

Abstract
During development, GABA exerts depolarizing action on immature neurons and, acting in synergy with glutamate, drives giant depolarizing potentials (GDPs) in the hippocampal network. Yet, blockade of the GABA(A) receptors transforms GDPs to epileptiform discharges suggesting dual, both excitatory and inhibitory, actions of GABA in the immature hippocampal network. However, the nature of this dualism in early GABA actions is poorly understood. Here we characterized the dynamics of synaptic currents mediated by GABA(A) and glutamate receptors through an estimation of the changes in their conductance and driving forces in neonatal rat CA3 pyramidal cells during GDPs. We found that depolarizing GABAergic and glutamatergic currents act in synergy at the GDPs' onset. However, during the peak of the population discharge, the inward synaptic current was essentially mediated by glutamate receptors whereas GABA currents transiently switched their direction from depolarizing to hyperpolarizing as a result of neuronal depolarization above the GABA(A) reversal potential. Thus, the action of GABA on CA3 pyramidal cells dynamically changes during GDPs from excitatory at the GDPs' onset to inhibitory at the GDPs' peak. We propose that the dynamic changes in GABA actions occurring during GDPs enable GABAergic interneurons not only to initiate the discharge of pyramidal cells but also to control excitation in the recurrent CA3 network preventing epileptiform synchronization.
SIGNIFICANCE STATEMENT During development GABA exerts a depolarizing action on immature neurons. However, at the network level the effects of GABA are complex involving both excitatory and inhibitory actions. Here we show that GABA actions critically depend on the network state. Although GABA depolarizes neurons at rest and at the onset of population bursts, it transiently becomes hyperpolarizing at the peak of the population bursts. These dynamic changes in GABA actions enable GABAergic interneurons not only to initiate the network discharge but also to control excitation to prevent epileptiform synchronization.
Keywords: development, GABA, giant depolarizing potentials, hippocampus, network, synchronization
Introduction
During development, activity in neuronal networks is characterized by particular patterns of activity that are thought to participate in the formation of neuronal circuits (Katz and Shatz, 1996; Ben-Ari, 2001; Khazipov and Luhmann, 2006; Blankenship and Feller, 2010; Colonnese and Khazipov, 2012). In the developing hippocampus, giant depolarizing potentials (GDPs) are a ubiquitous network activity pattern observed both in neonatal rodent and fetal primate hippocampal slices and in the intact rat hippocampus in vitro (Ben-Ari et al., 1989; Khalilov et al., 1997a; Leinekugel et al., 1998; Khazipov et al., 2001; for reviews, see Ben-Ari et al., 2007; Blankenship and Feller, 2010; Cherubini et al., 2011). GDPs are generated in the CA3 hippocampal region and are characterized by quasi-periodic (circa 0.3 Hz) synchronous depolarization and firing of CA3 pyramidal cells and interneurons within a time window of few hundred milliseconds. GDPs propagate from CA3 to CA1, dentate gyrus, and entorhinal cortex, they underlie synchronous neuronal calcium oscillations and support plasticity in the developing hippocampal networks (Leinekugel et al., 1997; Garaschuk et al., 1998; Menendez et al., 1998; Kasyanov et al., 2004; Mohajerani and Cherubini, 2006). In their generation, GDPs are mechanistically linked to depolarizing and excitatory actions of GABA. Initially GDPs were described in CA3 pyramidal cells as purely GABA driven events because their reversal potential is close to the reversal potential of the chloride currents mediated by the GABA(A) receptors and their sensitivity to the GABA(A) receptor antagonists (Ben-Ari et al., 1989). However, GDPs are also suppressed by glutamate receptor antagonists, and intracellular blockade of GABA(A) receptors reveals glutamatergic synaptic conductance activated during GDPs (Khazipov et al., 1997; Leinekugel et al., 1997; Bolea et al., 1999; Lamsa et al., 2000; Sipilä et al., 2005; Khalilov et al., 2014). These findings have led to a model where neuronal excitation during GDPs is brought about by synergistic excitatory actions of depolarizing GABA and glutamate (Ben-Ari et al., 1997; Khazipov et al., 1997; Leinekugel et al., 1997; Bolea et al., 1999; Cherubini et al., 2011). On the other hand, blockade of GABA(A) receptors induces hypersynchronous epileptiform discharges indicating that GABA also exerts inhibitory actions in the CA3 network (Khalilov et al., 1997b, 1999; Khazipov et al., 1997; Lamsa et al., 2000) and suggesting that depolarizing GABA exerts dual, both excitatory and inhibitory actions in the immature network. The nature of this dualism in the early GABA actions remains unclear, however.
Although the activation of both GABAergic and glutamatergic synaptic currents during GDPs have been well documented, the dynamics and relative contributions of GABAergic and glutamatergic synaptic currents during GDPs remain unknown. The amplitude and direction of a transmembrane current (I) depends on the conductance (G) and on the driving force (DF) acting on ions passing through this conductance (I = DF × G). Previous studies indicated that Ggaba significantly exceeds Gglu during GDPs and also reported that the onset of Ggaba activation precedes that of Gglu (Khazipov et al., 1997; Leinekugel et al., 1997; Bolea et al., 1999; Lamsa et al., 2000; Mohajerani and Cherubini, 2005; Cherubini et al., 2011; Khalilov et al., 2014). However, the DF values for both conductances and their dynamic changes during GDPs remain unknown; this precludes the estimation of currents. In the present study, we characterized the time course and relative contributions of the currents through GABA(A) and glutamate receptors during GDPs in keeping with the dynamic changes in DFgaba and DFglu measured during gramicidin perforated patch recordings. Our main finding is that during the time course of GDPs, GABA transiently switches its action from excitatory at the GDPs' onset to inhibitory at the GDPs' peak, where synaptic excitation is primarily brought about by glutamatergic synaptic currents. We propose that the dynamic changes in GABA actions during GDPs enable GABAergic interneurons not only to initiate the discharge of pyramidal cells, but also to control excitation in the recurrent glutamatergic network to prevent epileptiform synchronization.
Materials and Methods
Ethical approval.
All animal-use protocols followed the guidelines of the French National Institute of Health and Medical Research (INSERM, protocol N007.08.01) and the Kazan Federal University on the use of laboratory animals (ethical approval by the Institutional Animal Care and Use Committee of Kazan State Medical University N9-2013).
Brain slice preparation.
Acute horizontal brain slices were prepared from P5–P6 Wistar rats of either sex. P0 was the day of birth. The animals were decapitated and the brain was rapidly removed and placed into oxygenated (95% O2–5% CO2) ice-cold (2–5°C) artificial CSF (ACSF) of the following composition (in mm): 126 NaCl, 3.5 KCl, 2 CaCl2, 1.3 MgCl2, 25 NaHCO3, 1.2 NaH2PO4, and 11 glucose 11, pH 7.4. Four hundred to 500-μm-thick horizontal slices were cut using a Vibratome (VT 1000E; Leica). Slices were kept in oxygenated ACSF at room temperature (20°–22°C) for at least 1 h before use. For recordings, slices were placed into a submerged chamber and superfused with oxygenated ACSF at 30°–32°C at a flow rate of 2–4 ml/min.
Electrophysiological recordings.
Extracellular recordings of the local field potentials (LFPs) and multiple unit activity (MUA) were performed in the CA3 region of hippocampus using extracellular 50 μm tungsten electrodes (California Fine Wire) or with glass electrodes filled with ACSF (2–3 MΩ) placed in the pyramidal cell layer to record LFPs and MUA. The signals from extracellular recordings were amplified and filtered (10,000×; 0.1 Hz–10 kHz) using a DAM80i amplifier, digitized at 10 kHz, and saved on a PC for post hoc analysis.
Visual patch-clamp recordings were performed from CA3 pyramidal cells located in the vicinity of the extracellular electrode (separation distance <100 μm) using Axopatch 200B or MultiClamp700B (Molecular Devices) amplifiers. Patch electrodes were made from borosilicate glass capillaries (GC150F-15, Clark Electromedical Instruments) and had a resistance of 4–7 MΩ. The patch pipette solution for gramicidin perforated patch recording contained the following (in mm):150 KCl and 10 HEPES, buffered to pH 7.2 with Tris-OH. Gramicidin was first dissolved in DMSO to prepare a stock solution of 40 mg/ml and then diluted to a final concentration of 80 μg/ml in the pipette solution. The gramicidin containing solution was prepared and sonicated <1 h before the experiment. To facilitate cell-attached formation (4–10 GΩ) the tip of pipette was filled with a gramicidin-free solution. At 20–30 min after cell-attached formation, series resistance (Rs) decreased and stabilized at 12–61 MΩ. Series resistance was monitored for the duration of the recording session. At the end of each recording, negative pressure was applied to break the membrane and establish the whole-cell configuration. This was associated with a shift of the reversal potential of the GABA(A) receptor mediated responses to near 0 mV. The membrane potential values were corrected for series resistance off-line as V (corrected) = V (holding) − IRs. A picospritzer (General Valve Corporation) was used to puff-apply isoguvacine (100 μm in ACSF) from a glass pipette in stratum radiatum at a distance of about 100 μm from the soma. The pressure was from 10 to 20 psi, and the duration of the puff varied from 10 to 20 ms. Synaptic responses were evoked by electrical stimulation in the stratum radiatum via a theta glass electrode or via a bipolar nickel–chrome electrode.
Whole-cell recordings for the intracellular blockade of GABA(A) receptor mediated synaptic currents were performed using pipette solutions of the following composition (mm): 140 CsF, 1 CaCl2, 10 HEPES, 10 EGTA, pH 7.3. Recordings of the glutamate receptor mediated component of GDPs were performed after >15 min of dialysis, at this time the inwardly directed GABA(A) receptor mediated currents are completely blocked (Leinekugel et al., 1997; Bolea et al., 1999; Khalilov et al., 2014). The GABA(A) receptor mediated component of GDPs was recorded at the reversal potential of the glutamate receptor mediated currents using a pipette solution containing the following (in mm): 135 Cs-gluconate or -methansulfonate, 2 MgCl2, 0.1 CaCl2, 1 EGTA, and 10 HEPES, pH 7.25. Extracellular and patch-clamp recordings were digitized at 10 kHz with a Digidata 1400 interface card (Molecular Devices) and analyzed off-line using MATLAB (MathWorks) routines.
Data analysis.
Raw data were preprocessed using custom-written functions in MATLAB (MathWorks). Raw data from extracellular recordings were explored to detect MUA, following which the raw data were down-sampled to 1000 Hz. MUA was detected at a band-passed signal (>400 and <4000 Hz), where all negative events exceeding 3.5 SDs were considered to be spikes. Further analysis of extracellular units and LFP data were performed using custom-written, MATLAB-based programs. Because our analysis was based on comparisons of patch-clamp recordings through different cells and conditions, special care was taken to determine the reference time point of GDPs. First we explored the possibility of detecting GDPs from LFP deflections in the pyramidal cell layer that are typically associated with GDPs (Ben-Ari et al., 1989). However, the LFP signals associated with GDPs significantly varied in their duration and waveform between slices, which prevented us from using LFP signals for determining GDPs time reference. On the other hand, population MUA bursts that were associated with GDPs showed much more homogenous distribution between slices and animals and therefore MUA bursts were used for the detection and analysis of GDPs. MUA in the CA3 pyramidal cell layer was binned in 20 ms windows and smoothed using moving average with a span of 10 bins. After filtering at low-pass 50 Hz, all MUA frequency peaks exceeding 3.5 SDs above the mean were considered as MUA bursts. MUA bursts were further visually inspected to exclude false bursts caused by artifacts. The times of MUA peaks (Time = 0, hereafter referred to as “GDP time”) were further used for processing of the concomitant patch-clamp recordings through averaging within a −1000 to 2000 ms time window. Statistical analysis was based on Jackknife estimates of the mean and SDs with the significance level set at p < 0.05.
Results
In the present study, we used patch-clamp and extracellular recordings from the CA3 pyramidal cell layer to characterize the dynamics of the currents flowing through GABA(A) and glutamate receptors (Igaba and Iglu) during GDPs. To this end, we determined the dynamic changes in driving forces (DFgaba and DFglu) and conductances (Ggaba and Gglu) and further calculated dynamic current values for each conductance as I = DF × G.
Membrane potential in CA3 pyramidal cells during GDPs
GDPs were observed as recurrent polysynaptic events associated with MUA bursts during extracellular recordings occurring at 0.3 ± 0.1 Hz in all hippocampal slices of P5–P6 rats (n = 22 slices; Fig. 1). During GDPs, MUA attained the peak value of 158 ± 3 spikes/s and overall GDPs synchronized 87.2 ± 2.9% of total neuronal firing in CA3 pyramidal cell layer. Field GDPs were also associated with transient negative LFP deflections of −54.8 ± 14 μV. On average, GDPs half-duration measured at half-maximal MUA level was of 266.6 ± 20.6 ms (n = 1567 GDPs from 22 P5–P6 slices). The peak in MUA during GDPs was taken as a T = 0 reference point for further analysis of the data obtained using concomitant patch-clamp recordings from individual CA3 pyramidal cells.
Figure 1.

Extracellular and gramicidin perforated patch recordings of GDPs. A, Example traces of cell-attached recordings from a CA3 pyramidal cell and field potential recordings from the CA3 pyramidal cell layer with a corresponding MUA frequency plot below. Note that the cell fires one to three action potentials indicated with vertical bars during each GDP (marked by asterisk). A GDP outlined by a dashed box is shown on the right. B, Raster plot of the action potentials in cell-attached recordings aligned against field GDPs. C, Normalized average crosscorrelogram of the action potentials during cell-attached recordings versus MUA. D, Gramicidin perforated patch recordings in current-clamp mode from a CA3 pyramidal cell (same as A). Note that GDPs are associated with neuronal depolarization and action potential firing, which is similar to the firing recorded in cell-attached mode. A GDP outlined by a dashed box is shown on the right. E, Raster plot of the action potentials in perforated patch recordings aligned against field GDPs. F, Normalized average crosscorrelogram of the action potentials during gramicidin perforated patch recordings versus MUA (group data from n = 7 CA3 pyramidal cells, which fired action potentials during GDPs; P5–P6; shaded area shows Jackknife deviations).
Gramicidin perforated patch recordings were obtained from 11 CA3 pyramidal cells in P5–P6 rat hippocampal slices. Typical behavior of a CA3 pyramidal neuron in relation to population activity is shown on Figure 1. In cell-attached recordings before gramicidin perforation the cell fired one to three action potentials during GDPs and most of the spikes highly correlated with MUA recorded from the CA3 pyramidal cell layer in the vicinity of the patched cell (Fig. 1A,B). After gramicidin perforation, the pyramidal cell displayed GDPs occurring in synchrony with field GDPs and its firing in association with MUA bursts was maintained indicating that the membrane perforation did not affect cell excitability (Fig. 1D,E). Similar results were obtained in seven cells which fired action potentials during GDPs (Fig. 1C,F). Dynamic changes in membrane potential during GDPs in CA3 pyramidal cells referenced to the peak of MUA are shown in Figure 2A (average of n = 379 GDPs from n = 11 cells; shaded areas indicate Jackknife confidence intervals at p = 0.05). On average, neurons depolarized during GDPs from the membrane potential of −73.8 ± 1.2 mV (measured at an interval from −1000 to −500 ms from the MUA peak) to the peak value of −57.7 ± 1.7 mV attained at −10 ± 25 ms from the MUA peak (n = 11 cells). Because of a slight time jitter in the peak depolarization values between cells, the average GDP attained a slightly more hyperpolarized value of −59.7 ± 2.0 mV (Fig. 2A), but this difference was not significant. Following transition to whole-cell mode, GDPs showed several-fold increase in amplitude and an increase in firing with spikes occurring primarily before and after, but not at the peak of GDPs (data not shown), that is typical for recordings with a high-chloride pipette solution (Ben-Ari et al., 1989).
Figure 2.

Membrane potential dynamics during GDPs and Egaba in CA3 pyramidal cells. A, Group data analysis of the membrane potential during GDPs in CA3 pyramidal cells (379 GDPs from 11 P5–P6 CA3 pyramidal cells). Shaded areas indicate Jackknife deviations. Each open triangle corresponds to an individual cell, and the closed triangle shows an average ± Jackknife deviations. T = 0 corresponds to the MUA peak of GDPs detected from extracellular recordings. Inset, Top right, Egaba values in P5–P6 pyramidal cells (n = 11). Each open circle corresponds to an individual cell, closed circle shows an average ± Jackknife deviations. Below are shown corresponding frequency plots of MUA (B) and spikes from gramicidin perforated patch recordings in current-clamp mode (CC; C), and LFP average trace (D). E, Example traces of the responses evoked by brief local application of the GABA(A) receptor agonist during gramicidin perforated patch recordings from a P5 CA3 pyramidal cell at different holding potentials. F, The current–voltage relationships of the isoguvacine-evoked responses and their exponential fit revealing an Egaba value of −65 mV in this neuron.
GABAergic and glutamatergic conductances during GDPs
We further estimated Ggaba and Gglu in CA3 pyramidal cells during the time course of GDPs using whole-cell recordings and concomitant extracellular recordings of MUA from the CA3 pyramidal cell layer. Ggaba was determined during whole-cell voltage-clamp recordings with a low chloride based pipette solution at the reversal potential of glutamatergic currents (0 mV) as Ggaba = Igaba/DFgaba. Under these recording conditions, GDPs were characterized by large amplitude polysynaptic GABA(A) receptor mediated currents attaining the maximal value of 231.3 ± 86.4 pA that corresponds to a Ggaba value of 3.3 ± 1.1 nS (n = 10 P5–P6 cells; Figs. 3A,C, 4). Recordings of the glutamate receptor mediated component of GDPs using whole-cell recordings with a CsF-based solution to block GABA(A) receptors from inside the cell (Khazipov et al., 1997; Leinekugel et al., 1997; Khalilov et al., 2014) or with a low-chloride Cs-methanesulfonate based solution at the reversal potential of GABAergic currents, revealed Gglu during GDPs attaining maximal values of 0.11 ± 0.02 nS (n = 24, P5–P6 cells; Figs. 3B,C, 4). Application of the ionotropic glutamate receptor antagonists CNQX (20 μm) and D-APV (40 μm) completely suppressed glutamatergic postsynaptic currents and blocked both spontaneous and evoked GDPs (n = 5 cells, P4–P6; data not shown) in keeping with the results of previous studies (Khazipov et al., 1997; Leinekugel et al., 1997; Bolea et al., 1999; Lamsa et al., 2000; Sipilä et al., 2005; Khalilov et al., 2014). Thus, Ggaba largely dominates over Gglu during GDPs with a Ggaba/Gglu ratio of ∼30.
Figure 3.
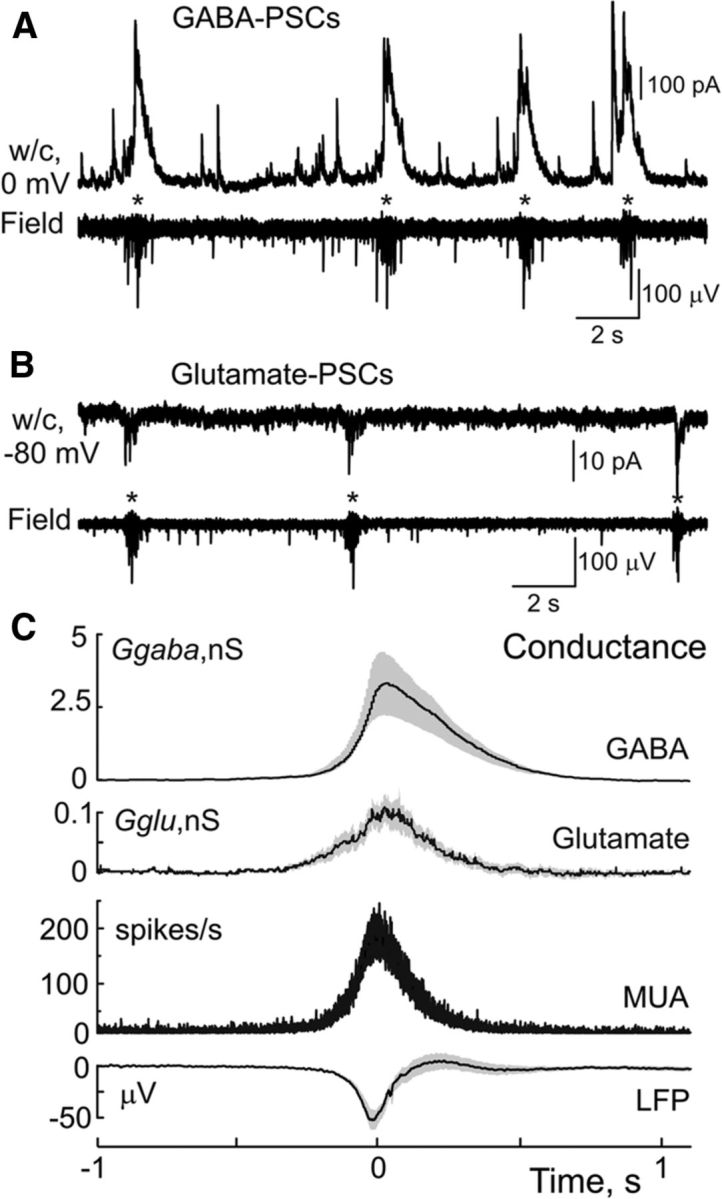
Dynamics of GABAergic and glutamatergic conductances activated during GDPs. A, B, Example traces of whole-cell recordings of GABA(A) (A) and glutamate (B) receptor mediated currents in CA3 pyramidal cells and field potential recordings from CA3 pyramidal cell layer with GDPs marked by asterisks. C, Average GABA(A) and glutamate receptor mediated conductances during GDPs. Group data for Ggaba were obtained from 10 P5–P6 CA3 pyramidal cells (total 351 GDPs) and for Gglu from 24 P5–P6 CA3 pyramidal cells (total 837 GDPs). Shaded areas indicate Jackknife confidence intervals.
Figure 4.

Dynamic changes in GABAergic and glutamatergic currents in CA3 pyramidal cells during GDPs. A, Average membrane potential during GDPs obtained during gramicidin perforated patch recordings (average of 379 GDPs from 11 P5–P6 CA3 pyramidal cells). B, Driving forces for glutamate and GABA(A) receptor mediated synaptic currents during GDPs (average of 837 and 351 GDPs from 24 and 10 P5–P6 CA3 pyramidal cells, respectively). C, Conductances mediated by GABA(A) and glutamate receptors during GDPs. D, Real currents through GABA(A) and glutamate receptors during GDPs. E, Average perievent CA3 MUA histogram obtained from 21 hippocampal slices of P5–P6 rats (n = 1567 GDPs). Values presented in A–E are aligned against the peak in MUA frequency during GDPs. Onset, catharsis, and termination phases of the GDP are outlined by vertical green dashed lines. F, Color-coded charge transfer mediated by GABAergic (circles) and glutamatergic (pyramids) synaptic currents during different phases of GDPs. Note that at GDPs' onset inward current is mediated by depolarizing GABA and glutamate. The catharsis phase, characterized by maximal discharge of CA3 neurons, is associated with the glutamatergic inward current, whereas GABAergic currents transiently switch their direction to hyperpolarizing. This phase is also characterized by maximal value of GABA(A) conductance also suggesting shunting inhibition. Termination of GDPs is associated with neuronal repolarization.
Driving forces for the GABAergic and glutamatergic currents during GDPs
The reversal potential of currents through GABA(A) receptors (Egaba) in P5–P6 CA3 pyramidal cells was measured during gramicidin perforated patch recordings from the current–voltage relationships of the GABA(A) receptor mediated responses, evoked by brief local application of the GABA(A) agonist isoguvacine or synaptic GABA(A)-PSCs evoked by electrical stimulation in the presence of the glutamate receptor antagonists CNQX (20 μm) and D-APV (40 μm; Fig. 2A,E,F). Both types of measurements gave similar results and therefore the data were pooled together providing Egaba values of −61.3 ± 1.8 mV (n = 11, P5–P6 CA3 pyramidal cells). These values are similar to those reported with gramicidin perforated patch recordings previously and to the values obtained using cell-attached measurements of the resting membrane potential and DFgaba values from the current–voltage relationships of currents through single NMDA and GABA(A) channels, respectively, (Tyzio et al., 2006, 2007, 2008).
In the following calculations of dynamic changes of DFgaba (DFgaba = Egaba − Em) during GDPs, we used the Em and Egaba values obtained during gramicidin perforated patch recordings. We found that DFgaba undergoes dramatic changes during GDPs (Fig. 4B). Before GDPs DFgaba was depolarizing (+12.5 ± 1.2 mV). However, at the GDPs' onset DFgaba progressively reduced and at the peak of GDPs even transiently switched polarity to hyperpolarizing attaining a value of −1.6 ± 1.9 mV at the peak of the average GDP, and to −3.5 ± 1.7 mV after correction of time jitter of the peak depolarization values during GDPs between individual cells. Assuming that Eglu = 0 mV (thus, DFglu = −Em), DFglu showed only small reduction from +73.8 ± 1.2 mV to +59.7 ± 2.0 mV at the peak of GDPs (Fig. 4B). Thus, DFgaba and DFglu undergo cardinally different changes during GDPs: whereas DFglu shows only a small reduction, DFgaba displays a V-shape change transiently losing its depolarizing power and switching to slightly hyperpolarizing direction at the GDPs' peak.
Real GABAergic and glutamatergic currents during GDPs
Knowing the values of driving forces and conductances, we made a further estimation of the currents flowing through the GABA(A) and glutamate receptors at different time points of GDP. These estimations were based on the results obtained from 11 CA3 pyramidal cells during gramicidin perforated current-clamp recordings, 10 cells during estimations of Ggaba, and 24 cells during estimations of Gglu. All data were referenced to the peak of the population GDP from the extracellular MUA recordings with a distinction of three phases based on MUA thresholds >3 SDs from the baseline and 50% of the peak MUA as an onset phase (−250 to −80 ms from the GDP peak), catharsis phase (−80 to +88 ms from the GDP peak) and termination phase (+88 to +350 ms from the GDP peak; Fig. 4E). As illustrated in Figure 4D, real GABAergic and glutamatergic currents showed very different dynamics during the time course of GDPs. At the GDPs' onset both Igaba and Iglu were inward attaining maximal values of 5.8 ± 1.5 and 3.8 ± 1.1 pA at the onset phase, respectively. Total charge transfer values for GABA and glutamate during the onset phase were of 0.67 ± 0.23 and 0.43 ± 0.16 pC. During the catharsis phase, Iglu continued to rise attaining a value of 6.5 ± 1.4 pA at GDPs' peak. Yet Igaba showed a reduction and a switch in polarity to attain −3.0 ± 1.0 pA at the GDPs' peak and the most negative value of −5.3 ± 1.7 pA at the time point of +20 ms after GDPs' peak. Total charge transfer values for GABAergic and glutamatergic currents during the catharsis phase were of −0.14 ± 0.06 pC and 0.90 ± 0.21 pC, respectively. During the termination phase, Iglu decayed in parallel with a decrease in MUA, whereas Igaba transiently returned to inward, attaining a second maximal level of 9.5 ± 2.6 pA at 330 ms after the GDP's peak. Total charge transfer during this phase for GABA and glutamate was of 1.57 ± 0.42 and 0.64 ± 0.21 pC, respectively.
Discussion
The principal finding of the present study is that GABAergic and glutamatergic synaptic currents dynamically change during GDPs as a result of changes in driving forces and conductances. At the GDPs' onset, the inward current is driven by depolarizing GABA and glutamate currents. However, at the peak of GDPs, Igaba transiently switches its direction from depolarizing to hyperpolarizing as a result of neuronal depolarization above Egaba values. The inward synaptic current at the peak of GDPs is primarily driven by glutamate. We propose that the dynamic changes in postsynaptic GABA actions during GDPs enable GABAergic interneurons not only to initiate the collective discharge of pyramidal cells during GDPs but also to control excitation in the recurrent glutamatergic CA3 network and to prevent epileptiform synchronization.
Previous studies indicated that Ggaba significantly exceeds Gglu during GDPs and also reported that the onset of Ggaba activation precedes that of Gglu (Khazipov et al., 1997; Leinekugel et al., 1997; Bolea et al., 1999; Lamsa et al., 2000; Mohajerani and Cherubini, 2005; Cherubini et al., 2011; Khalilov et al., 2014). Although our results are in agreement with these previous studies, we found that the currents through GABA(A) receptors during GDPs follow a very different trajectory than Ggaba and that this difference is entirely due to the dynamic changes in DFgaba. Indeed, although Igaba essentially contributed to the onset of GDPs as a result of high Ggaba and strong DFgaba, at the peak of GDPs inward Igaba transiently collapsed and then transiently switched to hyperpolarizing direction, and this transient change in Igaba was entirely due to DFgaba shifts. The dynamic change in Igaba during GDPs is probably even stronger than reported here, however. First, in our estimations of DFgaba we used the average GDP trace. However, due to a time jitter in GDPs between individual cells the level of the neuronal depolarization during average GDPs provided smaller values than the average of the peaks of depolarization. Moreover, even within the same neuron there was a jitter in membrane potential dynamics between GDPs. Second, although we assumed that Egaba does not change during GDPs, the inward electrical current carried by chloride ions flowing out of the cell through GABA(A) channels at the onset of GDPs suggests a depletion of intracellular chloride. Indeed, a reduction of intracellular chloride concentration has been demonstrated in immature hippocampal neurons during activation of GABA(A) receptors using the fluorescent chloride sensor MQAE (N-6-methoxyquinolinium acetoethylester; Marandi et al., 2002). Transient reduction in intracellular chloride has also been observed during GDP-like spontaneous population bursts in the embryonic chick spinal cord preparation (Chub et al., 2006). If similar changes in intracellular chloride also occur during hippocampal GDPs, one would expect a transient negative shift in Egaba to occur during the onset phase of GDPs, and thus, even more robust negative Igaba at the peak of GDPs than reported here.
In keeping with the dynamic changes in synaptic currents and population activity our findings support a three-phasic model of GDPs in CA3 pyramidal cells (Fig. 4).
Phase 1: onset
Excitation of CA3 pyramidal cells at the onset of GDP is driven by depolarizing GABAergic and glutamatergic currents that act in synergy during this phase. Interestingly, the contribution of Iglu to the integral inward current is comparable to that of Igaba despite much smaller Gglu than Ggaba values because of a much stronger driving force. Neuronal depolarization by inward GABA and glutamate currents is supported by low-threshold voltage gated noninactivating sodium current (Inap) mediating “depolarization sags” that may attain the action potential threshold and thus trigger spikes (Sipilä et al., 2005, 2006; Valeeva et al., 2010). Recurrent excitation during this phase grows as reflected by an increase in MUA and provides an avalanche-like increase in synaptic release of GABA and glutamate resulting in an increase of Ggaba and Gglu.
Phase 2: catharsis
Progressive build up of excitation in the CA3 network is supported by glutamatergic recurrent excitation which attains its maximum at the peak of GDP, when both maximal firing of CA3 cells and Gglu are observed. At the peak of GDPs, the inward current is primarily provided by Iglu, whose driving force is only minimally affected during GDP, because of highly positive Eglu values (0 mV). At the same time, Igaba collapses as a result of a drop in DFgaba and changes direction from inward to outward at the very peak of GDPs. Ggaba values are also maximal at the peak of GDPs. Therefore, at the peak of GDPs, GABA exerts a hyperpolarizing and shunting inhibitory effect on glutamatergic excitation thus preventing over synchronization in the CA3 network. This is in agreement with the effects of GABA(A) receptor blockers which strongly increase neuronal synchronization within CA3 and accelerate propagation of GDPs at low antagonist concentrations inducing partial blockade of GABA(A) receptors (Valeeva et al., 2010) and induce hypersynchronous epileptiform discharges when GABA(A) receptors are completely blocked (Khalilov et al., 1997b, 1999; Khazipov et al., 1997; Bolea et al., 1999; Lamsa et al., 2000).
Phase 3: termination
Upon neuronal repolarization, Iglu ceases as a result of reduction in Gglu but inward Igaba comes back as a result of an increase in DFgaba and still high Ggaba values. However, return of depolarizing Igaba has little effect on neuronal firing as at this terminal phase of GDPs, synaptic conductances are efficiently counterbalanced by potassium conductance, which is presumably mediated by slow calcium-activated potassium channels (Sipilä et al., 2006).
Thus, GABA action in the neonatal CA3 hippocampus dynamically oscillates between depolarizing during the resting network state and the initiation phase of GDPs to hyperpolarizing at the peak of network activation. We propose that the dynamic changes in GABA actions during GDPs enable GABAergic interneurons not only to initiate, acting in synergy with glutamatergic currents, the collective discharge of pyramidal cells during GDPs but also to control excitation in the recurrent glutamatergic CA3 network to prevent epileptiform synchronization. In future studies it would be of interest to determine whether similar dynamic changes in GABA actions during GDPs also occur in hippocampal interneurons. Previous studies indicated that GABA actions on neonatal hippocampal interneurons at resting state are diverse, ranging from depolarizing and excitatory to essentially isoelectric-shunting (Leinekugel et al., 1995; Khazipov et al., 1997; Verheugen et al., 1999; Banke and McBain, 2006; Tyzio et al., 2008; Sauer and Bartos, 2010). Large GABAergic conductance activated during GDPs indicates that interneurons depolarize and discharge during GDPs. However, behavior of different types of interneurons during GDPs remains unknown, except for stratum-radiatum interneurons, in which excitation during GDPs is primarily driven by depolarizing actions of GABA and glutamate (Khazipov et al., 1997). It is conceivable that the dynamic reduction of DFgaba occurs during GDPs in all types of interneurons; however, switch in polarity of GABA responses from depolarizing to hyperpolarizing will probably depend on the level of depolarization during GDPs and Egaba value in each individual interneuron. For example, it is likely to happen in stratum lucidum interneurons, where GABA is shunting at rest (Banke and McBain, 2006) and less likely for the parvalbumin-interneurons, where Egaba is strongly depolarizing (Sauer and Bartos, 2010). In future studies, it would be also of interest to determine whether similar dynamic changes in polarity of GABAergic currents also occur during other types of physiological and pathological network activities including epileptiform discharges (Cohen et al., 2002; Khalilov et al., 2003; Huberfeld et al., 2007; Nardou et al., 2009; Dzhala et al., 2010), post-traumatic and posthypoxic tissue (van den Pol et al., 1996; Nabekura et al., 2002; Payne et al., 2003; Price et al., 2009; Dzhala et al., 2012), where GABA exerts depolarizing actions at the resting network state (e.g., during down-states or interictal periods; Staley and Mody, 1992; Gulledge and Stuart, 2003; Szabadics et al., 2006; Khirug et al., 2008) but may change its direction to hyperpolarizing during the active state (up-states or population bursts).
Footnotes
This work was supported by INSERM (LIA to R.K.), the grant to leading scientists (11.G34.31.0075 to R.K.), the program of competitive growth of Kazan Federal University and RFBR (Grant 14-04-01457 to M. Mukhtarov). We thank Enrico Cherubini, Richard Miles, Jean-Luc Gaiarsa, and our colleagues at Inmed for helpful discussions.
The authors declare no competing financial interests.
References
- Banke TG, McBain CJ. GABAergic input onto CA3 hippocampal interneurons remains shunting throughout development. J Neurosci. 2006;26:11720–11725. doi: 10.1523/JNEUROSCI.2887-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Ari Y, Gaiarsa JL, Tyzio R, Khazipov R. GABA: a pioneer transmitter that excites immature neurons and generates primitive oscillations. Physiol Rev. 2007;87:1215–1284. doi: 10.1152/physrev.00017.2006. [DOI] [PubMed] [Google Scholar]
- Ben-Ari Y. Developing networks play similar melody. Trends Neurosci. 2001;24:353–360. doi: 10.1016/S0166-2236(00)01813-0. [DOI] [PubMed] [Google Scholar]
- Ben-Ari Y, Cherubini E, Corradetti R, Gaïarsa JL. Giant synaptic potentials in immature rat CA3 hippocampal neurones. J Physiol. 1989;416:303–325. doi: 10.1113/jphysiol.1989.sp017762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Ari Y, Khazipov R, Leinekugel X, Caillard O, Gaïarsa JL. GABAA, NMDA and AMPA receptors: a developmentally regulated 'ménage a trois'. Trends Neurosci. 1997;20:523–529. doi: 10.1016/S0166-2236(97)01147-8. [DOI] [PubMed] [Google Scholar]
- Blankenship AG, Feller MB. Mechanisms underlying spontaneous patterned activity in developing neural circuits. Nat Rev Neurosci. 2010;11:18–29. doi: 10.1038/nrn2759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolea S, Avignone E, Berretta N, Sanchez-Andres JV, Cherubini E. Glutamate controls the induction of GABA-mediated giant depolarizing potentials through AMPA receptors in neonatal rat hippocampal slices. J Neurophysiol. 1999;81:2095–2102. doi: 10.1152/jn.1999.81.5.2095. [DOI] [PubMed] [Google Scholar]
- Cherubini E, Griguoli M, Safiulina V, Lagostena L. The depolarizing action of GABA controls early network activity in the developing hippocampus. Mol Neurobiol. 2011;43:97–106. doi: 10.1007/s12035-010-8147-z. [DOI] [PubMed] [Google Scholar]
- Chub N, Mentis GZ, O'Donovan MJ. Chloride-sensitive MEQ fluorescence in chick embryo motoneurons following manipulations of chloride and during spontaneous network activity. J Neurophysiol. 2006;95:323–330. doi: 10.1152/jn.00162.2005. [DOI] [PubMed] [Google Scholar]
- Cohen I, Navarro V, Clemenceau S, Baulac M, Miles R. On the origin of interictal activity in human temporal lobe epilepsy in vitro. Science. 2002;298:1418–1421. doi: 10.1126/science.1076510. [DOI] [PubMed] [Google Scholar]
- Colonnese M, Khazipov R. Spontaneous activity in developing sensory circuits: implications for resting state fMRI. Neuroimage. 2012;62:2212–2221. doi: 10.1016/j.neuroimage.2012.02.046. [DOI] [PubMed] [Google Scholar]
- Dzhala VI, Kuchibhotla KV, Glykys JC, Kahle KT, Swiercz WB, Feng G, Kuner T, Augustine GJ, Bacskai BJ, Staley KJ. Progressive NKCC1-dependent neuronal chloride accumulation during neonatal seizures. J Neurosci. 2010;30:11745–11761. doi: 10.1523/JNEUROSCI.1769-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dzhala V, Valeeva G, Glykys J, Khazipov R, Staley K. Traumatic alterations in GABA signaling disrupt hippocampal network activity in the developing brain. J Neurosci. 2012;32:4017–4031. doi: 10.1523/JNEUROSCI.5139-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garaschuk O, Hanse E, Konnerth A. Developmental profile and synaptic origin of early network oscillations in the CA1 region of rat neonatal hippocampus. J Physiol. 1998;507:219–236. doi: 10.1111/j.1469-7793.1998.219bu.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulledge AT, Stuart GJ. Excitatory actions of GABA in the cortex. Neuron. 2003;37:299–309. doi: 10.1016/S0896-6273(02)01146-7. [DOI] [PubMed] [Google Scholar]
- Huberfeld G, Wittner L, Clemenceau S, Baulac M, Kaila K, Miles R, Rivera C. Perturbed chloride homeostasis and GABAergic signaling in human temporal lobe epilepsy. J Neurosci. 2007;27:9866–9873. doi: 10.1523/JNEUROSCI.2761-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasyanov AM, Safiulina VF, Voronin LL, Cherubini E. GABA-mediated giant depolarizing potentials as coincidence detectors for enhancing synaptic efficacy in the developing hippocampus. Proc Natl Acad Sci U S A. 2004;101:3967–3972. doi: 10.1073/pnas.0305974101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz LC, Shatz CJ. Synaptic activity and the construction of cortical circuits. Science. 1996;274:1133–1138. doi: 10.1126/science.274.5290.1133. [DOI] [PubMed] [Google Scholar]
- Khalilov I, Esclapez M, Medina I, Aggoun D, Lamsa K, Leinekugel X, Khazipov R, BenAri Y. A novel in vitro preparation: the intact hippocampal formation. Neuron. 1997a;19:743–749. doi: 10.1016/S0896-6273(00)80956-3. [DOI] [PubMed] [Google Scholar]
- Khalilov I, Khazipov R, Esclapez M, Ben-Ari Y. Bicuculline induces ictal seizures in the intact hippocampus recorded in vitro. Eur J Pharmacol. 1997b;319:R5–R6. doi: 10.1016/S0014-2999(96)00964-8. [DOI] [PubMed] [Google Scholar]
- Khalilov I, Dzhala V, Ben-Ari Y, Khazipov R. Dual role of GABA in the neonatal rat hippocampus. Dev Neurosci. 1999;21:310–319. doi: 10.1159/000017380. [DOI] [PubMed] [Google Scholar]
- Khalilov I, Holmes GL, Ben-Ari Y. In vitro formation of a secondary epileptogenic mirror focus by interhippocampal propagation of seizures. Nat Neurosci. 2003;6:1079–1085. doi: 10.1038/nn1125. [DOI] [PubMed] [Google Scholar]
- Khalilov I, Leinekugel X, Mukhtarov M, Khazipov R. Intracellular blockade of GABA(A) receptors in the rat hippocampal neurons. Biochemistry (Mosc) 2014;8:162–168. doi: 10.1134/S1990747814010061. [DOI] [Google Scholar]
- Khazipov R, Luhmann HJ. Early patterns of electrical activity in the developing cerebral cortex of humans and rodents. Trends Neurosci. 2006;29:414–418. doi: 10.1016/j.tins.2006.05.007. [DOI] [PubMed] [Google Scholar]
- Khazipov R, Leinekugel X, Khalilov I, Gaïarsa JL, Ben-Ari Y. Synchronization of GABAergic interneuronal network in CA3 subfield of neonatal rat hippocampal slices. J Physiol. 1997;498:763–772. doi: 10.1113/jphysiol.1997.sp021900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khazipov R, Esclapez M, Caillard O, Bernard C, Khalilov I, Tyzio R, Hirsch J, Dzhala V, Berger B, Ben-Ari Y. Early development of neuronal activity in the primate hippocampus in utero. J Neurosci. 2001;21:9770–9781. doi: 10.1523/JNEUROSCI.21-24-09770.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khirug S, Yamada J, Afzalov R, Voipio J, Khiroug L, Kaila K. GABAergic depolarization of the axon initial segment in cortical principal neurons is caused by the Na-K-2Cl cotransporter NKCC1. J Neurosci. 2008;28:4635–4639. doi: 10.1523/JNEUROSCI.0908-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamsa K, Palva JM, Ruusuvuori E, Kaila K, Taira T. Synaptic GABA(A) activation inhibits AMPA-kainate receptor-mediated bursting in the newborn (P0–P2) rat hippocampus. J Neurophysiol. 2000;83:359–366. doi: 10.1152/jn.2000.83.1.359. [DOI] [PubMed] [Google Scholar]
- Leinekugel X, Tseeb V, Ben-Ari Y, Bregestovski P. Synaptic GABAA activation induces Ca2+ rise in pyramidal cells and interneurons from rat neonatal hippocampal slices. J Physiol. 1995;487:319–329. doi: 10.1113/jphysiol.1995.sp020882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leinekugel X, Medina I, Khalilov I, Ben-Ari Y, Khazipov R. Ca2+ oscillations mediated by the synergistic excitatory actions of GABAA and NMDA receptors in the neonatal hippocampus. Neuron. 1997;18:243–255. doi: 10.1016/S0896-6273(00)80265-2. [DOI] [PubMed] [Google Scholar]
- Leinekugel X, Khalilov I, Ben-Ari Y, Khazipov R. Giant depolarizing potentials: the septal pole of the hippocampus paces the activity of the developing intact septohippocampal complex in vitro. J Neurosci. 1998;18:6349–6357. doi: 10.1523/JNEUROSCI.18-16-06349.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marandi N, Konnerth A, Garaschuk O. Two-photon chloride imaging in neurons of brain slices. Pflugers Arch. 2002;445:357–365. doi: 10.1007/s00424-002-0933-7. [DOI] [PubMed] [Google Scholar]
- Menendez de la Prida L, Bolea S, Sanchez-Andres JV. Origin of the synchronized network activity in the rabbit developing hippocampus. Eur J Neurosci. 1998;10:899–906. doi: 10.1046/j.1460-9568.1998.00097.x. [DOI] [PubMed] [Google Scholar]
- Mohajerani MH, Cherubini E. Spontaneous recurrent network activity in organotypic rat hippocampal slices. Eur J Neurosci. 2005;22:107–118. doi: 10.1111/j.1460-9568.2005.04198.x. [DOI] [PubMed] [Google Scholar]
- Mohajerani MH, Cherubini E. Role of giant depolarizing potentials in shaping synaptic currents in the developing hippocampus. Crit Rev Neurobiol. 2006;18:13–23. doi: 10.1615/CritRevNeurobiol.v18.i1-2.30. [DOI] [PubMed] [Google Scholar]
- Nabekura J, Ueno T, Okabe A, Furuta A, Iwaki T, Shimizu-Okabe C, Fukuda A, Akaike N. Reduction of KCC2 expression and GABAA receptor-mediated excitation after in vivo axonal injury. J Neurosci. 2002;22:4412–4417. doi: 10.1523/JNEUROSCI.22-11-04412.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nardou R, Ben-Ari Y, Khalilov I. Bumetanide, an NKCC1 antagonist, does not prevent formation of epileptogenic focus but blocks epileptic focus seizures in immature rat hippocampus. J Neurophysiol. 2009;101:2878–2888. doi: 10.1152/jn.90761.2008. [DOI] [PubMed] [Google Scholar]
- Payne JA, Rivera C, Voipio J, Kaila K. Cation-chloride co-transporters in neuronal communication, development and trauma. Trends Neurosci. 2003;26:199–206. doi: 10.1016/S0166-2236(03)00068-7. [DOI] [PubMed] [Google Scholar]
- Price TJ, Cervero F, Gold MS, Hammond DL, Prescott SA. Chloride regulation in the pain pathway. Brain Res Rev. 2009;60:149–170. doi: 10.1016/j.brainresrev.2008.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauer JF, Bartos M. Recruitment of early postnatal parvalbumin-positive hippocampal interneurons by GABAergic excitation. J Neurosci. 2010;30:110–115. doi: 10.1523/JNEUROSCI.4125-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sipilä ST, Huttu K, Soltesz I, Voipio J, Kaila K. Depolarizing GABA acts on intrinsically bursting pyramidal neurons to drive giant depolarizing potentials in the immature hippocampus. J Neurosci. 2005;25:5280–5289. doi: 10.1523/JNEUROSCI.0378-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sipilä ST, Huttu K, Voipio J, Kaila K. Intrinsic bursting of immature CA3 pyramidal neurons and consequent giant depolarizing potentials are driven by a persistent Na current and terminated by a slow Ca-activated K current. Eur J Neurosci. 2006;23:2330–2338. doi: 10.1111/j.1460-9568.2006.04757.x. [DOI] [PubMed] [Google Scholar]
- Staley KJ, Mody I. Shunting of excitatory input to dentate gyrus granule cells by a depolarizing GABAA receptor-mediated postsynaptic conductance. J Neurophysiol. 1992;68:197–212. doi: 10.1152/jn.1992.68.1.197. [DOI] [PubMed] [Google Scholar]
- Szabadics J, Varga C, Molnár G, Oláh S, Barzó P, Tamás G. Excitatory effect of GABAergic axo-axonic cells in cortical microcircuits. Science. 2006;311:233–235. doi: 10.1126/science.1121325. [DOI] [PubMed] [Google Scholar]
- Tyzio R, Cossart R, Khalilov I, Minlebaev M, Hübner CA, Represa A, Ben-Ari Y, Khazipov R. Maternal oxytocin triggers a transient inhibitory switch in GABA signaling in the fetal brain during delivery. Science. 2006;314:1788–1792. doi: 10.1126/science.1133212. [DOI] [PubMed] [Google Scholar]
- Tyzio R, Holmes GL, Ben-Ari Y, Khazipov R. Timing of the developmental switch in GABA(A) mediated signalling from excitation to inhibition in CA3 rat hippocampus using gramicidin perforated patch and extracellular recordings. Epilepsia. 2007;48:96–105. doi: 10.1111/j.1528-1167.2007.01295.x. [DOI] [PubMed] [Google Scholar]
- Tyzio R, Minlebaev M, Rheims S, Ivanov A, Jorquera I, Holmes GL, Zilberter Y, Ben-Ari Y, Khazipov R. Postnatal changes in somatic gamma-aminobutyric acid signalling in the rat hippocampus. Eur J Neurosci. 2008;27:2515–2528. doi: 10.1111/j.1460-9568.2008.06234.x. [DOI] [PubMed] [Google Scholar]
- Valeeva G, Abdullin A, Tyzio R, Skorinkin A, Nikolski E, Ben-Ari Y, Khazipov R. Temporal coding at the immature depolarizing GABAergic synapse. Front Cell Neurosci. 2010;4:17. doi: 10.3389/fncel.2010.00017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Pol AN, Obrietan K, Chen G. Excitatory actions of GABA after neuronal trauma. J Neurosci. 1996;16:4283–4292. doi: 10.1523/JNEUROSCI.16-13-04283.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verheugen JA, Fricker D, Miles R. Noninvasive measurements of the membrane potential and GABAergic action in hippocampal interneurons. J Neurosci. 1999;19:2546–2555. doi: 10.1523/JNEUROSCI.19-07-02546.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]