073/pnas.83.12.4134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geary C, Rothemund PWK, Andersen ES. 2014. A single-stranded architecture for cotranscriptional folding of RNA nanostructures. Science 345: 799–804. 10.1126/science.1253920 [DOI] [PubMed] [Google Scholar]
- Gilbert LA, Larson MH, Morsut L, Liu Z, Brar GA, Torres SE, Stern-Ginossar N, Brandman O, Whitehead EH, Doudna JA, et al. 2013. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell 154: 442–451. 10.1016/j.cell.2013.06.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopal V, Brieba LG, Guajardo R, McAllister WT, Sousa R. 1999. Characterization of structural features important for T7 RNAP elongation complex stability reveals competing complex conformations and a role for the non-template strand in RNA displacement. J Mol Biol 290: 411–431. 10.1006/jmbi.1999.2836 [DOI] [PubMed] [Google Scholar]
- Guajardo R, Sousa R. 1999. Characterization of the effects of Escherichia coli replication terminator protein (Tus) on transcription reveals dynamic nature of the Tus block to transcription complex progression. Nucleic Acids Res 27: 2814–2824. 10.1093/nar/27.13.2814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu JH, Miller SM, Geurts MH, Tang W, Chen L, Sun N, Zeina CM, Gao X, Rees HA, Lin Z, et al. 2018. Evolved Cas9 variants with broad PAM compatibility and high DNA specificity. Nature 556: 57–63. 10.1038/nature26155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Sousa R. 2000. T7 RNA polymerase elongation complex structure and movement. J Mol Biol 303: 347–358. 10.1006/jmbi.2000.4150 [DOI] [PubMed] [Google Scholar]
- Ji Z, Sharrocks AD. 2015. Proteomic analyses reveal distinct chromatin-associated and soluble transcription factor complexes. Mol Syst Biol 11: 775 10.15252/msb.20145504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. 2012. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337: 816–821. 10.1126/science.1225829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M, Jiang F, Taylor DW, Sternberg SH, Kaya E, Ma E, Anders C, Hauer M, Zhou K, Lin S, et al. 2014. Structures of Cas9 endonucleases reveal RNA-mediated conformational activation. Science 343: 1247997 10.1126/science.1247997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kassavetis GA, Kaya KM, Chamberlin MJ. 1978. Escherichia coli RNA polymerase-rifampicin complexes bound at promoter sites block RNA chain elongation by Escherichia coli RNA polymerase and T7-specific RNA polymerase. Biochemistry 17: 5798–5804. 10.1021/bi00619a029 [DOI] [PubMed] [Google Scholar]
- Kleinstiver BP, Prew MS, Tsai SQ, Nguyen NT, Topkar VV, Zheng Z, Joung JK. 2015a. Broadening the targeting range of Staphylococcus aureus CRISPR-Cas9 by modifying PAM recognition. Nat Biotechnol 33: 1293–1298. 10.1038/nbt.3404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinstiver BP, Prew MS, Tsai SQ, Topkar VV, Nguyen NT, Zheng Z, Gonzales APW, Li Z, Peterson RT, Yeh J-RJ, et al. 2015b. Engineered CRISPR-Cas9 nucleases with altered PAM specificities. Nature 523: 481–485. 10.1038/nature14592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X-T, Jun Y, Erickstad MJ, Brown SD, Parks A, Court DL, Jun S. 2016. tCRISPRi: tunable and reversible, one-step control of gene expression. Sci Rep 6: 39076 10.1038/srep39076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Johnson-Buck A, Yang YR, Shih WM, Yan H, Walter NG. 2018. Exploring the speed limit of toehold exchange with a cartwheeling DNA acrobat. Nat Nanotechnol 13: 723–729. 10.1038/s41565-018-0130-2 [DOI] [PubMed] [Google Scholar]
- Lorch Y, LaPointe JW, Kornberg RD. 1987. Nucleosomes inhibit the initiation of transcription but allow chain elongation with the displacement of histones. Cell 49: 203–210. 10.1016/0092-8674(87)90561-7 [DOI] [PubMed] [Google Scholar]
- Ma H, Tu L-C, Naseri A, Huisman M, Zhang S, Grunwald D, Pederson T. 2016. Multiplexed labeling of genomic loci with dCas9 and engineered sgRNAs using CRISPRainbow. Nat Biotechnol 34: 528–530. 10.1038/nbt.3526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakade S, Yamamoto T, Sakuma T. 2017. Cas9, Cpf1 and C2c1/2/3—What's next? Bioengineered 8: 265–273. 10.1080/21655979.2017.1282018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman AJ. 1997. The role of U5 snRNP in pre-mRNA splicing. EMBO J 16: 5797–5800. 10.1093/emboj/16.19.5797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimasu H, Ran FA, Hsu PD, Konermann S, Shehata SI, Dohmae N, Ishitani R, Zhang F, Nureki O. 2014. Crystal structure of Cas9 in complex with guide RNA and target DNA. Cell 156: 935–949. 10.1016/j.cell.2014.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimasu H, Shi X, Ishiguro S, Gau L, Hirano S, Okazaki S, Noda T, Abudayyeh OO, Gootenberg JS, Mori H, et al. 2018. Engineered CRISPR-Cas9 nuclease with expanded targeting space. Science 361: 1259–1262. 10.1126/science.aas9129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Keefe RT, Newman AJ. 1998. Functional analysis of the U5 snRNA loop 1 in the second catalytic step of yeast pre-mRNA splicing. EMBO J 17: 565–574. 10.1093/emboj/17.2.565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Keefe RT, Norman C, Newman AJ. 1996. The invariant U5 snRNA loop 1 sequence is dispensable for the first catalytic step of pre-mRNA splicing in yeast. Cell 86: 679–689. 10.1016/S0092-8674(00)80140-3 [DOI] [PubMed] [Google Scholar]
- Pavco PA, Steege DA. 1990. Elongation by Escherichia coli RNA polymerase is blocked in vitro by a site-specific DNA binding protein. J Biol Chem 265: 9960–9969. [PubMed] [Google Scholar]
- Pavco PA, Steege DA. 1991. Characterization of elongating T7 and SP6 RNA polymerases and their response to a roadblock generated by a site-specific DNA binding protein. Nucleic Acids Res 19: 4639–4646. 10.1093/nar/19.17.4639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi LS, Larson MH, Gilbert LA, Doudna JA, Weissman JS, Arkin AP, Lim WA. 2013. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 152: 1173–1183. 10.1016/j.cell.2013.02.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson CD, Ray GJ, DeWitt MA, Curie GL, Corn JE. 2016. Enhancing homology-directed genome editing by catalytically active and inactive CRISPR-Cas9 using asymmetric donor DNA. Nat Biotechnol 34: 339–344. 10.1038/nbt.3481 [DOI] [PubMed] [Google Scholar]
- Shao Y, Guan Y, Wang L, Qiu Z, Liu M, Chen Y, Wu L, Li Y, Ma X, Liu M, et al. 2014. CRISPR/Cas-mediated genome editing in the rat via direct injection of one-cell embryos. Nat Protoc 9: 2493–2512. 10.1038/nprot.2014.171 [DOI] [PubMed] [Google Scholar]
- Sherwood AV, Henkin TM. 2016. Riboswitch-mediated gene regulation: novel RNA architectures dictate gene expression responses. Annu Rev Microbiol 70: 361–374. 10.1146/annurev-micro-091014-104306 [DOI] [PubMed] [Google Scholar]
- Singh D, Sternberg SH, Fei J, Doudna JA, Ha T. 2016. Real-time observation of DNA recognition and rejection by the RNA-guided endonuclease Cas9. Nat Commun 7: 12778 10.1038/ncomms12778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sternberg SH, Redding S, Jinek M, Greene EC, Doudna JA. 2014. DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature 507: 62–67. 10.1038/nature13011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valero J, Pal N, Dhakal S, Walter NG, Famulok M. 2018. A bio-hybrid DNA rotor–stator nanoengine that moves along predefined tracks. Nat Nanotechnol 13: 496–503. 10.1038/s41565-018-0109-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigouroux A, Oldewurtel E, Cui L, Bikard D, van Teeffelen S. 2018. Tuning dCas9's ability to block transcription enables robust, noiseless knockdown of bacterial genes. Mol Syst Biol 14: e7899 10.15252/msb.20177899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watters KE, Strobel EJ, Yu AM, Lis JT, Lucks JB. 2016. Cotranscriptional folding of a riboswitch at nucleotide resolution. Nat Struct Mol Biol 23: 1–10. 10.1038/nsmb.3316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Lewis PJ. 2014. The interaction between bacterial transcription factors and RNA polymerase during the transition from initiation to elongation. Transcription 1: 66–69. 10.4161/trns.1.2.12791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang DY, Seelig G. 2011. Dynamic DNA nanotechnology using strand-displacement reactions. Nature 3: 103–113. 10.1038/nchem.957 [DOI] [PubMed] [Google Scholar]
- Zhou Y, Martin CT. 2006. Observed instability of T7 RNA polymerase elongation complexes can be dominated by collision-induced “bumping”. J Biol Chem 281: 24441–24448. 10.1074/jbc.M604369200 [DOI] [PubMed] [Google Scholar]