

Abstract
Dysregulated proinflammatory cytokine release has been implicated in the pathogenesis of several life-threatening acute lung illnesses such as pneumonia, sepsis, and acute respiratory distress syndrome. Suppressors of cytokine signaling proteins, particularly SOCS2, have recently been described as antiinflammatory mediators. However, the regulation of SOCS2 protein has not been described. Here we describe a mechanism of SOCS2 regulation by the action of the ubiquitin E3 ligase KIAA0317. KIAA0317-mediated degradation of SOCS2 exacerbated inflammation in vitro, and depletion of KIAA0317 in vivo ameliorated pulmonary inflammation. KIAA0317-knockout mice exhibited resistance to LPS-induced pulmonary inflammation, while KIAA03017 reexpression mitigated this effect. We uncovered a small molecule inhibitor of KIAA0317 protein (BC-1365) that prevented SOCS2 degradation and attenuated LPS- and P. aeruginosa–induced lung inflammation in vivo. These studies show KIAA0317 to be a critical mediator of pulmonary inflammation through its degradation of SOCS2 and a potential candidate target for therapeutic inhibition.
Keywords: Inflammation, Pulmonology
Keywords: Innate immunity, Ubiquitin-proteosome system
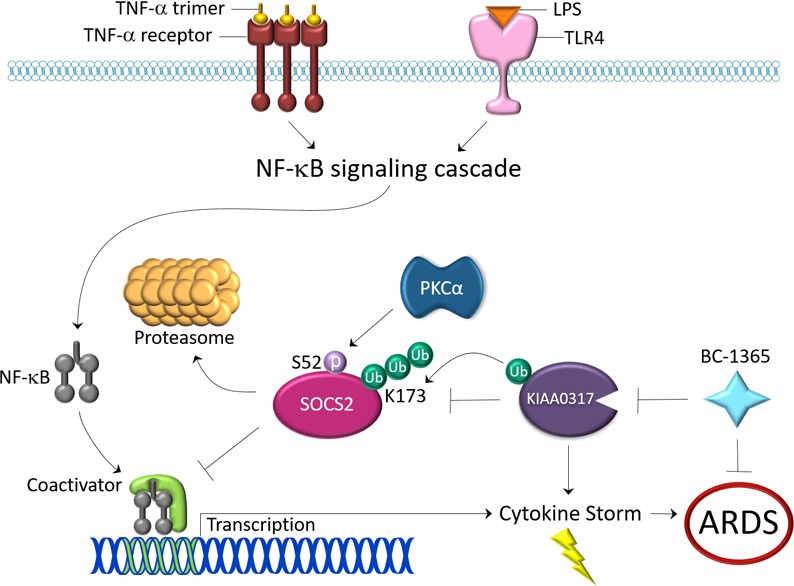
Ubiquitin E3 ligase KIAA0317 targets and ubiqutinates SOCS2 for proteasomal degradation, regulating cytokine release and pulmonary inflammation.
Introduction
The inflammatory response in the lung is a critical response to infection and for maintaining pulmonary homeostasis. Pathogen sensing by the host initiates inflammation via cytokine signaling cascades (1), which facilitates the activation of the immune system and recruitment of leukocytes to the afflicted area. However, uncontrolled pulmonary inflammation can be deleterious, as several acute pathologies including acute respiratory distress syndrome (ARDS) are associated with high proinflammatory cytokine levels (2). In the case of ARDS, dysregulated pulmonary inflammation results in capillary damage, disruption of the alveolar epithelium, alveolar damage caused by leukocytes, and the influx of edema fluid rich in connective tissue and protein (3). ARDS annually affects almost 0.25 million patients, with greater than 40% mortality (3–5).
Pulmonary inflammation is directly associated with proinflammatory cytokines such as IL-1, IL-6, and TNF (6, 7). Alveolar macrophages from ARDS patients show more activated NF-κB (8), and subsequent research has shown that the NF-κB signaling pathway promotes the transcription of cytokines involved in ARDS pathogenesis (8–11). Several families of proteins are tasked with suppressing cytokine signal transduction, including the SOCS proteins (12, 13). The protein SOCS2 regulates the fate of many key signal transduction proteins, such as the protein kinases FLT3 and p38, the growth hormone receptor, and even other SOCS proteins (14–17). Research suggests that SOCS2 suppresses the NF-κB pathway, similarly to other SOCS family members such as SOCS1 and SOCS3 (18). Further, SOCS2 was observed to mediate the degradation of the kinase STK38, which is known to enhance the activity of NF-κB signaling (19). Indeed, SOCS2 expression directly blunted NF-κB transactivation (20). In addition, SOCS2 was observed to regulate the stability of TRAF6, leading to inhibition of NF-κB pathways (21). Deletion of SOCS2 in mice leads to rampant systemic proinflammatory cytokine production following challenge with Toxoplasma gondii or LPS (22). Despite these studies, there is little research into the role of SOCS2 in pulmonary inflammation and injury. Previous research suggests that the stability of key proteins that regulate pulmonary inflammation influences lung pathology, such as the ubiquitin proteasome–dependent degradation of TLR2, PIAS1, and FBXL2 (23–25). To better understand the role of SOCS2 in pulmonary inflammation, we investigated the regulation of SOCS2 protein stability and its effects on pulmonary inflammation.
Here we report the ubiquitin-mediated degradation of SOCS2 protein and its effect on pulmonary inflammation. We characterized SOCS2 ubiquitination, which was dependent on PKCα-mediated phosphorylation and stimulated by LPS exposure, leading to SOCS2 protein degradation through the proteasome. Through affinity mass spectrometry, we identified the ubiquitin E3 ligase KIAA0317 (also known as AREL1) as an interacting partner with SOCS2 and elucidated a mechanism by which KIAA0317 potently regulated SOCS2 protein stability and signaling. Kiaa0317 knockdown in vivo increased SOCS2 protein level and lessened the severity of bacterially induced lung inflammation. Subsequently, we developed CRISPR/Cas9 Kiaa0317-knockout mice, which were resistant to LPS-induced pulmonary inflammation. Lentivirus rescue experiments with Kiaa0317-knockout mice ablated the protection from LPS-induced inflammation, which supported this putative mechanism. Finally, we screened and developed a small molecule inhibitor of KIAA0317, which showed efficacy in vitro and in vivo in preserving SOCS2 protein and ameliorating pulmonary inflammation in both bacterial and LPS-driven models of lung inflammation. Taken together, these experiments describe a mechanism controlling pulmonary inflammation and a potential avenue for therapeutic intervention.
Results
SOCS2 is downregulated during pulmonary distress.
To test the in vivo significance of SOCS2 in pulmonary inflammation, we analyzed its protein expression by immunoblotting lysate from circulating white blood cells from 7 control subjects and 10 patients with ARDS, as defined by Berlin criteria and agreed upon by a minimum of 3 members of an expert clinical panel (Table 1). Deidentified human plasma samples were obtained from the University of Pittsburgh Acute Lung Injury Biospecimen Repository. Following immunoblotting, we observed that patients with ARDS had significantly less immunoreactive SOCS2 in leukocyte lysate versus control subjects, suggesting SOCS2 protein is depleted during pulmonary inflammation (Figure 1A). We next sought to explore the role of SOCS2 in lung tissue types. We conducted single-cell RNA sequencing of normal human tissue to characterize SOCS2 cellular expression. Normal control lung tissue was obtained from organ donors, after rejection of the lungs for transplant prior to processing for RNA sequencing (data deposited in the NCBI’s Gene Expression Omnibus [GEO], GSE128169) (26). We observed SOCS2 to be predominantly expressed in pulmonary epithelia cells, specifically type II pneumocytes (Figure 1B). Lung epithelia play a critical role in pathogen detection and the initiation of inflammation during lung infection and stress (27, 28). We hypothesized that SOCS2 protein may be decreased in these cells in response to inflammatory stimuli such as LPS. We observed that SOCS2 protein had a half-life between 2 and 4 hours in murine lung epithelial (MLE) cells, which was prolonged with proteasomal inhibitor MG132 treatment (Figure 1C). Further, Socs2 protein showed a dose-dependent decrease upon expression of ubiquitin in MLE cells (Figure 1D), suggesting that Socs2 protein stability is regulated via the ubiquitin-proteasome system. We performed UbiCRest analysis to characterize Socs2 ubiquitination and observed that Socs2 predominantly assembled ubiquitin chains with K48 linkages (29) (Figure 1E). Further, we expressed several ubiquitin constructs with all lysines mutated except for a specific one, thus impairing linkage-specific polyubiquitin chains. We observed that ubiquitin with K48 alone was the only construct sufficient to generate high-molecular-weight Socs2 polyubiquitination protein signal (Figure 1F). Finally, treatment of MLE cells with LPS led to decreased Socs2 protein signal and an increase in polyubiquitinated Socs2, suggesting that SOCS2 ubiquitination and degradation are stimulated by LPS (Figure 1G). LPS treatment of MLE cells did not result in a significant change in Socs2 mRNA (Figure 1H).
Table 1. Clinical parameters of ARDS patients.

Figure 1. SOCS2 protein is ubiquitinated and degraded during pulmonary inflammation.

(A) Immunoblot analysis of leukocyte pellets from ARDS patients or control serum samples. Data represent mean ± SEM, (n = 7–10; ***P < 0.001 compared with control, Mann-Whitney U test). (B) Cell type clustering results of control lung tissue cells from single-cell RNA sequencing of SOCS2 transcript. (C) Immunoblotting following CHX (50 μg/mL), MG132 (20 μM), or leupeptin (50 μg/mL) treatment for the indicated times with ectopic expression of V5-tagged SOCS2 in MLE cells. (D) Immunoblotting following overexpression of ubiquitin (Ubi) in MLE cells. (E) UbiCRest analysis of SOCS2 ubiquitination following immunoprecipitation from MLE cells; digestion supernatant is shown below, with deubiquitinase enzymes indicated by asterisks. Ctrl, control; CD, catalytic domain. (F) Immunoblot analysis following expression of SOCS2-V5 and ubiquitin lysine-to-arginine mutants. (G) Immunoblotting analysis of MLE cells following SOCS2-HIS and HA-ubiquitin expression. Following HIS PD, eluate was immunoblotted for HA ubiquitin signal. (H) qPCR analysis of MLE cells treated with LPS for the indicated times. Data represent mean values ± SEM (n = 3; NS, P > 0.05 compared with 0 hours; Student’s 2-tailed unpaired t test). (C–G) Data are representative of n = 2–3 independent experiments.
KIAA0317 targets SOCS2 for ubiquitination.
The process of substrate ubiquitination is facilitated by substrate-engaging ubiquitin E3 ligases. We utilized recombinant GST-tagged SOCS2 as bait during incubation with epithelial cell BEAS-2B lysate prior to SOCS2 pull-down (PD) (Figure 2A). Following PD, we subjected the eluate to mass spectrometry analysis through MS BioWorks services. Analysis uncovered several differentially detected peptides among SOCS2 PD relative to control GST PD. Among these, we observed the protein KIAA0317 (79 peptides detected), which has been previously described as homologous to the E6-AP carboxyl terminus (HECT) domain ubiquitin E3 ligase (30–33). Expression of KIAA0317 in MLE cells accelerated SOCS2 degradation in half-life studies, while shRNA silencing of Kiaa0317 prolonged SOCS2 stability (Figure 2, B–E). KIAA0317 overexpression specifically decreased SOCS2 in a dose-dependent manner; a randomly selected HECT E3 ligase, UBE3B, was also tested as a negative control (Figure 2, F and G). KIAA0317 enhanced SOCS2 polyubiquitination in an in vivo ubiquitination assay (Figure 2H), and this effect was also observed through in vitro ubiquitination assays (Figure 2I). Expression of His-tagged SOCS2 in addition to LPS exposure led to an increased polyubiquitination signal upon His-SOCS2 PD. This signal was enhanced upon coexpression of KIAA0317 (Figure 2J), suggesting that LPS enhances KIAA0317-induced polyubiquitination of SOCS2. As SOCS2 is suggested to suppress NF-κB signaling (20), we tested the role of KIAA0317 in this signaling. Silencing of Kiaa0317 significantly reduced NF-κB promoter activity following stimulation with LPS or TNF (Figure 2, K and L). These data suggested that KIAA0317 is a candidate E3 ligase regulating SOCS2 ubiquitination and degradation.
Figure 2. KIAA0317 ubiquitinates and degrades SOCS2 in response to bacterial insult.

(A) Protein staining of eluate from control GST and SOCS2-GST bait and capture of BEAS-2B lysate prior to mass spectrometry analysis. (B) Immunoblotting following CHX treatment (50 μg/mL) for the indicated times with ectopic expression of empty plasmid or V5-tagged KIAA0317 in MLE cells. (C) SOCS2 protein densitometry (normalized to actin) for B (n = 2). (D) Immunoblotting following CHX treatment (50 μg/mL) for the indicated times with ectopic expression of Ctrl shRNA or Kiaa0317 shRNA in MLE cells. (E) SOCS2 protein densitometry (normalized to actin) for D (n = 2). (F and G) SOCS2 immunoblotting following (F) KIAA0317 and (G) UBE3B expression in MLE cells. (H) In vivo ubiquitination assay and SOCS2 immunoprecipitation following coexpression with KIAA0317 and ubiquitin. (I) In vitro ubiquitination assay of SOCS2 protein. (J) Immunoblot analysis of SOCS2 PD following KIAA0317 expression and LPS exposure. (K and L) NF-κB promoter activity assays in 293T cells transfected with control shRNA (CON) or KIAA0317 shRNA and exposed to (K) LPS (10 μg/mL) or (L) TNF (10 ng/mL) treatment for the indicated times. Data represent mean values ± SEM (n = 4; *P < 0.05; ****P < 0.0001; NS, P > 0.05 compared with the indicated treatment, with control shRNA 6-hour treatment (†), or with control shRNA 18-hour treatment (#). Two-way ANOVA with Bonferroni’s multiple-comparisons test. (F–J) Data are representative of n = 2–3 independent experiments.
LPS stimulates site-specific SOCS2 phosphorylation and KIAA0317-mediated ubiquitination and degradation.
Substrates are ubiquitinated often by conjugation to critical lysine sites. We mapped the putative ubiquitin acceptor site and the KIAA0317 binding site within SOCS2 (Figure 3A). To uncover the critical ubiquitin acceptor site within SOCS2, we constructed several SOCS2 lysine-to-arginine mutants and assayed their protein half-lives (Figure 3B). Of the SOCS2 point mutants tested, only SOCS2 K173R exhibited an extended half-life (Figure 3B). We also probed the KIAA0317 binding site within SOCS2. KIAA0317 protein was first immunoprecipitated from cell lysates and coupled to protein A/G resin. We then incubated KIAA0317 resin with in vitro synthesized SOCS2-V5 mutants. After washing and elution, we analyzed via immunoblotting. We determined that KIAA0317 binds within the N-terminus of SOCS2 (Figure 3C).
Figure 3. KIAA0317 targets SOCS2 phosphodegron for binding and ubiquitination.

(A) Schematic of SOCS2 deletional or point mutants used in mechanistic studies. (B) Immunoblot analysis of MLE cells expressing SOCS2 lysine mutants followed by CHX (50 μg/mL) treatment for the indicated times. (C) Deletion mapping of SOCS2 site for binding KIAA0317. (D) Immunoblot analysis of MLE cells expressing SOCS2 S52N followed by CHX (50 μg/mL) treatment for the indicated times. (E) Immunoblot analysis of SOCS2 S52N binding KIAA0317. (F) Immunoblotting of MLE cells cotransfected with WT SOCS2 or the S52N or K173R mutant without or with KIAA0317. The lanes were run on the same gel but were noncontiguous. (G) Schematic of KIAA0317 deletional or point mutants used in mechanistic studies. (H and I) Deletion and point mapping of KIAA0317 site for binding SOCS2. (J) KIAA0317 P779L binding assay with SOCS2. (K) KIAA0317 Peptide binding assay. (L) Immunoblot analysis of MLE cells expressing KIAA0317 WT and P779L at increasing doses. In B–F and H–L, data are representative of n = 2 independent experiments.
We hypothesized that this binding region within SOCS2 contained a critical moiety for recognition by KIAA0317. Ubiquitinated substrates are often posttranslationally modified (e.g., phosphorylation), and these sites function as “degrons” for E3 ligase targeting (34). Thus, we set out to determine whether there existed a key phosphodegron within SOCS2 for KIAA0317 recognition. Database analysis indicated that SOCS2 S52 is a candidate phosphorylation site within its N-terminus (NetPhos 2.0 software prediction [ref. 35]). Additionally, there is a naturally occurring missense polymorphism (S52N) (rs3741676) within this codon, and the SOCS2 S52N mutant exhibited an extended half-life (Figure 3D) and displayed lessened binding to KIAA0317 (Figure 3E). Finally, we observed that SOCS2 S52N as well as the K173R mutant were resistant to degradation when coexpressed with KIAA0317 (Figure 3F).
We sought to determine the kinase responsible for SOCS2 phosphorylation at S52. We screened for kinases via SOCS2 immunoprecipitation and immunoblot analysis. Of the kinases probed, only PKCα was detected in the SOCS2 immunoprecipitate (Supplemental Figure 1A; supplemental material available online with this article; https://doi.org/10.1172/jci.insight.129110DS1). We further observed that PKCα directly phosphorylated SOCS2 in an in vitro kinase assay (Supplemental Figure 1B). PKCα affects SOCS2 protein stability, as PKCα expression decreased the protein half-life of SOCS2 in a cycloheximide (CHX) chase (Supplemental Figure 1, C and D). Similarly, silencing of Pkca drastically stabilized SOCS2 protein (Supplemental Figure 1, E and F). Increasing duration and dose of LPS treatment promoted SOCS2 association with PKCα and KIAA0317, as well as the emergence of a phosphoserine signal corresponding to the predicted size of SOCS2 (~25 kDa) (Supplemental Figure 1, G and H). This signal was reduced when Pkca was silenced (Supplemental Figure 1I). Further, LPS increased KIAA0317 protein levels and decreased SOCS2 protein levels in both a time- and dose-dependent manner, as well as increasing the association between the 2 proteins (Supplemental Figure 1, G and H). To better understand the mechanism of KIAA0317 targeting of SOCS2, we embarked on a candidate mapping approach to identify the critical site within KIAA0317 for SOCS2 binding (Figure 3G). Through in vitro protein binding assays, we determined that KIAA0317 binds to SOCS2 through the KIAA0317 C-terminus (Figure 3, H and I). Interestingly, previous mapping studies have suggested that KIAA0317 binds to substrates through specific C-terminal residues (such as P779) (33). Additionally, KIAA0317 P779 contains a naturally occurring polymorphism (rs371610162) resulting in P779L missense mutation. We observed that KIAA0317 P779L mutant protein has weakened binding with SOCS2 (Figure 3J), as does a peptide corresponding to the P779L sequence (Figure 3K). Finally, SOCS2 protein is insensitive to dose-dependent overexpression of P779L relative to WT (Figure 3L). Together, these results suggest that in response to LPS exposure, KIAA0317 facilitates the ubiquitination and degradation of SOCS2 protein, mediated by PKCα-mediated phosphorylation. This is consistent with the results of previous studies that suggested PKCα plays a role in inducing proinflammatory signaling, specifically through the NF-κB pathway (36–39). We hypothesized that this interaction plays a role in mechanisms of pulmonary inflammation.
Kiaa0317 knockdown ameliorates Pseudomonas-induced lung injury in vivo.
To explore the role of Kiaa0317 in pulmonary inflammation, we pursued in vivo knockdown studies. Mice were first infected with lentivirus encoding empty shRNA or Kiaa0317 shRNA for 144 hours (1 × 107 PFU/mouse, intratracheally [i.t.]) and then challenged with PA103 (1 × 104 CFU/mouse, i.t.) for an additional 18 hours (Figure 4A). Mice were sacrificed and samples were processed for metrics of pulmonary inflammation. Kiaa0317 knockdown significantly decreased lavage protein concentrations, lavage cell counts, and cell infiltrates without affecting bacterial load (Figure 4, B, C, D, and I). Further, Kiaa0317 knockdown significantly decreased lavage cytokine levels of IL-6 and TNF (Figure 4, E and F), and improved survival (Figure 4G). Pulmonary parenchymal tissue from Kiaa0317-silenced mice showed higher SOCS2 protein levels (Figure 4H) and fewer inflammatory infiltrates (Figure 4I).
Figure 4. Kiaa0317 knockdown ameliorates Pseudomonas-induced lung injury in vivo.

(A) Treatment strategy for lentiviral infection (1 × 107 PFU/mouse) and PA103 exposure (1 × 104 CFU/mouse). Mice were euthanized, and lungs were lavaged with saline and harvested. Con, control. (B–D) Protein concentration, cell count, and bacterial count measurements in BALF from treated mice (n = 6–8 mice per group). In D, box represents interquartile range; line, median; and whiskers, highest and lowest values. (E and F) ELISA analysis of BALF cytokines IL-6 and TNF. (G) Survival studies of mice exposed to PA103 (i.t. 1 × 105 CFU/mouse, n = 8 mice per group). Mice were monitored over time; moribund, preterminal animals were immediately euthanized and recorded as deceased. Kaplan-Meier survival curves were generated and compared; data represent mean values (n = 8 mice per group). (H) Immunoblot analysis from representative murine lung homogenate; n = 4 per group. (I) Histology of murine lungs following H&E staining; scale bar: 100 μm. **P < 0.01, ***P < 0.001, ****P < 0.0001, NS: P > 0.05 compared with control shRNA, 2-tailed unpaired Student’s t test (B–F) or compared with control shRNA, log-rank (Mantel-Cox) test (G).
Kiaa0317–/– mice are resistant to LPS-induced lung inflammation.
As transient silencing of KIAA0317 in C57BL6/J mice conferred a protective phenotype against PA103-induced pulmonary inflammation, we extended these studies by generating Kiaa0317-deficient mice. Kiaa0317-knockout mice were generated using CRISPR/Cas9 technology using previously described techniques (40). An sgRNA targeting exon 13 of KIAA0317 introduced a double-stranded break, and subsequent NHEJ repair resulted in a 91-bp deletion in the genomic DNA sequence, with no effects in mouse morphology or the predicted off-targeting regions (Supplemental Figure 2, A–D). Sequencing of the cDNA of the generated knockout revealed that the genomic DNA deletion produces a cDNA transcript missing exon 13. Splicing of exon 12–14 generates a frameshift in exon 14 at amino acid 500 resulting in the introduction of a stop codon at position 503 of the knockout transcript (Supplemental Figure 2, A and B).
We next sought to explore the role of Kiaa0317 genetic deletion in experimental pulmonary inflammation. We observed decreased Kiaa0317 transcript signal from Kiaa0317+/– mice and transcript depletion in Kiaa0317–/– mice (Figure 5, A and B). To test the role of Kiaa0317 knockout in pulmonary inflammation, we challenged Kiaa0317+/+, Kiaa0317+/–, and Kiaa0317–/– mice with LPS (3 mg/kg) for 18 hours. Mice were euthanized prior to analysis of parameters of inflammatory injury. Relative to WT, Kiaa0317+/– and Kiaa0317–/– mice showed significant and stepwise decreases in indicators of inflammatory injury, such as bronchoalveolar lavage fluid (BALF) protein concentration (Figure 5C) and cell count (Figure 5D), as well as decreases in proinflammatory cytokine release (Figure 5, E–G). Histological analysis revealed fewer inflammatory infiltrates among Kiaa0317+/– and Kiaa0317–/– mice (Figure 5H). These data suggest that Kiaa0317 deletion confers protection against experimental pulmonary inflammation.
Figure 5. Kiaa0317 knockout confers protection from LPS-induced lung inflammation.

(A) sgRNA target site amplification from Kiaa0317+/+, Kiaa0317+/–, and Kiaa0317–/– mice. (B) Kiaa0317 transcript levels among Kiaa0317+/+, Kiaa0317+/–, and Kiaa0317–/– mice. Data represent mean values ± SEM (n = 3 mice). (C–G) Kiaa0317+/+, Kiaa0317+/–, and Kiaa0317–/– mice were i.t. inoculated with LPS (3 mg/kg) for 18 hours. Data represent mean values ± SEM (n = 3 mice per group). (C and D) Protein concentration and cell count from BALF. (E) BALF IL-6 concentration. (F) BALF TNF concentration. (G) BALF IL-1β concentration. (H) Histology of murine lungs following H&E staining; scale bar: 100 μm. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 compared with the indicated group or to sex-specific +/+ group; 1-way ANOVA with Tukey’s multiple-comparisons (B) or 1-way ANOVA with Dunnett’s multiple-comparisons test (C–G).
KIAA0317 reexpression sensitizes Kiaa0317–/– to LPS-induced pulmonary inflammation.
To further validate the role of Kiaa0317 in pulmonary inflammation, we conducted a series of lentiviral rescue experiments utilizing WT KIAA0317 and the binding-deficient P779L mutant, and with LPS challenge (Figure 6). Kiaa0317–/– mice with empty lentivirus showed lower BALF protein concentration, cell counts, and cytokine release relative to Kiaa0317+/+ mice (Figure 6, A–E). However, an inflammatory phenotype was observed upon lentiviral expression of WT KIAA0317 in Kiaa0317–/– mice. Interestingly, KIAA0317 P779L Kiaa0317–/– mice phenocopied control Kiaa0317–/– mice. Further, the distinct effect of expression of KIAA0317 WT and P779L was observed in BALF leukocyte differential counts (Figure 6, F–H). BALF from both Kiaa0317+/+ and Kiaa0317–/– mice expressing KIAA0317 showed a large count and proportion of neutrophils. However, BALF from Kiaa0317–/– mice expressing empty or P779L-encoded lentivirus contained fewer leukocytes and a smaller proportion of neutrophils (Figure 6G). Histological staining showed less inflammatory infiltration among Kiaa0317–/– mice expressing empty and KIAA0317 P779L–encoding lentivirus (Figure 6I). From these data, we conclude that the resistance to pulmonary inflammation by Kiaa0317–/– mice is due to KIAA0317.
Figure 6. Reexpression of KIAA0317 in Kiaa0317–/– mice ablates resistance to inflammation.

Mice were infected i.t. with empty lentivirus or lentivirus encoding KIAA0317 WT or KIAA0317 P779L (1 × 107 PFU/mouse) prior to LPS exposure (3 mg/kg). Following exposure, mice were euthanized, and lungs were lavaged with saline and harvested. (A and B) Protein concentration and cell count measurements from BALF. (C–E) BALF cytokine concentrations. (F–H) BALF leukocyte differential. (A–H) Data represent mean values ± SEM (n = 4–7 mice; *P < 0.05; **P < 0.01; ***P < 0.001 compared with Kiaa0317+/+ mice, 1-way ANOVA with Dunnett’s multiple comparisons). (I) Histology of murine lungs following H&E staining; scale bar: 200 μm. *P < 0.05, **P < 0.01, ***P < 0.001 compared with Kiaa0317+/+ mice; 1-way ANOVA with Dunnett’s multiple-comparisons test (A–H).
Resistance of Kiaa0317–/– mice to pulmonary inflammation is ablated upon depletion of SOCS2.
Next, we wanted to explore the role of SOCS2 in Kiaa0317–/– mice. To study this, we conducted lentiviral SOCS2 silencing experiments and LPS challenge with Kiaa0317+/+ and Kiaa0317–/– mice (Figure 7). We recapitulated previous results showing the susceptibility of SOCS2-silenced WT (Kiaa0317+/+) mice to LPS-induced inflammation and extended these observations to a lung inflammation model (22). BALF from these SOCS2-silenced Kiaa0317+/+ mice showed higher protein and cell concentrations, cytokine secretion, and leukocyte recruitment relative to control shRNA–infected mice (Figure 7, A–H). Similar to the observations described above (Figure 5), control Kiaa0317–/– mice showed resistance to LPS-induced pulmonary inflammation. However, SOCS2-silenced Kiaa0317–/– mice phenocopied SOCS2-silenced WT (Kiaa0317+/+) mice. We hypothesize that silencing of the putative substrate of KIAA0317, SOCS2, removes the protection of KIAA0317 knockout. Finally, histological analysis showed enhanced inflammatory infiltration among both SOCS2-silenced Kiaa0317+/+ and Kiaa0317–/– mice (Figure 7I). These rescue experiments suggest that Kiaa0317 is both necessary and sufficient for LPS-induced pulmonary inflammation. Further, this inflammatory phenotype operates through a KIAA0317/SOCS2 axis, as depletion of SOCS2 enhances inflammation, even in the background of Kiaa0317 genetic knockout. Collectively, the above studies suggest that Kiaa0317 plays an integral role in mediating cytokine-driven inflammation via the SOCS2/cytokine axis.
Figure 7. Protected phenotype of Kiaa0317–/– mice requires SOCS2.

Mice were infected i.t. with lentivirus encoding control shRNA or Socs2 shRNA (1 × 107 PFU/mouse) prior to LPS exposure (3 mg/kg). Following exposure, mice were euthanized, and lungs were lavaged with saline and harvested. (A and B) Protein concentration and cell count measurements from BALF. (C–E) BALF cytokine concentrations. (F–H) BALF leukocyte differential. In A–H, data represent mean values ± SEM (n = 5–6 mice). (I) Histology of murine lungs following H&E staining; scale bar: 200 μm. *P < 0.05, **P < 0.01, ***P < 0.001,****P < 0.0001 compared with control shRNA-Kiaa0317+/+ mice or as indicated; 1-way ANOVA with Tukey’s multiple-comparisons test.
KIAA0317 domain analysis and inhibitor screening.
KIAA0317 harbors a conserved HECT domain within its C-terminus (41, 42). Since the HECT domain carries out the E3 ligase activity of transferring ubiquitin to the substrate, we hypothesized that small molecule inhibition of the HECT domain would disrupt KIAA0317 substrate targeting. We constructed an in silico homology model using the NEDD4 HECT domain (43, 44) (2XBF.pdb) (Figure 8A) to assay potential ligands on the KIAA0317-HECT domain 3D structure (Figure 8B). We used molecular docking analysis and score-ranking operations through the LibDock program (Discovery Studio 3.5) to virtually screen 5 × 105 small molecules. The 10 highest-scoring molecules were selected and evaluated further using in vitro experiments (data not shown). In this model, the inhibitors interacted with distinct residues within the HECT domain (Figure 8C). We tested one of the selected compounds, termed BC-1365, in an in vitro binding assay, which resulted in potent inhibition of the KIAA0317-SOCS2 interaction (Figure 8D). We also tested this compound in MLE cells and observed increased SOCS2 protein levels, while KIAA0317 protein was unaffected (Figure 8E). BC-1365 titration also did not alter mRNA levels of Socs2 (Figure 8F). To probe the effect of BC-1365 on the activity of other HECT E3 ligases, we treated MLE and BEAS-2B cells with BC-1365 and immunoblotted for known HECT E3 ligase substrate protein levels (Table 2). We did not observe increases in any other HECT E3 ligase substrate relative to the increase in SOCS2 (Supplemental Figure 3, A and B). Additionally, we conducted target validation studies using RNAi knockdown of Kiaa0317 prior to treatment with BC-1365 and LPS (Supplemental Figure 3, C–E). We observed that BC-1365 treatment of MLE cells increased SOCS2 protein levels, but not to a greater extent in the background of Kiaa0317 silencing (Supplemental Figure 3C). We also tested the supernatant of these cells for proinflammatory cytokine release and observed decreased IL-6 and CXCL-1 during Kiaa0317 depletion compared with control; however, we observed no difference upon BC-1365 treatment among Kiaa0317-depleted cells (Supplemental Figure 3, D and E). Finally, we assayed the effect of BC-1365 on primary human PBMCs. We observed a dose-dependent effect of BC-1365 in decreasing the secretion of the proinflammatory cytokines IL-6, TNF, and IL-1β from human primary PBMCs exposed to LPS (Supplemental Figure 4, A–C). These data suggest that chemically inhibiting KIAA0317 protein prevents SOCS2 degradation and dampens proinflammatory signaling.
Figure 8. Chemical inhibition of KIAA0317 prevents SOCS2 degradation and inflammation in vitro.

(A) Structural analysis of the KIAA0317 HECT domain revealed a major cavity within the C-terminus of the HECT domain. (B and C) Docking study of the candidate inhibitor BC-1365 with the KIAA0317-HECT domain. (D) BC-1365 competition assay. SOCS2 protein was incubated with KIAA0317-bound resin and a titration of BC-1365 prior to immunoblot analysis. The relative amounts of SOCS2 detected in the PDs was normalized to vehicle and quantified. Data represent mean values ± SEM (n = 3). (E) Immunoblot analysis of MLE cells following exposure to a titration of BC-1365. Data represent mean values ± SEM (n = 4). (F) MLE cells were exposed to a titration of BC-1365 for 18 hours before qPCR analysis. Data represent mean values ± SEM (n = 4). NS: P > 0.05, *P < 0.05, **P < 0.01 compared with vehicle treatment; 1-way ANOVA with Dunnett’s multiple-comparisons test (E and F).
Table 2. HECT E3 ligase homology to KIAA0317.

Antiinflammatory activity of a KIAA0317 small molecule inhibitor in vivo.
To assess in vivo antiinflammatory activity of BC-1365, we exposed C57BL/6J mice to Pseudomonas aeruginosa (PA103) and BC-1365. Briefly, PA103 was administered i.t. (1 × 104 CFU), which was immediately followed by BC-1365 delivery i.p. (10 mg/kg) for 18-hour exposure. BC-1365 did not affect BALF bacterial counts (Figure 9A); however, BC-1365–treated mice displayed significantly decreased lavage protein concentrations, cell counts, and cell infiltrates relative to vehicle-treated mice (Figure 9, B, C, and H). Further, BC-1365 significantly decreased lavage cytokine levels (Figure 9, D–F), increased SOCS2 protein levels (Figure 9G), and reduced histological infiltrates (Figure 9H). As an alternative approach, we tested BC-1365 in vivo using an LPS model (Supplemental Figure 4, D–H). Briefly, LPS (3 mg/kg, i.t.) and BC-1365 (10 mg/kg, i.p.) were coadministered to C57BL/6J mice. Following 18 hours, mice were euthanized, and lungs were lavaged with saline. BC-1365 treatment led to significantly decreased lavage protein concentrations and cell counts, as well as a reduction in cell infiltrates in LPS-stimulated mice (Supplemental Figure 4, D, E, and H). Further, BC-1365 treatment significantly decreased lavage cytokine levels (Supplemental Figure 4, F and G). Hence, small molecule targeting of the KIAA0317/SOCS2 pathway reduced the severity of cytokine-driven lung inflammation (Supplemental Figure 5).
Figure 9. KIAA0317 small molecule inhibitor is antiinflammatory in vivo.

(A–E) C57BL/6J mice were exposed to PA103 and treated with BC-1365 for 18 hours. Following exposure, mice were sacrificed, and lungs were lavaged with saline and harvested. Box represents the interquartile range; line, the median; and whiskers, highest and lowest values. (A–C) Bacterial count, protein concentration, and cell count measurements from BALF. (D–F) BALF cytokine concentrations. Veh, vehicle. In A–F, data represent mean values ± SEM; n = 4–8 mice. NS: P > 0.05, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 compared with control mice or as indicated; 1-way ANOVA with Tukey’s multiple-comparisons test. (G) Immunoblotting of mouse lung homogenate for Kiaa0317 and Socs2 protein signal; n = 3 mice per treatment. (H) Representative histology of murine lungs following H&E staining; scale bar: 100 μm.
Discussion
These studies have established a mechanism of SOCS2 protein regulation leading to the control of pulmonary inflammation. We showed that the ubiquitin E3 ligase KIAA0317 targets SOCS2 for ubiquitination and degradation by the proteasome and modulates proinflammatory signaling. In mouse models of LPS and PA-103 exposure, KIAA0317 depletion and deletion reduced markers of pulmonary inflammation and rescued SOCS2 protein. Reintroduction of KIAA0317 protein into these models removed any protective phenotype; however, reintroduction of a binding-deficient KIAA0317 mutant preserved the protection from experimental inflammation. We also uncovered a small molecule inhibitor of KIAA0317 activity, BC-1365, that preserved SOCS2 protein from degradation in vitro and lessened cytokine release from primary tissue samples. BC-1365–treated mice also displayed resistance to experimental pulmonary inflammation.
SOCS2 mediates the degradation of a variety of proteins and functions as a feedback inhibitor to cytokine signaling (44). While SOCS2 has been shown to regulate several cytokine-related pathways (22, 45), recently SOCS2 has been observed to directly regulate NF-κB signaling (20, 21). Similarly, the silencing of KIAA0317 weakened LPS- and TNF-induced NF-κB promoter activity in HEK293 cells (Figure 2, K and L). LPS has been shown to induce cellular ubiquitination (46, 47), and we have previously observed E3 ligase induction and substrate association by such insult (23, 25). We observed that LPS exposure promoted SOCS2 association with KIAA0317, and accelerated SOCS2 protein ubiquitination and degradation (Supplemental Figure 1, H and I). LPS also increased SOCS2 polyubiquitin signal, which was further increased by coexpression with KIAA0317 (Figure 2J). These data show that KIAA0317 functions as a proinflammatory E3 ligase.
Mapping studies elucidated the critical regions of SOCS2-KIAA0317 binding. We observed that SOCS2 requires S52 for binding with KIAA0317, potentially functioning as a phosphodegron (Figure 3, C–F). Interestingly, there exists a naturally occurring polymorphism causing an S52N mutation (rs3741676). S52N SOCS2 protein was unable to bind KIAA0317 and resisted KIAA0317-mediated degradation (Figure 3, D–F). PKCA was found to associate with SOCS2, facilitate its phosphorylation, and regulate its protein stability (Supplemental Figure 1). This finding is interesting given that previous research showed that PKCA upregulates NF-κB signaling (38, 48), potentially through the regulation of SOCS2 protein stability. Further, we observed that the C-terminal region, and P779, within KIAA0317 was critical for SOCS2 binding (Figure 3, G–L). This residue itself is part of a naturally occurring polymorphism resulting in P779L mutation (rs371610162). KIAA0317 with P779L mutation was unable to degrade SOCS2 protein (Figure 3L). Additionally, P779L-expressing Kiaa0317–/– mice were resistant to LPS-induced pulmonary inflammation, compared with WT KIAA0317–expressed Kiaa0317–/– mice (Figure 6). These data further underscore the protective phenotype conferred by P779L mutation, potentially through the inability to degrade SOCS2 protein. Studies into the prevalence of these polymorphisms among populations and patient cohorts are needed to investigate the pathological implications in humans.
Inflammatory lung diseases such as ARDS are often triggered by opportunistic infection, including Gram-negative bacteria such as P. aeruginosa, leading to runaway inflammation and cytokine storm. We showed that shRNA knockdown of Kiaa0317 in a P. aeruginosa mouse model led to reduced lung inflammation and less cytokines and immune infiltrates compared with control, without changing P. aeruginosa bacteria levels in a mouse model. We further explored this relationship through CRISPR/Cas9-mediated deletion of Kiaa0317 in C57BL/6 mice (Figure 5). Kiaa0317+/– and Kiaa0317–/– mice showed increasing protection from pulmonary inflammation relative to WT mice. This effect could be due to the difference in effector protein abundance, as we observed depletion in KIAA0317 transcript through quantitative PCR (qPCR) analysis (Figure 5B). Cytokine signal transduction pathways related to inflammation and innate immunity undergo significant cross-talk and pleiotropy, which impairs the generalizability of reductionist studies. To address this, we reexpressed our deleted KIAA0317 protein in Kiaa0317–/– mice (Figure 6). As we observed a similar proinflammatory phenotype among KIAA0317-rescued compared with WT mice, we hypothesize that KIAA0317 protein is the driving effector in this model. Additionally, E3 ligases often regulate multiple substrates, specifically seen in the fellow HECT E3 ligase ITCH (49). To test the link between SOCS2 and KIAA0317, we silenced SOCS2 in Kiaa0317–/– mice prior to LPS challenge (Figure 7). By silencing SOCS2 expression in the background of KIAA0317, we questioned whether the protection conferred by KIAA0317 deletion occurred in the absence of SOCS2. However, SOCS2-silenced Kiaa0317–/– mice were susceptible to LPS-induced pulmonary inflammation and phenocopied SOCS2-silenced WT mice. This shows that the protective effect of KIAA0317 silencing is dependent on SOCS2. This interplay suggests the criticality of the 2 proteins in experimental lung inflammation. We suspect that this KIAA0317/SOCS2 axis plays an essential role in regulating pulmonary innate immunity.
There are limited therapeutic options for treating diseases of pulmonary inflammation. Previous efforts utilized systemic corticosteroids or inhibition of immune cell surface receptors or secreted cytokines (50–2). These broad-spectrum agents such as corticosteroids have shown adverse effects in clinical trials that outweigh any potential benefit (53). Chemical inhibition of ubiquitin E3 ligases has been shown to alleviate experimental lung inflammation (25, 54). We screened a chemical library in silico against the C-terminus of KIAA0317 and uncovered a chemical that we termed BC-1365 (Figure 8, A–C). BC-1365 abrogated SOCS2-KIAA0317 binding in vitro, restored SOCS2 protein levels in cells (Figure 8, D–F), and reduced the abundance of proinflammatory cytokines secreted from PBMCs (Supplemental Figure 4, A–C). Based on these data, we tested BC-1365 in both P. aeruginosa– and LPS-induced murine models of ARDS. BC-1365 was also effective in vivo, as BAL protein concentrations, BAL cell counts, lavage cytokine levels, and histological evidence of lung injury were all decreased in both models upon BC-1365 treatment (Figure 9). Small molecule inhibition of the KIAA0317-SOCS2 interaction reduced the severity of NF-κB–driven, cytokine-induced lung inflammation by modifying the abundance of SOCS2. These results suggest that KIAA0317 may be a promising new therapeutic target in the treatment of pulmonary inflammation. Future studies focusing on evaluating the safety profile, distribution, elimination, and metabolism of this antagonist are needed to fully understand the effects of this treatment, but the discovery of a therapy specific to a critical mechanism of pulmonary inflammation would be a crucial advancement in this field.
Methods
Further information can be found in Supplemental Methods, available online with this article.
Reagents and materials
Rabbit polyclonal anti-SOCS2 (catalog ab3692, RRID:AB_304007) and recombinant human PKCα protein (ab55672) were from Abcam. Recombinant human SOCS-2 protein (H00008835-P01) was from Abnova. WesternBright Sirius HRP substrate (K-12043) was from Advansta. QuikChange II XL Site-Directed Mutagenesis Kit (catalog 200521) was from Agilent Technologies. AnaSed (xylazine sterile solution) (catalog 59399-110-20) was from Akorn. PureLink RNA Mini Kit (catalog 12183025) was from Ambion. High-Capacity RNA-to-cDNA Kit (catalog 4387406) and SYBR Select Master Mix for CFX (catalog 4472942) were from Applied Biosystems. PA-103 (catalog 29260), MLE-12 (catalog CRL-2110, RRID:CVCL_3751), HEK293T (catalog CCL-2, RRID:CVCL_0030), and BEAS2-B (catalog CRL-9609, RRID:CVCL_0168) cells were from ATCC. Life ECO Thermal Cycler (BYQ6078) was from BIOER. Goat anti-rabbit HRP (Bio-Rad, catalog 170-6515, RRID:AB_11125142), goat anti-mouse HRP (catalog 170-6516, RRID:AB_11125547), rabbit anti-goat HRP (catalog 172-1034, RRID:AB_11125144), DC Protein Assay Kit (catalog 5000111), CFX96 Touch Real-Time PCR Detection System (catalog 1855196), Personal Molecular Imager (PMI) System (catalog 1709400), ChemiDoc XRS+ System (catalog 1708265), and TC20 Automated Cell Counter (catalog 1450102) were from Bio-Rad. UbiCRest assay kit (K-400) was from Boston Biochem. Mouse monoclonal anti–HA Tag (clone 6E2) (2367S, RRID:AB_10691311), rabbit polyclonal anti-SOCS1 (clone A156) (3950S, RRID:AB_2192983), and mouse monoclonal anti-phosphothreonine (9386S, RRID:AB_331239) were from Cell Signaling Technology. Lenti-X Packaging System (catalog 631276) was from Clontech. Human TNF-α recombinant protein (catalog 14-8329-81) and Mouse IL-6 ELISA Ready-SET-Go! (88-7064-22, RRID:AB_2574986) were from eBioscience. LPS, E. coli O111:B4 (LPS25) was from EMD Millipore. CHX (BML-GR310) and ubiquitinylation kit (BML-UW9920-0001) were from Enzo. TNF alpha Mouse ELISA Kit (BMS607-3TEN), IL-1 beta Mouse ELISA Kit (KMC0011), pcDNA3.1D-V5-HIS-TOPO (K490001), and pcDNA3.1D-V5-HIS-Empty (K490001) were from Invitrogen. C57BL/6J Mus musculus (RRID:IMSR_JAX:000664) mice were from JAX. Mouse monoclonal anti-ubiquitin (VU-1) (VU101) was from Life Sensors. ATP, [γ-32P]- 3000Ci/mmol 10mCi/ml (BLU002A) was from PerkinElmer. Streptavidin Magnetic Beads (catalog 88816) were from Pierce. TnT T7 Quick Coupled Transcription/Translation (L1170) was from Promega. Cignal NFkB Pathway Reporter Assay Kit (LUC) (CCS-013L) was from QIAGEN. X-tremeGENE 9 (XTG9-RO) (XTG9-RO) was from Roche. Rabbit polyclonal anti-SOCS3 (clone H-103) (sc-9023, RRID:AB_2193305), mouse monoclonal anti-CaMKI (clone H-8) (sc-137225, RRID:AB_2069999), rabbit polyclonal anti-ERK1 (clone C-16) (sc-93 RRID:AB_631453), rabbit polyclonal anti-MAPK14 (p38α) (clone C-20) (sc-535, RRID:AB_632138), mouse monoclonal anti-PKCα (clone H-7) (sc-8393, RRID:AB_628142), goat polyclonal anti–p-PKCα (S657) (sc-12356, RRID:AB_2168557), mouse monoclonal anti-phosphoserine (sc-81516, RRID:AB_1128626), and mouse monoclonal anti-PKCζ (sc-17781, RRID:AB_628148) were from Santa Cruz Biotechnology. Leupeptin (L2884), ATP (A2383), mouse monoclonal anti–β-actin (clone AC-15) (A5441, RRID:AB_476744), and rabbit polyclonal anti-KIAA0317 (SAB2108763) were from Sigma-Aldrich. Recombinant glutathione S-transferase protein (11213-HNAB) was from SinoBiological. Mouse monoclonal anti–V5 Tag (R960-25, RRID:AB_2556564), Protein A/G Magnetic Beads (catalog 88802), E. coli One Shot TOP10 (C404003), and pcDNA3.1 Directional TOPO Expression Kit (K490001) were from Thermo Fisher Scientific. pRK5-HA-Ubiquitin-WT (17608, RRID:Addgene17608) was a gift from Ted Dawson (55).
Contact for reagent and resource sharing
Further information and requests for reagents may be directed to and will be fulfilled by the corresponding author.
Experimental model and subject details
Human studies (ARDS).
Adults aged 18–90 years admitted to the Medical ICU in UPMC Presbyterian Hospital with acute respiratory failure requiring mechanical ventilation via endotracheal tube were enrolled as patients if the onset of acute respiratory failure was associated with established risk factors for developing acute lung injury (sepsis, pneumonia, aspiration, blood transfusion, pancreatitis, or trauma) or if intubation was performed for airway protection in the setting of nonpulmonary critical illness. Enrollment took place within 48 hours of the initiation of mechanical ventilation. After enrollment, patients were retrospectively subclassified by an expert clinical panel into categories: (i) “ARDS,” according to the Berlin Definition and agreed upon by a minimum of 3 members of a clinical panel, or (ii) “ventilated control,” as defined by intubation and mechanical ventilation for nonpulmonary critical illness without risk factor for ARDS. Cells were purified from whole blood, and white cells were pelleted for further study. The panel consisted of pulmonary faculty members and fellows at the University of Pittsburgh (JE, BM, and several others) who met semimonthly to classify subjects. Participation was voluntary among pulmonary faculty and fellows. Subclassifications were assigned if at least 5 faculty/fellows were present and 3 were in agreement about the subclassification (ARDS or airway control). For more details, see ref. 56.
Mouse experiments.
Mice were from the Jackson Laboratory. For lentiviral studies, mice were confirmed as anesthetized prior to i.t. instillation of 0.1 % (w/v) lysophosphatidylcholine (LysoPC) (specifically palmitoyl:stearoyl C16:C18) as an adjuvant. Following 1 hour, mice were i.t. administered empty VSV-G–pseudotyped lentivirus or encoding KIAA0317 WT or KIAA0317 P779L (pLVX); or control shRNA or Kiaa0317 shRNA (pLKO.1) (1 × 107 PFU/mouse). Plasmid was expressed for 144 hours prior to treatment. Mice were i.t. inoculated with LPS (3 mg/kg) or given P. aeruginosa (strain PA103, 1 × 104 CFU) for 18 hours. Following exposure, mice were euthanized, and lungs were lavaged with saline and harvested and subjected to analysis. Survival studies were performed on mice that were given P. aeruginosa (strain PA103, 1 × 105 CFU per mouse, i.t.). For drug studies, mice were deeply anesthetized as above. PA103 (1 × 104 CFU), or LPS (3 mg/kg) was administered i.t. before BC-1382 (10 mg/kg) was administered to the mice through i.p. injection. Eighteen hours later, animals were euthanized and analyzed as above.
Details of methods
Plasmid transfection.
For protein overexpression in MLE-12 and BEAS-2B cells, cells were nucleofected using Nucleofector 2B (Amaxa), program T-13. For protein overexpression in HEK293T cells, X-tremeGENE transfection reagents were used following the manufacturer’s protocol (Sigma-Aldrich). Expression was confirmed via Western blotting.
Immunoblotting.
Cell sample lysates were collected and digested in buffer A (150 mM NaCl, 50 mM Tris, 1.0 mM EDTA, 2 mM DTT, 0.025% sodium azide, and 1 mM phenylmethylsulfonyl fluoride) on ice. Lysates were prepared by brief sonication at 4°C. Insoluble cellular debris was precipitated through centrifugation at 15,000 g for 10 minutes at 4°C. Lysate supernatant was normalized for protein concentration and diluted in denaturing loading buffer, with a final 1× formulation of 50 mM Tris HCl pH 6.8, 2% SDS, 10% glycerol, and 100 mM DTT. Protein concentration was determined by Lowry assay, and samples were normalized for content. Samples were resolved via SDS-PAGE prior to immunoblotting. Signal was detected via chemiluminescence on a Kodak Imaging Station.
Chemical and compound treatment.
MLE-12 and other cell lines were seeded to a density of 0.20 × 106 cells/mL in polystyrene 6-wells dishes treated by vacuum gas plasma in media supplemented with 2% FBS for 18 hours. LPS, MG-132 (10 μM), leupeptin (10 μM), CHX (50 μg/mL), or BC compound was added to cells at the indicated doses for the indicated time periods prior to analysis.
In vitro ubiquitin conjugation assays.
The assay was performed in a volume of 20 μL containing 50 mM Tris, pH 7.6, 5 mM MgCl2, 0.6 mM DTT, 2 mM ATP, 400 μM MG132, 50 nM Ubiquitin activating enzyme, 0.5 μM UbcH5, 0.5 μM UbcH7, 2 μM ubiquitin, and 1 μM ubiquitin aldehyde. TnT-coupled (Promega) reticulocyte in vitro–synthesized tagless KIAA0317 and SOCS2-V5 proteins were purified via HisPur Resin (Thermo Fisher Scientific), and reaction products were processed for V5 immunoblotting.
Promoter assays.
HEK293 cells were cotransfected with Cignal NFkB luciferase reporter assay plasmids and empty, KIAA0317, SOCS2, control, or KIAA0317 shRNA for 24–48 hours before exposure to LPS (10 μg/mL) and TNF (10 ng/mL) for an additional 6 or 18 hours. Cells were then collected and assayed for luciferase activity according to the manufacturer’s instructions.
In vitro protein binding assays.
KIAA0317 protein was immunoprecipitated from 1 mg MLE cell lysate using a 1:100 dilution of KIAA0317 antibody (rabbit) for 4 hours at 25°C in IP buffer (50 mM Tris HCl pH 7.6, 150 mM NaCl, 0.25 % v/v Triton X-100) and coupled to protein A/G agarose resin for an additional hour. V5-tagged SOCS2 deletion and point mutants (50 μL) were in vitro synthesized using TnT translation kits for 90 minutes at 30°C. KIAA0317-bound resin was then incubated with the in vitro synthesized bait protein for 18 hours at 4°C. Following binding, resin was washed with IP buffer and eluted in 1× Laemmli buffer at 88°C for 5 minutes prior to immunoblot analysis. Similarly, SOCS2 protein was immunoprecipitated from MLE cell lysate subjected to binding against V5-tagged KIAA0317 mutants prior to immunoblot analysis, as described above.
Immunoprecipitation assays.
MLE cells were cultured and treated as indicated prior to collection. Cell pellets were lysed with IP buffer (0.25% Triton X-100 in 1× PBS, pH 7.6, 0.025% sodium azide, and 1 mM phenylmethylsulfonyl fluoride) on ice. Lysates were prepared by brief sonication at 4°C. Insoluble cellular debris were precipitated through centrifugation at 15,000 g for 10 minutes at 4°C. Lysate supernatant was normalized for protein concentration. Supernatants were exposed to a 1:50 dilution of the indicated antibody for 3 hours at 25°C. Immunoprecipitated protein was captured with 10 μL magnetic protein A/G resin per 1 mL lysate for 1 hour prior to 2 rounds of washing with IP buffer (1 mL). Protein was eluted by dilution in denaturing loading buffer, with a final 1× formulation of 50 mM Tris HCl pH 6.8, 2% SDS, 10% glycerol, and 100 mM DTT and boiling for 5 minutes. Eluted samples were resolved via SDS-PAGE and subjected to immunoblotting.
shRNA knockdown.
For gene silencing studies in cells, shRNAs were derived from the RNAi Consortium (TRC-Hs1.0, human) and purchased from GE Dharmacon. pLKO.1 plasmids encoding control or gene-specific shRNA were transfected using the plasmid transfection protocol described above. Following 24 hours, medium was exchanged for 0% serum–containing medium. After another 24 hours, cells were collected for subsequent analysis.
UbiCRest assay.
UbiCRest Assay was performed on immunoprecipitated SOCS2 protein according to the manufacturer’s instructions (Boston Biochem) prior to immunoblot analysis.
ELISA.
ELISAs were conducted on BALF samples from treated mice. BALF IL-1, IL-6, and TNF was detected by following the manufacturer’s instructions, and absorbance read using a microplate reader.
Tissue staining.
Murine lung samples were fixed in 10% neutral buffered formalin, embedded in paraffin, and sectioned. Sections were stained with H&E. Images were acquired from 20× lens from random fields from each section.
Molecular docking studies.
The docking experiments were carried out using Discovery Studio 3.5 (BIOVIA). KIAA0317 domain structural analysis revealed a major drug binding cavity within the C-terminal region. The binding cavity was adopted into the LibDock algorithm to screen for the optimal inhibitor. A library containing 3 million approved or experimental drugs was first used to screen potential ligands for KIAA0317. Based on the docking score and best-fit analysis of suitable ligands, 10 compounds were selected and tested.
In vitro drug-binding assays.
KIAA0317 protein was His purified from KIAA0317 expressed in 293T cells using TALON metal affinity resin. Resins were then extensively washed before exposure to BC-1365 at the indicated concentrations. Purified recombinant SOCS2 protein was then incubated with drug-bound KIAA0317 resins overnight. Resins were washed, and proteins were eluted and resolved on SDS-PAGE. The relative amounts of SOCS2 detected in the PDs were normalized to loading and quantified.
GST bait and PD.
Recombinant GST-SOCS2 protein was used as bait for BEAS-2B lysate incubation prior to PD and analysis, as previously described (57). Briefly, 1 μg GST-SOCS2 protein or recombinant GST protein was immobilized onto 0.5 mL Glutathione Sepharose 4B resin in binding buffer (1× PBS + 0.2% v/v Triton X-100) for 2 hours at 4°C with agitation. Following capture, the bait and resin were incubated with 1 mg BEAS-2B cell lysate for 4 hours at 4°C. The resin was washed 5 times (1 mL) with binding buffer prior to freezing of resin and shipment to MS Bioworks for analysis.
Statistics
Protein signal densitometry was quantified via ImageJ (NIH). Statistical comparisons were performed through Prism (GraphPad) utilizing 2-tailed t tests, Mann-Whitney U test, or 1-way ANOVA with Tukey’s or Dunnett’s multiple-comparisons test where described or by tests as described in the text, and with P < 0.05 indicative of significance.
Study approval
The University of Pittsburgh Institutional Review Board approved procedures involving human samples. Normal control lung tissue was obtained from organ donors, after rejection of the lungs for transplant. All procedures were approved by the University of Pittsburgh Institutional Animal Care and Use Committee (protocols 17030095 and 18114128). C57BL/6J males aged 8–10 weeks were acclimated to new housing for at least 3 days following shipping. Each house contained 4–5 mice with access to food and drink ad libitum in a room with a 12-hour light/12-hour dark cycle. We confirmed that mice were between 25 and 30 g before initiating any procedures. Mice were deeply anesthetized using a ketamine/xylazine mixture (80–100 mg/kg i.p.) prior to experimentation. Mice were carefully monitored over time; moribund, preterminal animals were immediately euthanized and recorded as deceased. Deidentified human plasma samples were obtained from the University of Pittsburgh Acute Lung Injury Biospecimen Repository (IRB PRO10110387). Written informed consent was provided by all participants or their surrogates in accordance with the Declaration of Helsinki. Leukocytes were obtained from patient blood according to a protocol approved by the University of Pittsburgh Investigational Review Board.
Author contributions
BBC designed and directed the study. BBC, RG, SG, and MJS oversaw and created the mouse models. TBL, ACM, JWE, and BBC analyzed the data, prepared the figures, and wrote the manuscript. TBL, ACM, JWE, SR, TAC, SRD, JDL, CLB, KCL, MJJ, and YL performed all experiments and assisted with animal experiments. BJM, YZ, and JWE assisted with human studies. EV and RL assisted with single-cell RNA sequencing data. TBL, JWE, RL, YL, and BBC provided funding for the studies.
Supplementary Material
Acknowledgments
This work was supported by NIH grants to TBL (5F31HL143843), JWE (1K08HL144820), RL and BBC (P50AR060780), YL and MJJ (1R01DK119627), YL (5R01HL142777), and BBC (5R35HL139860 and 5R01HL133184) and the University of Pittsburgh Aging Institute seed fund to BBC and YL.
Version 1. 10/03/2019
Electronic publication
Funding Statement
To Bill Chen
To Travis Lear
To John Evankovich
To Robert Lafyatis and Bill Chen
To Yuan Liu
To Yuan Liu
To Bill Chen
Footnotes
Conflict of interest: The work described in this study is protected by US Patent 10,344,003 to BBC covering KIAA0317 inhibitors and additional modifications. The patent was filed through the University of Pittsburgh of the Commonwealth System of Higher Education.
Copyright: © 2019, American Society for Clinical Investigation.
Reference information: JCI Insight. 2019;4(19):e129110.https://doi.org/10.1172/jci.insight.129110.
Contributor Information
Travis B. Lear, Email: TRL34@pitt.edu.
Shristi Rajbhandari, Email: shristi_2003@hotmail.com.
Bryan J. McVerry, Email: mcverrybj@upmc.edu.
Yingze Zhang, Email: zhanyx@upmc.edu.
Eleanor Valenzi, Email: valenzie@upmc.edu.
Sebastien Gingras, Email: sgingras@pitt.edu.
Karina C. Lockwood, Email: kcl26@pitt.edu.
Michael J. Jurczak, Email: jurczakm@pitt.edu.
Robert Lafyatis, Email: lafyatis@pitt.edu.
Yuan Liu, Email: yul119@pitt.edu.
References
- 1.Weitnauer M, Mijošek V, Dalpke AH. Control of local immunity by airway epithelial cells. Mucosal Immunol. 2016;9(2):287–298. doi: 10.1038/mi.2015.126. [DOI] [PubMed] [Google Scholar]
- 2.Moldoveanu B, et al. Inflammatory mechanisms in the lung. J Inflamm Res. 2009;2:1–11. [PMC free article] [PubMed] [Google Scholar]
- 3.Ware LB, Matthay MA. The acute respiratory distress syndrome. N Engl J Med. 2000;342(18):1334–1349. doi: 10.1056/NEJM200005043421806. [DOI] [PubMed] [Google Scholar]
- 4.Rubenfeld GD, et al. Incidence and outcomes of acute lung injury. N Engl J Med. 2005;353(16):1685–1693. doi: 10.1056/NEJMoa050333. [DOI] [PubMed] [Google Scholar]
- 5.Pham T, Rubenfeld GD. Fifty years of research in ARDS. The epidemiology of acute respiratory distress syndrome. a 50th birthday review. Am J Respir Crit Care Med. 2017;195(7):860–870. doi: 10.1164/rccm.201609-1773CP. [DOI] [PubMed] [Google Scholar]
- 6.Rahman A, Fazal F. Blocking NF-κB: an inflammatory issue. Proc Am Thorac Soc. 2011;8(6):497–503. doi: 10.1513/pats.201101-009MW. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zimmerman GA, et al. Endothelial activation in ARDS. Chest. 1999;116(1 suppl):18S–24S. doi: 10.1378/chest.116.suppl_1.18s. [DOI] [PubMed] [Google Scholar]
- 8.Schwartz MD, et al. Nuclear factor-kappa B is activated in alveolar macrophages from patients with acute respiratory distress syndrome. Crit Care Med. 1996;24(8):1285–1292. doi: 10.1097/00003246-199608000-00004. [DOI] [PubMed] [Google Scholar]
- 9.Blackwell TS, Christman JW. The role of nuclear factor-kappa B in cytokine gene regulation. Am J Respir Cell Mol Biol. 1997;17(1):3–9. doi: 10.1165/ajrcmb.17.1.f132. [DOI] [PubMed] [Google Scholar]
- 10.Lawrence T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb Perspect Biol. 2009;1(6):a001651. doi: 10.1101/cshperspect.a001651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Orfanos SE, Mavrommati I, Korovesi I, Roussos C. Pulmonary endothelium in acute lung injury: from basic science to the critically ill. In: Hedenstierna G, Mancebo J, Brochard L, Pinsky MR eds. Applied Physiology in Intensive Care Medicine. Heidelberg, Germany: Springer Berlin Heidelberg; 2009:215–27. [Google Scholar]
- 12.Yoshimura A, Naka T, Kubo M. SOCS proteins, cytokine signalling and immune regulation. Nat Rev Immunol. 2007;7(6):454–465. doi: 10.1038/nri2093. [DOI] [PubMed] [Google Scholar]
- 13.Trengove MC, Ward AC. SOCS proteins in development and disease. Am J Clin Exp Immunol. 2013;2(1):1–29. [PMC free article] [PubMed] [Google Scholar]
- 14.Kazi JU, Rönnstrand L. Suppressor of cytokine signaling 2 (SOCS2) associates with FLT3 and negatively regulates downstream signaling. Mol Oncol. 2013;7(3):693–703. doi: 10.1016/j.molonc.2013.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gan L, Liu Z, Zhang Z, Yang X, Liu J, Sun C. SOCS2 inhibited mitochondria biogenesis via inhibiting p38 MAPK/ATF2 pathway in C2C12 cells. Mol Biol Rep. 2014;41(2):627–637. doi: 10.1007/s11033-013-2901-z. [DOI] [PubMed] [Google Scholar]
- 16.Tannahill GM, Elliott J, Barry AC, Hibbert L, Cacalano NA, Johnston JA. SOCS2 can enhance interleukin-2 (IL-2) and IL-3 signaling by accelerating SOCS3 degradation. Mol Cell Biol. 2005;25(20):9115–9126. doi: 10.1128/MCB.25.20.9115-9126.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vesterlund M, et al. The SOCS2 ubiquitin ligase complex regulates growth hormone receptor levels. PLoS ONE. 2011;6(9):e25358. doi: 10.1371/journal.pone.0025358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Park SH, Kim KE, Hwang HY, Kim TY. Regulatory effect of SOCS on NF-kappaB activity in murine monocytes/macrophages. DNA Cell Biol. 2003;22(2):131–139. doi: 10.1089/104454903321515931. [DOI] [PubMed] [Google Scholar]
- 19.Shi DD, Shi H, Lu D, Li R, Zhang Y, Zhang J. NDR1/STK38 potentiates NF-κB activation by its kinase activity. Cell Biochem Funct. 2012;30(8):664–670. doi: 10.1002/cbf.2846. [DOI] [PubMed] [Google Scholar]
- 20.Paul I, et al. The ubiquitin ligase Cullin5SOCS2 regulates NDR1/STK38 stability and NF-κB transactivation. Sci Rep. 2017;7:42800. doi: 10.1038/srep42800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McBerry C, Gonzalez RM, Shryock N, Dias A, Aliberti J. SOCS2-induced proteasome-dependent TRAF6 degradation: a common anti-inflammatory pathway for control of innate immune responses. PLoS ONE. 2012;7(6):e38384. doi: 10.1371/journal.pone.0038384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Machado FS, et al. Anti-inflammatory actions of lipoxin A4 and aspirin-triggered lipoxin are SOCS-2 dependent. Nat Med. 2006;12(3):330–334. doi: 10.1038/nm1355. [DOI] [PubMed] [Google Scholar]
- 23.Chen BB, et al. A combinatorial F box protein directed pathway controls TRAF adaptor stability to regulate inflammation. Nat Immunol. 2013;14(5):470–479. doi: 10.1038/ni.2565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McKelvey AC, et al. RING finger E3 ligase PPP1R11 regulates TLR2 signaling and innate immunity. Elife. 2016;5:e18496. doi: 10.7554/eLife.18496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Coon TA, et al. The proinflammatory role of HECTD2 in innate immunity and experimental lung injury. Sci Transl Med. 2015;7(295):295ra109. doi: 10.1126/scitranslmed.aab3881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Valenzi E, et al. Single-cell analysis reveals fibroblast heterogeneity and myofibroblasts in systemic sclerosis-associated interstitial lung disease. Ann Rheum Dis. 2019;78(10):1379–1387. doi: 10.1136/annrheumdis-2018-214865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hippenstiel S, Opitz B, Schmeck B, Suttorp N. Lung epithelium as a sentinel and effector system in pneumonia — molecular mechanisms of pathogen recognition and signal transduction. Respir Res. 2006;7:97. doi: 10.1186/1465-9921-7-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Manicone AM. Role of the pulmonary epithelium and inflammatory signals in acute lung injury. Expert Rev Clin Immunol. 2009;5(1):63–75. doi: 10.1586/1744666X.5.1.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hospenthal MK, Mevissen TET, Komander D. Deubiquitinase-based analysis of ubiquitin chain architecture using Ubiquitin Chain Restriction (UbiCRest) Nat Protoc. 2015;10(2):349–361. doi: 10.1038/nprot.2015.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim JB, et al. Identification of a novel anti-apoptotic E3 ubiquitin ligase that ubiquitinates antagonists of inhibitor of apoptosis proteins SMAC, HtrA2, and ARTS. J Biol Chem. 2013;288(17):12014–12021. doi: 10.1074/jbc.M112.436113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kristariyanto YA, et al. Assembly and structure of Lys33-linked polyubiquitin reveals distinct conformations. Biochem J. 2015;467(2):345–352. doi: 10.1042/BJ20141502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Michel MA, et al. Assembly and specific recognition of k29- and k33-linked polyubiquitin. Mol Cell. 2015;58(1):95–109. doi: 10.1016/j.molcel.2015.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lear T, et al. Ubiquitin E3 ligase FIEL1 regulates fibrotic lung injury through SUMO-E3 ligase PIAS4. J Exp Med. 2016;213(6):1029–1046. doi: 10.1084/jem.20151229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Skaar JR, Pagan JK, Pagano M. Mechanisms and function of substrate recruitment by F-box proteins. Nat Rev Mol Cell Biol. 2013;14(6):369–381. doi: 10.1038/nrm3582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Blom N, Gammeltoft S, Brunak S. Sequence and structure-based prediction of eukaryotic protein phosphorylation sites. J Mol Biol. 1999;294(5):1351–1362. doi: 10.1006/jmbi.1999.3310. [DOI] [PubMed] [Google Scholar]
- 36.Kim H, Zamel R, Bai XH, Liu M. PKC activation induces inflammatory response and cell death in human bronchial epithelial cells. PLoS ONE. 2013;8(5):e64182. doi: 10.1371/journal.pone.0064182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kontny E, Ziółkowska M, Ryzewska A, Maśliński W. Protein kinase c-dependent pathway is critical for the production of pro-inflammatory cytokines (TNF-alpha, IL-1beta, IL-6) Cytokine. 1999;11(11):839–848. doi: 10.1006/cyto.1998.0496. [DOI] [PubMed] [Google Scholar]
- 38.Loegering DJ, Lennartz MR. Protein kinase C and Toll-like receptor signaling. Enzyme Res. 2011;2011:537821. doi: 10.4061/2011/537821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pelletier S, Gingras S, Green DR. Mouse genome engineering via CRISPR-Cas9 for study of immune function. Immunity. 2015;42(1):18–27. doi: 10.1016/j.immuni.2015.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rotin D, Kumar S. Physiological functions of the HECT family of ubiquitin ligases. Nat Rev Mol Cell Biol. 2009;10(6):398–409. doi: 10.1038/nrm2690. [DOI] [PubMed] [Google Scholar]
- 41.Huang L, et al. Structure of an E6AP-UbcH7 complex: insights into ubiquitination by the E2-E3 enzyme cascade. Science. 1999;286(5443):1321–1326. doi: 10.1126/science.286.5443.1321. [DOI] [PubMed] [Google Scholar]
- 42.Umadevi N, Kumar S, Narayana N. Crystallization and preliminary X-ray diffraction studies of the WW4 domain of the Nedd4-2 ubiquitin-protein ligase. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2005;61(pt 12):1084–1086. doi: 10.1107/S174430910503767X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kamadurai HB, et al. Insights into ubiquitin transfer cascades from a structure of a UbcH5B approximately ubiquitin-HECT(NEDD4L) complex. Mol Cell. 2009;36(6):1095–1102. doi: 10.1016/j.molcel.2009.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Krebs DL, Hilton DJ. SOCS proteins: negative regulators of cytokine signaling. Stem Cells. 2001;19(5):378–387. doi: 10.1634/stemcells.19-5-378. [DOI] [PubMed] [Google Scholar]
- 45.Letellier E, Haan S. SOCS2: physiological and pathological functions. Front Biosci (Elite Ed) 2016;8:189–204. doi: 10.2741/E760. [DOI] [PubMed] [Google Scholar]
- 46.Qureshi N, Morrison DC, Reis J. Proteasome protease mediated regulation of cytokine induction and inflammation. Biochim Biophys Acta. 2012;1823(11):2087–2093. doi: 10.1016/j.bbamcr.2012.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hu MM, et al. TRIM38 negatively regulates TLR3/4-mediated innate immune and inflammatory responses by two sequential and distinct mechanisms. J Immunol. 2015;195(9):4415–4425. doi: 10.4049/jimmunol.1500859. [DOI] [PubMed] [Google Scholar]
- 48.Zhen-jin Z, Peng L, Fa-yu L, Liyan S, Chang-fu S. PKCα take part in CCR7/NF-κB autocrine signaling loop in CCR7-positive squamous cell carcinoma of head and neck. Mol Cell Biochem. 2011;357(1-2):181–187. doi: 10.1007/s11010-011-0888-0. [DOI] [PubMed] [Google Scholar]
- 49.Melino G, et al. Itch: a HECT-type E3 ligase regulating immunity, skin and cancer. Cell Death Differ. 2008;15(7):1103–1112. doi: 10.1038/cdd.2008.60. [DOI] [PubMed] [Google Scholar]
- 50.Humphreys IR, Walzl G, Edwards L, Rae A, Hill S, Hussell T. A critical role for OX40 in T cell-mediated immunopathology during lung viral infection. J Exp Med. 2003;198(8):1237–1242. doi: 10.1084/jem.20030351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Marshall RP, et al. Angiotensin II and the fibroproliferative response to acute lung injury. Am J Physiol Lung Cell Mol Physiol. 2004;286(1):L156–L164. doi: 10.1152/ajplung.00313.2002. [DOI] [PubMed] [Google Scholar]
- 52.Gelinck LB, et al. The effect of anti-tumour necrosis factor alpha treatment on the antibody response to influenza vaccination. Ann Rheum Dis. 2008;67(5):713–716. doi: 10.1136/ard.2007.077552. [DOI] [PubMed] [Google Scholar]
- 53.Sweeney DA, Danner RL, Eichacker PQ, Natanson C. Once is not enough: clinical trials in sepsis. Intensive Care Med. 2008;34(11):1955–1960. doi: 10.1007/s00134-008-1274-6. [DOI] [PubMed] [Google Scholar]
- 54.Mallampalli RK, et al. Targeting F box protein Fbxo3 to control cytokine-driven inflammation. J Immunol. 2013;191(10):5247–5255. doi: 10.4049/jimmunol.1300456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lim KL, et al. Parkin mediates nonclassical, proteasomal-independent ubiquitination of synphilin-1: implications for Lewy body formation. J Neurosci. 2005;25(8):2002–2009. doi: 10.1523/JNEUROSCI.4474-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kitsios GD, et al. Respiratory microbiome profiling for etiologic diagnosis of pneumonia in mechanically ventilated patients. Front Microbiol. 2018;9:1413. doi: 10.3389/fmicb.2018.01413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Datta A, Kamthan A, Kamthan M. A simple protocol to detect interacting proteins by GST pull down assay coupled with MALDI or LC-MS/MS analysis. Protocol Exchange Web Site. doi(4): 10.1038/protex.2015.093. Posted October 12, 2015. Accessed September 20, 2019. [Google Scholar]
- 58.Kuratomi G, et al. NEDD4-2 (neural precursor cell expressed, developmentally down-regulated 4-2) negatively regulates TGF-beta (transforming growth factor-beta) signalling by inducing ubiquitin-mediated degradation of Smad2 and TGF-beta type I receptor. Biochem J. 2005;386(pt 3):461–470. doi: 10.1042/BJ20040738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cao XR, et al. Nedd4 controls animal growth by regulating IGF-1 signaling. Sci Signal. 2008;1(38):ra5. doi: 10.1126/scisignal.1160940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Torrino S, et al. The E3 ubiquitin-ligase HACE1 catalyzes the ubiquitylation of active Rac1. Dev Cell. 2011;21(5):959–965. doi: 10.1016/j.devcel.2011.08.015. [DOI] [PubMed] [Google Scholar]
- 61.Marchese A, Raiborg C, Santini F, Keen JH, Stenmark H, Benovic JL. The E3 ubiquitin ligase AIP4 mediates ubiquitination and sorting of the G protein-coupled receptor CXCR4. Dev Cell. 2003;5(5):709–722. doi: 10.1016/S1534-5807(03)00321-6. [DOI] [PubMed] [Google Scholar]
- 62.Xu H, et al. WWP2 promotes degradation of transcription factor OCT4 in human embryonic stem cells. Cell Res. 2009;19(5):561–573. doi: 10.1038/cr.2009.31. [DOI] [PubMed] [Google Scholar]
- 63.Zhong Q, Gao W, Du F, Wang X. Mule/ARF-BP1, a BH3-only E3 ubiquitin ligase, catalyzes the polyubiquitination of Mcl-1 and regulates apoptosis. Cell. 2005;121(7):1085–1095. doi: 10.1016/j.cell.2005.06.009. [DOI] [PubMed] [Google Scholar]
- 64.Miyazaki K, et al. NEDL1, a novel ubiquitin-protein isopeptide ligase for dishevelled-1, targets mutant superoxide dismutase-1. J Biol Chem. 2004;279(12):11327–11335. doi: 10.1074/jbc.M312389200. [DOI] [PubMed] [Google Scholar]
- 65.Zhu H, Kavsak P, Abdollah S, Wrana JL, Thomsen GH. A SMAD ubiquitin ligase targets the BMP pathway and affects embryonic pattern formation. Nature. 1999;400(6745):687–693. doi: 10.1038/23293. [DOI] [PubMed] [Google Scholar]
- 66.Krishnamoorthy V, Khanna R, Parnaik VK. E3 ubiquitin ligase HECW2 mediates the proteasomal degradation of HP1 isoforms. Biochem Biophys Res Commun. 2018;503(4):2478–2484. doi: 10.1016/j.bbrc.2018.07.003. [DOI] [PubMed] [Google Scholar]
- 67.Lin X, Liang M, Feng XH. Smurf2 is a ubiquitin E3 ligase mediating proteasome-dependent degradation of Smad2 in transforming growth factor-beta signaling. J Biol Chem. 2000;275(47):36818–36822. doi: 10.1074/jbc.C000580200. [DOI] [PubMed] [Google Scholar]
- 68.Seo SR, et al. The novel E3 ubiquitin ligase Tiul1 associates with TGIF to target Smad2 for degradation. EMBO J. 2004;23(19):3780–3792. doi: 10.1038/sj.emboj.7600398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yu Y, Hayward GS. The ubiquitin E3 ligase RAUL negatively regulates type i interferon through ubiquitination of the transcription factors IRF7 and IRF3. Immunity. 2010;33(6):863–877. doi: 10.1016/j.immuni.2010.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cheon S, et al. The ubiquitin ligase UBE3B, disrupted in intellectual disability and absent speech, regulates metabolic pathways by targeting BCKDK. Proc Natl Acad Sci USA. 2019;116(9):3662–3667. doi: 10.1073/pnas.1818751116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mishra A, Godavarthi SK, Jana NR. UBE3A/E6-AP regulates cell proliferation by promoting proteasomal degradation of p27. Neurobiol Dis. 2009;36(1):26–34. doi: 10.1016/j.nbd.2009.06.010. [DOI] [PubMed] [Google Scholar]
- 72.Hochrainer K, Pejanovic N, Olaseun VA, Zhang S, Iadecola C, Anrather J. The ubiquitin ligase HERC3 attenuates NF-κB-dependent transcription independently of its enzymatic activity by delivering the RelA subunit for degradation. Nucleic Acids Res. 2015;43(20):9889–9904. doi: 10.1093/nar/gkv1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.