

Abstract
Enzyme replacement therapy (ERT) is a treatment option for lysosomal storage disorders (LSDs) caused by deficiencies of soluble lysosomal enzymes. ERT depends on receptor-mediated transport of intravenously injected recombinant enzyme to lysosomes of patient cells. The blood–brain barrier (BBB) prevents efficient transfer of therapeutic polypeptides from the blood to the brain parenchyma and thus hinders effective treatment of LSDs with CNS involvement. We compared the potential of five brain-targeting peptides to promote brain delivery of the lysosomal enzyme arylsulfatase A (ASA). Fusion proteins between ASA and the protein transduction domain of the human immunodeficiency virus TAT protein (Tat), an Angiopep peptide (Ang-2), and the receptor-binding domains of human apolipoprotein B (ApoB) and ApoE (two versions, ApoE-I and ApoE-II) were generated. All ASA fusion proteins were enzymatically active and targeted to lysosomes when added to cultured cells. In contrast to wild-type ASA, which is taken up by mannose-6-phosphate receptors, all chimeric proteins were additionally endocytosed via mannose-6-phosphate-independent routes. For ASA-Ang-2, ASA-ApoE-I, and ASA-ApoE-II, uptake was partially due to the low-density lipoprotein receptor-related protein 1. Transendothelial transfer in a BBB cell culture model was elevated for ASA-ApoB, ASA-ApoE-I, and ASA-ApoE-II. Brain delivery was, however, increased only for ASA-ApoE-II. ApoE-II was also superior to wild-type ASA in reducing lysosomal storage in the CNS of ASA-knock-out mice treated by ERT. Therefore, the ApoE-derived peptide appears useful to treat metachromatic leukodystrophy and possibly other neurological disorders more efficiently.
Keywords: arylsulfatase A, blood–brain barrier, enzyme replacement therapy, lysosomal storage disease, metachromatic leukodystrophy, peptide vectors
Introduction
Lysosomal storage diseases (LSDs) are inherited metabolic disorders that have in common that a defective lysosomal function leads to the accumulation of undegradable macromolecules within the lysosomal compartment (Greiner-Tollersrud and Berg, 2005). LSDs are progressive diseases, most of which become apparent during childhood and lead to mental retardation, high morbidity, and premature death. Although individually rare, they represent a relevant subgroup among pediatric neurological disorders because they have a combined prevalence of 1 in 7000–8000 live births (Hopwood, 2012).
Most LSDs are caused by functional deficiencies of soluble lysosomal enzymes. Because cells express surface receptors that bind exogenous lysosomal enzymes and deliver them to lysosomes, such disorders can theoretically be treated by administration of the recombinantly expressed counterpart of the defective enzyme (Desnick and Schuchman, 2012). Actually, enzyme replacement therapy (ERT) based on repeated and lifelong intravenous injection of active enzyme has been clinically approved for six LSDs. More than 20 years of clinical experience have revealed, however, that ERT has clear limitations. The blood–brain barrier (BBB) is largely impermeable for lysosomal enzymes. Accordingly, therapeutic effects are restricted to peripheral organs. Similar to other neurological diseases, then, the delivery of the therapeutic molecules from the circulation to the brain parenchyma remains a highly challenging issue (Gabathuler, 2010).
In the present study, we compared different peptide vectors to improve the brain delivery of the lysosomal enzyme arylsulfatase A (ASA, E.C. 3.1.6.8). For this purpose, five peptides that were shown previously to promote transcytosis of cargo molecules across the BBB were individually fused to the C terminus of ASA: (1) the protein transduction domain of the transactivator of transcription (TAT) protein of the human immunodeficiency virus 1 (Frankel and Pabo, 1988), (2) the low-density lipoprotein receptor (LDLR)-binding domain of apolipoprotein B (APOB; Spencer and Verma, 2007), (3) and (4) two overlapping peptides derived from apolipoprotein E (APOE) which bind to the LDLR and the LDLR-related protein 1 (LRP1; Wang et al., 1997; Clayton et al., 1999), and (5) the synthetic peptide Angiopep-2, which was derived from the LRP1-ligand aprotinin (Demeule et al., 2008). The peptides are here designated as Tat, ApoB, ApoE-I, ApoE-II, and Ang-2, respectively. Tat was chosen because it facilitated brain delivery of fused β-galactosidase and erythropoietin when injected into the blood circulation of mice (Schwarze et al., 1999; Zhang et al., 2010). ApoB promoted brain delivery of fused glucocerebrosidase, green fluorescent protein, and the metalloprotease neprilysin (Spencer and Verma, 2007; Spencer et al., 2011), whereas the receptor-binding domain of APOE has been exploited to deliver β-galactosidase and antibodies to the CNS (Sarkar et al., 2011). The vector function of Ang-2 has been validated by efficient brain targeting of conjugated nanoparticles and therapeutic agents of low molecular weight such as paclitaxel, doxorubicin, and etoposide (Gabathuler, 2010). The biochemical and pharmacological properties of the five ASA fusion proteins and wild-type ASA were compared in various in vitro and in vivo assays. The therapeutic potential of the most promising fusion protein was evaluated in a preclinical enzyme replacement trial.
Materials and Methods
DNA restriction and modifying enzymes were from Thermo Scientific. Oligonucleotides were from MWG Biotech. The following cell types were used: (1) mouse embryonic fibroblasts (MEFs) from a C57BL/6 wild-type mouse, (2) MEFs deficient for the insulin-like growth factor II/mannose 6-phosphate receptor, MPR300 (MEF-mpr−/−; Pohlmann et al., 1995), (3) MEFs deficient for ASA (MEF-asa−/−; Hess et al., 1996), (4) primary porcine brain capillary endothelial cells (PBCECs; von Wedel-Parlow et al., 2009), (5) mouse brain endothelial cells (bEND.3; ATCC number CRL-2299), (6) Chinese hamster ovary cells (CHO-K1; ATCC number CCL-61), and (7) CHO-K1 cells that lack LRP1 (CHO-lrp−/−; kindly provided by Claus Pietrzik, Mainz, Germany).
Expression vectors.
Synthetic DNA sequences encoding the invariant linker LGGGGSGGGGSGGGGSGGGGS and one of the following peptides were inserted downstream from the full-length ASA cDNA (Stein et al., 1989) cloned into the plasmid pcDNA3 (Life Technologies): (1) aa 47–57 of the TAT protein (Frankel and Pabo, 1988), (2) aa 3371–3409 of human APOB (Spencer and Verma, 2007), (3) aa 130–152 of human APOE (Wang et al., 1997), (4) aa 141–149 of human APOE as tandem repeat (Clayton et al., 1999), and (5) Angiopep-2 (Demeule et al., 2008). The fusion proteins are shown schematically in Figure 1A.
Figure 1.
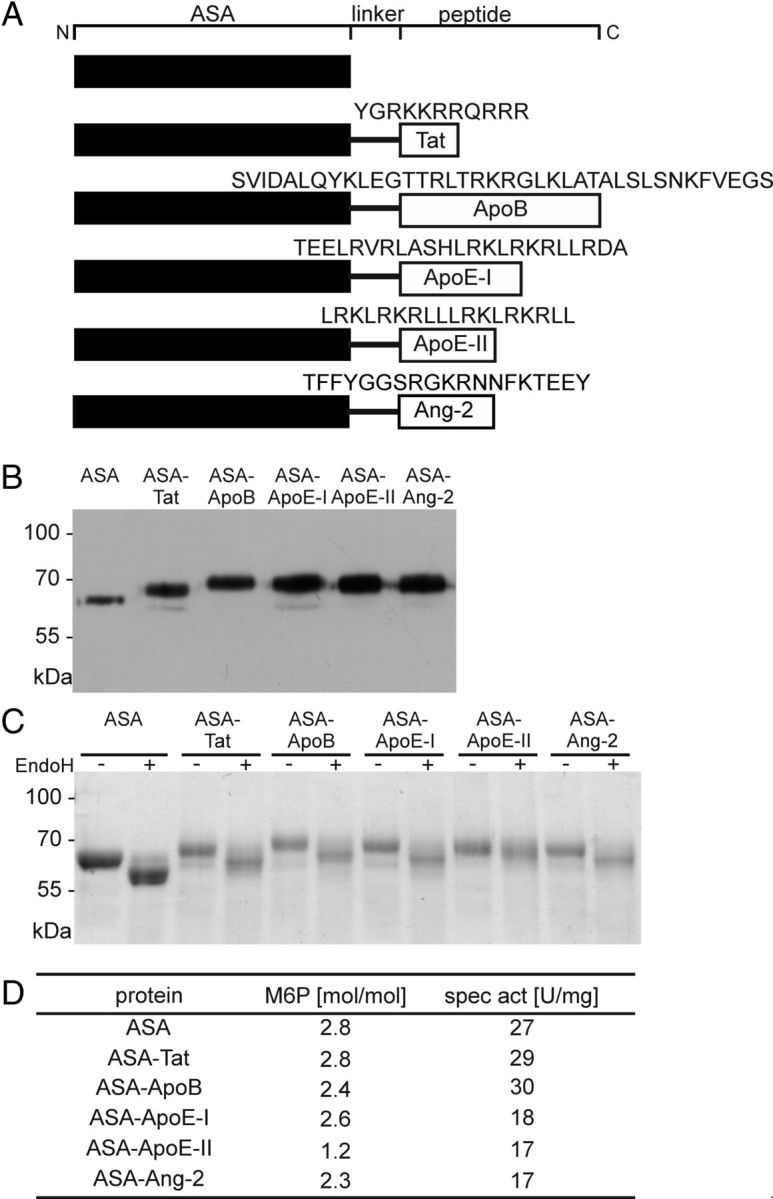
Biochemical characterization of ASA fusion proteins. A, Schemes of ASA fusion proteins. Amino acid sequences of the peptide vectors are given in the one-letter code. Domains are not drawn to scale. B, Western blot analysis. Fifty nanograms of purified ASA and 2.5–10 μl of conditioned medium from CHO-S cells stably expressing ASA fusion proteins were loaded per lane and reacted with a polyclonal anti-human ASA antibody. C, Deglycosylation of ASA fusion proteins with EndoH. Digested and undigested protein was separated by SDS-PAGE and stained with PageBlue Protein Staining solution (Thermo Scientific). D, M6P contents and specific activities as indicated.
Production of ASA fusion proteins.
CHO-suspension (CHO-S) cells (Life Technologies) were transfected with the pcDNA3 constructs using a Bio-Rad gene pulser II at 25 μF and 750 V. Single clones with resistance to G418 were selected by standard methods and screened for high ASA activities in the conditioned medium (Baum et al., 1959). Producer cell clones were cultured in miniPERM bioreactors (Sarstedt) using serum-free ExCell302 medium with HT supplement (both from Sigma), 2 mm l-glutamine, 100 units/ml penicillin, and 100 μg/ml streptomycin (all from Life Technologies). ASA fusion proteins were purified by immunoaffinity chromatography as described previously (Sommerlade et al., 1994).
Biochemical characterization.
Western blotting, analysis of M6P concentrations, and activity measurements were as described previously (Baum et al., 1959; Schröder et al., 2010). Proteins were deglycosylated either with 100 mU/μg N-glycosidase F according to the manufacturer's recommendations (PNGaseF; Roche Applied Science) or with endoglycosidase H (EndoH; Matthes et al., 2011).
Cellular uptake studies.
Cells were incubated with 2.5 μg/ml ASA or ASA fusion protein for 24 h at 37 C. Glucose 6-phosphate (G6P, 7.5 mm; Sigma) and mannose 6-phosphate (M6P, 7.5 mm, Sigma) were added as indicated. After incubation, recombinant protein bound to cell surfaces was removed with two washes of 50 mm glycine, 150 mm NaCl, pH 3.0. Cells were lysed in 1× TBS, pH 7.4, 0.5% Triton N-101 and the protein concentration of the lysate was measured with the Dc assay (Bio-Rad). The intracellular level and total input of recombinant protein was determined by a sandwich ELISA specific for the human ASA (Matzner et al., 2000). For the analysis of the intracellular sorting, MEF-asa−/− cells were stained as described previously (Klein et al., 2009).
BBB cell culture model.
The transendothelial transfer of ASA fusion proteins was determined with PBCECs cultured on microporous polycarbonate filters (von Wedel-Parlow et al., 2009). For each ASA fusion protein, control dishes were incubated with wild-type ASA under identical conditions. To correct for possible variations in the apical concentrations, the basolateral level of ASA fusion protein was normalized to its apical concentration in the same dish. To express the differences between transfer rates, normalized values for ASA fusion proteins were related to the mean of the corresponding ASA controls, which was regarded as 100%.
Animal studies.
All mouse studies were approved by the local animal care and use committee (LANUV reference number 8.87–50.10.35.09.079). ASA fusion proteins were injected into the tail vein of ASA-knock-out mice (Hess et al., 1996) or APOE-knock-out mice (B6.129P2-Apoetm1Unc/Crl from Charles River) at a dose of 20 mg/kg body weight. Age-matched control mice were injected with the same dose of wild-type ASA. Female mice were used for all experiments. For pharmacokinetic studies, a blood sample of ∼20 μl was taken immediately after injection. Mice were deeply anesthetized 110 min after injection, a blood sample was taken 120 min after injection and mice were processed as described previously (Matzner et al., 2005). The serum half-life of the injected proteins was calculated from the ASA concentrations measured immediately after injection and 120 min after injection assuming a monoexponential decline (Matzner et al., 2005). ASA tissue levels were normalized on the protein concentration of the homogenate and on the ASA serum level immediately after injection. Normalized tissue levels of ASA fusion proteins are expressed as percentages of mean wild-type ASA levels measured in the same tissue of the control mice. For the ERT trial, 12-month-old ASA-knock-out mice were treated with four doses of 20 mg/kg recombinant protein given in weekly intervals and lipid profiles were analyzed as described previously (Matzner et al., 2005).
Results
Biochemical characterization
The ASA fusion proteins and wild-type ASA (Fig. 1A) were purified from the secretions of stably transfected CHO-S cells and had the predicted molecular weights (Fig. 1B). Treatment with EndoH, an endoglycosidase that cleaves high-mannose-type N-glycans, reduced the molecular weights of ASA, ASA-Tat, ASA-ApoB, ASA-ApoE-I, and ASA-Ang-2 by 3.4–4.9 kDa (Fig. 1C). In contrast, a size shift of only 1.1 kDa was seen for ASA-ApoE-II. When ASA-ApoE-II was treated with PNGase F, which cleaves all three types of N-linked oligosaccharides (high-mannose, hybrid, complex), the size decrement was 7.1 kDa (data not shown).
Wild-type ASA exhibited 2.8 M6P residues per molecule, reproducing previous results (Matthes et al., 2011). The mean M6P concentration of ASA fusion proteins ranged between 82% and 100% of the wild-type level (Fig. 1D). The only exception was ASA-ApoE-II, which showed <50% of the normal M6P content.
To determine possible adverse effects of the fused peptides on the enzymatic function of the ASA domain, ASA activities of purified fusion proteins were determined with an artificial substrate (Baum et al., 1959). The specific activities of the chimeric proteins ranged between 63% and 111% of the wild-type ASA (Fig. 1D).
Uptake of wild-type ASA by endothelial and nonendothelial cells
In initial experiments, uptake of wild-type ASA by two cell types of brain endothelial origin (bEND.3, PBCEC) and two cell types of nonendothelial origin (CHO-K1, MEF) was compared. Uptake by CHO-K1 and MEF cells was substantially higher than that by the brain endothelial cells. CHO-K1 cells, for example, internalized 59- and 37-fold more ASA on average than bEND.3 cells (Fig. 2A) and PBCECs (data not shown), respectively. M6P (7.5 mm) reduced the efficient uptake by CHO-K1 and MEF cells by >99%, whereas the low internalization by bEND.3 cells and PBCEC was diminished by only 47% (Fig. 2A) and 59% (data not shown), respectively. Notably, the resulting residual uptake rates of endothelial cells exceeded those of the nonendothelial cells (Fig. 2A; data not shown for PBCECs).
Figure 2.
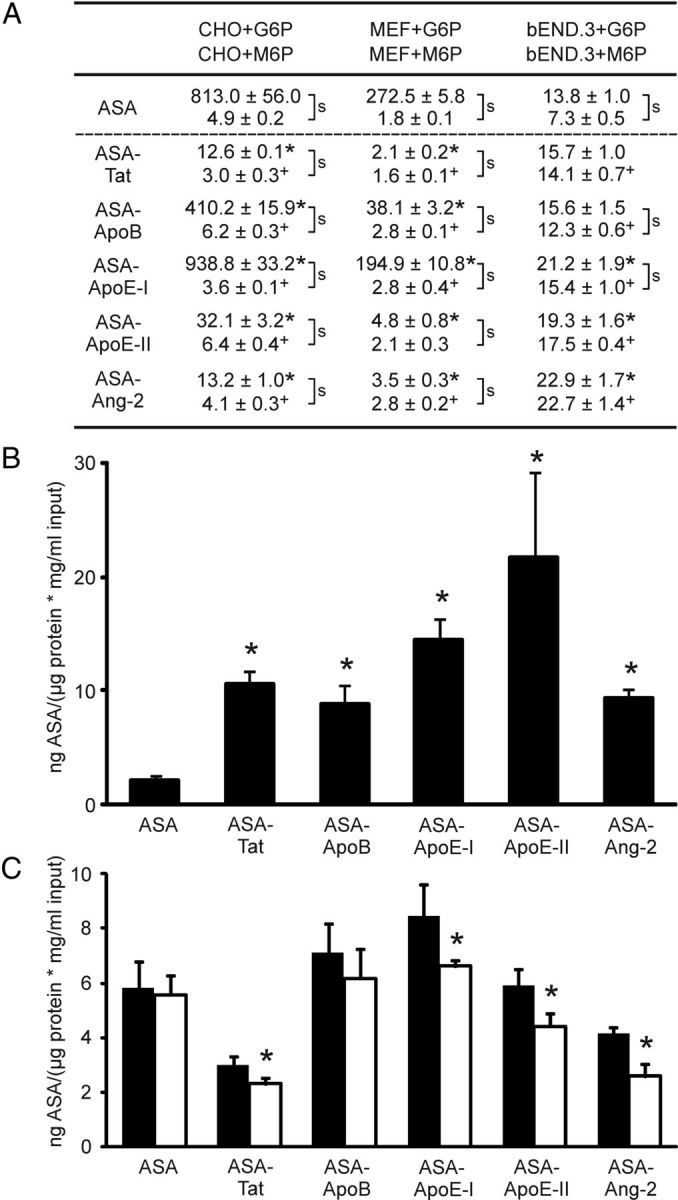
Cellular uptake of ASA fusion proteins. A, Uptake by CHO-K1, MEF, and bEND.3 cells as indicated. Cells were either coincubated with 7.5 mm M6P to block MPR300-mediated endocytosis (lower values) or, as a control, with 7.5 mm G6P (upper values). Uptake is expressed as nanograms of intracellular ASA per microgram of input ASA and micrograms of extractable protein. Numbers represent mean ± SD of n = 3 wells per condition. S-signs denote statistically significant differences between M6P and G6P feedings (Student's t test, p < 0.05). Asterisks and plus signs indicate statistical differences between an ASA fusion protein and the ASA control in the presence of G6P and M6P, respectively. B, Uptake by MEF-mpr−/− cells (deficient for MPR300). Bars represent mean ± SD of n = 4–6 wells per condition. Asterisks indicate statistically significant differences from wild-type ASA (Student's t test, p < 0.05). C, LRP1-dependent endocytosis. CHO-K1 wild-type cells (closed bars) and CHO-lrp−/− cells (deficient for LRP1; open bars) were incubated with 7.5 mm M6P to block M6P-dependent endocytosis. Bars represent mean ± SD of n = 3 wells per condition. Asterisks indicate a statistically significant difference between LRP1-deficient and wild-type CHO-K1 cells (Student's t test, p < 0.05).
M6P-dependent and M6P-independent cellular uptake of ASA fusion proteins
CHO-K1, MEF, and bEND.3 cells were used to analyze the cellular uptake of ASA fusion proteins. In the absence of M6P, internalization rates differed considerably between the fusion proteins. For CHO-K1 cells, for example, the uptake of ASA-Tat, ASA-ApoE-II, and ASA-Ang-2 was >95% lower than for wild-type ASA, whereas endocytosis of ASA-ApoE-I was 15% above normal (Fig. 2A). Similar peptide-specific differences were seen when MEF cells were fed. In contrast to CHO-K1 and MEF cells, bEND.3 cells showed a low endocytic activity and there was a general trend for a more efficient uptake of ASA fusion proteins compared with wild-type ASA. The internalization of ASA-ApoE-I, ASA-ApoE-II, and ASA-Ang-2, for example, was significantly increased by 54%, 40%, and 66%, respectively.
M6P (7.5 mm) reduced the uptake of all five ASA fusion proteins. Internalization of ASA-Tat and ASA-ApoE-I by CHO-K1 cells, for example, was diminished by 76% and >99%, respectively. Effects of M6P on bEND.3 cells were less dramatic. Depending on the fusion protein, uptake was diminished by 1–47% and reductions for ASA-Tat, ASA-ApoE-II, and ASA-Ang-2 failed to reach statistical significance. Furthermore, M6P aggravated the tendency for increased uptake of ASA fusion proteins by bEND.3 cells and compared with wild-type ASA, 1.7- to 3.1-fold more of the ASA fusion proteins were internalized.
To avoid possible unspecific side effects of high M6P concentrations, the M6P-independent uptake was also analyzed using MPR300-deficient MEF cells (Pohlmann et al., 1995). Just as bEND.3 cells coincubated with M6P, MPR300-deficient MEF cells endocytosed all five ASA fusion proteins more efficiently than wild-type ASA (Fig. 2B). The mean uptake was increased by factors of 3.9–9.5.
We compared uptake rates of LRP1-expressing and LRP1-deficient CHO-K1 cells to determine whether ASA fusion proteins are internalized via LRP1. Competitive amounts of M6P were added to all dishes to block MPR300. Uptake of wild-type ASA was indistinguishable between LRP1-deficient and LRP1-expressing cells and no significant difference was seen for ASA-ApoB. Endocytosis of the other four fusion proteins was, however, reduced in LRP1-deficient cells (Fig. 2C).
To analyze subcellular trafficking of endocytosed ASA fusion proteins, ASA-deficient MEF cells (Hess et al., 1996) were fed with recombinant enzyme. Costaining of Lamp-2 and ASA indicated an exclusive endosomal/lysosomal localization of ASA and ASA fusion proteins (Fig. 3).
Figure 3.
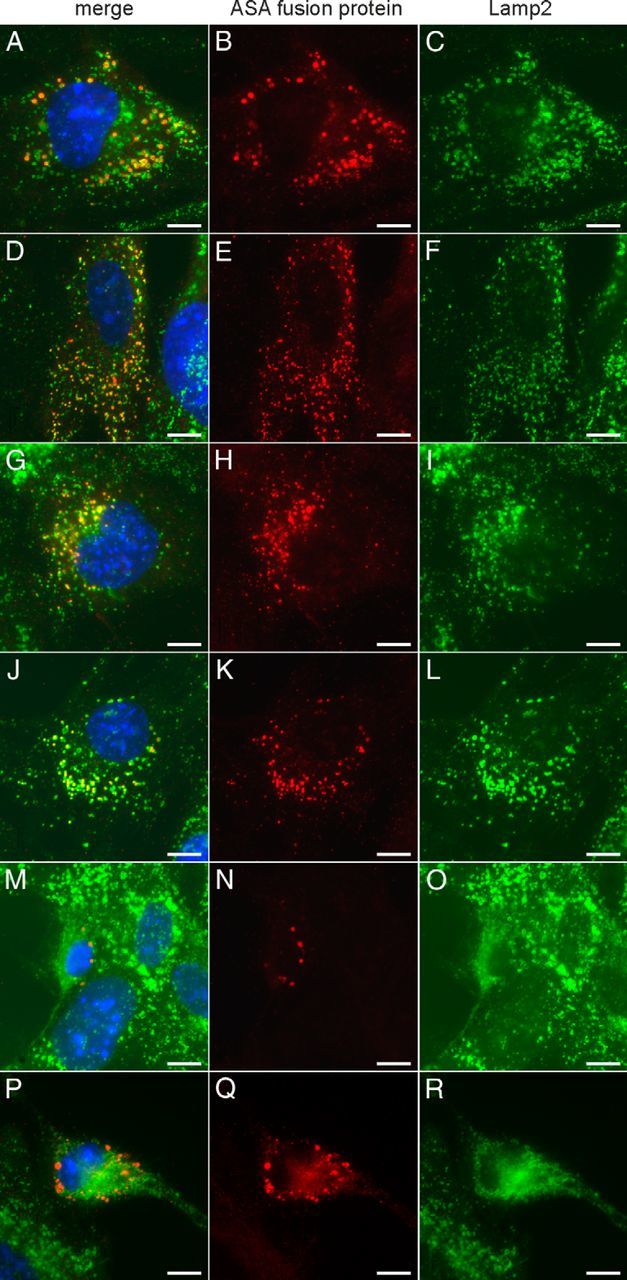
Immunofluorescence staining of MEF-asa−/− (ASA-deficient) cells after feeding of ASA fusion proteins. Cells were fed with wild-type ASA (A–C), ASA-Tat (D–F), ASA-ApoB (G–I), ASA-ApoE-I (J–L), ASA-ApoE-II (M–O), and ASA-Ang-2 (P–R), respectively. Recombinant enzyme and LAMP-2 were stained in red (middle) and green (right), respectively. Merge panels (left) also show nuclei counterstained with DAPI (blue). Scale bars, 10 μm.
Basolateral transfer of ASA fusion proteins in an in vitro BBB model
The transendothelial transfer rate of ASA fusion proteins was evaluated in vitro using an established cell culture model of the BBB (von Wedel-Parlow et al., 2009). This consists of PBCECs grown on microporous cell culture inserts separating an apical from a basolateral compartment representing the blood and brain side of the capillary endothelium, respectively (Fig. 4A). Within 24 h, ∼0.02% of the apically added wild-type ASA was transferred to the basolateral medium, reproducing data from previous transport studies (Matthes et al., 2011). Although the basolateral transfer rates of ASA-Tat and ASA-Ang-2 were indistinguishable from that of wild-type ASA, those of ASA-ApoB, ASA-ApoE-I, and ASA-ApoE-II were significantly increased by factors of 2.2, 2.0, and 1.5, respectively (Fig. 4B).
Figure 4.

Basolateral transfer of ASA fusion proteins in an in vitro BBB model. A, Scheme of the experimental setup. B, Basolateral concentrations of ASA (closed bars) and ASA fusion proteins (open bars) were measured 24 h after the addition of 0.1 mg/ml purified protein to the apical medium. Bars represent mean ± SD of n = 3–5 wells per condition. Asterisks indicate statistically significant differences between ASA and ASA fusion proteins (Student's t test, p < 0.05).
Treatment of ASA-knock-out mice
Intravenously injected wild-type ASA was cleared from the blood of ASA-knock-out mice (Hess et al., 1996) with an apparent serum half-life of 11 min (data not shown). ASA-Tat, ASA-ApoB, ASA-ApoE-I, and ASA-Ang-2 showed similar half-lives of 13–16 min, whereas ASA-ApoE-II had an ∼2-fold prolonged serum half-life of ∼30 min.
ASA-Tat and ASA-Ang-2 showed the same tissue distribution as wild-type ASA, whereas significant differences were seen for ASA-ApoB, ASA-ApoE-I, and ASA-ApoE-II (Fig. 5A–E). The brain levels of ASA-ApoB, for example, were 43% lower than those of wild-type ASA on average (Fig. 5B). In contrast, brain levels of ASA-ApoE-II were increased by 54% (p < 0.05; Fig. 5D). To test the hypothesis that delivery of ASA-ApoE-II to the brain is hindered by endogenously expressed APOE competing for APOE receptors at the BBB, we treated also APOE-knock-out mice with ASA-ApoE-II. In this experiment, brain levels of ASA-ApoE-II were 62% higher than that of wild-type ASA. The small difference between brain levels of ApoE- and ASA-knock-out mice was, however, not significant and, for the other tissues, essentially the same levels were obtained (Fig. 5F).
Figure 5.
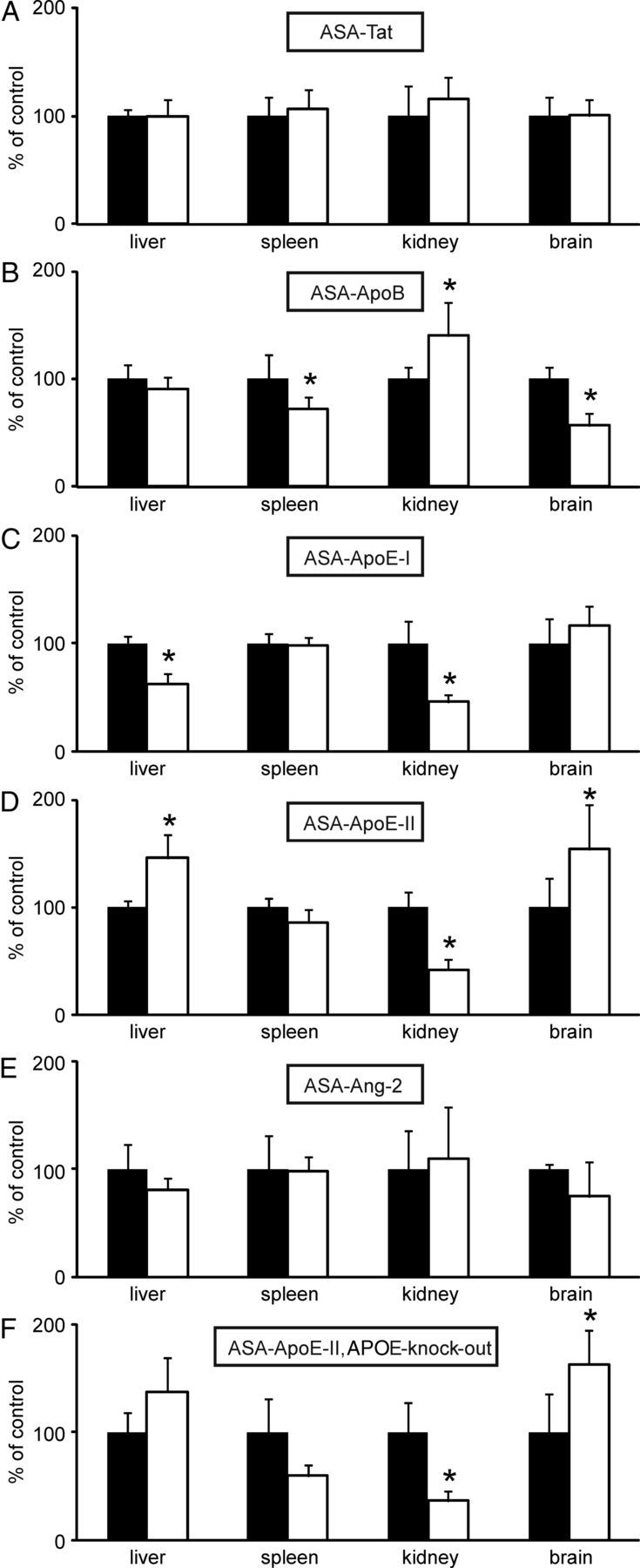
Tissue distribution of ASA fusion proteins. ASA-knock-out mice were injected with ASA-Tat (A), ASA-ApoB (B), ASA-ApoE-I (C), ASA-ApoE-II (D), and ASA-Ang-2 (E), respectively (open bars). In F, APOE-knock-out mice were injected with ASA-ApoE-II. For each fusion protein, a separate set of control mice was treated with ASA (closed bars). Bars represent mean ± SD of n = 4–5 mice. Asterisks indicate statistically significant differences between wild-type ASA and ASA fusion proteins (Student's t test, p < 0.05).
To compare the therapeutic potential of ASA-ApoE-II and wild-type ASA, ASA-knock-out mice were treated by four injections of 20 mg/kg recombinant enzyme given in weekly intervals. Treatment with wild-type ASA reduced sulfatide storage in both kidney and brain by 22% (p < 0.05, Student's t test; Fig. 6). ASA-ApoE-II was more effective and diminished sulfatide storage by 37% and 38%, respectively. Therefore, the fusion protein cleared 1.7-fold more sulfatide from the two tissues than wild-type ASA.
Figure 6.

Reduction of sulfatide storage by ERT. Sulfatide and cholesterol levels were quantified in kidney (A) and total brain (B) of ASA-knock-out mice (ASA−/−) treated with vehicle (mock), wild-type ASA (wt ASA), and ASA-ApoE-II, respectively. Untreated wild-type mice (ASA+/+) were used as controls. Sulfatide levels were normalized on cholesterol levels and are expressed as the fold increase above mean levels of wild-type tissues. Bars represent mean ± SD of n = 3 mice per group. ASA-ApoE-II reduced sulfatide storage more efficiently than wild-type ASA (Student's t test, p-values indicated).
Discussion
A number of peptides have been reported to surmount the BBB and to promote delivery of cargo molecules to the brain (Gabathuler, 2010). In an attempt to optimize ERT in mouse models of metachromatic leukodystrophy (MLD; Matzner et al., 2005, 2009; Matthes et al., 2012), five of them were individually fused to the human ASA polypeptide and analyzed (Fig. 1A).
The N-glycosylation and M6P content of ASA-Tat, ASA-ApoB, ASA-ApoE-I, and ASA-Ang-2 was similar to that of wild-type ASA (Fig. 1C,D). ASA-ApoE-II, however, exhibited less M6P residues and an increased concentration of complex and/or hybrid type N-glycans. This suggests that the ApoE-II domain interferes with the N-acetylglucosamine-1-phosphotransferase, which catalyzes the first step in the formation of M6P residues (Coutinho et al., 2012). Phosphorylation of high-mannose-type N-glycans precedes oligosaccharide remodeling and hampers their maturation to hybrid and complex type structures (Miller et al., 1993). Therefore, the shift toward hybrid and/or complex type oligosaccharides might be secondary to the low mannosyl phosphorylation of ASA-ApoE-II.
Wild-type ASA, used as a control, was efficiently endocytosed by CHO-K1 and MEF cells and soluble M6P reduced internalization by >99% (Fig. 2A). MPR300-mediated endocytosis appears, therefore, to be the primary entry route for wild-type ASA into these and other nonendothelial cells (Hartmann et al., 1992; Schmitz et al., 1995; Muschol et al., 2002; Matzner et al., 2008). Brain endothelial cells (bEND.3, PBCECs) endocytose up to 59-fold less ASA than nonendothelial cells (CHO-K1, MEFs). Therefore, only minute amounts of enzyme will be routed to brain endothelial cells when they compete with nonendothelial cells for intravenously injected ASA. This wass clearly confirmed by measurements of ASA tissue levels after injection of wild-type ASA into ASA-knock-out mice. Although mean levels of liver, spleen, and kidney ASA were 3250, 1200, and 37 ng/mg, respectively, only 0.6 ng/mg was found in the brain (n = 25, data not shown). After the addition of soluble M6P, the high endocytic activity of nonendothelial cells dropped below the levels of bEND.3 cells and PBCECs (Fig. 2A and data not shown). This indicates that the discrepancy in the uptake rates is entirely due to a very low endocytic activity of the MPR300 in brain endothelial cells. Because the MPR300 has a low activity at the BBB and acts as a sink for lysosomal enzymes in the periphery, delivery of ASA to the brain might be favored by two basically independent strategies: (1) by reducing the MPR300-dependent bulk flow of injected ASA to peripheral tissues and (2) by targeting the enzyme to M6P-independent receptors at the BBB.
When the MPR300 was blocked, bEND.3 cells endocytosed ASA fusion proteins 2- to 3-fold faster than wild-type ASA, with the highest uptake rates detectable for ASA-ApoE-I, ASA-ApoE-II, and ASA-Ang-2 (Fig. 2A). LRP1 contributed to the M6P-independent uptake of these three constructs (Fig. 2C). Another observation was that all five peptides impaired the MPR300-mediated endocytosis by CHO-K1 and/or MEF cells (for ASA-ApoE-I only in MEF cells). Uptake of ASA-Tat, for example, was >98% lower than for wild-type ASA (Fig. 2A). Therefore, the fused peptides target new receptor system(s) at the BBB and concomitantly reduce the affinity of the ASA to the MPR300; that is, they comply with both requirements proposed to favor ASA delivery to the brain (see last paragraph).
Preclinical ERT trials revealed that the observed retargeting from MPR300- to M6P-independent receptors indeed had the potential to alter the biodistribution of ASA fusion proteins in vivo (Fig. 5). In view of the endocytosis experiments and increased transendothelial transfer rates observed in vitro (Fig. 4), it was, however, surprising that, among the five ASA fusion proteins, only ASA-ApoE-II acquired significantly higher brain levels than wild-type ASA. The ApoB-peptide, for example, mediated the highest transcytotic rate in the BBB model, but it did not increase—and in fact reduced— the delivery of ASA to the brain. This suggests that even though receptors for ApoB exist at the BBB, such receptors might preponderate in peripheral tissues and cause, under competitive conditions, a redistribution of the fusion protein to the disfavor of the brain.
The most important result of our study is that the ApoE-II peptide not only increased brain delivery of ASA, but also improved the therapeutic potential of ERT in a mouse model of MLD. Compared with wild-type ASA, the brain level and sulfatide clearance was elevated by similar factors of 1.6 and 1.7, respectively. Progressive demyelination is a hallmark of MLD. However, not only oligodendrocytes, but also astrocytes, microglial cells, and subpopulations of neurons store sulfatide (von Figura et al., 2001). The ApoE-II peptide might also affect the dissemination of ASA between these cell types. A redistribution of ASA-ApoE-II between cell types may explain the unexpected result for kidney, where the sulfatide clearance is improved (Fig. 6A) even though the tissue level is diminished (Fig. 5D). Possibly, a smaller amount of ASA is more effective there because the ApoE-II peptide targets ASA to cells that are the most affected by storage.
The potential of ApoE-II to target ASA across the BBB confirms a recent gene therapy approach in which a fusion protein between α-l-iduronidase and an APOE peptide reached 5-fold higher brain levels than wild-type α-l-iduronidase (Wang et al., 2013). However, not all peptides derived from the receptor-binding domain of APOE are equivalent. In our study, ASA-ApoE-I did not reach the elevated brain levels of ASA-ApoE-II and this was correlated with a decreased M6P content (Fig. 1D), a lower uptake rate by CHO K1 cells, and a higher serum half-life of ASA-ApoE-II. Therefore, brain targeting of ASA-ApoE-II via LRP1 and/or other receptors might be advanced by higher levels of enzyme staying for longer periods of time in the circulation of the CNS. Sly and coworkers were the first to demonstrate that a prolongation of the plasma half-life can increase the delivery of a lysosomal enzyme to the brain parenchyma (Grubb et al., 2008; Huynh et al., 2012). These investigators modified the N-glycans of the lysosomal enzyme β-glucuronidase to eliminate its uptake by MPR300 and other carbohydrate-dependent receptors. Compared with unmodified enzyme, modified β-glucuronidase had a >100-fold prolonged plasma half-life, acquired 6-fold higher brain levels, and eliminated brain storage in β-glucuronidase-deficient mice more efficiently.
In summary, we have shown here that a peptide derived from the receptor-binding domain of APOE is effective in delivering ASA to the brain of mice. We demonstrated binding of the fusion protein to LRP1, a high-capacity transport system that mediates rapid transcytosis of proteins from blood to brain (Gabathuler, 2010). We have shown previously that treatment of MLD mice with high doses of intravenously injected wild-type ASA improves the CNS histopathology and function due to small amounts of ASA reaching the brain parenchyma (Matzner et al., 2005, 2009; Matthes et al., 2012). In the present study, we have demonstrated that ASA-ApoE-II is superior to wild-type ASA in reducing sulfatide storage in brain and kidney. Minor variations in the residual enzyme activity between 0% and 5% of the normal ASA activity determine the entire spectrum of clinical manifestations of MLD from early-onset forms to healthy individuals (von Figura et al., 2001). It is therefore likely that the observed increase in brain delivery and sulfatide clearance may significantly improve the therapeutic benefit of ERT. Future long-term studies in ASA-knock-out mice and MLD patients are required to test this notion.
Footnotes
This work was supported by the Seventh Framework Programme for research, technological development, and demonstration activities of the European Community LeukoTreat (Grant 241622 to V.G.) and by the European Leukodystrophy Association (Grant 2009-031I5A to U.M.). We thank Claus Pietrzik (Mainz, Germany) and Heidi Simonis (Bonn, Germany) for kindly providing LRP1-deficient CHO-K1 cells and for excellent technical assistance, respectively.
The authors declare no competing financial interests.
References
- Baum H, Dogson KS, Spencer B. The assay of arylsulphatases A and B in human urine. Clin Chim Acta. 1959;4:453–455. doi: 10.1016/0009-8981(59)90119-6. [DOI] [PubMed] [Google Scholar]
- Clayton D, Brereton IM, Kroon PA, Smith R. NMR studies of the low-density lipoprotein receptor-binding peptide of apolipoprotein E bound to dodecylphosphocholine micelles. Protein Sci. 1999;8:1797–1805. doi: 10.1110/ps.8.9.1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coutinho MF, Prata MJ, Alves S. Mannose-6-phosphate pathway: a review on its role in lysosomal function and dysfunction. Mol Genet Metab. 2012;105:542–550. doi: 10.1016/j.ymgme.2011.12.012. [DOI] [PubMed] [Google Scholar]
- Demeule M, Régina A, Ché C, Poirier J, Nguyen T, Gabathuler R, Castaigne JP, Béliveau R. Identification and design of peptides as a new drug delivery system for the brain. J Pharmacol Exp Ther. 2008;324:1064–1072. doi: 10.1124/jpet.107.131318. [DOI] [PubMed] [Google Scholar]
- Desnick RJ, Schuchman EH. Enzyme replacement therapy for lysosomal diseases: lessons from 20 years of experience and remaining challenges. Annu Rev Genomics Hum Genet. 2012;13:307–335. doi: 10.1146/annurev-genom-090711-163739. [DOI] [PubMed] [Google Scholar]
- Frankel AD, Pabo CO. Cellular uptake of the tat protein from human immunodeficiency virus. Cell. 1988;55:1189–1193. doi: 10.1016/0092-8674(88)90263-2. [DOI] [PubMed] [Google Scholar]
- Gabathuler R. Approaches to transport therapeutic drugs across the blood-brain barrier to treat brain diseases. Neurobiol Dis. 2010;37:48–57. doi: 10.1016/j.nbd.2009.07.028. [DOI] [PubMed] [Google Scholar]
- Greiner-Tollersrud OK, Berg T. Lysosomal storage disorders. In: Saftig P, editor. Lysosomes. Georgetown: Landes Bioscience; 2005. pp. 60–73. [Google Scholar]
- Grubb JH, Vogler C, Levy B, Galvin N, Tan Y, Sly WS. Chemically modified beta-glucuronidase crosses blood-brain barrier and clears neuronal storage in murine mucopolysaccharidosis VII. Proc Natl Acad Sci U S A. 2008;105:2616–2621. doi: 10.1073/pnas.0712147105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann H, Meyer-Alber A, Braulke T. Metabolic actions of insulin-like growth factor II in cultured adult rat hepatocytes are not mediated through the insulin-like growth factor II receptor. Diabetologia. 1992;35:216–223. doi: 10.1007/BF00400920. [DOI] [PubMed] [Google Scholar]
- Hess B, Saftig P, Hartmann D, Coenen R, Lüllmann-Rauch R, Goebel HH, Evers M, von Figura K, D'Hooge R, Nagels G, De Deyn P, Peters C, Gieselmann V. Phenotype of arylsulfatase A-deficient mice: relationship to human metachromatic leukodystrophy. Proc Natl Acad Sci U S A. 1996;93:14821–14826. doi: 10.1073/pnas.93.25.14821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopwood JJ. Genetics of lysosomal storage disorders and counselling. In: Mehta A, Winchester B, editors. Lysosomal storage disorders. Hoboken, NJ: Wiley-Blackwell; 2012. pp. 29–36. [Google Scholar]
- Huynh HT, Grubb JH, Vogler C, Sly WS. Biochemical evidence for superior correction of neuronal storage by chemically modified enzyme in murine mucopolysaccharidosis VII. Proc Natl Acad Sci U S A. 2012;109:17022–17027. doi: 10.1073/pnas.1214779109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein D, Yaghootfam A, Matzner U, Koch B, Braulke T, Gieselmann V. Mannose 6-phosphate receptor-dependent endocytosis of lysosomal enzymes is increased in sulfatide-storing kidney cells. Biol Chem. 2009;390:41–48. doi: 10.1515/BC.2009.009. [DOI] [PubMed] [Google Scholar]
- Matthes F, Wölte P, Böckenhoff A, Hüwel S, Schulz M, Hyden P, Fogh J, Gieselmann V, Galla HJ, Matzner U. Transport of arylsulfatase A across the blood-brain barrier in vitro. J Biol Chem. 2011;286:17487–17494. doi: 10.1074/jbc.M110.189381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthes F, Stroobants S, Gerlach D, Wohlenberg C, Wessig C, Fogh J, Gieselmann V, Eckhardt M, D'Hooge R, Matzner U. Efficacy of enzyme replacement therapy in an aggravated mouse model of metachromatic leukodystrophy declines with age. Hum Mol Genet. 2012;21:2599–2609. doi: 10.1093/hmg/dds086. [DOI] [PubMed] [Google Scholar]
- Matzner U, Harzer K, Learish RD, Barranger JA, Gieselmann V. Long-term expression and transfer of arylsulfatase A into brain of arylsulfatase A-deficient mice transplanted with bone marrow expressing the arylsulfatase A cDNA from a retroviral vector. Gene Ther. 2000;7:1250–1257. doi: 10.1038/sj.gt.3301232. [DOI] [PubMed] [Google Scholar]
- Matzner U, Herbst E, Hedayati KK, Lüllmann-Rauch R, Wessig C, Schröder S, Eistrup C, Möller C, Fogh J, Gieselmann V. Enzyme replacement improves nervous system pathology and function in a mouse model for metachromatic leukodystrophy. Hum Mol Genet. 2005;14:1139–1152. doi: 10.1093/hmg/ddi126. [DOI] [PubMed] [Google Scholar]
- Matzner U, Matthes F, Weigelt C, Andersson C, Eistrup C, Fogh J, Gieselmann V. Non-inhibitory antibodies impede lysosomal storage reduction during enzyme replacement therapy of a lysosomal storage disease. J Mol Med (Berl) 2008;86:433–442. doi: 10.1007/s00109-008-0309-3. [DOI] [PubMed] [Google Scholar]
- Matzner U, Lüllmann-Rauch R, Stroobants S, Andersson C, Weigelt C, Eistrup C, Fogh J, D'Hooge R, Gieselmann V. Enzyme replacement improves ataxic gait and central nervous system histopathology in a mouse model of metachromatic leukodystrophy. Mol Ther. 2009;17:600–606. doi: 10.1038/mt.2008.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller AL, Norton V, Robertson R, Jenks M, Yeh RY, Wright D. Light and heavy lysosomes: characterization of N-acetyl-beta-d-hexosaminidase isolated from normal and I-cell disease lymphoblasts. Glycobiology. 1993;3:313–318. doi: 10.1093/glycob/3.4.313. [DOI] [PubMed] [Google Scholar]
- Muschol N, Matzner U, Tiede S, Gieselmann V, Ullrich K, Braulke T. Secretion of phosphomannosyl-deficient arylsulphatase A and cathepsin D from isolated human macrophages. Biochem J. 2002;368:845–853. doi: 10.1042/BJ20020249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pohlmann R, Boeker MW, von Figura K. The two mannose 6-phosphate receptors transport distinct complements of lysosomal proteins. J Biol Chem. 1995;270:27311–27318. doi: 10.1074/jbc.270.45.27311. [DOI] [PubMed] [Google Scholar]
- Sarkar G, Curran GL, Mahlum E, Decklever T, Wengenack TM, Blahnik A, Hoesley B, Lowe VJ, Poduslo JF, Jenkins RB. A carrier for non-covalent delivery of functional beta-galactosidase and antibodies against amyloid plaques and IgM to the brain. PLoS On, 2011;6:e28881. doi: 10.1371/journal.pone.0028881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz F, Bresciani R, Hartmann H, Braulke T. Effect of insulin-like growth factor II on uptake of arylsulfatase A by cultured rat hepatocytes and Kupffer cells. J Hepatol. 1995;22:356–363. doi: 10.1016/0168-8278(95)80290-8. [DOI] [PubMed] [Google Scholar]
- Schröder S, Matthes F, Hyden P, Andersson C, Fogh J, Müller-Loennies S, Braulke T, Gieselmann V, Matzner U. Site-specific analysis of N-linked oligosaccharides of recombinant lysosomal arylsulfatase A produced in different cell lines. Glycobiology. 2010;20:248–259. doi: 10.1093/glycob/cwp171. [DOI] [PubMed] [Google Scholar]
- Schwarze SR, Ho A, Vocero-Akbani A, Dowdy SF. In vivo protein transduction: delivery of a biologically active protein into the mouse. Science. 1999;285:1569–1572. doi: 10.1126/science.285.5433.1569. [DOI] [PubMed] [Google Scholar]
- Sommerlade HJ, Hille-Rehfeld A, von Figura K, Gieselmann V. Four monoclonal antibodies inhibit the recognition of arylsulphatase A by the lysosomal enzyme phosphotransferase. Biochem J. 1994;297:123–130. doi: 10.1042/bj2970123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer BJ, Verma IM. Targeted delivery of proteins across the blood-brain barrier. Proc Natl Acad Sci U S A. 2007;104:7594–7599. doi: 10.1073/pnas.0702170104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer B, Marr RA, Gindi R, Potkar R, Michael S, Adame A, Rockenstein E, Verma IM, Masliah E. Peripheral delivery of a CNS targeted, metalo-protease reduces aβ toxicity in a mouse model of Alzheimer's disease. PLoS One. 2011;6:e16575. doi: 10.1371/journal.pone.0016575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein C, Gieselmann V, Kreysing J, Schmidt B, Pohlmann R, Waheed A, Meyer HE, O'Brien JS, von Figura K. Cloning and expression of human arylsulfatase A. J Biol Chem. 1989;264:1252–1259. [PubMed] [Google Scholar]
- von Figura K, Gieselmann V, Jaeken J. Metachromatic leukodystrophy. In: Scriver CR, Beaudet AL, Sly WS, Valle D, Childs B, Kinzler KW, Vogelstein B, editors. The metabolic and molecular bases of inherited disease. New York: McGraw-Hill; 2001. pp. 3695–3724. [Google Scholar]
- von Wedel-Parlow M, Wölte P, Galla HJ. Regulation of major efflux transporters under inflammatory conditions at the blood-brain barrier in vitro. J Neurochem. 2009;111:111–118. doi: 10.1111/j.1471-4159.2009.06305.x. [DOI] [PubMed] [Google Scholar]
- Wang D, El-Amouri SS, Dai M, Kuan CY, Hui DY, Brady RO, Pan D. Engineering a lysosomal enzyme with a derivative of receptor-binding domain of apoE enables delivery across the blood-brain barrier. Proc Natl Acad Sci U S A. 2013;110:2999–3004. doi: 10.1073/pnas.1222742110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Ciraolo G, Morris R, Gruenstein E. Identification of a neuronal endocytic pathway activated by an apolipoprotein E (apoE) receptor binding peptide. Brain Res. 1997;778:6–15. doi: 10.1016/S0006-8993(97)00877-9. [DOI] [PubMed] [Google Scholar]
- Zhang F, Xing J, Liou AK, Wang S, Gan Y, Luo Y, Ji X, Stetler RA, Chen J, Cao G. Enhanced delivery of erythropoietin across the blood-brain barrier for neuroprotection against ischemic neuronal injury. Transl Stroke Res. 2010;1:113–121. doi: 10.1007/s12975-010-0019-3. [DOI] [PMC free article] [PubMed] [Google Scholar]