

Abstract
Changes in expression of the neural cell adhesion molecule 2 (NCAM2) have been proposed to contribute to neurodevelopmental disorders in humans. The role of NCAM2 in neuronal differentiation remains, however, poorly understood. Using genetically encoded Ca2+ reporters, we show that clustering of NCAM2 at the cell surface of mouse cortical neurons induces submembrane [Ca2+] spikes, which depend on the L-type voltage-dependent Ca2+ channels (VDCCs) and require activation of the protein tyrosine kinase c-Src. We also demonstrate that clustering of NCAM2 induces L-type VDCC- and c-Src-dependent activation of CaMKII. NCAM2-dependent submembrane [Ca2+] spikes colocalize with the bases of filopodia. NCAM2 activation increases the density of filopodia along neurites and neurite branching and outgrowth in an L-type VDCC-, c-Src-, and CaMKII-dependent manner. Our results therefore indicate that NCAM2 promotes the formation of filopodia and neurite branching by inducing Ca2+ influx and CaMKII activation. Changes in NCAM2 expression in Down syndrome and autistic patients may therefore contribute to abnormal neurite branching observed in these disorders.
Keywords: calcium, cell adhesion molecule, neurite outgrowth, neurons, voltage-dependent Ca2+ channel
Introduction
Neuronal differentiation during brain development involves the growth and extensive branching of axons and dendrites to enable the formation of numerous contacts with other neurons required for the integration of neurons into the neuronal networks in the brain. Neuronal differentiation is highly regulated by cell adhesion molecules expressed at the neuronal cell surface, which activate a plethora of intracellular signaling cascades required for growth (Aricescu and Jones, 2007; Dalva et al., 2007; Maness and Schachner, 2007; Gahmberg et al., 2009; Togashi et al., 2009; Cavallaro and Dejana, 2011).
Neural cell adhesion molecule 2 (NCAM2), also known as the olfactory cell adhesion molecule, belongs to the Ig superfamily of cell adhesion molecules (Alenius and Bohm, 1997; Paoloni-Giacobino et al., 1997; Yoshihara et al., 1997) and mediates homophilic adhesion (Yoshihara et al., 1997; Rasmussen et al., 2008; Kulahin et al., 2011). NCAM2 is expressed predominantly in the brain, and is detectable already at postnatal day 1 (Paoloni-Giacobino et al., 1997; Yoshihara et al., 1997). Expression of NCAM2 in the developing brain and similarity to its paralog NCAM, which plays an important role in neuronal differentiation (Leshchyns'ka et al., 2003; Santuccione et al., 2005; Bodrikov et al., 2008), suggest that NCAM2 also plays a role in the regulation of neuronal development (Winther et al., 2012). In humans, NCAM2 is encoded by a gene located on chromosome 21, and its abnormal expression was therefore proposed to contribute to Down syndrome (Paoloni-Giacobino et al., 1997; Makino and McLysaght, 2010). Single nucleotide polymorphisms and deletions involving the NCAM2 gene have also been linked to autism (Molloy et al., 2005; Haldeman-Englert et al., 2010; Hussman et al., 2011).
Although NCAM2 is widely expressed in different brain regions (Paoloni-Giacobino et al., 1997), the research has mostly focused on the olfactory bulb, where NCAM2 has been shown to play a role in the compartmental organization of axons and dendrites and target selection (Alenius and Bohm, 2003; Walz et al., 2006; Borisovska et al., 2011). The role of NCAM2 in regulation of neuronal development outside of the olfactory system remains, however, poorly investigated.
Among signaling cascades activated by cell adhesion molecules, changes in intracellular Ca2+ levels play a prominent role (Doherty et al., 2000; Hansen et al., 2008; Sheng et al., 2013). Ca2+ serves as a secondary signaling messenger, which has critical roles in the regulation of neuronal motility and neurite elongation in developing neurons (Bedlack et al., 1992; Rehder and Kater, 1992; Henley and Poo, 2004; Henley et al., 2004; Gomez and Zheng, 2006; Zheng and Poo, 2007). In this study, we used genetically encoded Ca2+ reporters to investigate changes in intracellular [Ca2+] in response to NCAM2 activation in developing cortical neurons. We show that activation of NCAM2 induces submembrane [Ca2+] spikes along neurites, and particularly at the bases of filopodia. We also demonstrate that these Ca2+ changes depend on activation of the protein tyrosine kinase c-Src and Ca2+ influx via L-type voltage-dependent Ca2+ channels (VDCCs), and result in calcium/calmodulin-dependent protein kinase II (CaMKII) activation and increased formation of filopodia and neurite branching.
Materials and Methods
Antibodies, inhibitors, and toxins.
Mouse monoclonal antibodies against NCAM2 (sc-136328) from Santa Cruz Biotechnology were used for the treatment of living neurons and isolated growth cones, immunocytochemistry (IC), immunoprecipitation (IP), and Western blot (WB). We extensively tested the antibodies and confirmed that they specifically recognize the recombinant extracellular domain of NCAM2 by Western blot and NCAM2 overexpressed in transfected CHO cells by immunocytochemistry (data not shown) and Western blot (see Fig. 2B). Mouse monoclonal antibodies against CaMKIIα (sc-13141, Santa Cruz Biotechnology) were used in WB, rabbit polyclonal antibodies against CaMKIIα phosphorylated at Thr-286 (sc-12886-R, Santa Cruz Biotechnology) were used in WB, rabbit polyclonal antibodies against NCAM1 (sc-10735, Santa Cruz Biotechnology) were used in WB, mouse monoclonal antibodies against c-Src (sc-8056, Santa Cruz Biotechnology) were used in WB, rabbit polyclonal antibodies against c-Src phosphorylated at Tyr-416 (#2101, Cell Signaling Technology) were used in WB, and rabbit polyclonal antibodies against L-type Ca2+ channels (sc-16229-R, Santa Cruz Biotechnology) were used in IC. Nonimmune mouse IgG used as control for the treatment of living neurons and in IP were from Santa Cruz Biotechnology. Horseradish peroxidase (HRP)-conjugated whole IgG-specific anti-rabbit secondary antibodies used in WB and Cy2- or Cy3-conjugated anti-rabbit and anti-mouse secondary antibodies used in IC were purchased from Jackson ImmunoResearch Laboratories. Biotin-conjugated Fab-specific anti-mouse secondary antibodies from Sigma-Aldrich were used in WB to prevent detection of the heavy chain of primary antibodies used for IP or growth cone stimulation and were visualized using HRP-conjugated NeutrAvidin protein from Thermo Scientific. Alexa Fluor 546 phalloidin was from Life Technologies. The inhibitor of the Src family of protein tyrosine kinases, PP2, and ionomycin (calcium salt) were from Cayman Chemical. Nifedipine and BAPTA-AM were from Sigma-Aldrich.
Figure 2.
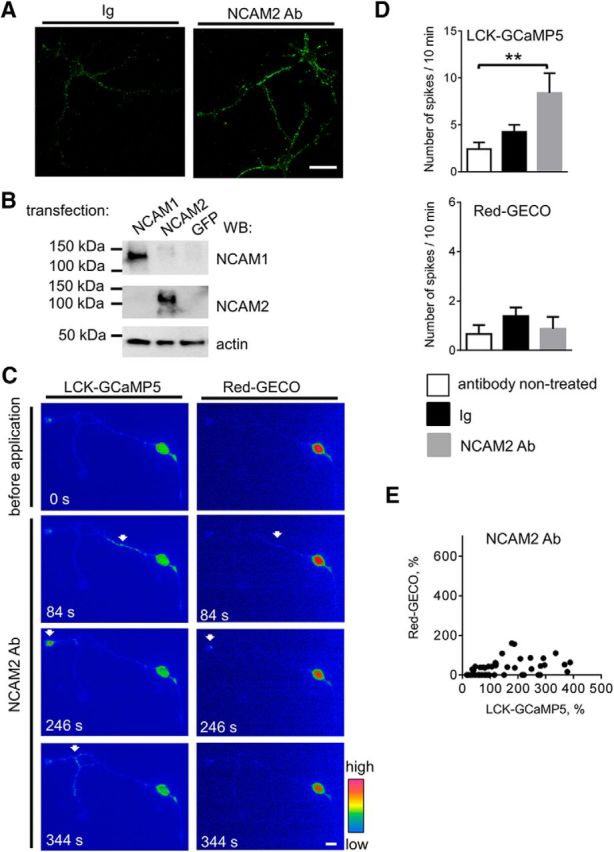
NCAM2 activation increases the frequency of submembrane [Ca2+] spikes in young cortical neurons. A, Representative images of cultured 2-d-old cortical neurons incubated for 10 min with nonimmune Igs or antibodies against the extracellular domain of NCAM2 (NCAM2 Ab) and fixed and labeled for NCAM2 are shown. Note that NCAM2 antibodies enhance clustering of NCAM2 at the neuronal cell surface. Scale bar, 20 μm. B, CHO cells transfected with NCAM1 (NCAM140 isoform), NCAM2, or GFP were analyzed by Western blot with antibodies against the extracellular domain of NCAM2, NCAM1, or actin used as the loading control. Note that NCAM2 antibodies recognize overexpressed NCAM2, but not NCAM1. C, Pseudocolored images of a cultured 2-d-old cortical neuron cotransfected with LCK-GCaMP5 and Red-GECO are shown. Images were taken before and after application of NCAM2 antibodies. Arrows show examples of transient LCK-GCaMP5 and Red-GECO fluorescence intensity increases (spikes) along neurites and in the growth cone. Scale bar, 10 μm. D, Graphs show the number of LCK-GCaMP5 or Red-GECO fluorescence intensity spikes in nontreated neurons and in neurons treated with nonimmune Igs or NCAM2 antibodies (NCAM2 Ab) for 10 min (mean + SEM; n > 15 neurons in each group). **p < 0.01, one-way ANOVA with Tukey's multiple-comparison test. Note that application of NCAM2 antibodies induces an increase in the frequency of submembrane [Ca2+] spikes. E, The graph shows the amplitude of Red-GECO fluorescence intensity increases at the sites of LCK-GCaMP5 spikes as a function of the amplitude of LCK-GCaMP5 fluorescence intensity increase in neurons treated with NCAM2 antibodies. Note the LCK-GCaMP5 fluorescence spikes, which are not accompanied by cytosolic Red-GECO spikes. n = 12 neurons analyzed.
Animals.
Brain tissues from 1- to 3-d-old C57BL/6 mice of either sex were used to prepare cultures of primary neurons and for biochemical experiments. Experiments were approved by the Animal Care and Ethics Committee of the University of New South Wales (permit 12/135B).
DNA constructs and siRNAs.
DNA constructs coding for red cytosolic (Red-GECO1, Addgene plasmid 32444), red nucleus-localized (NLS-Red-GECO1, Addgene plasmid 32462), and green plasma membrane-localized (LCK-GCaMP5G, Addgene plasmid 34924) intensiometric Ca2+ indicators were from Addgene and as described previously (Zhao et al., 2011; Akerboom et al., 2012). Control siRNA (sc-37007) and siRNAs designed to knock down expression of mouse c-Src (sc-29859), CaMKIIα (sc-29901), or CaMKIIβ (sc-38952) were from Santa Cruz Biotechnology. The efficiency of the knockdown in cultured cortical neurons has been confirmed by immunocytochemical labeling with respective antibodies, which showed an ∼50% reduction of expression of c-Src, CaMKIIα, and CaMKIIβ at 24 h after transfection.
Cell culture and transfection.
Mouse cortical neurons were prepared as described previously (Li et al., 2013). Neurons were maintained in Neurobasal A medium (Life Technologies) supplemented with 2% B-27 (Life Technologies), glutamine (Life Technologies), and FGF-2 (2 ng/ml; Life Technologies) on glass coverslips coated with poly-d-lysine (100 μg/ml). Neurons were transfected before plating by electroporation using a Neon transfection system (Life Technology).
Live cell imaging and quantification.
Cortical neurons were used for live cell imaging at 24–72 h after transfection. Unless indicated otherwise, recordings were performed in 4 mm K+ buffer [containing (in mm) 150 NaCl, 4 KCl, 2 MgCl2, 10 glucose, 10 HEPES, 2 CaCl2]. Coverslips with transfected neurons were placed into glass-bottom Petri dishes (MatTek Corporation) and kept at 37°C. Recordings were performed using an Eclipse TiE fluorescence microscope (Nikon), a Plan Apo VC 60× water-immersion objective (Nikon; numerical aperture, 1.2), a pE-2 CoolLED excitation light source, and NIS-elements software (version 4.0; Nikon). Images were captured every 2 s. Each experiment included recording of spontaneous activity (baseline) for 10 min, followed by application of either NCAM2 antibodies (2 μg/ml) or control nonimmune Igs (2 μg/ml) and recording of the induced activity for 10 min. Next, 90 mm K+ buffer [containing (in mm) 64 NaCl, 90 KCl, 2 MgCl2, 10 glucose, 10 HEPES, 2 CaCl2] was added to the neurons to induce depolarization-induced Ca2+ influx, and the neurons were recorded for an additional 10 min. When indicated, after recording baseline activity, neurons were incubated with nifedipine (10 μm) or PP2 (10 μm) for 10 min before application of NCAM2 antibodies or control nonimmune Igs.
In experiments aimed to compare the sensitivity of LCK-GCaMP5 and Red-GECO, during time-lapse recordings, neurons were treated with 20 μm ionomycin in 4 mm K+ buffer without Ca2+ to induce Ca2+ release from internal stores. In another set of experiments, neurons were incubated for 10 min in 4 mm K+ buffer without Ca2+ containing 1 mm EGTA to deplete internal Ca2+ stores. The buffer was then exchanged for 4 mm K+ buffer without Ca2+ supplemented with 4 μm ionomycin, and changes in the fluorescence intensity of the reporters were monitored during stepwise increases in extracellular Ca2+ concentration achieved by addition of the respective amounts of 2 mm CaCl2.
The fluorescence intensity of the signals was quantified in manually outlined somata, neurites, and bases of filopodia using ImageJ (National Institutes of Health). The bases of filopodia were defined as ∼3-μm-long segments of neurites wherefrom filopodia originated. Spikes were defined as transient increases in the fluorescence intensity of the LCK-GCaMP5 reporter with the amplitude at least 15% over the baseline level. Numbers and duration of spikes were calculated by analyzing time-lapse recordings in ImageJ. The amplitudes of LCK-GCaMP5 and Red-GECO fluorescence intensities within the spikes were calculated by manually outlining neurite regions comprising the spike and measuring the fluorescence intensities of the reporters in consecutive images acquired before and during the spike in ImageJ.
Immunofluorescence labeling.
Neurons were incubated with NCAM2 antibodies (2 μg/ml) or control nonimmune Igs (2 μg/ml) for 10 min at 37°C in a CO2 incubator. Unless indicated otherwise, all further procedures were performed at room temperature. Neurons were washed with PBS (pH 7.2) and then fixed for 15 min in 4% paraformaldehyde in PBS. Immunofluorescence labeling was performed essentially as described previously (Sytnyk et al., 2002, 2006). Briefly, neurons were washed with PBS and treated with 0.25% Triton X-100 in PBS for 5 min to permeabilize cell membranes. When indicated in the text, the permeabilization step was omitted to label NCAM2 at the cell surface only. Nonspecific binding was blocked by incubating neurons with 5% donkey serum in PBS for 20 min. Neurons were incubated overnight at 4°C with primary antibodies diluted in 1% donkey serum in PBS. Neurons were then washed and incubated with corresponding secondary antibodies diluted in 1% donkey serum in PBS for 1 h. After washing with PBS, neurons were embedded in Aqua-Poly/Mount (Polysciences). Immunofluorescence images were captured at room temperature using a confocal laser scanning microscope (C1si; Nikon), NIS-element software (version 4.0; Nikon), and a Plan Apo VC 60× oil-based objective (Nikon; numerical aperture, 1.4).
Analysis of numbers of filopodia, neurite length, and branching.
Analysis of the effect of NCAM2 activation on the neuronal morphology was performed essentially as described previously (Tian et al., 2012). NCAM2 antibodies (2 μg/ml) or control nonimmune Igs (2 μg/ml) were applied to neurons immediately after plating. When indicated, NCAM2 antibodies and nonimmune Igs were applied together with nifedipine (10 μm) or an inhibitor of CaMKII, KN62 (10 μm). Neurons were maintained for 24 or 48 h at 37°C in the 5% CO2 environment, then fixed with 4% paraformaldehyde in PBS applied for 15 min at room temperature and labeled with Alexa Fluor 546 phalloidin in accordance with the manufacturer's instructions. After washing with PBS, neurons were embedded in Aqua-Poly/Mount. Immunofluorescence images were captured at room temperature using a confocal laser scanning microscope C1si (Nikon), NIS-element software (version 4.0; Nikon), and a 60× oil-based objective (Nikon). Lengths of neurites were measured by manually tracing neurites in ImageJ. Numbers of filopodia and neurite branches were also counted in ImageJ. Branches were defined as protrusions with a diameter of >50% of the diameter of the parent neurite and with a length of >10 μm. All other protrusions were counted as filopodia.
Isolation of growth cones.
Brain homogenates of 1- to 3-d-old mice were prepared using a Potter homogenizer in HOMO buffer (containing 1 mm MgCl2, 1 mm CaCl2, 5 mm Tris, pH 7.4, 0.1 mm PMSF, EDTA-free protease inhibitor cocktail, and 0.32 m sucrose). Growth cones were then isolated essentially as described previously (Westphal et al., 2010; Chernyshova et al., 2011). Brain homogenates were centrifuged at 1660 × g for 15 min at 4°C. The supernatant was collected and centrifuged on a discontinuous density gradient of 0.75/1.0/2.66 m sucrose at 242,000 × g for 30 min at 4°C. The interface between the load and 0.75 m sucrose, containing growth cones, was collected, resuspended in 10 ml of HOMO buffer, and pelleted by centrifugation at 100,000 × g for 40 min at 4°C to obtain purified growth cones.
Treatment of the isolated growth cones.
Treatments were performed essentially as described previously (Westphal et al., 2010; Chernyshova et al., 2011). Freshly isolated growth cones (1 mg of total protein) were resuspended in 150 μl of HEPES buffer [containing (in mm) 150 NaCl, 4 KCl, 2 MgCl2, 10 glucose, 10 HEPES, and 2 CaCl2, pH 7.4]; incubated for 30 min at room temperature with PP2 (10 μm), BAPTA-AM (30 μm), nifedipine (10 μm), or vehicle (DMSO) used to prepare stock solutions of inhibitors; and then incubated with antibodies against NCAM2 or control nonimmune mouse Igs for 15 min at 37°C with constant gentle shaking. Samples were then loaded onto 0.5 ml cushions of 0.4 m sucrose in Tris-buffered saline (TBS, pH 7.5) and centrifuged at 100,000 × g at 4°C for 1 h. Pellets were resuspended in 150 μl Laemmli buffer and used for Western blot analysis.
Coimmunoprecipitation.
Coimmunoprecipitation was performed essentially as described previously (Shetty et al., 2013). Homogenates of brains from 1- to 3-d-old mice containing 1 mg of total protein were lysed in lysis buffer (50 mm Tris-HCl, pH 7.5, 150 mm NaCl, 1% v/v Nonidet P-40, 1 mm Na4P2O7, 1 mm NaF, 2 mm Na3VO4, EDTA-free protease inhibitor cocktail, 0.1 mm PMSF) for 30 min at 4°C. Lysates were centrifuged for 15 min at 16,000 × g at 4°C. The supernatant was precleared with protein A/G agarose beads (Santa Cruz Biotechnology) for 3 h at 4°C and then incubated with NCAM2 antibodies or control nonimmune Igs overnight at 4°C. Protein complexes were precipitated with protein A/G agarose beads applied for 3 h at 4°C. The beads were washed twice with lysis buffer and once with TBS. Protein complexes were eluted with Laemmli buffer for 10 min at 70°C and analyzed by Western blot. The approximate percentage of c-Src molecules bound to NCAM2 in brain homogenates was calculated as described previously (Santuccione et al., 2005).
Western blot analysis.
Proteins were separated using 4–12% Bis-Tris Bolt mini gels (Life Technologies). Molecular weight markers were prestained protein standards from Bio-Rad. Proteins were then electroblotted onto Immobilon-P PVDF transfer membranes (0.45 μm; Millipore). Membranes were washed with TBS (pH 7.4) and blocked for 1 h at room temperature with either 1% BSA in TBS when phosphospecific antibodies were used or 5% w/v skim milk in TBS when other antibodies were used. Membranes were then incubated with the appropriate primary antibodies overnight at room temperature followed by incubation with the corresponding HRP- or biotin-conjugated secondary antibodies for 1 h at room temperature. Biotin was then detected with HRP-conjugated NeutrAvidin applied for 1 h at room temperature. Luminata Forte Western HRP substrate (Merck Millipore) was sprayed onto the membranes to visualize the labeling. Images were captured using MicroChemi 4.2 (DNR Bio-Imaging Systems). The intensity of protein bands was measured using ImageJ.
Results
Spontaneous changes in intracellular Ca2+ levels in developing cortical neurons include submembrane [Ca2+] spikes, which can be accompanied by cytosolic [Ca2+] increases
Before analyzing the effect of NCAM2 activation on intracellular Ca2+ levels, we first investigated the levels and types of spontaneous changes in intracellular Ca2+ levels in different subcellular compartments occurring in growing cultured cortical neurons. To analyze changes in Ca2+ levels in different subcellular compartments, neurons were transfected with genetically encoded Ca2+ reporters LCK-GCaMP5 or NLS-Red-GECO, containing subcellular localization signals that target them to the neuronal plasma membrane and nucleus, respectively, or Red-GECO, which does not contain a localization signal and is targeted to the cytosol (Zhao et al., 2011; Akerboom et al., 2012). In nontreated 2- to 3-d-old neurons, the three reporters showed faint fluorescence and expected distribution, with LCK-GCaMP5 and Red-GECO detectable throughout all neurites and in soma, in agreement with their membrane and cytosolic localization, and NLS-Red-GECO fluorescence restricted to the somata in accordance with its nuclear localization (Fig. 1A). To confirm the functionality of the reporters, neurons were time-lapse recorded before and after application of 90 mm K+ used to depolarize membranes and induce Ca2+ influx via VDCCs. Application of 90 mm K+ resulted in a sharp increase in the fluorescence intensity of all three reporters in all neurons analyzed (n > 20; Fig. 1A), indicating that all three reporters can detect changes in Ca2+ levels in these neurons. Interestingly, these results also indicate that depolarization-induced Ca2+ influx at the cell surface plasma membrane detectable with LCK-GCaMP5 was efficiently translated into the Ca2+ increase in the cytosol and nuclei detectable with Red-GECO and NLS-Red-GECO, respectively.
Figure 1.
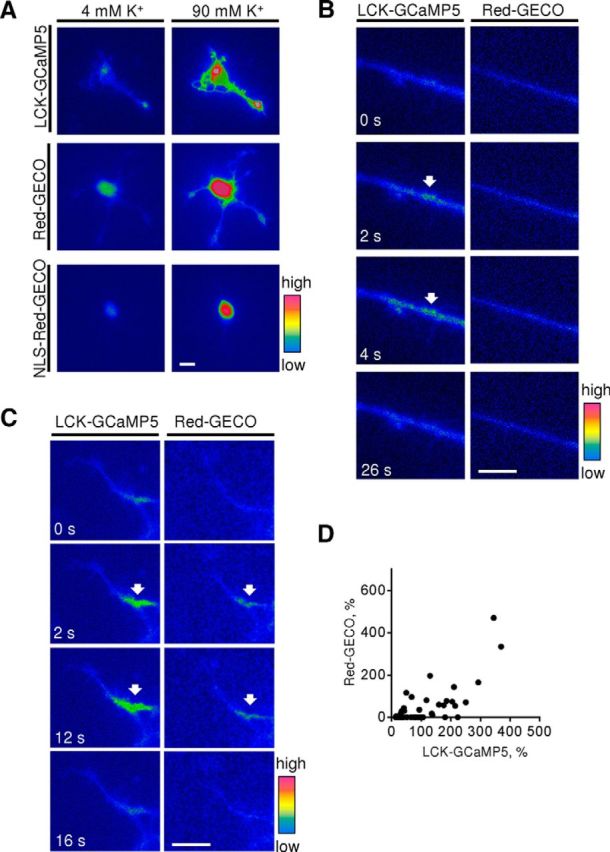
Genetically encoded Ca2+ reporters detect depolarization-induced and spontaneous changes of intracellular [Ca2+] in cortical neurons. A, Cultured 1- to 3-d-old cortical neurons were transfected with membrane-localized LCK-GCaMP5, cytosolic Red-GECO, or nucleus-localized NLS-Red-GECO. Pseudocolored images of neurons at resting conditions (4 mm K+) and 8 s after application of 90 mm K+ are shown. Note that application of 90 mm K+ induced an increase in LCK-GCaMP5, Red-GECO, and NLS-Red-GECO fluorescence intensities reflecting Ca2+ influx to the respective subcellular compartments. Similar responses were observed in n > 20 neurons in each group. B, C, Time-lapse recordings of neurites of neurons cotransfected with LCK-GCaMP5 and Red-GECO are shown. Note submembrane [Ca2+] spikes reflected by transient increases in intensity of LCK-GCaMP5 fluorescence (arrows). These spikes are either not accompanied (B) or accompanied (C, arrows) by increases in Red-GECO fluorescence intensity. Scale bars, 10 μm. D, The graph shows the amplitude of Red-GECO fluorescence intensity increases at the sites of LCK-GCaMP5 spikes as a function of the amplitude of LCK-GCaMP5 fluorescence intensity increase. Note that the amplitude of the Red-GECO fluorescence intensity increase correlates with the amplitude of the LCK-GCaMP5 fluorescence intensity increase. n = 10 neurons analyzed.
Time-lapse recordings of live, nontreated, transfected neurons showed baseline activity in neurons characterized by transient local increases in the fluorescence intensity of LCK-GCaMP5 and Red-GECO in growth cones, along neurites, or in soma of neurons. To analyze the relationship between these spikes in submembrane and cytosolic Ca2+ levels, both reporters were cotransfected into the neurons and subjected to time-lapse recordings, which showed that spikes in the fluorescence intensity of the submembrane LCK-GCaMP5 reporter were often but not always accompanied by simultaneous increases in the fluorescence intensity of the cytosolic Red-GECO reporter (Fig. 1B,C). The amplitude of Red-GECO fluorescence spikes correlated with the amplitude of LCK-GCaMP5 fluorescence spikes (Fig. 1D). Interestingly, whereas ∼80% of LCK-GCaMP5 fluorescence spikes with an amplitude over 100% of the baseline level were accompanied by Red-GECO fluorescence spikes, ∼75% of LCK-GCaMP5 fluorescence spikes with an amplitude below 100% of the baseline level were not accompanied by Red-GECO fluorescence spikes (Fig. 1D), suggesting that submembrane Ca2+ levels have to reach a certain threshold to be translated into a detectable increase in cytosolic Ca2+ levels. No changes in Red-GECO fluorescence intensity were detectable in the absence of LCK-GCaMP5 fluorescence spikes, indicating that submembrane [Ca2+] spikes trigger cytosolic [Ca2+] increases under baseline conditions. No detectable spontaneous transient changes in the fluorescence intensity of NLS-Red-GECO were observed under baseline conditions, indicating that overall nuclear Ca2+ levels remained unchanged.
NCAM2 activation induces an increase in the frequency of submembrane [Ca2+] spikes along growing neurites of cortical neurons
Immunofluorescence labeling of 2-d-old detergent nonpermeabilized cultured cortical neurons with antibodies against the extracellular domain of NCAM2 showed that NCAM2 was distributed diffusely or in small clusters at the cell surface of somata, neurites, and growth cones of neurons (Fig. 2A). To analyze whether NCAM2 activation induces changes in Ca2+ levels in neurons, we increased clustering of NCAM2 at the neuronal cell surface (Fig. 2A) using antibodies specifically recognizing the extracellular domain of NCAM2 (Fig. 2B), a protocol widely used to activate cell adhesion molecules (Westphal et al., 2010; Sheng et al., 2013). Fluorescence images of live 2- to 3-d-old neurons transfected with LCK-GCaMP5 and Red-GECO or NLS-Red-GECO were time-lapse recorded before and after application of the antibodies against NCAM2. Time-lapse recordings showed that NCAM2 antibodies induced an approximately threefold increase in the frequency of LCK-GCaMP5 fluorescence spikes observed either in somata, along neurites, or in growth cones compared to the basal activity levels before NCAM2 antibody application (Fig. 2C,D). In contrast, treatment of neurons with control nonimmune Igs did not significantly affect the frequency of LCK-GCaMP5 spikes (Fig. 2D). NCAM2 activation did not affect the frequency of Red-GECO fluorescence spikes (Fig. 2D). This resulted in reduced correlation between the amplitudes of LCK-GCaMP5 fluorescence spikes and concomitant Red-GECO fluorescence increases measured at the center of LCK-GCaMP5 fluorescence spike in neurons treated with NCAM2 antibodies (Pearson correlation coefficient, r = 0.43; Fig. 2E) compared to control neurons (r = 0.71; Fig. 1D), suggesting that NCAM2 activation predominantly influences submembrane Ca2+ levels.
To further confirm that both reporters can detect inducible changes in Ca2+ levels in neurites and somata, we compared changes in the fluorescence intensity of LCK-GCaMP5 and Red-GECO in neurons treated with ionomycin. Application of ionomycin in Ca2+-free buffer induced concomitant increases in LCK-GCaMP5 and Red-GECO fluorescence intensities in somata and neurites of neurons (Fig. 3A,B), indicating that both reporters can detect Ca2+ released from internal stores by ionomycin (Di Virgilio et al., 1984). To compare changes in the fluorescence intensity of both reporters in response to ionophore-mediated delivery of extracellular Ca2+, neurons were preincubated in Ca2+-free medium with EGTA to deplete internal Ca2+ stores (Di Virgilio et al., 1984) and then treated with different concentrations of Ca2+ in the presence of ionomycin to analyze the responses of the reporters to defined Ca2+ levels (Lew et al., 1986). These experiments showed that the fluorescence intensities of both LCK-GCaMP5 and Red-GECO were not changed when Ca2+ concentration was increased to 0.4 mm Ca2+, but rapidly increased in somata and neurites when Ca2+ concentration was increased to 0.6 mm Ca2+ (n = 11; Fig. 4A,B). These observations indicate that LCK-GCaMP5 and Red-GECO have similar lower detection limits and the sensitivity of both reporters is sufficient to detect concomitant changes in submembrane and cytosolic Ca2+ levels. The amplitude of the Red-GECO fluorescence intensity increase was ∼2.5 times lower compared to the amplitude of the LCK-GCaMP5 fluorescence intensity increase (Figs. 3C, 4B), indicating lower sensitivity of Red-GECO compared to LCK-GCaMP5, in accordance with previous reports (Zhao et al., 2011; Akerboom et al., 2012).
Figure 3.

LCK-GCaMP5 and Red-GECO detect ionomycin-induced increases in submembrane and cytosolic [Ca2+] in somata and neurites of cortical neurons. A, Pseudocolored images of a 2-d-old cortical neuron cotransfected with LCK-GCaMP5 and Red-GECO are shown. Images were taken before and after application of ionomycin. Note an increase in submembrane and cytosolic [Ca2+] after application of ionomycin. Scale bar, 10 μm. B, Graphs show changes in fluorescence intensities of LCK-GCaMP5 and Red-GECO in neurites of cultured 1- to 3-d-old cortical neurons before and after application of ionomycin. An individual recording (top) and the average of n = 11 recordings (mean ± SEM) are shown. Fluorescence intensities were normalized to the mean fluorescence intensities before application of ionomycin, set to 0. C, Graphs show mean + SEM amplitudes of LCK-GCaMP5 and Red-GECO fluorescence intensity increases in response to ionomycin in somata and neurites of neurons (n = 11 neurons in each group were analyzed).
Figure 4.
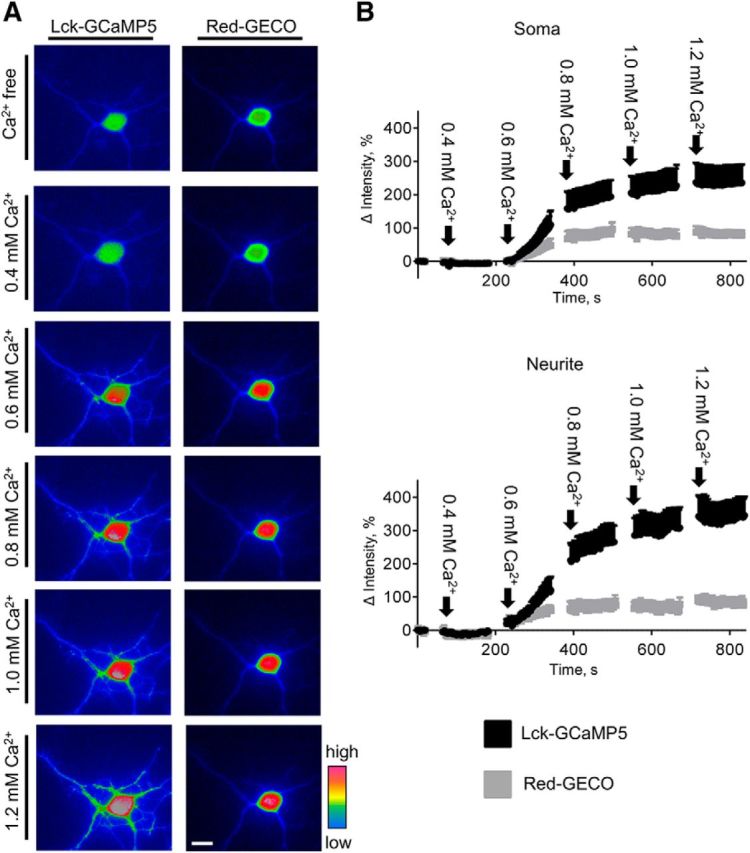
LCK-GCaMP5 and Red-GECO have similar activation thresholds. A, Pseudocolored images of a 3-d-old cortical neuron cotransfected with LCK-GCaMP5 and Red-GECO are shown. The neuron was preincubated in the Ca2+-free buffer containing 1 mm EGTA to deplete internal Ca2+ stores, placed in the Ca2+-free buffer, and imaged in the presence of 3 μm ionomycin and indicated Ca2+ concentrations. Note the increases in LCK-GCaMP5 and Red-GECO fluorescence intensity at Ca2+ concentrations above 0.6 mm. Scale bar, 10 μm. B, Graphs show fluorescence intensities of LCK-GCaMP5 and Red-GECO in somata and neurites of cultured 1- to 3-d-old cortical neurons at indicated concentrations of Ca2+. Mean ± SEM values from n = 11 recordings are shown. Fluorescence intensities were normalized to the fluorescence intensities in the Ca2+-free buffer, set to 0.
Our combined observations thus indicate that NCAM2 activation induces predominantly submembrane Ca2+ increases, although we cannot exclude that small NCAM2-dependent increases in cytosolic Ca2+ levels remained below the detection limit.
NCAM2 activation induces a steady increase in submembrane and cytosolic [Ca2+] in cortical neurons with high levels of baseline activity
Interestingly, in a small subpopulation of neurons (4 neurons of 60 analyzed), application of NCAM2 antibodies induced a continuous steady increase in LCK-GCaMP5 fluorescence intensity, with an amplitude of 129.4 ± 37.6% in somata and 128.4 ± 59.7% in neurites, within 51.2 ± 11.2 s after antibody application, which was observed till the end of time-lapse recordings, i.e., with a duration of over 500 s (Fig. 5A,C). In contrast to neurons showing an NCAM2-dependent increase in the number of LCK-GCaMP5 fluorescence spikes, the steady increase in LCK-GCaMP5 fluorescence was accompanied by an increase in Red-GECO fluorescence intensity, with an amplitude of 37 ± 11.5% in somata and 62.0 ± 26.2% in neurites (Fig. 5A,C), indicating an increase in cytosolic Ca2+ levels. None of the neurons treated with control nonimmune Ig showed this steady increase in submembrane and cytosolic [Ca2+], indicating the specificity of the effect. In neurons showing a steady increase in Ca2+ levels in response to NCAM2 antibodies, numbers of LCK-GCaMP5 and Red-GECO fluorescence spikes during baseline recordings, i.e., before application of NCAM2 antibodies, were higher (n = 25 ± 15 spikes per 10 min) compared to those in other neurons (n = 2.4 ± 0.7 spikes per 10 min), indicating that NCAM2 activation induces a steady increase in Ca2+ levels in spontaneously highly active neurons.
Figure 5.
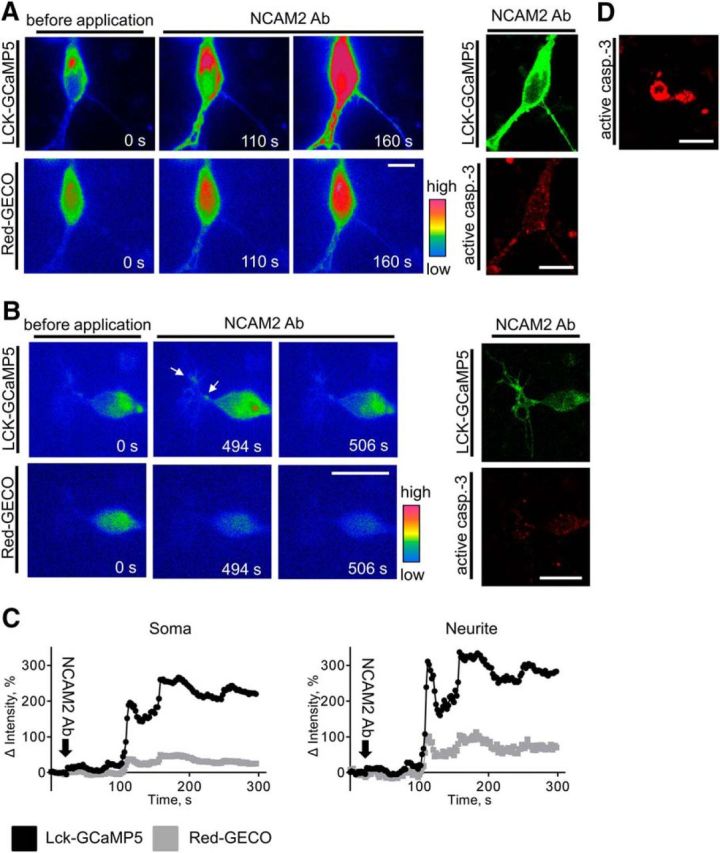
NCAM2 activation induces a steady increase in submembrane and cytosolic [Ca2+] in a subpopulation of highly active cortical neurons. A, B, Pseudocolored images of 2-d-old cortical neurons cotransfected with LCK-GCaMP5 and Red-GECO are shown. Images were taken before and after application of antibodies against NCAM2. Note a steady increase in submembrane and cytosolic [Ca2+] after application of NCAM2 antibodies in the neuron shown in A and submembrane [Ca2+] spikes in the neuron shown in B. At the end of recordings, neurons were fixed and colabeled with antibodies against active caspase-3 (right). Note the higher levels of the active caspase-3 in the neuron in A. C, Graphs show changes in fluorescence intensities of LCK-GCaMP5 and Red-GECO in the soma and neurites of the neuron shown in A. Fluorescence intensities were normalized to the mean fluorescence intensities before application of NCAM2 antibodies, set to 0. D, An example of an active caspase-3-positive apoptotic neuron is shown. Scale bars, 10 μm.
Chronically increased levels of intracellular Ca2+ have been reported to result in activation of the apoptotic pathways (Bezprozvanny and Mattson, 2008). To analyze whether NCAM2-induced steady increases in intracellular Ca2+ levels can affect neuronal viability, at the end of time-lapse recordings, neurons were fixed and colabeled with antibodies against active caspase-3. Although neurons showing the NCAM2-dependent steady increase in Ca2+ levels were observed very rarely, we were able to identify and analyze two neurons showing this behavior. Analysis of the immunofluorescence images showed that in both neurons, levels of active caspase-3 were increased (Fig. 5A; active caspase-3 immunofluorescence intensity levels in soma, 781.5 ± 14.27 AU, where AU is defined as gray scale values of the 16 bit pixel) compared to neurons that showed an increase in the number of submembrane Ca2+ spikes but no steady increase in intracellular Ca2+ level (Fig. 5B; active caspase-3 immunofluorescence intensity levels in soma, 430.1 ± 18.94 AU). The levels of active caspase-3 in neurons with the steady increase of Ca2+ levels were still much lower compared to the levels of active caspase-3 in apoptotic cells identified by their degenerated morphology (Fig. 5D; active caspase-3 immunofluorescence intensity levels in soma, 2697 ± 141.5 AU). Since recordings were performed for 10 min only, we then analyzed numbers of apoptotic neurons in cultures incubated with NCAM2 antibodies for 24 h. This analysis showed that numbers of apoptotic neurons were slightly but statistically significantly increased from 6.5 ± 1.1% in control Ig-treated neurons to 10.8 ± 1.8% in NCAM2 antibody-treated neurons (p < 0.05, unpaired t test; n > 30 image fields containing at least 10 neurons analyzed). Our data thus suggest that a continuous NCAM2-dependent increase in Ca2+ levels predisposes neurons to apoptosis.
NCAM2-dependent Ca2+ influx depends on L-type VDCCs
The increased frequency of submembrane [Ca2+] spikes in NCAM2 antibody-treated neurons suggests that NCAM2 activation influences the activity of cell surface Ca2+ channels. Since L-type VDCCs have been implicated in Ca2+ signaling induced by another member of the NCAM family, NCAM (for review, see Sheng et al., 2013), we analyzed whether NCAM2 colocalizes with L-type VDCCs in developing cortical neurons. Neurons treated either with control nonimmune Ig or antibodies against NCAM2 to induce clustering of NCAM2 were fixed and colabeled for NCAM2 and L-type VDCCs. Analysis of the immunofluorescence images showed that accumulations of L-type VDCCs at the neuronal cell surface, defined as regions with L-type VDCC immunofluorescence intensity at least twofold higher than in the adjacent regions, often colocalized with clusters of NCAM2 (Fig. 6A). Application of NCAM2 antibodies increased levels of NCAM2 in L-type VDCC accumulations by ∼30% (Fig. 6A; mean ± SEM of NCAM2 immunofluorescence intensity in L-type VDCC accumulation, 25.2 ± 1.3 vs 32.6 ± 1.5 AU in control Ig- and NCAM2 antibody-treated neurons, respectively, where AU is defined as gray scale values of 8 bit pixels; p < 0.001, t test; n > 65 neurites analyzed), further suggesting a functional link between NCAM2 activation and L-type VDCC activity.
Figure 6.

NCAM2-dependent submembrane [Ca2+] spikes are L-type VDCC dependent. A, Neurites of cultured cortical neurons treated with nonimmune Igs or NCAM2 antibodies, fixed, and colabeled with antibodies against NCAM2 and L-type VDCCs are shown. Note clusters of NCAM2 colocalized with accumulations of L-type VDCCs (arrows). Scale bar, 10 μm. B, The graph shows mean + SEM numbers of submembrane and cytosolic [Ca2+] spikes in neurons cotransfected with LCK-GCaMP5 and Red-GECO over the 10 min recording time (n > 13 neurons were analyzed in each group). Neurons were either not treated or treated with control nonimmune Igs or NCAM2 antibodies in the presence of nifedipine, an inhibitor of L-type VDCCs, or vehicle (DMSO), used to prepare the stock solution of nifedipine. Note that the NCAM2-dependent increase in the number of submembrane [Ca2+] spikes detectable with the LCK-GCaMP5 reporter is abolished by nifedipine. *p < 0.05, one-way ANOVA with Dunnett's multiple-comparison test.
To confirm this link, we analyzed the effect of nifedipine, an L-type VDCC inhibitor, on the NCAM2-dependent increase in the frequency of [Ca2+] spikes. The NCAM2-dependent increase in the number of LCK-GCaMP5 fluorescence spikes was blocked in the presence of nifedipine (Fig. 6B), indicating that L-type VDCCs are required for NCAM2-dependent Ca2+ influx in developing cortical neurons. Application of nifedipine did not, however, affect the frequency of LCK-GCaMP5 and Red-GECO fluorescence spikes in neurons during baseline recordings in the absence of NCAM2 activation (Fig. 6B).
NCAM2 activation does not increase depolarization-induced Ca2+ influx
To analyze whether an increase in the number of submembrane [Ca2+] spikes in response to NCAM2 activation is related to an increase of the overall conductivity of cell surface VDCCs, we compared depolarization-induced Ca2+ influx in neurons incubated with NCAM2 antibodies or control nonimmune Ig. To this aim, immediately after recording the effect of control nonimmune Ig and NCAM2 antibodies on the levels of baseline activity (Fig. 2C,D), the extracellular solution was exchanged for the buffer containing 90 mm K+ to depolarize neuronal membranes (Fig. 7A). Time-lapse recordings of neurons before and during application of 90 mm K+ showed a sharp increase in the fluorescence intensity of Ca2+ reporters followed by a slow decline in their fluorescence intensity (Fig. 7A–C), indicating Ca2+ influx followed by channel inactivation and Ca2+ clearance. Analysis of LCK-GCaMP5 fluorescence dynamics in neurites and somata of neurons showed that the amplitude of the 90 mm K+-induced fluorescence increase in soma and neurites of neurons treated with NCAM2 antibodies was slightly lower compared to that in neurons treated with control nonimmune Igs (Fig. 7B–E), thereby excluding the possibility that activation of NCAM2 results in overall increased conductivity of VDCCs. This reduction in the amplitude of the 90 mm K+-induced response rather suggested partial inactivation of VDCCs, possibly due to increased spontaneous activity in response to NCAM2 antibodies over the 10 min incubation time preceding the application of 90 mm K+ (Fig. 2C,D). The rate of decay of the LCK-GCaMP5 fluorescence intensity was not significantly affected in neurons treated with NCAM2 antibodies compared to neurons treated with control Igs (Fig. 7B,C). The amplitudes of the 90 mm K+-induced increases in the fluorescence intensities of Red-GECO and NLS-Red-GECO and the rates of their fluorescence decay were not significantly affected in neurons treated with NCAM2 antibodies compared to neurons treated with control Igs (Fig. 7B–E).
Figure 7.
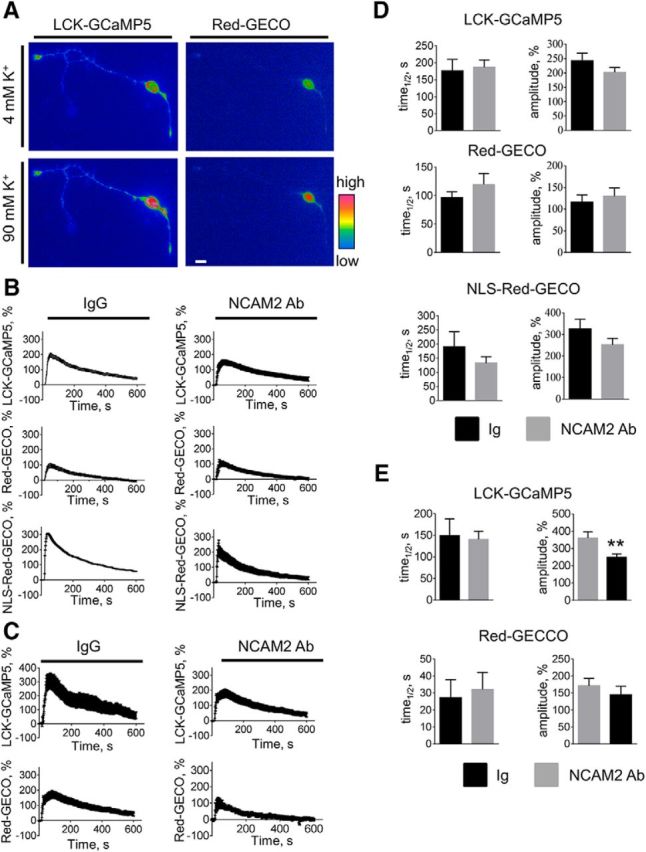
NCAM2 activation does not potentiate the depolarization-induced Ca2+ influx in young cultured cortical neurons. A, Pseudocolored images of a 2-d-old cultured cortical neuron cotransfected with LCK-GCaMP5 and Red-GECO and treated with 90 mm K+ to analyze depolarization-induced Ca2+ influx are shown. The 90 mm K+–containing buffer was applied after recording the spontaneous activity shown in Figure 2C. Images shown were captured immediately before and at 10 s after application of 90 mm K+. Note an increase in the fluorescence intensity of the reporters observed after the application of 90 mm K+. Scale bar, 10 μm. B, C, The graphs show mean ± SEM fluorescence intensities of LCK-GCaMP5, Red-GECO, and NLS-Red-GECO in somata (B) and fluorescence intensities of LCK-GCaMP5 and Red-GECO in neurites (C) of cultured 1- to 3-d-old cortical neurons before (first 10 s) and after application of the 90 mm K+–containing buffer. Fluorescence intensities were normalized to the mean fluorescence intensities before application of 90 mm K+. Neurons were pretreated for 10 min with nonimmune Igs or NCAM2 antibodies. Note that 90 mm K+-induced changes in the fluorescence intensities of Ca2+ reporters are not increased in NCAM2 antibody-treated versus Ig-treated neurons. D, E, Graphs show mean + SEM amplitudes of LCK-GCaMP5, Red-GECO, and NLS-GECO fluorescence intensity increases in response to 90 mm K+ and the half-life (time1/2) of LCK-GCaMP5, Red-GECO, and NLS-GECO fluorescence intensity decay after reaching the peak in somata (D) and neurites (E) of neurons pretreated with Ig or NCAM2 antibodies (n = 10–15 neurons analyzed in each group). Note that the amplitude of the 90 mm K+-induced increase in LCK-GCaMP5 fluorescence is reduced in neurites of neurons treated with NCAM2 antibodies compared to neurons treated with Ig. **p < 0.01, t test.
NCAM2-dependent submembrane [Ca2+] spikes require activation of the tyrosine protein kinase c-Src
Tyrosine protein kinase c-Src has been implicated previously as a major kinase regulating the frequency of [Ca2+] spikes in nonneuronal cell types (Gulia et al., 2013). We thus tested whether NCAM2 influences c-Src activity. To this aim, growth cones isolated from the brain tissue of 1- to 3-d-old mice were treated with control nonimmune Igs or antibodies against NCAM2 to induce clustering of NCAM2 at the cell surface of growth cones. Growth cones were then analyzed by Western blot with antibodies against total and activated autophosphorylated forms of c-Src. This analysis showed that levels of activated c-Src were significantly increased in growth cones treated with NCAM2 antibodies compared to growth cones treated with control Igs (Fig. 8A). NCAM2-dependent c-Src activation was blocked by a c-Src kinase inhibitor, PP2 (Fig. 8A). NCAM2-dependent c-Src activation was reduced but not blocked by nifedipine or the membrane-permeable Ca2+ chelator BAPTA-AM (Fig. 8A). Our observations thus indicate that NCAM2 activates c-Src in a Ca2+-independent manner and that this effect is enhanced by Ca2+ influx via L-type VDCCs.
Figure 8.

NCAM2 activates c-Src and induces submembrane [Ca2+] spikes in a c-Src-dependent manner. A, Growth cones isolated from the brain tissue of 1- to 3-d-old mice and treated with nonimmune Igs or NCAM2 antibodies in the presence or absence of nifedipine, BAPTA-AM, or c-Src-kinase family inhibitor PP2 were analyzed by Western blot with antibodies against total and autophosphorylated activated c-Src. Note that NCAM2 antibodies induce an increase in the level of activated c-Src, and that this increase is reduced by BAPTA-AM and nifedipine and is blocked by PP2. The graph shows the quantification of the blots (mean + SEM; n = 6), with signals in growth cones treated with Ig in the absence of inhibitors set to 1. B, NCAM2 immunoprecipitates from the brain lysates of 1- to 3-d-old mice were probed by Western blot with antibodies against NCAM2 and c-Src. Mock immunoprecipitation with nonimmune Igs served as a control. Note that c-Src coimmunoprecipitated with NCAM2. C, The graph shows mean + SEM numbers of submembrane and cytosolic [Ca2+] spikes in neurons cotransfected with LCK-GCaMP5 and Red-GECO over a 10 min recording time (n > 13 neurons analyzed in each group). Neurons were either not treated or treated with control nonimmune Igs or NCAM2 antibodies in the presence of PP2 or vehicle (DMSO) used to prepare the stock solution of PP2. Note that the NCAM2-dependent increase in the number of submembrane [Ca2+] spikes detectable with the LCK-GCaMP5 reporter is abolished by PP2. D, The graph shows mean + SEM numbers of submembrane and cytosolic [Ca2+] spikes in neurons cotransfected with LCK-GCaMP5, Red-GECO, and control siRNA or Src siRNA over a 10 min recording time (n > 18 neurons analyzed in each group). Neurons were analyzed before and after application of NCAM2 antibodies. Note that the NCAM2-dependent increase in the number of submembrane [Ca2+] spikes detectable with the LCK-GCaMP5 reporter is abolished by Src siRNA. *p < 0.05, ***p < 0.001, one-way ANOVA with Dunnett's multiple-comparison test.
To investigate whether NCAM2 and c-Src form a molecular complex, NCAM2 was immunoprecipitated from lysates of brain tissue from 1- to 3-d-old mice. Western blot analysis of NCAM2 immunoprecipitates with antibodies against c-Src showed that c-Src coimmunoprecipitated with NCAM2 (Fig. 8B). Approximately 15.6 ± 1.6% of all c-Src was found in a complex with NCAM2, indicating that the interaction is physiologically significant.
We then analyzed whether c-Src activity is required for the NCAM2-dependent increase in the frequency of [Ca2+] spikes in cultured cortical neurons. The NCAM2-dependent increase in the number of LCK-GCaMP5 fluorescence spikes was blocked in the presence of PP2 (Fig. 8C). Application of PP2 did not, however, affect the frequency of LCK-GCaMP5 and Red-GECO fluorescence spikes in neurons during baseline recordings in the absence of NCAM2 activation (Fig. 8C). The NCAM2-dependent increase in the number of LCK-GCaMP5 fluorescence spikes was also blocked in neurons transfected with c-Src siRNA (Fig. 8D). Our combined observations thus indicate that NCAM2 activates c-Src to induce [Ca2+] spikes.
NCAM2 activation increases submembrane [Ca2+] spikes at the bases of filopodia
To identify the functional role that NCAM2-induced [Ca2+] spikes play in developing neurons, we next analyzed the subcellular localization of submembrane [Ca2+] spikes. Analysis of the time-lapse recordings of neurons transfected with LCK-GCaMP5 showed that 57.6 ± 11.5% of all spikes in NCAM2 antibody-treated neurons (n = 12 neurons analyzed) and 23.9 ± 9.6% of all spikes in control Ig-treated neurons (n = 14 neurons analyzed, p < 0.05 compared to NCAM2 antibody-treated neurons, Mann–Whitney test) were colocalized with the bases of filopodia (Fig. 9A). In spikes colocalizing with filopodia, the highest increase in LCK-GCaMP5 fluorescence intensity during the spike occurred at the center of the filopodial base, with more moderate increases observed in the vicinity of the filopodial base (Fig. 9B). The overall number of submembrane [Ca2+] spikes at filopodial bases and their amplitudes and durations were increased in NCAM2 antibody-treated neurons compared to control Ig-treated neurons or to basal activity levels before NCAM2 antibody application (Fig. 9B,C). These effects were blocked by nifedpine and PP2 (Fig. 9C), indicating that L-type VDCCs and c-Src are required for NCAM2-dependent [Ca2+] spikes at filopodia.
Figure 9.

NCAM2 activation induces increased submembrane [Ca2+] spikes at the bases of filopodia. A, Examples of submembrane [Ca2+] spikes along a neurite of a 2-d-old cultured cortical neuron cotransfected with LCK-GCaMP5. Images were taken before and after application of NCAM2 antibodies and are shown in pseudocolor. Note the [Ca2+] increase at the bases of filopodia. Scale bar, 5 μm. B, The graph shows the changes of LCK-GCaMP5 fluorescence intensity during the spike (mean + SEM, n > 20 spikes analyzed) along neurites in the vicinity of the bases of filopodia. The dashed line denotes the center of the filopodial base. Spikes in neurons treated with control nonimmune Igs or NCAM2 antibodies were analyzed. Note that the highest increases in LCK-GCaMP5 fluorescence are observed at the bases of filopodia. The increase is higher in NCAM2 Ab-treated neurons. *p < 0.05, t test for measurements within 2 μm of the center of filopodial base. C, Graphs show the numbers, durations, and amplitudes (mean + SEM) of submembrane [Ca2+] spikes observed at the bases of filopodia in neurons before application of antibodies (nontreated) and after application of control nonimmune Igs or NCAM2 antibodies. When indicated, neurons were preincubated with nifedipine (nif) or PP2. *p < 0.05, ***p < 0.001, one-way ANOVA with Tukey's multiple-comparison test (n > 11 neurons analyzed in each group). Note the increased amplitudes and durations of the submembrane [Ca2+] spikes in neurons treated with NCAM2 antibodies.
NCAM2 activation promotes the formation of filopodia and neurite branching
To analyze whether the NCAM2-dependent increase in submembrane [Ca2+] spikes correlates with changes in the numbers of filopodia, numbers of filopodia were analyzed in cultured cortical neurons incubated with antibodies against NCAM2 or control nonimmune Igs either for 24 or 48 h. This analysis showed that the number of filopodia along neurites was increased by ∼40% in neurons treated with NCAM2 antibodies compared to neurons treated with control Igs both at 24 and 48 h after antibody application, indicating that NCAM2 activation promotes the formation of filopodia (Fig. 10A,B). An increase in numbers of filopodia in NCAM2 antibody-treated neurons was accompanied by increased branching of neurites (Fig. 10B,C) and an overall increase in neurite lengths (Fig. 10C). Since similar results were obtained at both time points, further experiments were performed with neurons treated for 24 h.
Figure 10.
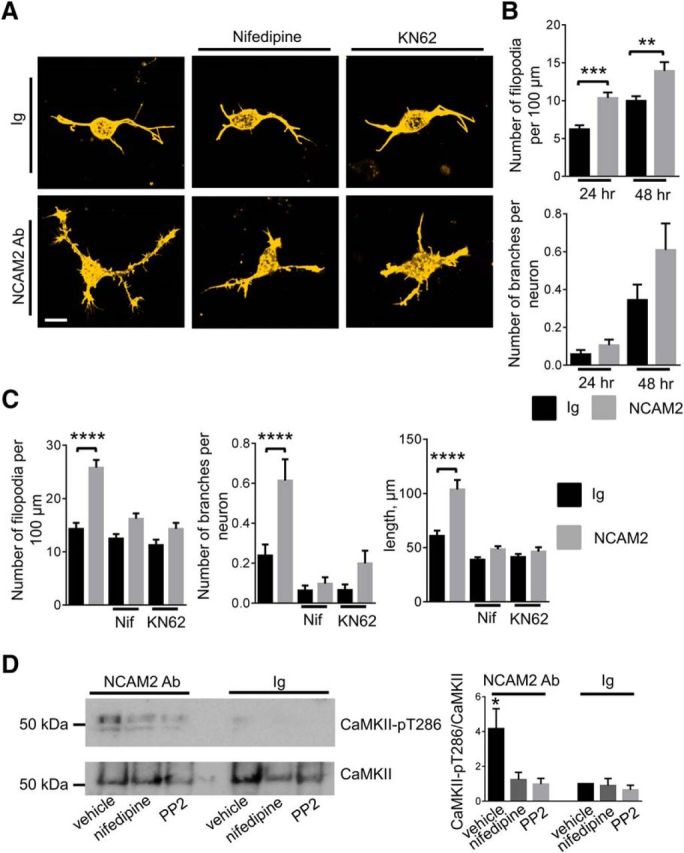
NCAM2 activation promotes the formation of filopodia and neurite branching in an L-type VDCC-, c-Src-, and CaMKII-dependent manner. A, Images of cortical neurons incubated for 24 h with control nonimmune Igs or NCAM2 antibodies in the absence or presence of the L-type VDCC inhibitor (nifedipine) or CaMKII inhibitor (KN62). Neurons were labeled with fluorescent phalloidin to detect filamentous actin and visualize filopodia. Note the higher density of filopodia and branches along neurites of neurons treated with NCAM2 antibodies in the absence of inhibitors compared to neurons incubated with Ig. Note that this effect is blocked by the inhibitors. Scale bar, 10 μm. B, Graphs show the numbers of filopodia per 100 μm of neurite length and numbers of branching neurites per neuron (mean + SEM) in neurons treated with nonimmune Igs or NCAM2 antibodies for 24 h (n > 100 neurons analyzed) or 48 h (n > 60 neurons analyzed). **p < 0.01, ***p < 0.001, one-way ANOVA with Tukey's multiple-comparison test. C, Graphs show numbers of filopodia per 100 μm of neurite length, numbers of branching neurites per neuron, and total lengths of all neurites per neuron in neurons treated for 24 h with nonimmune Igs or NCAM2 antibodies in the absence or presence of nifedipine or KN-62. Mean + SEM values are shown. ****p < 0.0001, one-way ANOVA with Tukey's multiple-comparison test (n > 100 neurons analyzed in each group). D, Growth cones isolated from the brain tissue of 1- to 3-d-old mice and treated with nonimmune Igs or NCAM2 antibodies in the presence or absence of nifedipine or PP2 were analyzed by Western blot with antibodies against total and autophosphorylated activated CaMKII. Note that NCAM2 antibodies induce an increase in the level of activated CaMKII, and that this increase is reduced by PP2 and nifedipine. The graph shows quantification of the blots (mean + SEM, n = 3 experiments). *p < 0.05, one-way ANOVA with Dunnett's multiple-comparison test. Signals in growth cones treated with Ig in the absence of inhibitors were set to 1.
NCAM2-induced formation of filopodia was inhibited in the presence of nifedipine (Fig. 10A,C), indicating that Ca2+ influx via L-type VDCCs is required. Among Ca2+-activated enzymes, activation of CaMKII, and particularly its two isoforms CaMKIIα and CaMKIIβ, was shown previously to induce formation of filopodia and neurite branching and outgrowth in cultured hippocampal neurons (Fink et al., 2003; Bodrikov et al., 2008). KN62, a CaMKII inhibitor, also blocked the NCAM2-dependent formation of filopodia and neurite branching and outgrowth (Fig. 10A,C), indicating that CaMKII is required for NCAM2-dependent morphogenesis and suggesting that NCAM2 activates CaMKII.
To test whether NCAM2 activates CaMKII, growth cones isolated from brain tissue of 1- to 3-d-old mice were treated with control nonimmune Igs or antibodies against NCAM2 to induce clustering of NCAM2 at the cell surface of growth cones. Growth cones were then analyzed by Western blot with antibodies against total and activated autophosphorylated forms of CaMKII. This analysis showed that the levels of activated CaMKII were approximately threefold higher in growth cones treated with NCAM2 antibodies compared to growth cones treated with control Igs (Fig. 10D), indicating that NCAM2 activates CaMKII. This effect was inhibited by nifedipine and PP2 (Fig. 10D), indicating that c-Src activation and Ca2+ influx via L-type VDCCs are required for NCAM2-dependent CaMKII activation.
In agreement with these data, NCAM2-induced formation of filopodia and neurite branching and outgrowth were inhibited in neurons transfected c-Src, CaMKIIα, or CaMKIIβ siRNAs (Fig. 11A,B) further confirming the role of c-Src and CaMKII in NCAM2-dependent morphogenesis.
Figure 11.
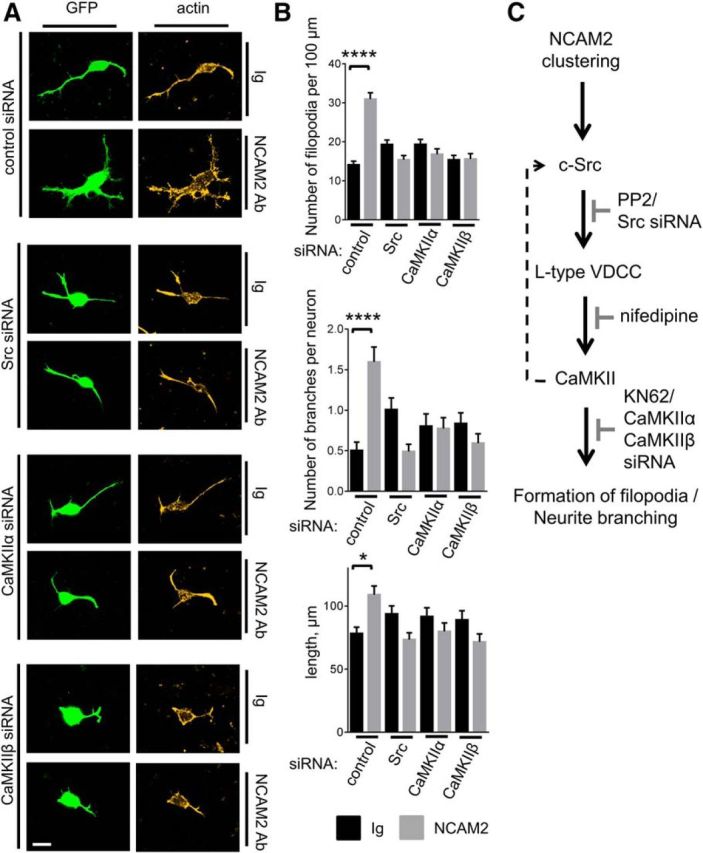
NCAM2-dependent formation of filopodia and neurite branching are inhibited in neurons transfected with c-Src, CaMKIIα, or CaMKIIβ siRNA. A, Images of cortical neurons cotransfected with GFP and control siRNA, Src siRNA, CaMKIIα siRNA, or CaMKIIβ siRNA and incubated for 24 h with control nonimmune Igs or NCAM2 antibodies. Neurons were labeled with fluorescent phalloidin to detect filamentous actin and visualize filopodia. Note the NCAM2-dependent increase in the density of filopodia and branches in neurons transfected with control siRNA, and that this effect is inhibited by c-Src, CaMKIIα, and CaMKIIβ siRNAs. Scale bar, 10 μm. B, Graphs show numbers of filopodia per 100 μm of neurite length, numbers of branching neurites per neuron, and total lengths of all neurites per neuron in neurons treated for 24 h with nonimmune Igs or NCAM2 antibodies. Neurons were transfected with control siRNA, Src siRNA, CaMKIIα siRNA, or CaMKIIβ siRNA. Mean + SEM values are shown. *p < 0.05, ****p < 0.0001, one-way ANOVA with Tukey's multiple-comparison test (n > 100 neurons analyzed in each group). C, Schematic representation of the proposed NCAM2-activated signaling pathway is shown. NCAM2 activation at the neuronal cell surface induces activation of c-Src, followed by activation of L-type VDCCs. Ca2+ influx via L-type VDCCs results in activation of CaMKII, which promotes formation of filopodia and neurite branching. A proposed CaMKII-mediated positive feedback loop that enhances c-Src activation after the initial Ca2+ influx is shown as a dashed line. Steps inhibited by the c-Src inhibitor PP2, Src siRNA, L-type VDCC blocker nifedipine, CaMKII inhibitor KN62, CaMKIIα siRNA, or CaMKIIβ siRNA are also indicated (gray).
Discussion
In this work, we have identified the cell adhesion molecule NCAM2 as a new regulator of filopodial formation and neurite branching. Since the intracellular molecular pathways activated by NCAM2 are poorly investigated, in the search for possible mechanisms of NCAM2-mediated morphogenesis, we used genetically encoded calcium reporters to analyze the effect of the activation of NCAM2 at the neuronal cell surface on the intracellular Ca2+ levels. Our results indicate that activation of NCAM2 at the neuronal cell surface of developing cortical neurons results in increased frequency of submembrane [Ca2+] spikes along neurites of neurons and particularly at the bases of filopodia.
Activation of cell adhesion molecules of different families including NCAM, another cell adhesion molecule of the Ig superfamily and a paralog of NCAM2, was shown previously to induce Ca2+ influx and an overall increase in intracellular Ca2+ levels in different neuronal types (for review, see Sheng et al., 2013). Interestingly, however, by using Ca2+ reporters targeted to the plasma membrane, cytosol, and nuclei of neurons, we found that NCAM2 activation predominantly influences submembrane Ca2+ levels, but does not significantly affect cytosolic and nuclear Ca2+ levels, although we cannot exclude that small increases in cytosolic Ca2+ levels remained below detection limits due to the lower sensitivity of Red-GECO and small amounts of cytosol in neurites. Furthermore, in contrast to NCAM, activation of which induces a continuous increase in overall Ca2+ levels (Kiryushko et al., 2006), only a small subgroup of spontaneously highly active neurons showed a continuous increase in submembrane and cytosolic [Ca2+] after NCAM2 activation. These observations thus indicate that NCAM2 activation induces a response that can be functionally distinct from the response activated by NCAM. This spatially restricted NCAM2-induced Ca2+ influx may be important for local activation of enzymes involved in the formation of filopodia. Our data also suggest that the tight regulation of Ca2+ influx may also be important to prevent cytotoxicity, since a continuous increase in submembrane and cytosolic [Ca2+] levels in response to NCAM2 activation in a small population of neurons correlated with increased activation of caspase-3, suggesting that it predisposes neurons to apoptosis. This idea is in agreement with previous studies showing that increases in cytosolic Ca2+ levels can be cytotoxic and may result in apoptosis (Bezprozvanny and Mattson, 2008). What molecular mechanisms control Ca2+ influx is an important question for future analysis.
Our observation that NCAM2 induces submembrane [Ca2+] spikes points to the cell surface Ca2+ channels as a major source of Ca2+ influx in response to NCAM2 activation (Fig. 11C). In agreement, we show that NCAM2-dependent [Ca2+] spikes are blocked by nifedipine, an inhibitor of L-type VDCCs, which are highly expressed in postnatal cortical neurons (Schlick et al., 2010). The fact that NCAM2 activation does not induce an overall increase in VDCC conductivity suggests that NCAM2 activation increases the open probability of L-type VDCCs, resulting in transient submembrane [Ca2+] spikes. L-type VDCCs are phosphorylated by c-Src in cerebellar granule neurons (Evans and Pocock, 1999; Bence-Hanulec et al., 2000), and c-Src activity is required for L-type VDCC-dependent [Ca2+] spikes in tsA-201 cells (Gulia et al., 2013). Our observation that c-Src coimmunoprecipitates with NCAM2 from brain lysates indicates that the two proteins are components of a molecular complex, suggesting that NCAM2 and c-Src associate with each other or interact via intermediate binding partners. The physiological significance of this interaction is confirmed by our data showing that clustering of NCAM2 induces activation of c-Src and that inhibition of c-Src activity or knock-down of c-Src expression using siRNA blocks NCAM2-dependent submembrane [Ca2+] spikes, which also indicates that c-Src plays a key role in NCAM2-dependent L-type VDCC activation (Fig. 11C).
As a downstream signaling enzyme in the NCAM2-activated signaling pathway, we have identified CaMKII (Fig. 11C), which has been shown previously to be tethered to L-type VDCCs and activated by Ca2+ entering through these channels (Hudmon et al., 2005; Wheeler et al., 2008; Lee et al., 2009). In agreement, NCAM2-dependent activation of CaMKII is blocked by the L-type VDCC inhibitor in our experiments. Among possible downstream substrates, CaMKII phosphorylates and activates RPTPα, a major activator of the c-Src family members, which also associates with NCAM1 (Bodrikov et al., 2005, 2008). Our observation that nifedipine and BAPTA also partially inhibit NCAM2-dependent c-Src activation suggests that CaMKII functions in a positive feedback loop, which enhances c-Src activation after the initial Ca2+ influx (Fig. 11C).
CaMKII has been shown previously to play an important role in regulation of neuronal morphogenesis (Fink et al., 2003). It is also required for neuronal differentiation induced in response to activation of cell adhesion molecules (Williams et al., 1995; Bodrikov et al., 2008). Interestingly, we found that NCAM2-dependent CaMKII activation results not only in overall increased neurite length, as reported previously for NCAM (Bodrikov et al., 2008), but also in a significant increase in the density of filopodia along neurites. Local [Ca2+] transients play a key role in the growth and stabilization of filopodia and their development into stable neurite branches (Lohmann et al., 2002, 2005; Ruthazer et al., 2006). NCAM2-dependent neuronal morphogenesis is therefore likely to be initiated at the level of NCAM2-induced [Ca2+] spikes at the bases of filopodia along neurites, promoting stabilization of the filopodia and branching of the neurites (Fig. 11C).
Our observations on the role of NCAM2 in the regulation of neurite growth and branching are in agreement with previous findings showing abnormal organization of axonal and dendritic compartments in olfactory glomeruli in NCAM2-deficient mice (Walz et al., 2006; Borisovska et al., 2011) and incorrect innervation of olfactory glomeruli in transgenic mice overexpressing NCAM2 (Alenius and Bohm, 2003). Single nucleotide polymorphisms and deletions of the NCAM2 gene have been linked to autism in humans (Molloy et al., 2005; Haldeman-Englert et al., 2010). As a gene located on chromosome 21 in humans, NCAM2 has also been suggested to play a role in Down syndrome (Paoloni-Giacobino et al., 1997; Akeson et al., 2001) and is overexpressed in brains of Down syndrome patients (Lockstone et al., 2007). Dendritic branching is reduced in autism and Down syndrome (Becker et al., 1986; Takashima et al., 1994; Raymond et al., 1996). Changes in NCAM2-mediated Ca2+ signaling may therefore contribute to pathological processes resulting in abnormal neuronal development in these disorders.
Footnotes
This work was supported by the National Health and Medical Research Council (V.S.) and the Rebecca L. Cooper Medical Research Foundation (V.S., I.L.).
The authors declare no competing financial interests.
References
- Akerboom J, Chen TW, Wardill TJ, Tian L, Marvin JS, Mutlu S, Calderón NC, Esposti F, Borghuis BG, Sun XR, Gordus A, Orger MB, Portugues R, Engert F, Macklin JJ, Filosa A, Aggarwal A, Kerr RA, Takagi R, Kracun S, et al. Optimization of a GCaMP calcium indicator for neural activity imaging. J Neurosci. 2012;32:13819–13840. doi: 10.1523/JNEUROSCI.2601-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akeson EC, Lambert JP, Narayanswami S, Gardiner K, Bechtel LJ, Davisson MT. Ts65Dn–localization of the translocation breakpoint and trisomic gene content in a mouse model for Down syndrome. Cytogenet Cell Genet. 2001;93:270–276. doi: 10.1159/000056997. [DOI] [PubMed] [Google Scholar]
- Alenius M, Bohm S. Identification of a novel neural cell adhesion molecule-related gene with a potential role in selective axonal projection. J Biol Chem. 1997;272:26083–26086. doi: 10.1074/jbc.272.42.26083. [DOI] [PubMed] [Google Scholar]
- Alenius M, Bohm S. Differential function of RNCAM isoforms in precise target selection of olfactory sensory neurons. Development. 2003;130:917–927. doi: 10.1242/dev.00317. [DOI] [PubMed] [Google Scholar]
- Aricescu AR, Jones EY. Immunoglobulin superfamily cell adhesion molecules: zippers and signals. Curr Opin Cell Biol. 2007;19:543–550. doi: 10.1016/j.ceb.2007.09.010. [DOI] [PubMed] [Google Scholar]
- Becker LE, Armstrong DL, Chan F. Dendritic atrophy in children with Down's syndrome. Ann Neurol. 1986;20:520–526. doi: 10.1002/ana.410200413. [DOI] [PubMed] [Google Scholar]
- Bedlack RS, Jr, Wei M, Loew LM. Localized membrane depolarizations and localized calcium influx during electric field-guided neurite growth. Neuron. 1992;9:393–403. doi: 10.1016/0896-6273(92)90178-G. [DOI] [PubMed] [Google Scholar]
- Bence-Hanulec KK, Marshall J, Blair LA. Potentiation of neuronal L calcium channels by IGF-1 requires phosphorylation of the alpha1 subunit on a specific tyrosine residue. Neuron. 2000;27:121–131. doi: 10.1016/S0896-6273(00)00014-3. [DOI] [PubMed] [Google Scholar]
- Bezprozvanny I, Mattson MP. Neuronal calcium mishandling and the pathogenesis of Alzheimer's disease. Trends Neurosci. 2008;31:454–463. doi: 10.1016/j.tins.2008.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodrikov V, Leshchyns'ka I, Sytnyk V, Overvoorde J, den Hertog J, Schachner M. RPTPalpha is essential for NCAM-mediated p59fyn activation and neurite elongation. J Cell Biol. 2005;168:127–139. doi: 10.1083/jcb.200405073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodrikov V, Sytnyk V, Leshchyns'ka I, den Hertog J, Schachner M. NCAM induces CaMKIIalpha-mediated RPTPalpha phosphorylation to enhance its catalytic activity and neurite outgrowth. J Cell Biol. 2008;182:1185–1200. doi: 10.1083/jcb.200803045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borisovska M, McGinley MJ, Bensen A, Westbrook GL. Loss of olfactory cell adhesion molecule reduces the synchrony of mitral cell activity in olfactory glomeruli. J Physiol. 2011;589:1927–1941. doi: 10.1113/jphysiol.2011.206276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavallaro U, Dejana E. Adhesion molecule signalling: not always a sticky business. Nat Rev Mol Cell Biol. 2011;12:189–197. doi: 10.1038/nrm3068. [DOI] [PubMed] [Google Scholar]
- Chernyshova Y, Leshchyns'ka I, Hsu SC, Schachner M, Sytnyk V. The neural cell adhesion molecule promotes FGFR-dependent phosphorylation and membrane targeting of the exocyst complex to induce exocytosis in growth cones. J Neurosci. 2011;31:3522–3535. doi: 10.1523/JNEUROSCI.3109-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalva MB, McClelland AC, Kayser MS. Cell adhesion molecules: signalling functions at the synapse. Nat Rev Neurosci. 2007;8:206–220. doi: 10.1038/nrn2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Virgilio F, Lew DP, Pozzan T. Protein kinase C activation of physiological processes in human neutrophils at vanishingly small cytosolic Ca2+ levels. Nature. 1984;310:691–693. doi: 10.1038/310691a0. [DOI] [PubMed] [Google Scholar]
- Doherty P, Williams G, Williams EJ. CAMs and axonal growth: a critical evaluation of the role of calcium and the MAPK cascade. Mol Cell Neurosci. 2000;16:283–295. doi: 10.1006/mcne.2000.0907. [DOI] [PubMed] [Google Scholar]
- Evans GJ, Pocock JM. Modulation of neurotransmitter release by dihydropyridine-sensitive calcium channels involves tyrosine phosphorylation. Eur J Neurosci. 1999;11:279–292. doi: 10.1046/j.1460-9568.1999.00427.x. [DOI] [PubMed] [Google Scholar]
- Fink CC, Bayer KU, Myers JW, Ferrell JE, Jr, Schulman H, Meyer T. Selective regulation of neurite extension and synapse formation by the beta but not the alpha isoform of CaMKII. Neuron. 2003;39:283–297. doi: 10.1016/S0896-6273(03)00428-8. [DOI] [PubMed] [Google Scholar]
- Gahmberg CG, Fagerholm SC, Nurmi SM, Chavakis T, Marchesan S, Grönholm M. Regulation of integrin activity and signalling. Biochim Biophys Acta. 2009;1790:431–444. doi: 10.1016/j.bbagen.2009.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez TM, Zheng JQ. The molecular basis for calcium-dependent axon pathfinding. Nat Rev Neurosci. 2006;7:115–125. doi: 10.1038/nrn1844. [DOI] [PubMed] [Google Scholar]
- Gulia J, Navedo MF, Gui P, Chao JT, Mercado JL, Santana LF, Davis MJ. Regulation of L-type calcium channel sparklet activity by c-Src and PKC-alpha. Am J Physiol Cell Physiol. 2013;305:C568–C577. doi: 10.1152/ajpcell.00381.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haldeman-Englert CR, Chapman KA, Kruger H, Geiger EA, McDonald-McGinn DM, Rappaport E, Zackai EH, Spinner NB, Shaikh TH. A de novo 8.8-Mb deletion of 21q21.1-q21.3 in an autistic male with a complex rearrangement involving chromosomes 6, 10, and 21. Am J Med Genet A. 2010;152A:196–202. doi: 10.1002/ajmg.a.33176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen SM, Berezin V, Bock E. Signaling mechanisms of neurite outgrowth induced by the cell adhesion molecules NCAM and N-cadherin. Cell Mol Life Sci. 2008;65:3809–3821. doi: 10.1007/s00018-008-8290-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henley J, Poo MM. Guiding neuronal growth cones using Ca2+ signals. Trends Cell Biol. 2004;14:320–330. doi: 10.1016/j.tcb.2004.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henley JR, Huang KH, Wang D, Poo MM. Calcium mediates bidirectional growth cone turning induced by myelin-associated glycoprotein. Neuron. 2004;44:909–916. doi: 10.1016/j.neuron.2004.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudmon A, Schulman H, Kim J, Maltez JM, Tsien RW, Pitt GS. CaMKII tethers to L-type Ca2+ channels, establishing a local and dedicated integrator of Ca2+ signals for facilitation. J Cell Biol. 2005;171:537–547. doi: 10.1083/jcb.200505155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussman JP, Chung RH, Griswold AJ, Jaworski JM, Salyakina D, Ma D, Konidari I, Whitehead PL, Vance JM, Martin ER, Cuccaro ML, Gilbert JR, Haines JL, Pericak-Vance MA. A noise-reduction GWAS analysis implicates altered regulation of neurite outgrowth and guidance in autism. Mol Autism. 2011;2:1. doi: 10.1186/2040-2392-2-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiryushko D, Korshunova I, Berezin V, Bock E. Neural cell adhesion molecule induces intracellular signaling via multiple mechanisms of Ca2+ homeostasis. Mol Biol Cell. 2006;17:2278–2286. doi: 10.1091/mbc.E05-10-0987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulahin N, Kristensen O, Rasmussen KK, Olsen L, Rydberg P, Vestergaard B, Kastrup JS, Berezin V, Bock E, Walmod PS, Gajhede M. Structural model and trans-interaction of the entire ectodomain of the olfactory cell adhesion molecule. Structure. 2011;19:203–211. doi: 10.1016/j.str.2010.12.014. [DOI] [PubMed] [Google Scholar]
- Lee SJ, Escobedo-Lozoya Y, Szatmari EM, Yasuda R. Activation of CaMKII in single dendritic spines during long-term potentiation. Nature. 2009;458:299–304. doi: 10.1038/nature07842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leshchyns'ka I, Sytnyk V, Morrow JS, Schachner M. Neural cell adhesion molecule (NCAM) association with PKCbeta2 via betaI spectrin is implicated in NCAM-mediated neurite outgrowth. J Cell Biol. 2003;161:625–639. doi: 10.1083/jcb.200303020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lew PD, Monod A, Waldvogel FA, Dewald B, Baggiolini M, Pozzan T. Quantitative analysis of the cytosolic free calcium dependency of exocytosis from three subcellular compartments in intact human neutrophils. J Cell Biol. 1986;102:2197–2204. doi: 10.1083/jcb.102.6.2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Leshchyns'ka I, Chernyshova Y, Schachner M, Sytnyk V. The neural cell adhesion molecule (NCAM) associates with and signals through p21-activated kinase 1 (Pak1) J Neurosci. 2013;33:790–803. doi: 10.1523/JNEUROSCI.1238-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lockstone HE, Harris LW, Swatton JE, Wayland MT, Holland AJ, Bahn S. Gene expression profiling in the adult Down syndrome brain. Genomics. 2007;90:647–660. doi: 10.1016/j.ygeno.2007.08.005. [DOI] [PubMed] [Google Scholar]
- Lohmann C, Finski A, Bonhoeffer T. Local calcium transients regulate the spontaneous motility of dendritic filopodia. Nat Neurosci. 2005;8:305–312. doi: 10.1038/nn1406. [DOI] [PubMed] [Google Scholar]
- Lohmann C, Myhr KL, Wong RO. Transmitter-evoked local calcium release stabilizes developing dendrites. Nature. 2002;418:177–181. doi: 10.1038/nature00850. [DOI] [PubMed] [Google Scholar]
- Makino T, McLysaght A. Ohnologs in the human genome are dosage balanced and frequently associated with disease. Proc Natl Acad Sci U S A. 2010;107:9270–9274. doi: 10.1073/pnas.0914697107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maness PF, Schachner M. Neural recognition molecules of the immunoglobulin superfamily: signaling transducers of axon guidance and neuronal migration. Nat Neurosci. 2007;10:19–26. doi: 10.1038/nn1827. [DOI] [PubMed] [Google Scholar]
- Molloy CA, Keddache M, Martin LJ. Evidence for linkage on 21q and 7q in a subset of autism characterized by developmental regression. Mol Psychiatry. 2005;10:741–746. doi: 10.1038/sj.mp.4001691. [DOI] [PubMed] [Google Scholar]
- Paoloni-Giacobino A, Chen H, Antonarakis SE. Cloning of a novel human neural cell adhesion molecule gene (NCAM2) that maps to chromosome region 21q21 and is potentially involved in Down syndrome. Genomics. 1997;43:43–51. doi: 10.1006/geno.1997.4782. [DOI] [PubMed] [Google Scholar]
- Rasmussen KK, Kulahin N, Kristensen O, Poulsen JC, Sigurskjold BW, Kastrup JS, Berezin V, Bock E, Walmod PS, Gajhede M. Crystal structure of the Ig1 domain of the neural cell adhesion molecule NCAM2 displays domain swapping. J Mol Biol. 2008;382:1113–1120. doi: 10.1016/j.jmb.2008.07.084. [DOI] [PubMed] [Google Scholar]
- Raymond GV, Bauman ML, Kemper TL. Hippocampus in autism: a Golgi analysis. Acta Neuropathol. 1996;91:117–119. doi: 10.1007/s004010050401. [DOI] [PubMed] [Google Scholar]
- Rehder V, Kater SB. Regulation of neuronal growth cone filopodia by intracellular calcium. J Neurosci. 1992;12:3175–3186. doi: 10.1523/JNEUROSCI.12-08-03175.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruthazer ES, Li J, Cline HT. Stabilization of axon branch dynamics by synaptic maturation. J Neurosci. 2006;26:3594–3603. doi: 10.1523/JNEUROSCI.0069-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santuccione A, Sytnyk V, Leshchyns'ka I, Schachner M. Prion protein recruits its neuronal receptor NCAM to lipid rafts to activate p59fyn and to enhance neurite outgrowth. J Cell Biol. 2005;169:341–354. doi: 10.1083/jcb.200409127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlick B, Flucher BE, Obermair GJ. Voltage-activated calcium channel expression profiles in mouse brain and cultured hippocampal neurons. Neuroscience. 2010;167:786–798. doi: 10.1016/j.neuroscience.2010.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng L, Leshchyns'ka I, Sytnyk V. Cell adhesion and intracellular calcium signaling in neurons. Cell Commun Signal. 2013;11:94. doi: 10.1186/1478-811X-11-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shetty A, Sytnyk V, Leshchyns'ka I, Puchkov D, Haucke V, Schachner M. The neural cell adhesion molecule promotes maturation of the presynaptic endocytotic machinery by switching synaptic vesicle recycling from adaptor protein 3 (AP-3)- to AP-2-dependent mechanisms. J Neurosci. 2013;33:16828–16845. doi: 10.1523/JNEUROSCI.2192-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sytnyk V, Leshchyns'ka I, Delling M, Dityateva G, Dityatev A, Schachner M. Neural cell adhesion molecule promotes accumulation of TGN organelles at sites of neuron-to-neuron contacts. J Cell Biol. 2002;159:649–661. doi: 10.1083/jcb.200205098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sytnyk V, Leshchyns'ka I, Nikonenko AG, Schachner M. NCAM promotes assembly and activity-dependent remodeling of the postsynaptic signaling complex. J Cell Biol. 2006;174:1071–1085. doi: 10.1083/jcb.200604145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takashima S, Iida K, Mito T, Arima M. Dendritic and histochemical development and ageing in patients with Down's syndrome. J Intellect Disabil Res. 1994;38:265–273. doi: 10.1111/j.1365-2788.1994.tb00394.x. [DOI] [PubMed] [Google Scholar]
- Tian N, Leshchyns'ka I, Welch JH, Diakowski W, Yang H, Schachner M, Sytnyk V. Lipid raft-dependent endocytosis of close homolog of adhesion molecule L1 (CHL1) promotes neuritogenesis. J Biol Chem. 2012;287:44447–44463. doi: 10.1074/jbc.M112.394973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Togashi H, Sakisaka T, Takai Y. Cell adhesion molecules in the central nervous system. Cell Adh Migr. 2009;3:29–35. doi: 10.4161/cam.3.1.6773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walz A, Mombaerts P, Greer CA, Treloar HB. Disrupted compartmental organization of axons and dendrites within olfactory glomeruli of mice deficient in the olfactory cell adhesion molecule, OCAM. Mol Cell Neurosci. 2006;32:1–14. doi: 10.1016/j.mcn.2006.01.013. [DOI] [PubMed] [Google Scholar]
- Westphal D, Sytnyk V, Schachner M, Leshchyns'ka I. Clustering of the neural cell adhesion molecule (NCAM) at the neuronal cell surface induces caspase-8- and -3-dependent changes of the spectrin meshwork required for NCAM-mediated neurite outgrowth. J Biol Chem. 2010;285:42046–42057. doi: 10.1074/jbc.M110.177147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler DG, Barrett CF, Groth RD, Safa P, Tsien RW. CaMKII locally encodes L-type channel activity to signal to nuclear CREB in excitation-transcription coupling. J Cell Biol. 2008;183:849–863. doi: 10.1083/jcb.200805048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams EJ, Mittal B, Walsh FS, Doherty P. A Ca2+/calmodulin kinase inhibitor, KN-62, inhibits neurite outgrowth stimulated by CAMs and FGF. Mol Cell Neurosci. 1995;6:69–79. doi: 10.1006/mcne.1995.1007. [DOI] [PubMed] [Google Scholar]
- Winther M, Berezin V, Walmod PS. NCAM2/OCAM/RNCAM: cell adhesion molecule with a role in neuronal compartmentalization. Int J Biochem Cell Biol. 2012;44:441–446. doi: 10.1016/j.biocel.2011.11.020. [DOI] [PubMed] [Google Scholar]
- Yoshihara Y, Kawasaki M, Tamada A, Fujita H, Hayashi H, Kagamiyama H, Mori K. OCAM: A new member of the neural cell adhesion molecule family related to zone-to-zone projection of olfactory and vomeronasal axons. J Neurosci. 1997;17:5830–5842. doi: 10.1523/JNEUROSCI.17-15-05830.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Araki S, Wu J, Teramoto T, Chang YF, Nakano M, Abdelfattah AS, Fujiwara M, Ishihara T, Nagai T, Campbell RE. An expanded palette of genetically encoded Ca(2)(+) indicators. Science. 2011;333:1888–1891. doi: 10.1126/science.1208592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng JQ, Poo MM. Calcium signaling in neuronal motility. Annu Rev Cell Dev Biol. 2007;23:375–404. doi: 10.1146/annurev.cellbio.23.090506.123221. [DOI] [PubMed] [Google Scholar]