

Abstract
The blood–CSF barrier (BCSFB) consists of a monolayer of choroid plexus epithelial (CPE) cells that maintain CNS homeostasis by producing CSF and restricting the passage of undesirable molecules and pathogens into the brain. Alzheimer's disease is the most common progressive neurodegenerative disorder and is characterized by the presence of amyloid β (Aβ) plaques and neurofibrillary tangles in the brain. Recent research shows that Alzheimer's disease is associated with morphological changes in CPE cells and compromised production of CSF. Here, we studied the direct effects of Aβ on the functionality of the BCSFB. Intracerebroventricular injection of Aβ1–42 oligomers into the cerebral ventricles of mice, a validated Alzheimer's disease model, caused induction of a cascade of detrimental events, including increased inflammatory gene expression in CPE cells and increased levels of proinflammatory cytokines and chemokines in the CSF. It also rapidly affected CPE cell morphology and tight junction protein levels. These changes were associated with loss of BCSFB integrity, as shown by an increase in BCSFB leakage. Aβ1–42 oligomers also increased matrix metalloproteinase (MMP) gene expression in the CPE and its activity in CSF. Interestingly, BCSFB disruption induced by Aβ1–42 oligomers did not occur in the presence of a broad-spectrum MMP inhibitor or in MMP3-deficient mice. These data provide evidence that MMPs are essential for the BCSFB leakage induced by Aβ1–42 oligomers. Our results reveal that Alzheimer's disease-associated soluble Aβ1–42 oligomers induce BCSFB dysfunction and suggest MMPs as a possible therapeutic target.
SIGNIFICANCE STATEMENT No treatments are yet available to cure Alzheimer's disease; however, soluble Aβ oligomers are believed to play a crucial role in the neuroinflammation that is observed in this disease. Here, we studied the effect of Aβ oligomers on the often neglected barrier between blood and brain, called the blood–CSF barrier (BCSFB). This BCSFB is formed by the choroid plexus epithelial cells and is important in maintaining brain homeostasis. We observed Aβ oligomer-induced changes in morphology and loss of BCSFB integrity that might play a role in Alzheimer's disease progression. Strikingly, both inhibition of matrix metalloproteinase (MMP) activity and MMP3 deficiency could protect against the detrimental effects of Aβ oligomer. Clearly, our results suggest that MMP inhibition might have therapeutic potential.
Keywords: Alzheimer's disease, amyloid β toxicity, blood–CSF barrier, choroid plexus, matrix metalloproteinases
Introduction
Alzheimer's disease is the most common progressive form of dementia, characterized by synaptic loss, neurodegeneration, and impairment of cognitive function. Overproduction and decreased clearance of amyloid β (Aβ) peptide, followed by formation of amyloid plaques, is believed to be the central player of Alzheimer's disease (Deane et al., 2009; Alvira-Botero and Carro, 2010). More than a decade ago, it was observed that progression of the disease is more strongly correlated with the presence of soluble Aβ than with the number of plaques (McLean et al., 1999). This Aβ molecular form, which is an intermediate between monomeric Aβ and insoluble amyloid plaques, is now recognized as important mediator in the pathology of Alzheimer's disease. Recently, it was shown that mice injected with oligomerized Aβ1–42 display neuronal cell loss, tau hyperphosphorylation, and impairment of hippocampus-dependent memory (Brouillette et al., 2012; Ledo et al., 2013). Similarly, injection of Aβ1–42 in the brain ventricles of zebrafish embryos leads to cognitive deficits and tau hyperphosphorylation (Nery et al., 2014).
The microenvironment in the brain is strictly maintained by two main barriers: the blood–brain barrier (BBB) in the brain parenchyma and the blood–CSF barrier (BCSFB) in the choroid plexus (CP). The BBB separates brain interstitial fluid from blood at the level of brain capillaries, which are formed by endothelial cells connected through tight junctions, restricting paracellular transport. In addition, pericytes and astrocytic end feet contribute to strict insulation of the BBB (Zlokovic, 2008). In contrast, capillaries in the CP are highly fenestrated due to the absence of tight junctions. Therefore, the BCSFB is formed by CP epithelial (CPE) cells that are firmly interconnected by tight junctions separating blood from CSF. CPE cells also contain many specific transport systems and receptors that provide them with an active role in the regulation of transport from blood to CSF and vice versa. In addition, the BCSFB acts as a relay station that senses inflammation signals from both the CNS (Batra et al., 2010; Sharma et al., 2010; Simard et al., 2011) and the periphery (Marques et al., 2007; Marques et al., 2009a; Marques et al., 2009b; Vandenbroucke et al., 2012). CPE cells respond to inflammatory stimuli by producing proinflammatory molecules, which is often associated with disturbance of barrier integrity and results in transmission of the inflammatory signals to the rest of the brain (Mitchell et al., 2009; Coisne and Engelhardt, 2011) and increased white blood cell influx into the brain (Demeestere et al., 2015).
Alzheimer's disease is associated with numerous changes in CP morphology and function, of which the most prominent are decreased CSF production, changes in metabolic activity, and reduced clearance of toxins, including Aβ (Serot et al., 2000; Serot et al., 2003; Emerich et al., 2005; Krzyzanowska and Carro, 2012; Serot et al., 2012; Marques et al., 2013; Spector and Johanson, 2013). In addition, the vascular system of the brain is compromised (Zlokovic, 2011). Recent studies suggest that one of the possible causes of BBB disruption in Alzheimer's disease is degeneration and loss of pericytes (Sagare et al., 2013; Winkler et al., 2014). Breakdown of the endothelial and epithelial barriers has been linked to increased activity of matrix metalloproteinases (MMPs), endopeptidases that are implicated in several inflammatory processes (Vandenbroucke and Libert, 2014) and are known to affect tight junction functionality and extracellular matrix composition, eventually aggravating brain inflammation (Zeni et al., 2007; Batra et al., 2010; Vandenbroucke et al., 2012). Several studies have suggested that MMPs play a role in Alzheimer's disease pathogenesis (Wang et al., 2014). For example, CSF and brain MMP levels are higher in Alzheimer's disease patients than in age-matched controls and MMPs have been implicated in degradation of Aβ (Guo et al., 2006; Yan et al., 2006; Yin et al., 2006; Hanzel et al., 2014; Kauwe et al., 2014; Mroczko et al., 2014). Here, we studied the direct toxic effects of Aβ1–42 oligomers on the functionality of the CPE.
Materials and Methods
Mice.
Female C57BL/6 mice (8–10 weeks old) were purchased from Janvier and housed in our specific pathogen-free (SPF) animal facility. MMP3-deficient mice (Mudgett et al., 1998) in the C57BL/6 background were bred in our SPF facility (MMP3−/−). Mice were housed in groups of 4–6/cage with ad libitum access to food and water and a 14 h light/10 h dark cycle. All experiments were approved by the ethics committee of the Faculty of Sciences of Ghent University.
Preparation of Aβ1–42 oligomers.
Oligomerized Aβ1–42 was prepared as described previously (Kuperstein et al., 2010; Brouillette et al., 2012). Briefly, Aβ1–42 (rPeptide; catalog #A-1163-1) or scrambled Aβ1–42 (rPeptide; catalog #A-1004-1) was dissolved at 1 mg/ml in hexafluoroisopropanol (HFIP; Sigma-Aldrich; catalog #105228), followed by HFIP removal in a SpeedVac vacuum concentrator. The resulting peptide film was resolved at 1 mg/ml in DMSO (Sigma-Aldrich; catalog #D4540). Next, the peptide was purified from DMSO on a 5 ml HiTrap desalting column (GE Healthcare; catalog #17-408-01) and eluted with Tris-EDTA buffer (50 mm Tris and 1 mm EDTA, pH 7.5). The resulting peptide concentration was determined using the Thermo Scientific–Pierce Micro BCA Protein Assay (catalog #23225) according to the manufacturer's instructions. Finally, the eluted peptide was allowed to aggregate for 2 h at room temperature and then diluted to 1 μg/ml in Tris-EDTA buffer. To confirm the oligomer formation, we characterized the freshly prepared Aβ1–42 oligomers by analyzing Thioflavin T incorporation into aggregates, performing dot blot analysis, atomic force microscopy analysis, Coomassie-stained SDS-PAGE gel analysis, and cell toxicity assays as described previously (data not shown; Brouillette et al., 2012).
Injection of Aβ1–42 oligomers and scrambled peptide in the cerebral ventricles.
Animals were anesthetized with isoflurane and placed in a stereotactic frame. Body temperature was maintained at 37°C using a heating pad. Injection coordinates were measured from the bregma (anteroposterior −0.07, mediolateral 0.1, dorsoventral −0.3) and were determined using the Franklin and Paxinos mouse brain atlas. A volume of 5 μl (1 μg/ml peptide) was injected in the right lateral cerebral ventricle using a Hamilton needle. To assess the role of MMPs, we used three treatments: Aβ1–42 oligomers (in Tris-EDTA buffer) combined with 1 μg of broad spectrum MMP inhibitor GM6001 (Merck; catalog #CC1100) dissolved in DMSO, Aβ1–42 oligomers (in Tris-EDTA buffer) combined with DMSO and vehicle for the control group.
BCSFB and BBB permeability.
BCSFB and BBB permeability were determined as described previously (Vandenbroucke et al., 2012). In brief, 4 kDa FITC-dextran (Sigma-Aldrich; catalog #46944) was injected intravenously 1 h before CSF collection. Two and 6 h after Aβ1–42 oligomer injection, mice were sedated with ketamine/xylazine and CSF was obtained from the fourth ventricle using the cisterna magna puncture method (Liu and Duff, 2008). Next, mice were transcardially perfused with d-PBS/heparin (0.2% heparin) and brain tissue was isolated. CSF samples were diluted 100-fold in sterile d-PBS and BCSFB leakage was determined by measurement of fluorescence at λex/λem = 488/520 nm. Brain samples were cut into small pieces, incubated overnight at 37°C in formamide while shaking, and supernatant was collected after centrifugation for 15 min at maximal speed. Brain fluid was diluted twofold in sterile d-PBS and BBB leakage was determined by measurement of fluorescence at λex/λem = 488/520 nm.
Tissue isolation.
For RNA and protein analysis, mice were transcardially perfused with d-PBS/heparin (0.2% heparin) supplemented with 0.5% bromophenol blue. Brain tissue was dissected out, CP was obtained from all four ventricles, and hippocampus was isolated. Isolated hippocampus was kept in RNAlater (Ambion; catalog #AM7020) and CP was snap frozen in liquid nitrogen. For immunohistochemical analysis, brain was immediately frozen in cryoprotectant (Thermo Scientific; catalog #4583) and stored at −80°C for cryosectioning or fixed in 4% paraformaldehyde (PFA) followed by paraffin embedding for paraffin sections.
Immunohistochemistry.
For Occludin and Claudin-5 immunostaining of cryosections, 30 μm sagittal sections were cut using a cryostat (Micron; catalog #HM500) and mounted on slides. After air drying for 2 h, sections were fixed with 1% PFA for 10 min, washed for 5 min in PBS, and permeabilized for 10 min with 0.1% NP-40. After 2 washes with PBS, samples were blocked for 1 h at room temperature with 5% BSA (Sigma-Aldrich; catalog #A2153) and then incubated with primary antibody (Life Technologies; Occl, 1:100, 33–1500; Cldn5, 1:50, 35–2500). After incubation overnight at 4°C, they were washed and incubated with secondary antibody (Thermo Scientific; goat anti-mouse-DyLight633 and goat anti-rabbit-DyLight633, 1:400) diluted in 5% BSA for 90 min at room temperature (RT). For E-cadherin and Iba1 staining, paraffin-embedded brains were sectioned at 4 μm in sagittal orientation, dewaxed, and rinsed in water and PBS before staining. Antigen retrieval was done using citrate buffer (Dako; catalog #S2031), followed by washing in PBS. For E-cadherin staining, slides were incubated overnight at 4°C with 0.01 m NaBH4 (Sigma-Aldrich; catalog #452882) to further reduce autofluorescence. After rinsing in PBS, permeabilization was done with 0.05% Tween-20 (Sigma-Aldrich; catalog #P1379) for 30 min at RT. Samples were blocked with 2% BSA for 30 min at RT, followed by incubation for 90 min at RT with primary (E-Cadherin, 1:500; BD Transduction Laboratories; catalog #610181) and secondary antibodies (Alexa Fluor 568 goat anti-mouse, 1:500; Life Technologies) diluted in 5% BSA. For Iba-1 staining, antigen retrieval was followed by overnight incubation at 4°C with primary antibody (Iba1, 1:1000; Wako; catalog #019 19741). The next day, slides were washed and incubated with LSAB2 System HRP (DAKO; catalog #K0672) and visualization was done using DAB chromogen. Finally, slides were dehydrated and xylene-based mounting medium was applied. Cells were visualized using Olympus BX51 microscope. Iba1-positive cells were counted in predefined area of the brain, including both cortex and hippocampus, using Fiji (http://fiji.sc/Fiji). Microglia were classified into resting and activated according to adopted criteria (Hains and Waxman, 2006).
Cytokine/chemokine measurements.
Cytokines and chemokines in CSF were measured using the Bio-Plex cytokine assays (Bio-Rad; catalog #M60009RDPD) according to the manufacturer's instructions.
RNA isolation.
Total RNA was isolated with the (mi)RNeasy kit (Qiagen; catalog #74106 and #217004). RNA concentration and purity were determined spectrophotometrically using the Nanodrop Technologies ND-1000.
Real-time qPCR.
After RNA isolation, cDNA was synthesized by using a cDNA Synthesis Kit (Bio-Rad; catalog #172-5038). Real-time qPCR was performed on the Light Cycler 480 system (Roche) using the Light-Cycler 480 SYBR Green I Master mix (Roche; catalog #04887352001) or the SensiFAST SYBR No-ROX Kit (Bioline; catalog #BIO-98002). Expression levels were normalized to the expression of the two or three most stable reference genes, as determined by the geNorm Housekeeping Gene Selection Software (Vandesompele et al., 2002): Hprt, Rpl, and Ubc for CP and Gapdh, Rpl, and Ubc for hippocampus. The sequences of the forward and reverse primers for the different genes are provided in Table 1.
Table 1.
Overview of the sequences of the forward and reverse primers used for qPCR analysis
| Gene | Forward | Reverse |
|---|---|---|
| Il1β | CACCTCACAAGCAGAGCACAAG | GCATTAGAAACAGTCCAGCCCATAC |
| Il6 | TAGTCCTTCCTACCCCAATTTCC | TTGGTCCTTAGCCACTCCTTC |
| Tnf | ACCCTGGTATGAGCCCATATAC | ACACCCATTCCCTTCACAGAG |
| Ocln | CCAGGCAGCGTGTTCCT | TTCTAAATAACAGTCACCTGAGGGC |
| Cldn5 | GCAAGGTGTATGAATCTGTGCT | GTCAAGGTAACAAAGAGTGCCA |
| Zo1 | AGGACACCAAAGCATGTGAG | GGCATTCCTGCTGGTTACA |
| Zo3 | ACCCTATGGCCTGGGCTTC | CCCGGGTACAACGTGTCC |
| Cldn1 | TCTACGAGGGACTGTGGATG | TCAGATTCAGCAAGGAGTCG |
| Ecdh | TCGGAAGACTCCCGATTCAAA | CGGACGAGGAAACTGGTCTC |
| Mmp1a | CCTTGATGAGACGTGGACCAA | ATGTGGTGTTGTTGCACCTGT |
| Mmp2 | AGATCTTCTTCTTCAAGGACCGGTT | GGCTGGTCAGTGGCTTGGGGTA |
| Mmp3 | AGTCTACAAGTCCTCCACAG | TTGGTGATGTCTCAGGTTCC |
| Mmp8 | ATTCCCAAGGAGTGTCCAAGC | TGATTGTCATATCTCCAGCACTGG |
| Mmp9 | CTGGACAGCCAGACACTAAAG | CTCGCGGCAAGTCTTCAGAG |
| Mmp13 | TTTATTGTTGCTGCCCATGA | GGTCCTTGGAGTGATCCAGA |
| Mmp14 | CAGTATGGCTACCTACCTCCAG | GCCTTGCCTGTCACTTGTAAA |
| Rpl | CCTGCTGCTCTCAAGGTT | TGGTTGTCACTGCCTGGTACTT |
| Ubc | AGGTCAAACAGGAAGACAGACGTA | TCACACCCAAGAACAAGCACA |
| Gapdh | TGAAGCAGGCATCTGAGGG | CGAAGGTGGAAGAGTGGGAG |
| Hprt | AGTGTTGGATACAGGCCAGAC | CGTGATTCAAATCCCTGAAGT |
| β2M | ATGCACGCAGAAAGAAATAGCAA | AGCTATCTAGGATATTTCCAATTTTTGAA |
| βact | GCTTCTAGGCGGACTGTTACTGA | GCCATGCCAATGTTGTCTCTTAT |
MMP activity.
MMP activity in the CSF was analyzed using the OmniMMP fluorogenic substrate kit according to manufacturer's guidelines (Enzo Life Sciences; catalog #BML-AK016). In brief, CSF samples were diluted 50-fold in assay buffer and MMP activity was determined by measuring the increase in fluorescence at λex/λem = 320/460 nm.
Western blot.
For Occludin Western blot analysis, CP samples from two mice were pooled and homogenized in 50 μl of lysis buffer containing 0.5% CHAPS (Sigma-Aldrich; catalog #C9426) and protease inhibitor complete tablet (Roche Applied Science; catalog #11 873 580 001). Protein concentration was determined using the Thermo Scientific–Pierce Micro BCA Protein Assay and 60 μg of proteins were loaded on a 12.5% SDS-PAGE gel. For MMP3 Western blot analysis, 2 μl of each sample, derived from scrambled and Aβ1–42 oligomer injected mice, was loaded on a 12.5% SDS-PAGE gel. Further protocol remained the same for both Occludin and MMP3 detection. Semidry transfer was performed for 1 h at constant current (45 mA) and membranes were blocked for 1 h using Odyssey blocking buffer (Li-Cor; catalog #927-40000), followed by overnight incubation with primary antibody at 4°C (anti-Occludin, 1:250, Life Technologies, catalog #33-1500; anti-MMP3, 1:100, Abcam, catalog #ac52915). Next, membranes were incubated with the secondary antibody (Thermo Scientific anti-mouse Dylight 680, 1:1000) for 2 h at RT and bands were visualized using Odyssey software.
Serial block-face scanning electron microscopy.
For serial block-face scanning electron microscopy (SBF-SEM), CP tissue was dissected and immediately transferred into fixation buffer (2% paraformaldehyde, Sigma-Aldrich; 2.5% glutaraldehyde, Electron Microscopy Sciences in 0.15 m cacodylate buffer, pH 7.4). After overnight fixation at 4°C, samples were washed 3 × 5 min in cacodylate buffer and subsequently osmicated in 2% osmium (EMS), 1.5% ferrocyanide, and 2 mm CaCl2 in cacodylate buffer for 1 h on ice, and then washed extensively in ultrapure water (UPW). This was followed by incubation in 1% thiocarbohydrazide (20 min), washes in UPW, and a second osmication in 2% osmium in UPW (30 min). The samples were washed 5 × 3 min in UPW and placed in 2% uranyl acetate at 4°C overnight. The following day, they were stained with Walton's lead aspartate stain for 30 min at 60°C. For this, a 30 mm l-aspartic acid solution was used to freshly dissolve lead nitrate (final concentration 20 mm, pH 5.5). The solution was filtered after incubation for 30 min at 60°C.
After the final washes, the samples were dehydrated using a series of ice-cold solutions of increasing ethanol concentration (30%, 50%, 70%, 90%, and twice 100%), followed by two dehydrations of 30 min in 100% acetone. Subsequent infiltration with resin (Durcupan; EMS) was done by first incubating the samples in 50% resin in acetone for 4 h, followed by at least 5 changes of fresh 100% resin (including 2 overnight incubations). Next, samples were embedded in fresh resin and cured in the oven at 65°C for 72 h.
For SBF-SEM, the resin-embedded samples were mounted on an aluminum specimen pin (Gatan) using conductive epoxy (Circuit Works). The specimens were trimmed in a pyramid shape using an ultramicrotome (Ultracut; Leica) and the block surface was trimmed until smooth and at least a small part of tissue was present at the block face. Next, samples were coated with 5 nm Pt in a Quorum Q 150T ES sputter coater (Quorum Technologies). The aluminum pins were placed in the Gatan 3View2XP in a Zeiss Merlin SEM for imaging at 1.6 kV with a Gatan Digiscan II ESB detector. The Gatan 3view2XP was set to section 300 sections of 70 nm. IMOD (http://bio3d.colorado.edu/imod/) and Fiji (Schindelin et al., 2012) were used for registration of the 3D image stack and conversion to TIFF file format. Representation of the cell in 3D movies and snapshots was done in Imaris (BitPlane). 3D modeling of CPE cells was performed using IMOD software.
Statistics.
Data were analyzed using GraphPad Prism and Student's t test and are presented as means ± SEM. Significance levels are indicated on the graphs as follows: *0.01 ≤ p < 0.05; **0.001 ≤ p < 0.01; ***0.0001 ≤ p < 0.001; and ****p < 0.0001.
Results
Intracerebroventricular administration of Aβ1–42 oligomers induces inflammation in brain, CP, and CSF
In Alzheimer's disease, Aβ and its deposits, along with neurofibrillary tangles, induce a chronic local upregulation of inflammatory mediators (Akiyama et al., 2000). We prepared Aβ1–42 oligomers by resuspending Aβ1–42 peptide film in Tris-EDTA buffer as described previously (Kuperstein et al., 2010), followed by incubation for 2 h to allow oligomerization. Next, C57BL/6 mice were injected intracerebroventricularly with Aβ1–42 oligomers (Brouillette et al., 2012) and the effect on brain inflammation was studied. Gene expression analysis of the hippocampus revealed that several inflammatory genes were significantly upregulated 6 h after Aβ1–42 oligomer injection: Il6, IL1β, and TNF levels are shown in Figure 1a–c, respectively. In contrast, Iba1 immunostaining did not reveal a significant increase in microglia activation in mice 6 h after intracerebroventricular injection of Aβ1–42 (Fig. 1e,f) compared with injection of scrambled peptide (Fig. 1d,f). The CP is one of the sites involved in the production, deposition, and clearance of Aβ in the brain (Crossgrove et al., 2005; Crossgrove et al., 2007; Krzyzanowska and Carro, 2012). To study the direct effect of Aβ1–42 oligomers on inflammation in the CP and hippocampus, CSF, CP, and hippocampus were isolated 2 and 6 h after intracerebroventricular injection of scrambled peptide or Aβ1–42 oligomers and total RNA was extracted from CP and hippocampus. Next, we determined mRNA levels of several proinflammatory cytokines (Fig. 2a–h). Il1β, Il6, Tnf, and Inos were significantly upregulated 2 h after intracerebroventricular injection of Aβ1–42 compared with scrambled peptide. The upregulation was even more pronounced 6 h after intracerebroventricular injection in CP (Fig. 2a–d). Similarly, we determined mRNA levels of the proinflammatory cytokines in the hippocampus (Fig. 2e–h). As observed in the CP, there was a significant increase in the Il1β, Il6, Tnf, and Inos mRNA expression at 2 h. With the exception of iNos, these effects were more pronounced at 6 h.
Figure 1.

Analysis of inflammation in the hippocampus after intracerebroventricular injection of oligomerized Aβ1–42 in the cerebral ventricles. a–c, mRNA expression analysis of Il1β (a), Il6 (b), and Tnf (c) in the hippocampus 6 h after intracerebroventricular injection of Aβ1–42 oligomers in C57BL/6 mice compared with control hippocampus samples (n = 4). d, e, Representative images of Iba1 staining of brain sections from C57BL/6 mice 6 h after intracerebroventricular injection with scrambled peptide (d) or Aβ1–42 oligomers (e). As indicated on the images, the region surrounding the lateral ventricle is represented. f, Quantification of the percentage of activated microglia in cortex and hippocampus of scrambled and Aβ1–42 oligomer injected mice (n = 5). Scale bar, 25 μm.
Figure 2.

Cytokine and chemokine analyses of CP, hippocampus, and CSF after intracerebroventricular injection of Aβ1–42 oligomers. a–d, mRNA expression analysis of Il1β (a), Il6 (b), Tnf (c), and Inos (d) in CP after intracerebroventricular injection of Aβ1–42 oligomers (gray) in C57BL/6mice compared with control CP samples (black) (n = 3–4). e–h, mRNA expression analysis of Il1β (e), Il6 (f), Tnf (g), and Inos (h), in the hippocampus after intracerebroventricular injection of Aβ1–42 oligomers (gray) in C57BL/6mice compared with control samples (black) (n = 3–4). i–p, Levels of cytokines (IL-1β, IL-6, TNFα, and IFNγ) and chemokines (MIP-1α, MIP-1β, MCP-1, and GM-CSF) in CSF isolated from C57BL/6 mice injected intracerebroventricularly with scrambled peptide (black) or Aβ1–42 oligomers (gray) (n = 4).
Next, to determine whether this upregulation in cytokine expression was accompanied by inflammatory protein expression, we evaluated cytokine levels in CSF using the BioPlex assay (Fig. 2i–l). The significantly increased presence of the proinflammatory cytokines IL-1β, IL-6, TNFα, and IFNγ in CSF at both time points correlated with the elevated mRNA levels observed in the CP: they were significantly increased 2 and 6 h after intracerebroventricular injection of Aβ1–42. In parallel, we measured several chemokines in the CSF: MIP-1α, MIP-1β, MCP-1, and GM-CSF were all upregulated at the tested time points in the presence of Aβ1–42 (Fig. 2m–p).
Our data show that Aβ1–42 oligomers induce a strong inflammatory response in the brain that is more pronounced in the CP than in the rest of the brain. This is reflected by increased cytokine and chemokine levels in the CSF.
Intracerebroventricular injection of Aβ1–42 oligomers induces morphological changes in the CP and loss of BCSFB integrity
The CP undergoes morphological alterations in Alzheimer's disease (Marques et al., 2013). Therefore, we injected C57BL/6 mice intracerebroventricularly with oligomerized Aβ1–42 and studied CPE morphology 6 h later using SBF-SEM. This technology uses a fully automated microtome installed in the SEM chamber to produce serial sections that makes 3D reconstructions of SEM samples (Denk and Horstmann, 2004) and this revealed that Aβ1–42 induces loss of the typical cuboidal structure of CPE cells (Fig. 3; Movies 1 and 2). To better visualize the differences in our SBF-SEM images, we performed 3D modeling of CPE cells to highlight the morphological differences. CPE cell outlines were drawn manually using IMOD software for ∼200 sections per sample. These sections were then merged to generate a 3D model of the cell shape. CPE cells retained typical cuboidal structure after scrambled peptide injection. In contrast, several cells in Aβ1–42 oligomer-injected mice lost their cuboidal shape and the overall cell volume was reduced compared with the scrambled injected CPE cells. In addition, CPE cells in Aβ1–42 oligomer-injected mice displayed reduced nucleus size (Fig. 3c,d). Next, we analyzed whether the severe morphological changes induced by Aβ1–42 oligomers affect BCSFB integrity. To compare blood–CSF permeability in control mice and in mice injected intracerebroventricularly with Aβ1–42 oligomers, we injected 4 kDa FITC-dextran intravenously 1 h before CSF isolation and measured fluorescence in CSF. The results revealed that Aβ1–42 oligomers induce permeability of the BCSFB 6 h after intracerebroventricular injection, demonstrating that oligomerized Aβ1–42 disturbs CPE barrier functionality (Fig. 4a).
Figure 3.

CP morphology analysis by SFB-SEM. a, b, Representative SBF-SEM images of CP cells of C57BL/6 mice injected intracerebroventricularly with scrambled peptide (a) or Aβ1–42 oligomers (b). Cell shape is outlined in green. c, d, 3D modeling (blue) based on merging ∼200 sections of CPE cells from scrambled (c) and Aβ1–42 oligomer (d) injected mice. Only cell shape was considered; basolateral labyrinth and microvilli were neglected while generating the 3D modeling. Mv, Microvilli; Nu, nucleus. Scale bar, 2 μm.
Figure 4.
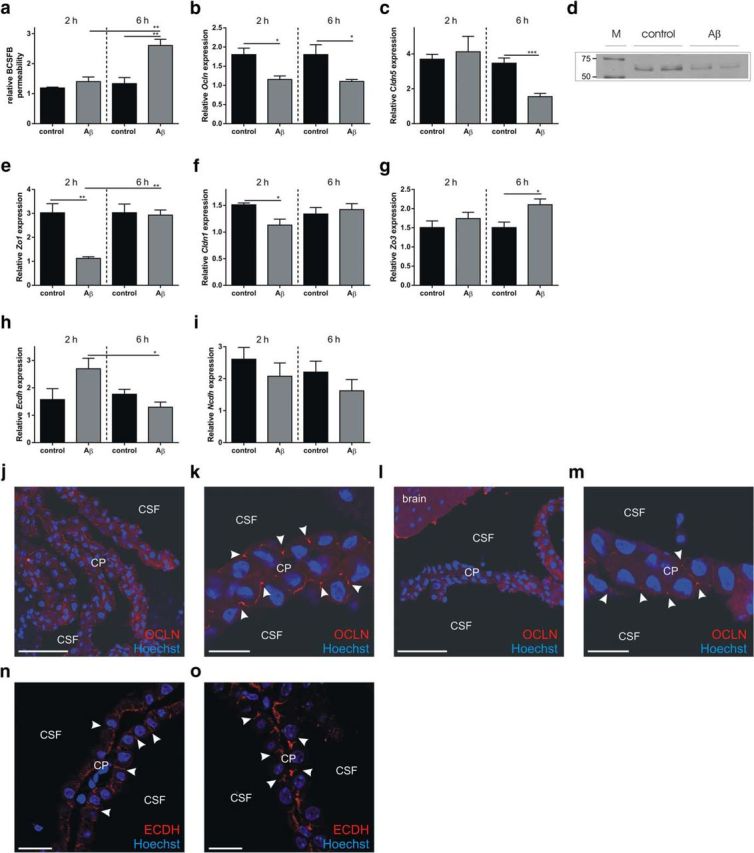
Analysis of Aβ1–42 oligomer-induced disruption of BCSFB integrity. a, Relative BCSFB permeability 2 and 6 h after intracerebroventricular injection of Aβ1–42 oligomers in the cerebral ventricles (gray) compared with control mice (black) (n = 4). b, c, Ocln (b) and Cldn5 gene (c) expression in CP tissue of C57BL/6 mice injected intracerebroventricularly with scrambled peptide (black) or Aβ1–42 oligomers (gray) (n = 4). d, Western blot analysis of Occludin in CP tissue from C57BL/6 control mice and mice injected intracerebroventricularly with Aβ1–42 oligomers. e–i, Zo1 (e), Cldn1 (f), Zo3 (g), Ecdh (h), and Ncdh (i) gene expression in CP tissue of C57BL/6 mice injected in the cerebral ventricles with Aβ1–42 oligomers (gray) or scrambled peptide (black) (n = 4). j–m, Representative confocal images of C57BL/6 control mice (j, k) and mice injected intracerebroventricularly with Aβ1–42 (l, m) oligomers for 6 h (red, Occludin; blue, Hoechst). The arrowheads point to the apically located tight junctions. Scale bars: e, g, 50 μm; f, h, 10 μm. n = 5. n, o, Representative confocal images of control (n) and Aβ1–42 (o) oligomers injected intracerebroventricularly in C57BL/6 mice stained for E-cadherin (ECDH; red) and Hoechst (blue). The arrowheads point to adherens junctions.
Subsequently, we used qPCR, Western blotting, and immunostaining to determine whether the alterations in BCSFB integrity induced by Aβ1–42 oligomers were correlated with changes in junctional proteins in the CP. Gene expression analysis revealed that several tight junction genes were affected by intracerebroventricular injection of Aβ1–42 oligomers. As shown in Figure 4, b and c, expression of Claudin-5 (Cldn5) and Occludin (Ocln) was significantly downregulated in response to Aβ1–42 oligomers. Notably, Ocln was significantly downregulated at both time points tested, whereas Cldn5 was significantly downregulated only 6 h after Aβ1–42 oligomer injection. Zona occludens-1 (Zo1) and Claudin-1 (Cldn1) were significantly downregulated 2 h after Aβ1–42 oligomer injection (Fig. 4e,f). In contrast, Zo3 showed no change in gene expression at that time point (Fig. 4g). Gene expression of the adherens junctional genes E-cadherin (Ecdh) and N-cadherin (Ncdh) was unaffected at both time points (Fig. 4h,i). OCLN, the tight junction protein that was most severely affected 6 h after intracerebroventricular injection, was also analyzed by Western blot. CP tissue was isolated from control mice and from mice injected intracerebroventricularly with Aβ1–42 oligomers and pooled samples were analyzed by SDS-PAGE. Immunodetection revealed that OCLN protein levels are downregulated by intracerebroventricular injection of Aβ1–42 oligomers (Fig. 4d). This was also studied by fluorescent immunostaining. Confocal imaging revealed that, in control mice, OCLN was enriched at the apical side of the CPE cells (Fig. 4j,k, white arrowheads). In agreement with the Western blot analysis, the OCLN signal was reduced after Aβ1–42 oligomer injection (Fig. 4l,m, white arrowheads). We also analyzed the adherens junction protein ECDH. Like OCLN, this protein was enriched at the apical side of CPE cells, but it was unaffected by intracerebroventricular injection of Aβ1–42 oligomers (Fig. 4n,o). Our data show that tight junction proteins are affected at the mRNA and/or protein level upon intracerebroventricular Aβ1–42 oligomer injection, which might explain the observed BCSFB leakage.
In parallel, we also studied the effect of Aβ1–42 oligomers on BBB integrity. As shown in Figure 5a, we did not observe a significant increase in BBB leakage 6 h after Aβ1–42 oligomer administration. In addition, also Ocln, Cldn1, and Zo1 gene expression was not affected in the hippocampus upon intracerebroventricular injection of Aβ1–42 oligomers, whereas Cldn5 did show a significant decrease in gene expression compared with scrambled peptide control (Fig. 5b–e). Clearly, intracerebroventricular injection of Aβ1–42 oligomers only modestly affects the BBB 6 h after the injection.
Figure 5.

Analysis of the effect of Aβ1–42 oligomers on BBB integrity. a, Relative permeability of the BBB 6 h after intracerebroventricular injection of Aβ1–42 oligomers (gray) compared with scrambled peptide injected mice (black) (n = 10–13). b–e, Relative gene expression of tight junction proteins Occludin (b), Claudin-5 (c), Zona occludens-1 (d), and Claudin-1 (e) in the hippocampus 6 h after intracerebroventricular injection of scrambled peptide (black) or Aβ1–42 oligomers (gray) (n = 5).
Aβ1–42 oligomer-induced BCSFB disruption is linked to increased MMP expression and activity
Previous studies have shown altered expression of MMPs in Alzheimer's disease patients (Wang et al., 2014) and MMPs have been implicated in BCSFB disruption in response to TNF (Zeni et al., 2007) and in several inflammatory conditions (Vandenbroucke and Libert, 2014) such as sepsis (Vandenbroucke et al., 2012) and stroke (Batra et al., 2010). To determine whether MMPs play a role in the Aβ1–42 oligomer-induced BCSFB leakage, we analyzed MMP gene expression in the CP and MMP activity in the CSF. Figure 6a shows the fold changes in gene expression of Mmp1a, Mmp2, Mmp3, Mmp8, Mmp9, Mmp13, and Mmp14 2 h (black) and 6 h (gray) after intracerebroventricular injection of Aβ1–42 oligomers. Mmp3 was significantly upregulated 2 h after intracerebroventricular injection and Mmp3, Mmp8, Mmp9, and Mmp13 at 6 h after injection. This was associated with an increase in MMP activity in the CSF 6 h after Aβ1–42 oligomer injection (Fig. 6b). Next, we coinjected Aβ1–42 oligomers intracerebroventricularly with the broad-spectrum MMP inhibitor GM6001. The results show that inhibition of MMP activity prevented the Aβ1–42 oligomer-induced BCSFB disruption. Indeed, comparison of BCSFB integrity of mice injected with Aβ1–42 oligomers alone or combined with MMP inhibitor revealed that MMP inhibition prevents oligomerized Aβ1–42 from inducing BCSFB leakage (Fig. 6c). As expected, this was associated with a decrease in MMP activity in the CSF (Fig. 6b). Clearly, inhibiting MMP activity diminishes the detrimental effects of Aβ1–42 oligomers. Based on the gene expression analysis displayed in Figure 6a, we also analyzed MMP3 protein level in the CSF. Western blot analysis clearly showed an increase in secreted MMP3 (Fig. 6d). Finally, we injected Aβ1–42 oligomers intracerebroventricularly in wild-type and MMP3-deficient (MMP3−/−) mice to confirm the suspected role of MMPs, especially MMP3. As shown in Figure 6e, absence of MMP3 significantly prevented BCSFB leakage induced by Aβ1–42 oligomers. In agreement with this, Aβ1–42 oligomers did not affect the morphology of the CPE cells in MMP3−/− mice (Fig. 7).
Figure 6.

Analysis of the role of MMPs in Aβ1–42 oligomer-induced disruption of BCSFB permeability. a, Fold change in Mmp gene expression in the CP 2 h (black) and 6 h (gray) after intracerebroventricular injection of Aβ1–42 oligomers in the cerebral ventricles compared with control samples (n = 3–4). b, Total MMP activity in CSF of C57BL/6 mice 6 h after intracerebroventricular injection of scrambled peptide (control), Aβ1–42 oligomers (Aβ), or Aβ1–42 oligomers together with GM6001 (Aβ + GM6001) (n = 6). c, BCSFB permeability of C57BL/6 mice 6 h after intracerebroventricular injection of Aβ1–42 oligomers alone (Aβ) or combined with the MMP inhibitor GM6001 (Aβ + GM6001) compared with scrambled peptide injected mice (control) (n = 6–7). d, Western blot analysis of MMP3 protein levels in CSF of control and Aβ1–42 oligomer intracerebroventricularly injected mice. e, Relative BCSFB permeability in wild-type (black) and MMP3−/− (gray) C57BL/6 mice 6 h after intracerebroventricular injection of scrambled control or Aβ1–42 oligomers (n = 4–10).
Figure 7.

CP morphology analysis by SFB-SEM of MMP3−/− mice. a, b, Representative SFB-SEM images of the CPE cells of scrambled peptide (a) and Aβ1–42 oligomer (b) intracerebroventricularly injected MMP3−/− mice. Cell shape is represented in green. Basolateral labyrinth and microvilli were neglected while drawing the cell outline. Mv, Microvilli; Nu, nucleus.
Discussion
Neuroinflammation occurs very early on in disease progression and in several brain regions affected by Alzheimer's disease. Abnormal molecules such as Aβ1–42, hyperphosphorylated tau protein, and damaged neurons induce the activation of cytokines, complement proteins, and chemokines, as well as other acute phase pathways, eventually leading to inflammation (Akiyama et al., 2000). In a 2004 human study, increased levels of proinflammatory cytokines were found in plasma before the clinical onset of the disease (Engelhart et al., 2004). In addition, cell culture studies and animal models of Alzheimer's disease demonstrated that inflammation precedes the appearance of pathological hallmarks of Alzheimer's disease such as senile plaques and neurofibrillary tangles (Floden et al., 2005; Wright et al., 2013). Some evidence indicates that soluble Aβ oligomers, an intermediate between monomeric Aβ and insoluble amyloid plaques, are an important mediator in the inflammation and pathology of Alzheimer's disease. Although the concentration of soluble Aβ1–42 was not uniform in different studies (Nitta et al., 1997; Kong et al., 2005; Marco and Skaper, 2006; Brouillette et al., 2012; Cetin et al., 2013), in most, Aβ1–42 was able to initiate a cascade of the events that recapitulate the key pathological hallmarks of Alzheimer's disease. In vitro incubation of macrophages, microglia, astrocytes, and oligodendrocytes with soluble Aβ induced inflammation (Johnstone et al., 1999; Lee et al., 2002; Smits et al., 2002). In vivo, microvessels forming the BBB isolated from the brains of Alzheimer's disease patients showed markedly increased levels of IL-1β, IL-6, and TNF compared with control subjects (Grammas and Ovase, 2001; Tripathy et al., 2007).
Here, we made use of Aβ1–42 oligomers prepared as described previously, which are known to induce neuronal cell loss, tau hyperphosphorylation, and impairment of hippocampus-dependent memory (Brouillette et al., 2012; Ledo et al., 2013; Nery et al., 2014). Injected Aβ1–42 levels were ∼fourfold compared with levels measured in transgenic APP/PS1 mice (Maia et al., 2013). In agreement with previous studies, intracerebroventricular injection of Aβ1–42 oligomers induced brain inflammation, as reflected by increased gene expression of Il6, Il1β, and TNF in the hippocampus, the area of the brain crucial for memory and among the first regions to be affected in Alzheimer's disease. Using the same mouse model as in our study, Kuperstein et al. (2010) found inhibition of new memory formation 24 h after Aβ1–42 oligomer injection. Because elevated cytokine levels in hippocampus are able to induce memory impairment in rats (Czerniawski and Guzowski, 2014), it could be speculated that elevated proinflammatory cytokine levels contribute to memory deterioration in this model of Alzheimer's disease.
Several studies have shown that Alzheimer's disease is associated with morphological and functional changes at the BCSFB (Serot et al., 2000; Serot et al., 2003; Emerich et al., 2005; Krzyzanowska and Carro, 2012; Serot et al., 2012; Marques et al., 2013; Spector and Johanson, 2013). However, it is not fully understood how the integrity of the BCSFB is affected in Alzheimer's disease and whether the CP is involved in triggering and/or enhancing the inflammatory response to Aβ. Therefore, we studied the effect of intracerebroventricular injection of Aβ1–42 oligomers on the BCSFB. The BCSFB is formed by a single continuous layer of modified cuboidal epithelium, the CPE cells, which are attached on their basal sides to the basal lamina (Wolburg and Paulus, 2010). CPE cells are large and have a centrally positioned spherical nucleus, copious cytoplasm, and plenty of apically positioned microvilli. In addition to their barrier function, CPE cells are responsible for the production and secretion of CSF, as well as other biologically active components involved in nutrition, endocrine function, and clearance of the brain areas (Johanson et al., 2008). Here, we show that the CP responds to intracerebroventricular injection of soluble Aβ1–42 oligomers by increasing its expression of the proinflammatory cytokines Il1β, Il6, Tnfα, and Inos. This was associated with significantly elevated levels of cytokines and chemokines in the CSF. One of the Alzheimer's disease theories proposes that monomeric and oligomeric Aβ activate microglia, which respond by secreting proinflammatory cytokines, leading to the spread of inflammation, cell death, and overall neurodegeneration (Maezawa et al., 2011; Solito and Sastre, 2012). However, despite the observed increased mRNA levels of proinflammatory cytokines in the hippocampus upon intracerebroventricular injection of Aβ1–42 oligomers, we did not observe significant microglial activation. This indicates that the inflammatory signal generated in the CPE in response to soluble Aβ1–42 oligomers might occur before microglia activation during the development of Alzheimer's disease. However, we do not exclude that the injected Aβ1–42 oligomers also affect other cell types, as noted in the past by other research groups (Kopec and Carroll, 1998; von Bernhardi and Eugenín, 2004).
Some of the reported Alzheimer's disease-associated changes involve CPE cell atrophy, accumulation of Biondi tangles and lipofuscin deposits, irregular basement thickening, decreased CSF production and secretion, and diminished capacity for clearance of unwanted molecules from CSF (Emerich et al., 2005). In our study, we observed that a single intracerebroventricular injection of Aβ1–42 oligomers affected the cuboidal morphology of CPE cells, suggesting that some of the observed morphological changes at the CPE in Alzheimer's disease occur early during disease development.
The ability of CPE cells to restrict paracellular passage of molecules depends on the presence of junctional complexes at their apical sides (Redzic, 2011). Several studies have shown a link between increased cytokine levels and decreased tight junction expression at the BBB and BCSFB (Minagar and Alexander, 2003; Barichello et al., 2011; Chai et al., 2014). Diminished BBB integrity, which leads to an increase in permeability and a decrease in tight junction expression, was found in both mouse and human Alzheimer's disease studies (Romanitan et al., 2010; Biron et al., 2011; Carrano et al., 2011; Hartz et al., 2012). In agreement with the Aβ1–42 oligomer-induced cytokine increase in the CSF, we observed that a single intracerebroventricular injection of Aβ1–42 oligomers affected the gene and protein expression levels of tight junctions. Furthermore, we observed increased leakage of fluorescently labeled dextran from blood into the CSF, indicating that Aβ1–42 injection leads to loss of BCSFB integrity. In contrast, we did not observe loss of BBB integrity 6 h after intracerebroventricular injection of Aβ1–42 oligomers.
MMPs have been recognized as important players in neurodegenerative diseases (Rosenberg, 2009). The large family of MMPs includes collagenases, gelatinases, stromelysins, and membrane-type MMPs, which are expressed by endothelial, epithelial, and inflammatory cells at the different brain barriers in response to inflammation (Vandenbroucke and Libert, 2014). The involvement of MMPs in BCSFB breakdown has been observed in various disease models (Zeni et al., 2007; Batra et al., 2010; Vandenbroucke et al., 2012). So far, there are no data implicating MMPs in the loss of BCSFB integrity during Alzheimer's disease. In our acute model of Alzheimer's disease, we observed increased mRNA levels of several MMPs associated with increased MMP activity in the CSF. The Aβ1–42 oligomer-induced increase was most prominent for Mmp3. Importantly, the broad-spectrum MMP inhibitor GM6001 prevented the detrimental effects of Aβ1–42 oligomer injection on the BCSFB, suggesting the involvement of MMPs in Aβ1–42 oligomer-induced BCSFB breakdown. Although we did not show this in our study, the observed BCSFB dysfunction might be due to direct cleavage of tight junction proteins, as was shown previously for several MMPs (Nava et al., 2013), or it might also be caused by cytokine activation or cleavage of extracellular matrix components, as we have shown previously (Vandenbroucke et al., 2012; Vandenbroucke et al., 2013).
MMP3 is believed to be a key player in inflammation and is secreted by CPE cells (Thouvenot et al., 2006). Indeed, we were able to detect secreted MMP3 in the CSF of Aβ1–42 oligomer-injected mice. Interestingly, several recent studies propose MMP3 as a potential biomarker for Alzheimer's disease because it is significantly upregulated in the brain, CSF, and plasma of Alzheimer's disease patients (Yoshiyama et al., 2000; Horstmann et al., 2010; Hanzel et al., 2014; Kauwe et al., 2014; Mroczko et al., 2014). A recent study in a mouse model of spinal cord injury showed that MMP3-deficient mice display reduced disruption of the blood–spinal cord barrier (Lee et al., 2014). To further study the role of MMP3, we injected MMP3-deficient mice intracerebroventricularly with Aβ1–42 oligomer and found that the absence of MMP3 strongly reduced the Aβ1–42 oligomer-induced BCSFB leakage. Moreover, Mmp8, Mmp9, and Mmp13 mRNA levels were elevated in the CP upon intracerebroventricular injection of Aβ1–42 oligomer, so these MMPs might also play a role in the observed loss of BCSFB integrity. In previous work, we showed that MMP8 contributes to inflammation-induced BCSFB leakage (Vandenbroucke et al., 2012) and MMP8 (Schubert-Unkmeir et al., 2009), MMP9 (Chiu and Lai, 2013), and MMP13 (Vandenbroucke et al., 2013) have been linked to tight junction dysregulation. Previously, MMP9 was shown to be involved in BBB breakdown mediated by ApoE4 (Halliday et al., 2013). Altogether, our data provide solid evidence for a detrimental role of MMPs in Aβ1–42 oligomer-induced BCSFB disruption.
As schematically depicted in Figure 8, our data indicate that Aβ1–42 oligomers, via CPE-derived cytokine and MMP secretion, might induce BCSFB barrier breakdown early during the development of Alzheimer's disease, thereby contributing to enhancement of neuroinflammation. Indeed, loss of brain barrier integrity might further aggravate neuroinflammation by entrance of undesirable molecules into the brain. It has been suggested that BBB dysfunction might contribute to Alzheimer's disease (Erickson and Banks, 2013). Based on our results, BCSFB dysfunction might be an even earlier event during disease development, so further research is needed to study whether restoring BCSFB integrity might reduce neuroinflammation during Alzheimer's disease.
Figure 8.

Schematic overview of direct effects of Aβ1–42 oligomers on the BCSFB. A monolayer of CPE cells, tightly connected by tight junctions, restricts entrance of molecules from the fenestrated capillaries into the CSF. Injection of oligomerized Aβ1–42 into the cerebral ventricles of mice (1) leads to secretion of MMPs, cytokines, and chemokines from CPE cells into the CSF (2), and this induces disruption of tight junctions (3), eventually resulting in BCSFB leakage.
In conclusion, our study shows that the presence of Aβ1–42 oligomers in the CSF induces disruption of the BCSFB via the production of proinflammatory cytokines and MMPs, which is linked to loss of tight junction functionality.
Footnotes
This work was supported by the Research Foundation–Flanders, the Concerted Research Actions of Ghent University, the Belgian Science Policy (Interuniversity Attraction Pools Grant IAP7/07), and the Ministry of Education, Science and Technological Development of the Republic of Serbia (Grant ON173056). The Zeiss Merlin with Gatan 3View2XP was acquired through a CLEM grant from Minister Ingrid Lieten to the VIB Bio-Imaging Core. We thank Amin Bredan for editing the manuscript and Joke Vanden Berghe and Sonia Bartunkova for technical assistance.
The authors declare no competing financial interests.
References
- Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, Cooper NR, Eikelenboom P, Emmerling M, Fiebich BL, Finch CE, Frautschy S, Griffin WS, Hampel H, Hull M, Landreth G, Lue L, Mrak R, Mackenzie IR, McGeer PL, et al. Inflammation and Alzheimer's disease. Neurobiol Aging. 2000;21:383–421. doi: 10.1016/S0197-4580(00)00124-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvira-Botero X, Carro EM. Clearance of amyloid-beta peptide across the choroid plexus in Alzheimer's disease. Curr Aging Sci. 2010;3:219–229. doi: 10.2174/1874609811003030219. [DOI] [PubMed] [Google Scholar]
- Barichello T, Pereira JS, Savi GD, Generoso JS, Cipriano AL, Silvestre C, Petronilho F, Dal-Pizzol F, Vilela MC, Teixeira AL. A kinetic study of the cytokine/chemokines levels and disruption of blood-brain barrier in infant rats after pneumococcal meningitis. J Neuroimmunol. 2011;233:12–17. doi: 10.1016/j.jneuroim.2010.10.035. [DOI] [PubMed] [Google Scholar]
- Batra A, Latour LL, Ruetzler CA, Hallenbeck JM, Spatz M, Warach S, Henning EC. Increased plasma and tissue MMP levels are associated with BCSFB and BBB disruption evident on post-contrast FLAIR after experimental stroke. J Cereb Blood Flow Metab. 2010;30:1188–1199. doi: 10.1038/jcbfm.2010.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biron KE, Dickstein DL, Gopaul R, Jefferies WA. Amyloid triggers extensive cerebral angiogenesis causing blood brain barrier permeability and hypervascularity in Alzheimer's disease. PLoS One. 2011;6:e23789. doi: 10.1371/journal.pone.0023789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brouillette J, Caillierez R, Zommer N, Alves-Pires C, Benilova I, Blum D, De Strooper B, Buée L. Neurotoxicity and memory deficits induced by soluble low-molecular-weight amyloid-beta1–42 oligomers are revealed in vivo by using a novel animal model. J Neurosci. 2012;32:7852–7861. doi: 10.1523/JNEUROSCI.5901-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrano A, Hoozemans JJ, van der Vies SM, Rozemuller AJ, van Horssen J, de Vries HE. Amyloid Beta induces oxidative stress-mediated blood-brain barrier changes in capillary amyloid angiopathy. Antioxid Redox Signal. 2011;15:1167–1178. doi: 10.1089/ars.2011.3895. [DOI] [PubMed] [Google Scholar]
- Cetin F, Yazihan N, Dincer S, Akbulut G. The effect of intracerebroventricular injection of beta amyloid peptide (1–42) on caspase-3 activity, lipid peroxidation, nitric oxide and NOS expression in young adult and aged rat brain. Turk Neurosurg. 2013;23:144–150. doi: 10.5137/1019-5149.JTN.5855-12.1. [DOI] [PubMed] [Google Scholar]
- Chai Q, He WQ, Zhou M, Lu H, Fu ZF. Enhancement of blood-brain barrier permeability and reduction of tight junction protein expression are modulated by chemokines/cytokines induced by rabies virus infection. J Virol. 2014;88:4698–4710. doi: 10.1128/JVI.03149-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu PS, Lai SC. Matrix metalloproteinase-9 leads to claudin-5 degradation via the NF-kappaB pathway in BALB/c mice with eosinophilic meningoencephalitis caused by Angiostrongylus cantonensis. PLoS One. 2013;8:e53370. doi: 10.1371/journal.pone.0053370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coisne C, Engelhardt B. Tight junctions in brain barriers during central nervous system inflammation. Antioxid Redox Signal. 2011;15:1285–1303. doi: 10.1089/ars.2011.3929. [DOI] [PubMed] [Google Scholar]
- Crossgrove JS, Li GJ, Zheng W. The choroid plexus removes beta-amyloid from brain cerebrospinal fluid. Exp Biol Med (Maywood) 2005;230:771–776. doi: 10.1177/153537020523001011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crossgrove JS, Smith EL, Zheng W. Macromolecules involved in production and metabolism of beta-amyloid at the brain barriers. Brain Res. 2007;1138:187–195. doi: 10.1016/j.brainres.2006.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czerniawski J, Guzowski JF. Acute neuroinflammation impairs context discrimination memory and disrupts pattern separation processes in hippocampus. J Neurosci. 2014;34:12470–12480. doi: 10.1523/JNEUROSCI.0542-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deane R, Bell RD, Sagare A, Zlokovic BV. Clearance of amyloid-beta peptide across the blood-brain barrier: implication for therapies in Alzheimer's disease. CNS Neurol Disord Drug Targets. 2009;8:16–30. doi: 10.2174/187152709787601867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demeestere D, Libert C, Vandenbroucke RE. Clinical implications of leukocyte infiltration at the choroid plexus in (neuro)inflammatory disorders. Drug discovery today. 2015 doi: 10.1016/j.drudis.2015.05.003. [DOI] [PubMed] [Google Scholar]
- Denk W, Horstmann H. Serial block-face scanning electron microscopy to reconstruct three-dimensional tissue nanostructure. PLoS Biol. 2004;2:e329. doi: 10.1371/journal.pbio.0020329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emerich DF, Skinner SJ, Borlongan CV, Vasconcellos AV, Thanos CG. The choroid plexus in the rise, fall and repair of the brain. Bioessays. 2005;27:262–274. doi: 10.1002/bies.20193. [DOI] [PubMed] [Google Scholar]
- Engelhart MJ, Geerlings MI, Meijer J, Kiliaan A, Ruitenberg A, van Swieten JC, Stijnen T, Hofman A, Witteman JC, Breteler MM. Inflammatory proteins in plasma and the risk of dementia: the rotterdam study. Arch Neurol. 2004;61:668–672. doi: 10.1001/archneur.61.5.668. [DOI] [PubMed] [Google Scholar]
- Erickson MA, Banks WA. Blood-brain barrier dysfunction as a cause and consequence of Alzheimer's disease. J Cereb Blood Flow Metab. 2013;33:1500–1513. doi: 10.1038/jcbfm.2013.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floden AM, Li S, Combs CK. Beta-amyloid-stimulated microglia induce neuron death via synergistic stimulation of tumor necrosis factor alpha and NMDA receptors. J Neurosci. 2005;25:2566–2575. doi: 10.1523/JNEUROSCI.4998-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grammas P, Ovase R. Inflammatory factors are elevated in brain microvessels in Alzheimer's disease. Neurobiol Aging. 2001;22:837–842. doi: 10.1016/S0197-4580(01)00276-7. [DOI] [PubMed] [Google Scholar]
- Guo S, Wang S, Kim WJ, Lee SR, Frosch MP, Bacskai BJ, Greenberg SM, Lo EH. Effects of apoE isoforms on beta-amyloid-induced matrix metalloproteinase-9 in rat astrocytes. Brain Res. 2006;1111:222–226. doi: 10.1016/j.brainres.2006.06.041. [DOI] [PubMed] [Google Scholar]
- Hains BC, Waxman SG. Activated microglia contribute to the maintenance of chronic pain after spinal cord injury. J Neurosci. 2006;26:4308–4317. doi: 10.1523/JNEUROSCI.0003-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halliday MR, Pomara N, Sagare AP, Mack WJ, Frangione B, Zlokovic BV. Relationship between cyclophilin a levels and matrix metalloproteinase 9 activity in cerebrospinal fluid of cognitively normal apolipoprotein e4 carriers and blood-brain barrier breakdown. JAMA Neurol. 2013;70:1198–1200. doi: 10.1001/jamaneurol.2013.3841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanzel CE, Iulita MF, Eyjolfsdottir H, Hjorth E, Schultzberg M, Eriksdotter M, Cuello AC. Analysis of matrix metallo-proteases and the plasminogen system in mild cognitive impairment and Alzheimer's disease cerebrospinal fluid. J Alzheimers Dis. 2014;40:667–678. doi: 10.3233/JAD-132282. [DOI] [PubMed] [Google Scholar]
- Hartz AM, Bauer B, Soldner EL, Wolf A, Boy S, Backhaus R, Mihaljevic I, Bogdahn U, Klünemann HH, Schuierer G, Schlachetzki F. Amyloid-beta contributes to blood-brain barrier leakage in transgenic human amyloid precursor protein mice and in humans with cerebral amyloid angiopathy. Stroke. 2012;43:514–523. doi: 10.1161/STROKEAHA.111.627562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horstmann S, Budig L, Gardner H, Koziol J, Deuschle M, Schilling C, Wagner S. Matrix metalloproteinases in peripheral blood and cerebrospinal fluid in patients with Alzheimer's disease. Int Psychogeriatr. 2010;22:966–972. doi: 10.1017/S1041610210000827. [DOI] [PubMed] [Google Scholar]
- Johanson CE, Duncan JA, 3rd, Klinge PM, Brinker T, Stopa EG, Silverberg GD. Multiplicity of cerebrospinal fluid functions: New challenges in health and disease. Cerebrospinal Fluid Res. 2008;5:10. doi: 10.1186/1743-8454-5-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnstone M, Gearing AJ, Miller KM. A central role for astrocytes in the inflammatory response to beta-amyloid; chemokines, cytokines and reactive oxygen species are produced. J Neuroimmunol. 1999;93:182–193. doi: 10.1016/S0165-5728(98)00226-4. [DOI] [PubMed] [Google Scholar]
- Kauwe JS, Bailey MH, Ridge PG, Perry R, Wadsworth ME, Hoyt KL, Staley LA, Karch CM, Harari O, Cruchaga C, Ainscough BJ, Bales K, Pickering EH, Bertelsen S, Bertelsen S, Fagan AM, Holtzman DM, Morris JC, Goate AM. Genome-wide association study of CSF levels of 59 alzheimer's disease candidate proteins: significant associations with proteins involved in amyloid processing and inflammation. PLoS Genet. 2014;10:e1004758. doi: 10.1371/journal.pgen.1004758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong LN, Zuo PP, Mu L, Liu YY, Yang N. Gene expression profile of amyloid beta protein-injected mouse model for Alzheimer disease. Acta Pharmacol Sin. 2005;26:666–672. doi: 10.1111/j.1745-7254.2005.00129.x. [DOI] [PubMed] [Google Scholar]
- Kopec KK, Carroll RT. Alzheimer's beta-amyloid peptide 1–42 induces a phagocytic response in murine microglia. J Neurochem. 1998;71:2123–2131. doi: 10.1046/j.1471-4159.1998.71052123.x. [DOI] [PubMed] [Google Scholar]
- Krzyzanowska A, Carro E. Pathological alteration in the choroid plexus of Alzheimer's disease: implication for new therapy approaches. Front Pharmacol. 2012;3:75. doi: 10.3389/fphar.2012.00075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuperstein I, Broersen K, Benilova I, Rozenski J, Jonckheere W, Debulpaep M, Vandersteen A, Segers-Nolten I, Van Der Werf K, Subramaniam V, Braeken D, Callewaert G, Bartic C, D'Hooge R, Martins IC, Rousseau F, Schymkowitz J, De Strooper B. Neurotoxicity of Alzheimer's disease Abeta peptides is induced by small changes in the Abeta42 to Abeta40 ratio. EMBO J. 2010;29:3408–3420. doi: 10.1038/emboj.2010.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledo JH, Azevedo EP, Clarke JR, Ribeiro FC, Figueiredo CP, Foguel D, De Felice FG, Ferreira ST. Amyloid-beta oligomers link depressive-like behavior and cognitive deficits in mice. Mol Psychiatry. 2013;18:1053–1054. doi: 10.1038/mp.2012.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JY, Choi HY, Ahn HJ, Ju BG, Yune TY. Matrix metalloproteinase-3 promotes early blood-spinal cord barrier disruption and hemorrhage and impairs long-term neurological recovery after spinal cord injury. Am J Pathol. 2014;184:2985–3000. doi: 10.1016/j.ajpath.2014.07.016. [DOI] [PubMed] [Google Scholar]
- Lee YB, Nagai A, Kim SU. Cytokines, chemokines, and cytokine receptors in human microglia. J Neurosci Res. 2002;69:94–103. doi: 10.1002/jnr.10253. [DOI] [PubMed] [Google Scholar]
- Liu L, Duff K. A technique for serial collection of cerebrospinal fluid from the cisterna magna in mouse. J Vis Exp. 2008;pii:960. doi: 10.3791/960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maezawa I, Zimin PI, Wulff H, Jin LW. Amyloid-beta protein oligomer at low nanomolar concentrations activates microglia and induces microglial neurotoxicity. J Biol Chem. 2011;286:3693–3706. doi: 10.1074/jbc.M110.135244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maia LF, Kaeser SA, Reichwald J, Hruscha M, Martus P, Staufenbiel M, Jucker M. Changes in amyloid-beta and Tau in the cerebrospinal fluid of transgenic mice overexpressing amyloid precursor protein. Sci Transl Med. 2013;5:194re192. doi: 10.1126/scitranslmed.3006446. [DOI] [PubMed] [Google Scholar]
- Marco S, Skaper SD. Amyloid beta-peptide1–42 alters tight junction protein distribution and expression in brain microvessel endothelial cells. Neurosci Lett. 2006;401:219–224. doi: 10.1016/j.neulet.2006.03.047. [DOI] [PubMed] [Google Scholar]
- Marques F, Sousa JC, Correia-Neves M, Oliveira P, Sousa N, Palha JA. The choroid plexus response to peripheral inflammatory stimulus. Neuroscience. 2007;144:424–430. doi: 10.1016/j.neuroscience.2006.09.029. [DOI] [PubMed] [Google Scholar]
- Marques F, Sousa JC, Coppola G, Falcao AM, Rodrigues AJ, Geschwind DH, Sousa N, Correia-Neves M, Palha JA. Kinetic profile of the transcriptome changes induced in the choroid plexus by peripheral inflammation. J Cereb Blood Flow Metab. 2009a;29:921–932. doi: 10.1038/jcbfm.2009.15. [DOI] [PubMed] [Google Scholar]
- Marques F, Sousa JC, Coppola G, Geschwind DH, Sousa N, Palha JA, Correia-Neves M. The choroid plexus response to a repeated peripheral inflammatory stimulus. BMC Neurosci. 2009b;10:135. doi: 10.1186/1471-2202-10-135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marques F, Sousa JC, Sousa N, Palha JA. Blood-brain-barriers in aging and in Alzheimer's disease. Mol Neurodegener. 2013;8:38. doi: 10.1186/1750-1326-8-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean CA, Cherny RA, Fraser FW, Fuller SJ, Smith MJ, Beyreuther K, Bush AI, Masters CL. Soluble pool of Abeta amyloid as a determinant of severity of neurodegeneration in Alzheimer's disease. Ann Neurol. 1999;46:860–866. doi: 10.1002/1531-8249(199912)46:6<860::AID-ANA8>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- Minagar A, Alexander JS. Blood-brain barrier disruption in multiple sclerosis. Mult Scler. 2003;9:540–549. doi: 10.1191/1352458503ms965oa. [DOI] [PubMed] [Google Scholar]
- Mitchell K, Yang HY, Berk JD, Tran JH, Iadarola MJ. Monocyte chemoattractant protein-1 in the choroid plexus: a potential link between vascular pro-inflammatory mediators and the CNS during peripheral tissue inflammation. Neuroscience. 2009;158:885–895. doi: 10.1016/j.neuroscience.2008.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mroczko B, Groblewska M, Zboch M, Kulczyńska A, Koper OM, Szmitkowski M, Kornhuber J, Lewczuk P. Concentrations of matrix metalloproteinases and their tissue inhibitors in the cerebrospinal fluid of patients with Alzheimer's disease. J Alzheimers Dis. 2014;40:351–357. doi: 10.3233/JAD-131634. [DOI] [PubMed] [Google Scholar]
- Mudgett JS, Hutchinson NI, Chartrain NA, Forsyth AJ, McDonnell J, Singer II, Bayne EK, Flanagan J, Kawka D, Shen CF, Stevens K, Chen H, Trumbauer M, Visco DM. Susceptibility of stromelysin 1-deficient mice to collagen-induced arthritis and cartilage destruction. Arthritis Rheum. 1998;41:110–121. doi: 10.1002/1529-0131(199801)41:1<110::AID-ART14>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- Nava P, Kamekura R, Nusrat A. Cleavage of transmembrane junction proteins and their role in regulating epithelial homeostasis. Tissue Barriers. 2013;1:e24783. doi: 10.4161/tisb.24783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nery LR, Eltz NS, Hackman C, Fonseca R, Altenhofen S, Guerra HN, Freitas VM, Bonan CD, Vianna MR. Brain intraventricular injection of amyloid-beta in zebrafish embryo impairs cognition and increases tau phosphorylation, effects reversed by lithium. PLoS One. 2014;9:e105862. doi: 10.1371/journal.pone.0105862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitta A, Fukuta T, Hasegawa T, Nabeshima T. Continuous infusion of beta-amyloid protein into the rat cerebral ventricle induces learning impairment and neuronal and morphological degeneration. Jpn J Pharmacol. 1997;73:51–57. doi: 10.1254/jjp.73.51. [DOI] [PubMed] [Google Scholar]
- Redzic Z. Molecular biology of the blood-brain and the blood-cerebrospinal fluid barriers: similarities and differences. Fluids Barriers CNS. 2011;8:3. doi: 10.1186/2045-8118-8-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romanitan MO, Popescu BO, Spulber S, Băjenaru O, Popescu LM, Winblad B, Bogdanovic N. Altered expression of claudin family proteins in Alzheimer's disease and vascular dementia brains. J Cell Mol Med. 2010;14:1088–1100. doi: 10.1111/j.1582-4934.2009.00999.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg GA. Matrix metalloproteinases and their multiple roles in neurodegenerative diseases. Lancet Neurol. 2009;8:205–216. doi: 10.1016/S1474-4422(09)70016-X. [DOI] [PubMed] [Google Scholar]
- Sagare AP, Bell RD, Zhao Z, Ma Q, Winkler EA, Ramanathan A, Zlokovic BV. Pericyte loss influences Alzheimer-like neurodegeneration in mice. Nat Commun. 2013;4:2932. doi: 10.1038/ncomms3932. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert-Unkmeir A, Slanina H, Frosch M. Mammalian cell transcriptome in response to meningitis-causing pathogens. Expert Rev Mol Diagn. 2009;9:833–842. doi: 10.1586/erm.09.68. [DOI] [PubMed] [Google Scholar]
- Serot JM, Béné MC, Foliguet B, Faure GC. Morphological alterations of the choroid plexus in late-onset Alzheimer's disease. Acta Neuropathol. 2000;99:105–108. doi: 10.1007/PL00007412. [DOI] [PubMed] [Google Scholar]
- Serot JM, Béné MC, Faure GC. Choroid plexus, aging of the brain, and Alzheimer's disease. Front Biosci. 2003;8:s515–s521. doi: 10.2741/1085. [DOI] [PubMed] [Google Scholar]
- Serot JM, Zmudka J, Jouanny P. A possible role for CSF turnover and choroid plexus in the pathogenesis of late onset Alzheimer's disease. J Alzheimers Dis. 2012;30:17–26. doi: 10.3233/JAD-2012-111964. [DOI] [PubMed] [Google Scholar]
- Sharma HS, Zimmermann-Meinzingen S, Johanson CE. Cerebrolysin reduces blood-cerebrospinal fluid barrier permeability change, brain pathology, and functional deficits following traumatic brain injury in the rat. Ann N Y Acad Sci. 2010;1199:125–137. doi: 10.1111/j.1749-6632.2009.05329.x. [DOI] [PubMed] [Google Scholar]
- Simard PF, Tosun C, Melnichenko L, Ivanova S, Gerzanich V, Simard JM. Inflammation of the choroid plexus and ependymal layer of the ventricle following intraventricular hemorrhage. Transl Stroke Res. 2011;2:227–231. doi: 10.1007/s12975-011-0070-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smits HA, Rijsmus A, van Loon JH, Wat JW, Verhoef J, Boven LA, Nottet HS. Amyloid-beta-induced chemokine production in primary human macrophages and astrocytes. J Neuroimmunol. 2002;127:160–168. doi: 10.1016/S0165-5728(02)00112-1. [DOI] [PubMed] [Google Scholar]
- Solito E, Sastre M. Microglia function in Alzheimer's disease. Front Pharmacol. 2012;3:14. doi: 10.3389/fphar.2012.00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spector R, Johanson CE. Sustained choroid plexus function in human elderly and Alzheimer's disease patients. Fluids Barriers CNS. 2013;10:28. doi: 10.1186/2045-8118-10-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thouvenot E, Lafon-Cazal M, Demettre E, Jouin P, Bockaert J, Marin P. The proteomic analysis of mouse choroid plexus secretome reveals a high protein secretion capacity of choroidal epithelial cells. Proteomics. 2006;6:5941–5952. doi: 10.1002/pmic.200600096. [DOI] [PubMed] [Google Scholar]
- Tripathy D, Thirumangalakudi L, Grammas P. Expression of macrophage inflammatory protein 1-alpha is elevated in Alzheimer's vessels and is regulated by oxidative stress. J Alzheimers Dis. 2007;11:447–455. doi: 10.3233/jad-2007-11405. [DOI] [PubMed] [Google Scholar]
- Vandenbroucke RE, Libert C. Is there new hope for therapeutic matrix metalloproteinase inhibition? Nat Rev Drug Discov. 2014;13:904–927. doi: 10.1038/nrd4390. [DOI] [PubMed] [Google Scholar]
- Vandenbroucke RE, Dejonckheere E, Van Lint P, Demeestere D, Van Wonterghem E, Vanlaere I, Puimège L, Van Hauwermeiren F, De Rycke R, Mc Guire C, Campestre C, López-Otin C, Matthys P, Leclercq G, Libert C. Matrix metalloprotease 8-dependent extracellular matrix cleavage at the blood-CSF barrier contributes to lethality during systemic inflammatory diseases. J Neurosci. 2012;32:9805–9816. doi: 10.1523/JNEUROSCI.0967-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandenbroucke RE, Dejonckheere E, Van Hauwermeiren F, Lodens S, De Rycke R, Van Wonterghem E, Staes A, Gevaert K, López-Otin C, Libert C. Matrix metalloproteinase 13 modulates intestinal epithelial barrier integrity in inflammatory diseases by activating TNF. EMBO Mol Med. 2013;5:932–948. doi: 10.1002/emmm.201202100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, Speleman F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002;3:RESEARCH0034. doi: 10.1186/gb-2002-3-7-research0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Bernhardi R, Eugenín J. Microglial reactivity to beta-amyloid is modulated by astrocytes and proinflammatory factors. Brain Res. 2004;1025:186–193. doi: 10.1016/j.brainres.2004.07.084. [DOI] [PubMed] [Google Scholar]
- Wang XX, Tan MS, Yu JT, Tan L. Matrix metalloproteinases and their multiple roles in Alzheimer's disease. BioMed Res Int. 2014;2014:908636. doi: 10.1155/2014/908636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler EA, Sagare AP, Zlokovic BV. The pericyte: a forgotten cell type with important implications for Alzheimer's disease? Brain Pathol. 2014;24:371–386. doi: 10.1111/bpa.12152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolburg H, Paulus W. Choroid plexus: biology and pathology. Acta Neuropathol. 2010;119:75–88. doi: 10.1007/s00401-009-0627-8. [DOI] [PubMed] [Google Scholar]
- Wright AL, Zinn R, Hohensinn B, Konen LM, Beynon SB, Tan RP, Clark IA, Abdipranoto A, Vissel B. Neuroinflammation and neuronal loss precede Abeta plaque deposition in the hAPP-J20 mouse model of Alzheimer's disease. PLoS One. 2013;8:e59586. doi: 10.1371/journal.pone.0059586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan P, Hu X, Song H, Yin K, Bateman RJ, Cirrito JR, Xiao Q, Hsu FF, Turk JW, Xu J, Hsu CY, Holtzman DM, Lee JM. Matrix metalloproteinase-9 degrades amyloid-beta fibrils in vitro and compact plaques in situ. J Biol Chem. 2006;281:24566–24574. doi: 10.1074/jbc.M602440200. [DOI] [PubMed] [Google Scholar]
- Yin KJ, Cirrito JR, Yan P, Hu X, Xiao Q, Pan X, Bateman R, Song H, Hsu FF, Turk J, Xu J, Hsu CY, Mills JC, Holtzman DM, Lee JM. Matrix metalloproteinases expressed by astrocytes mediate extracellular amyloid-beta peptide catabolism. J Neurosci. 2006;26:10939–10948. doi: 10.1523/JNEUROSCI.2085-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshiyama Y, Asahina M, Hattori T. Selective distribution of matrix metalloproteinase-3 (MMP-3) in Alzheimer's disease brain. Acta Neuropathol. 2000;99:91–95. doi: 10.1007/PL00007428. [DOI] [PubMed] [Google Scholar]
- Zeni P, Doepker E, Schulze-Topphoff U, Huewel S, Tenenbaum T, Galla HJ. MMPs contribute to TNF-alpha-induced alteration of the blood-cerebrospinal fluid barrier in vitro. Am J Physiol Cell Physiol. 2007;293:C855–C864. doi: 10.1152/ajpcell.00470.2006. [DOI] [PubMed] [Google Scholar]
- Zlokovic BV. The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron. 2008;57:178–201. doi: 10.1016/j.neuron.2008.01.003. [DOI] [PubMed] [Google Scholar]
- Zlokovic BV. Neurovascular pathways to neurodegeneration in Alzheimer's disease and other disorders. Nat Rev Neurosci. 2011;12:723–738. doi: 10.1038/nrn3114. [DOI] [PMC free article] [PubMed] [Google Scholar]