

Abstract
There is increased interest in whether bariatric surgeries such as Roux-en-Y gastric bypass (RYGB) achieve their profound weight-lowering effects in morbidly obese individuals through the brain. Hypothalamic inflammation is a well-recognized etiologic factor in obesity pathogenesis and so represents a potential target of RYGB, but clinical evidence in support of this is limited. We therefore assessed hypothalamic T2-weighted signal intensities (T2W SI) and fractional anisotropy (FA) values, 2 validated radiologic measures of brain inflammation, in relation to BMI and fat mass, as well as circulating inflammatory (C-reactive protein; CrP) and metabolic markers in a cohort of 27 RYGB patients at baseline and 6 and 12 months after surgery. We found that RYGB progressively increased hypothalamic T2W SI values, while it progressively decreased hypothalamic FA values. Regression analyses further revealed that this could be most strongly linked to plasma CrP levels, which independently predicted hypothalamic FA values when adjusting for age, sex, fat mass, and diabetes diagnosis. These findings suggest that RYGB has a major time-dependent impact on hypothalamic inflammation status, possibly by attenuating peripheral inflammation. They also suggest that hypothalamic FA values may provide a more specific radiologic measure of hypothalamic inflammation than more commonly used T2W SI values.
Keywords: Metabolism, Neuroscience
Keywords: Neuroimaging, Obesity
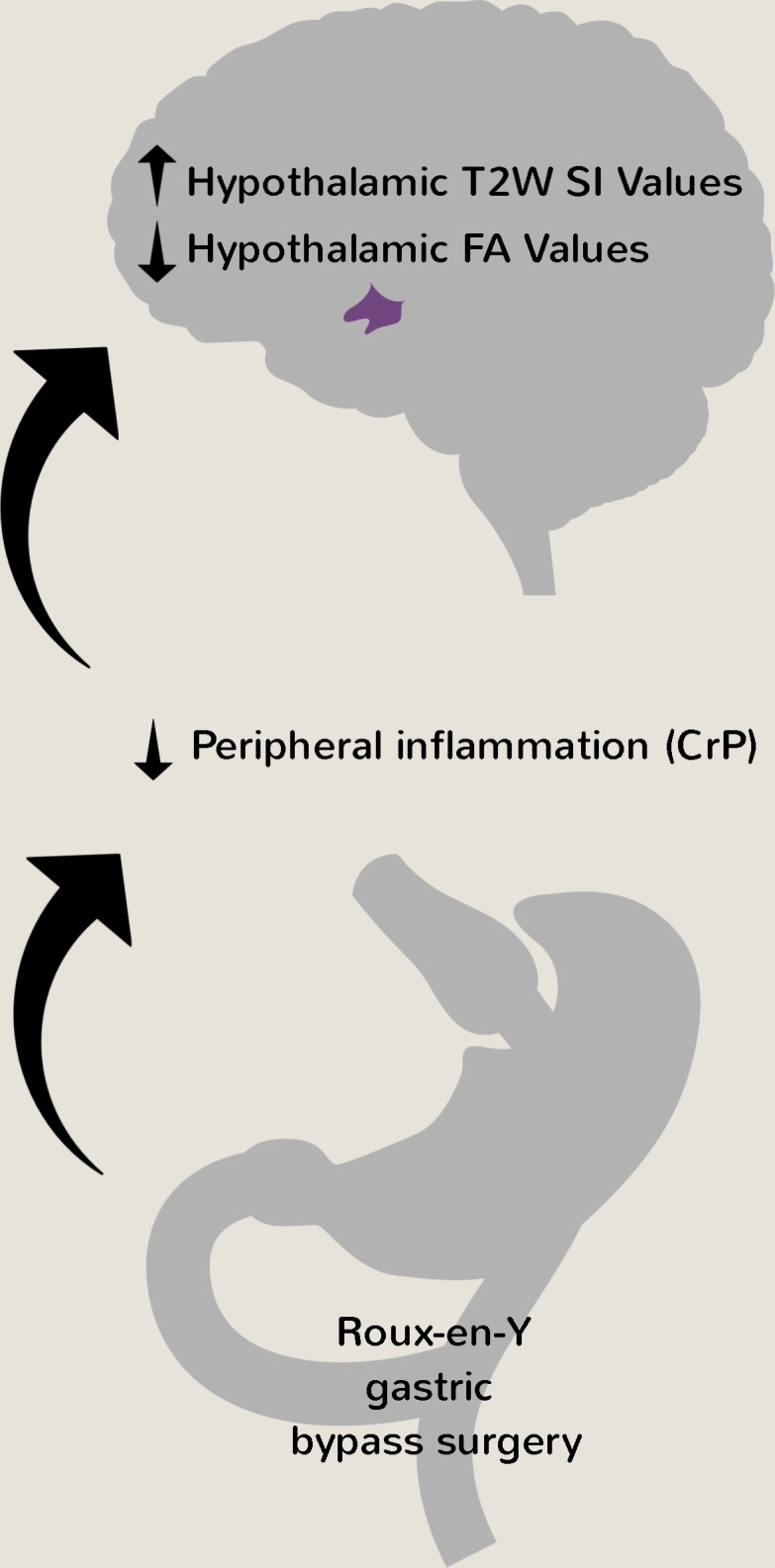
Gastric bypass has a major effect on hypothalamic inflammation status, which may be mediated through attenuation of peripheral inflammation.
Introduction
Historically, bariatric surgeries were first considered to induce striking and durable weight loss in morbidly obese individuals through a combination of restricting the intake and absorption of food (1). Studies using rodent models, in particular, have helped to modify this view, and instead, altered gut-brain communication has taken center-stage in the apparent reprogramming of whole-body energy balance set-point that occurs after surgery (2–8). Roux-en-Y gastric bypass (RYGB) is one of the most effective and frequently performed bariatric surgeries, and according to a recent estimate, as many as 72,000 procedures were carried out between 2014–2018 worldwide (9). RYGB reconfigures the gastrointestinal tract in such a way that more ingested food and digestive secretions reach distal parts of the small intestine in a shorter time frame during a meal, with major effects on gut hormone release and metabolic outcomes (10–12). Patients typically lose 30%–40% body weight in the first year after surgery — a total that is sustained for up to 12 years (13) — and also experience marked improvements in glucose and lipid metabolism (14). However, RYGB is not without its drawbacks, which include severe hypoglycemia (15), malnutrition (16), an increased risk of bone fractures (17), and — in some cases — esophagogastric cancers (18). A more detailed understanding of the mechanisms behind the beneficial effects of RYGB could therefore lead to the development of safer, noninvasive alternatives.
The causes of obesity are complex and multifactorial, and they include genetic, environmental, psychological, and sociological aspects (19). One prominent theory is that common obesity, in part, arises from hypothalamic inflammation, which is characterized by the activation and proliferation of microglia and astrocytes and by dysregulated neuronal structure and function (20). In rodents, the rise in circulating saturated free fatty acids from high-fat diet consumption is thought to trigger proinflammatory signaling cascades in all of these brain cell types through TLR4, leading to the release of proinflammatory cytokines, such as TNF-α and IL-1β (21–24). This then interferes with the healthy functioning of local leptin receptor–expressing neurons through autocrine and paracrine mechanisms, leading to central leptin resistance and weight gain (21). As such, molecular inactivation of the proinflammatory enzyme inhibitor of nuclear factor κ-B kinase subunit β (IKKβ) in hypothalamic microglia (25), astrocytes (26, 27), and neurons (21) preserves central leptin sensitivity and/or protects mice from high-fat diet–induced obesity.
Evidence that hypothalamic inflammation occurs in human obesity first came from a retrospective analysis of clinical T2-weighted (T2W) MRI data (28). Ex vivo radiologic and histologic assessment of postmortem brain samples then linked the higher hypothalamic T2W signal intensity (SI) that develops with increasing BMI to the severity of hypothalamic inflammation (29). Furthermore, circulating levels of the proinflammatory cytokine IL-6 and its downstream target in the liver C-reactive protein (CrP), has more recently been shown to correlate positively with hypothalamic T2W SI (30), suggesting that peripheral and central inflammatory processes are closely intertwined in humans. Diffusion-weighted (DW) MRI metrics can also provide a complementary radiologic measure of hypothalamic inflammation to T2W SI values (31, 32). DW MRI measures the channeled movement of water molecules in a certain direction (often represented as a diffusion ellipsoid or tensor) and, as such, is typically used for the study of major white matter tracts in the brain (33). Fractional anisotropy (FA) is a DW MRI metric that reflects the directionality of water movement in a given voxel as a function of 3 orthogonal eigenvalues of the diffusion tensor. FA values are high (close to 1) in structured brain regions such the optic chiasm and low (close to zero) in fluid-filled brain regions such as the third ventricle. Because of the loss of tissue microstructure that usually accompanies inflammation, hypothalamic principle eigenvalues have been shown to inversely correlate with BMI (32). Along with the independent confirmation that both hypothalamic microglial and astrocytic markers are increased in the obese human hypothalamus (30, 34), these findings collectively provide strong support for the argument that targeting hypothalamic inflammatory pathways represents a promising therapeutic approach in the fight against obesity (35).
In further support of the above statement, RYGB has been shown to reduce hypothalamic mRNA expression of several proinflammatory cytokines/chemokines, as well as microglial cell markers/numbers in diet-induced obese mice (36, 37). However, in a longitudinal T2W MRI analysis of 10 RYGB patients, no significant reduction in hypothalamic inflammation 10 months after surgery was found (30). A potential reason for this could be that the changes in tissue water content measured by T2W MRI may not be sufficient enough to signal subtle changes in hypothalamic inflammation status. This is especially because obesity is considered to be a chronic low-grade inflammatory state, in general (35). Other reasons could be the relatively low sample size used or that patients were scanned too soon after surgery before any beneficial effects on hypothalamic inflammation could occur (30). Therefore, to assess in more detail whether RYGB attenuates obesity-associated hypothalamic inflammation in the clinical setting, we performed analyses on T2W and DW MRI brain datasets of 27 morbidly obese patients obtained at baseline and 6 and 12 months after surgery (38). In a potentially novel approach, we further calculated hypothalamic T2W/T1-weighted (T1W) SI ratios on the basis that they may more sensitively reflect changes in hypothalamic inflammation status by exponentially enhancing water-based contrast (39, 40).
Results
Consistent with the profound weight loss that typically occurs following RYGB (13, 14), the BMI of patients in the present study dropped from 47.8 ± 5.5 kg/m2 at baseline to 37.5 ± 5.0 kg/m2 at 6 months and 35.2 ± 4.5 kg/m2 at 12 months after surgery (Figure 1A). This was accompanied with a decrease in total body fat mass from 53.3% ± 1.9% at baseline to 38.6% ± 2.2% at 6 months and 37.3% ± 2.3% at 12 months after surgery (Figure 1B). Correspondingly, plasma levels of leptin and adiponectin, 2 adipokines that inversely circulate in proportion to adiposity (41), decreased and increased, respectively (Figure 1, C and D). There were also significant improvements in the standard marker of peripheral inflammation CrP (Figure 1E) and systemic markers of glucose metabolism, such as glycated hemoglobin (HbA1c) and plasma insulin (Figure 1, F and G) causing homoeostatic model of insulin resistance (HOMA-IR) values to drop by approximately 75% by the 12-month time point after surgery (Figure 1H). Systemic markers of lipid metabolism such as plasma triglycerides (TG), cholesterol, and LDL also significantly improved after RYGB (Figures 1, I–K). However, plasma HDL, which usually increases with attenuation of hypercholesterolemia, did not significantly change from baseline at either the 6-month or 12-month time points after surgery (Figure 1L).
Figure 1. RYGB markedly improves BMI, body fat mass, and circulating inflammatory and metabolic markers in obese patients.

(A) BMI, (B) fat mass percentage (%), (C) plasma leptin, (D) adiponectin, (E) C-reactive peptide (CrP), (F) glycated hemoglobin (HbA1c), (G) insulin, (H) homeostatic model of insulin resistance (HOMA-IR), (I) triglycerides (TG), (J) cholesterol, (K) LDL, and (L) HDL in morbidly obese patients at baseline (n = 16–27), 6 months (n = 22–27), and 12 months (n = 17–27) after RYGB. Numbers in each graph represent mean ± SEM. Statistical significance was determined by mixed-effects model analysis with Tukey’s post hoc test. **** P < 0.0001, *** P < 0.001, ** P < 0.01, and * P < 0.05.
Having established the marked beneficial effects of RYGB on BMI, fat mass, and circulating inflammatory and metabolic markers in the present cohort of patients, we assessed potential changes in hypothalamic inflammation by analyzing brain structural MRI datasets. Representative coronal T1W, T2W, T2W/T1W, and DW FA MRI data with the mediobasal hypothalamic (MBH) and amygdala masks used for our analyses are shown in Figure 2A. Hypothalamic T2W SI values gradually increased after RYGB (P < 0.01) (Figure 2B), while hypothalamic T2W/T1W SI ratios more robustly increased (P < 0.0001) (Figure 2C). In opposing contrast, hypothalamic FA values gradually decreased after RYGB (Figure 2D). Similarly, amygdala T2W SI values gradually increased after RYGB (Figure 2E), although the increase in T2W/T1W SI ratios were not as robust as they were for the hypothalamus (Figure 2F). There were no significant changes in amygdala FA values at any time point after surgery (Figure 2G). Previous cross-sectional and longitudinal human brain T2W MRI studies assessing hypothalamic inflammation in obesity and after RYGB have normalized hypothalamic T2W SI values to those of the amygdala to control for scan-to-scan variability brought about by scanner hardware (28, 30). When doing so, we recorded a progressive decrease in hypothalamic T2WI SI values normalized to amygdala after RYGB that attained statistical significance at the 12-month time point (P < 0.05) (Figure 1H). Performing the same normalization procedure to hypothalamic T2W/T1W SI ratios and FA values, however, did not yield any significant changes at any time point after surgery (Figure 1, I and J).
Figure 2. RYGB progressively alters radiologic measures of hypothalamic inflammation in obese patients.

(A) Representative coronal T1-weighted (T1W), T2-weighted (T2W), and T2W/T1W and diffusion-weighted fractional anisotropy (DW FA) images with the mediobasal hypothalamic (MBH) and amygdala regions of interest colored in red and blue, respectively. (B–J) Mean MBH T2W SI (B), T2W/T1W SI ratios (C), and FA values (D); mean amygdala T2W SI (E), T2W/T1W SI ratios (F), and FA values (G); and mean MBH normalized to amygdala T2W SI (H), T2W/T1W SI ratios (I), and FA values (J) of RYGB patients at baseline (n = 24–27), 6 months (n = 20–27), and 12 months (n = 17–22) after surgery. Numbers in each graph represent mean ± SEM. Statistical significance was determined by mixed-effects model analysis with Tukey’s post hoc test. **** P < 0.0001, ** P < 0.01, and * P < 0.05.
To find a potential cause for the observed changes in radiologic measures of hypothalamic and amygdala inflammation after RYGB, we next performed univariate linear regression analysis with BMI, fat mass, and circulating inflammatory and metabolic markers as dependent variables. This revealed that hypothalamic T2W SI values, and particularly T2W/T1W SI ratios, negatively correlated with BMI (Figure 3, A and B), fat mass (Figure 3, D and E), and plasma leptin levels (data not shown). There were also negative correlations between hypothalamic T2W SI values and T2W/T1W SI ratios with plasma CrP (Figure 3, G and H) and insulin (Figure 3, J and K) levels, but not HOMA-IR values (Figure 3, M and N), although only the former survived correction for multiple comparisons. In line with the time point analysis, hypothalamic FA values exhibited the opposite pattern, with only plasma CrP values showing a positive correlation that remained significant after correction for multiple comparisons (Figure 3, C, F, I, L, and O). Similar to the hypothalamus, amygdala T2W SI values negatively correlated with BMI (Figure 4A), fat mass (Figure 4D), plasma leptin (data not shown), CrP (Figure 4G), and insulin (Figure 4J) levels, but not HOMA-IR values (Figure 4, M and N), although only the former 2 survived correction for multiple comparisons. There were also negative correlations between amygdala T2W/T1W SI ratios with BMI (Figure 4B), fat mass (Figure 4E), plasma leptin (data not shown), CrP (Figure 4H), and insulin (Figure 4K) levels, although only plasma CrP survived correction for multiple comparisons. Amygdala FA values did not correlate with any phenotypic measure or circulating inflammatory or metabolic marker (Figure 4, C, F, I, L, and O). We also did not find any significant correlation for individual changes in BMI, fat mass, or circulating inflammatory and metabolic markers with changes in hypothalamic and amygdala T2W SI values, T2W/T1W ratios, and FA values between baseline, 6 months after RYGB, and 12 months after RYGB (data not shown).
Figure 3. Relationships between BMI, body fat mass, and circulating inflammatory and metabolic markers with radiologic measures of hypothalamic inflammation in RYGB patients.

(A–O) Univariate linear regressions and Pearson correlations between BMI (A–C), fat mass % (D–F), plasma CrP (G–I), log plasma insulin (J–L), and HOMA-IR (M–O) with mean MBH T2W SI (A, D, G, J, and M), T2W/T1W SI ratios (B, E, H, K, and N), and FA values (C, F, I, L, and O) of RYGB patients at baseline (n = 14–25), 6 months (n = 14–25), and 12 months after surgery (n = 11–22). Solid regression lines indicate least squares fit of data, and dotted lines indicate 95% CI. P values were determined by 2-tailed Student’s t test. Asterisk signifies correlations that remained statistically significant after Bonferroni correction for multiple comparisons (P < 0.0014).
Figure 4. Relationships between BMI, body fat mass, circulating inflammatory and metabolic markers with radiologic measures of amygdala inflammation in RYGB patients.

(A–O) Univariate linear regressions and Pearson correlations between BMI (A–C), fat mass % (D–F), plasma CrP (G–I), log plasma insulin (J–L), and HOMA-IR (M–O) with mean amygdala T2W SI (A, D, G, J, and M), T2W/T1W SI ratios (B, E, H, K, and N), and FA values (C, F, I, L, and O) of RYGB patients at baseline (n = 14–24), 6 months (n = 15–23), and 12 months after surgery (n = 14–21). Solid regression lines indicate least squares fit of data, and dotted lines indicate 95% CI. P values were determined by 2-tailed Student’s t test. Asterisk signifies correlations that remained statistically significant after Bonferroni correction for multiple comparisons (P < 0.0014).
Prompted by the univariate linear regression analysis findings, we then performed multivariate linear regression analysis using the radiologic measures of hypothalamic and amygdala inflammation as dependent variables and plasma CrP levels as the independent variable, while also correcting for fat mass, age, sex, and diabetes diagnosis. This model revealed that fat mass independently predicted changes in hypothalamic T2W SI values and T2W/T1W SI ratios, while only plasma CrP independently predicted hypothalamic FA values (Table 1). On the other hand, fat mass and plasma CrP predicted amygdala T2W SI values and T2W/T1W SI ratios, respectively, while both of these independent variables predicted amygdala FA values (Table 2).
Table 1. Body fat mass, sex, and plasma CrP levels predict radiologic measures of hypothalamic inflammation in obese patients before and after RYGB.

Table 2. Body fat mass and plasma CrP levels predict radiologic measures of amygdala inflammation in obese patients before and after RYGB.

Discussion
In the present study, we asked whether RYGB attenuates obesity-associated hypothalamic inflammation in humans using established radiologic techniques (28–32, 42–46). We also used a potentially novel, more sensitive analysis method that entails a straightforward division of T2W by T1W images to enhance water-based contrast (39, 40). Unexpectedly, we found that hypothalamic T2W SI values, and particularly T2W/T1W SI ratios, gradually increased while hypothalamic FA values gradually decreased after RYGB. Furthermore, using univariate and multivariate linear regression analysis, we could link these changes in radiologic measures of hypothalamic inflammation to reductions in BMI, fat mass, and plasma CrP, with the latter being an independent predictor of hypothalamic FA values.
The relationships between hypothalamic T2W SI and FA values with BMI in the present study contrast with those previously found in individuals who had gained rather than lost weight (28–30, 32, 43, 44). Small-animal T2W MRI (31) and ex vivo T2W MRI of human postmortem brain samples (29) originally related the increased hypothalamic T2W SI in obesity to the proliferation and activation of microglia and astrocytes. The latter cell type accumulates water under inflammatory conditions through aquaporin-4 (47), explaining how their increased number/activity contributes to increased T2W SI, which is largely water based in biological tissue. In principle, during hypothalamic inflammation, water movement would become less restricted due to the loss of tissue microstructure, explaining the decrease in hypothalamic principle eigenvalue in obesity, as detected previously by DW MRI (32). The molecular and cellular bases of the changes in radiologic measures of hypothalamic inflammation in the present study are unclear and warrant further investigation, but it is noteworthy that — at least in mice — weight loss of a similar magnitude to RYGB paradoxically resulted in marked hypothalamic astrocyte but not microglial reactive gliosis (48). This was attributed to an increase in plasma free fatty acids, well recognized triggers of hypothalamic inflammation (23, 24), consequential to white adipose tissue lipolysis (48). Unfortunately, we did not measure plasma free fatty acids in the present study, so we could not determine if similar processes to those in mice after profound weight loss similarly occur in humans after RYGB. We did, however, find that plasma CrP associated with hypothalamic T2W SI values, T2W/T1W SI ratios, and FA values, confirming that peripheral and central inflammatory processes are closely intertwined in humans (30). Additionally, since the reductions in plasma CrP after RYGB independently predicted the reduction in hypothalamic FA but not T2W SI values, this radiologic measure may more selectively reflect hypothalamic inflammation. Indeed, hypothalamic T2W SI values have been shown to dissociate from changes in hypothalamic inflammation status in mice (42). The reduction in CrP release from the liver after RYGB could be due to the reduction in adipose tissue inflammation and IL-6 secretion from resident immune cells (49).
The increased hypothalamic T2W SI values after RYGB in the present study contrast with the findings of Kreutzer et al., in which there was a trend toward a decrease in hypothalamic T2W SI values normalized to those of the amygdala in the left hemisphere at the 10-month time point after surgery (30). However, when normalizing hypothalamic T2W SI values to that of amygdala, we found a progressive decrease after RYGB, which attained statistical significance at the 12-month time point. One potential explanation for these discrepant findings is that, unlike the patients in the study of Kreutzer et al., our patients showed marked reductions in peripheral inflammation after surgery, which — as mentioned above — may have a major bearing on hypothalamic inflammation status (30). Other reasons behind these discrepant findings could be the larger sample size used in our study or the fact that our patients were scanned at a slightly later time point after RYGB. Indeed, at the earlier 6-month time point, the reduction in hypothalamic T2W SI normalized to that of amygdala did not quite attain statistical significance. Nevertheless, caution should be taken when interpreting the normalized values, as changes in amygdala T2W SI values clearly caused a bias. Indeed, the amygdala itself has not only emerged as a brain feeding center with molecularly defined neuronal populations that increase (50–52) and decrease food intake (53), but it is also a site of high-fat diet–induced inflammation (54, 55). Furthermore, amygdala inflammation is a cause of insulin resistance (55), which may explain the strong relationships we found between plasma insulin with T2W SI values in this brain region. Human DW MRI findings have further revealed strong positive correlations between amygdala apparent diffusion coefficients, which are inversely proportional to FA values, and plasma fibrinogen (another marker of peripheral inflammation) in obesity (46). Despite the fact that we did not detect any significant changes in amygdala FA values after RYGB or any correlation with plasma CrP levels in our univariate linear regression analysis, this is in line with how plasma CrP predicted amygdala FA values in our multivariate linear regression analysis. Therefore, normalization of hypothalamic radiologic measures with that of amygdala in the context of obesity does not appear to be an ideal approach. Instead, calculating T2W/T1W SI ratios not only cancels out hardware-related issues, but also reduces variance, thereby increasing statistical power. This approach may also have more general diagnostic applications, especially in the early stages of brain inflammation before overt neurologic symptoms develop.
Taking all of the above into consideration, the findings from the present study can be interpreted in 2 ways. First, it is possible that RYGB actually aggravates hypothalamic inflammation, thereby contributing to weight loss through reduced food intake. Indeed, it has recently been shown that the small-molecule drug celastrol increases signaling of the proinflammatory cytokine receptor IL-1R1 in the hypothalamus of diet-induced obese mice to induce similar magnitudes of weight loss as RYGB through reduced food intake (56). This is in line with the proposition that hypothalamic glial cells play more complex roles in the regulation of whole-body energy balance than simply inducing central leptin resistance and causing weight gain (57). Alternatively, the fact that lower plasma CrP values associated with higher hypothalamic T2W SI and lower hypothalamic FA values suggests that RYGB attenuates hypothalamic inflammation. In doing so, RYGB would reduce overall hypothalamic glial cell but not neuronal numbers (30). The resulting void in cell mass would then conceivably lead to an increase in T2W SI and a decrease in FA values due to increases in hypothalamic water content and freer water movement, respectively. The enhanced hypothalamic function (58) and resting connectivity (59) previously reported for patients after RYGB supports the second interpretation. Moreover, CSF levels of the antiinflammatory cytokine IL-10 has been found to increase in patients after RYGB (59).
Whether the changes in radiologic measures of hypothalamic inflammation observed in the present study are specific to RYGB or secondary to weight loss is unclear. This can be addressed in future studies on patients after gastric banding (60). Interestingly, RYGB was recently shown to reduce hypothalamic mRNA expression of several proinflammatory cytokines/chemokines, as well as microglial markers/numbers in diet-induced obese mice, independently of weight loss and reductions in high-fat diet intake (36, 37). Because bile diversion to the ileum recapitulated most of the effects of RYGB in this context, it suggests that bile acids sufficiently attenuate hypothalamic inflammation (36). Future studies can determine whether the hypothalamic radiologic changes that take place after RYGB relate to the well-documented increase in plasma bile acids (61) in conjunction with the decrease in peripheral inflammation. Considering that the glucagon-like peptide 1 (GLP-1) analogue liraglutide similarly attenuates microglial reactive gliosis in diet-induced obese mice (62), it would be worth determining whether the increase in circulating GLP-1 after RYGB (63) also relates to the alteration in hypothalamic inflammation status.
While T2W SI and FA values have been shown to reflect changes in brain inflammation determined histologically (29, 31, 42, 45, 64), they at best represent indirect measures. Similar issues arise when interpreting functional MRI data, which reflect changes in blood oxygenation level–dependent signal (BOLD) rather than changes in neuronal activity per se (65). Therefore, the findings of the present study provide an indication of changes in hypothalamic inflammation status after RYGB. Since confirmation by immunohistochemical methods cannot be achieved in humans as in rodent studies, future work applying PET imaging with radiotracers that directly measure microglial activation (66) could complement our findings. It has to be kept in mind, however, that MRI offers higher spatial resolution than PET, so any subtle changes in the hypothalamus may be overlooked by the latter neuroimaging technique. Another shortcoming of the present analysis is that changes in each radiologic measure of hypothalamic inflammation after RYGB did not associate with changes in BMI or adiposity, arguing against causation. One way to confirm that attenuation of hypothalamic inflammation actively contributes to weight loss after RYGB is to chemically induce this in rodents and to assess changes in feeding and body weight (35). We also did not include a healthy-weight control in our analysis due to the lack of T2W brain MRI and plasma CrP, glucose, and insulin data. Future studies can formally reveal whether the changes in hypothalamic and amygdala T2W SI, T2W/T1W SI ratios, and FA values after RYGB represent a return to a healthy brain state or whether a new one is achieved.
In summary, we have provided radiologic evidence that hypothalamic inflammation status is dramatically, albeit progressively, changed by RYGB, possibly by attenuating peripheral inflammation. We also show that T2W/T1W SI ratios and FA values provide useful complementary radiologic metrics to T2W SI values in assessing brain inflammation. Our findings suggest that pharmacologic obesity treatments could be designed to target peripheral and/or central inflammatory processes to cause marked and durable weight loss with the associated metabolic benefits (35).
Methods
Subjects.
We reanalyzed previously published (38) MRI datasets obtained from 27 morbidly obese patients at baseline (6 males and 21 females, 14 with diabetes, age 51.1 ± 9.6 years), as well as 6 months (6 males and 21 females, 3 with diabetes, age 51.8 ±9.9 years) and 12 months (4 males and 18 females, 3 with diabetes, age 52.6 ± 8.5 years) after laparoscopic RYGB. Plasma samples were collected at each time point and analyzed for fasting leptin, adiponectin, glucose, HbA1c, insulin, CrP, TG, cholesterol, LDL, and HDL as previously described (38). HOMA-IR values were obtained by the multiplying plasma insulin levels (in μU/ml) by plasma glucose levels (in mmol/l) and dividing the output by 22.5 (67). Body fat mass percentage was determined using the Nutriguard M system (Data Input GmbH) together with the BodyComposition software, version 9.0 (MEDI CAL HealthCare GmbH).
Scanning parameters.
All imaging experiments were performed using a 3 Tesla (3T) VERIO MRI scanner (Siemens). For each session, we first acquired T2W fluid-attenuated inversion-recovery (FLAIR) images (28 axial slices; 4-mm thickness, inversion time [TI] = 2,500 ms; repetition time [TR] = 10,000 ms; echo time [TE] = 90 ms; readout pulse flip angle, α = 180°; bandwidth = 199 Hz/pixel; image matrix = 256 × 192; field of view [FOV] = 220 × 165 mm²; nominal spatial resolution = 4 × 0.85 × 0.85 mm³; 1 average). This was followed by acquiring T1W 3-dimensional magnetization-prepared rapid gradient echo (MP-RAGE) images with selective water excitation and linear phase encoding in sagittal slice orientation (TI = 900 ms; TR = 1,300 ms; TE = 2.98 ms; readout pulse flip angle, α = 9°; bandwidth = 238 Hz/pixel; image matrix = 256 × 240; FOV = 256 × 240 mm²; nominal spatial resolution = 1 × 1 × 1 mm³; 1 average). Finally, whole-brain DW images (twice-refocused spin echo-planar imaging sequence, 64 axial slices, 2-mm thickness, no gap, TE = 88 ms, TR = 12.4 s, α = 90°, bandwidth = 1,684 Hz/pixel, FOV = 220 × 220 mm2, 110 × 110 image matrix, Generalized Auto-calibrating Partially Parallel Acquisition (GRAPPA), acceleration factor 2) were acquired applying 60 diffusion-encoding gradient directions and a b value of 1,000 s/mm2. We acquired 7 images without diffusion weighting (b value 0 s/mm²) as an anatomical reference for offline motion correction at the beginning and end of each block of 10 images.
Data processing.
T1W and T2W MRI data were aligned onto the Montreal Neurological Institute (MNI) standard template using the Functional MRI of the Brain (FMRIB) Linear Registration Tool (FLIRT) application within the FMRIB Software Library (FSL) (68). This approach builds on the approach used to more sensitively assess gray matter myelination from dividing T1W images by T2W images, which exponentially increases fat-based contrast (39, 40). If water is represented by x in the T2W image and by 1/x in the T1W image, and if the receive bias field is represented by b in both images, then:
T2W/T1W = x*b / (1/x)*b = x2
This eliminates receive bias field and exponentially increases water-based contrast (39).
DW MRI data were first quality checked with DTIPrep (69) and then processed with FSL and Leipzig Image Processing and Statistical Inference Algorithms (LIPSIA). Images were motion corrected with rigid-body transformation, based on the anatomical reference images. This was combined with global registration to the T1W images. After skull stripping, images were coregistered to the standard MNI template. We applied rotation parameters to correct gradient direction of each image. We interpolated registered images, representing the individual anisotropy, using an isotropic voxel resolution of 1 mm. Finally, a diffusion tensor was fitted to each voxel, and FA was calculated from the eigenvalues of the diffusion tensor.
Since no standard mask for the MBH is currently available, bilateral MBH masks (2 mm3) were manually drawn on the aligned T1W, T2W, and FA images for each individual as in previous studies (28, 30). We located the hypothalamus in each 3-dimensional MRI volume with reference to clearly identifiable anatomical landmarks, such as the lateral ventricles, third ventricle and median eminence in the coronal plane, the optic nerves and suprachiasmatic nucleus in the transverse plane, and the pituitary stalk and pituitary gland in the sagittal plane. We also manually drew bilateral amygdala masks (4 mm3) on the same coronal plane as the MBH as in previous studies (28, 30). Since these masks are of a similar size and shape as those used in these previous studies (28, 30), this ensured our findings were comparable and that any differences were not simply due to different brain regions of interest being analyzed. Mean MBH and amygdala T2W SI values, T2W/T1W ratios, and FA values were then obtained using the fslstats application within FSL.
Statistics.
Longitudinal data was tested for normality of distribution using the Shapiro-Wilk test. Since not all patients underwent MRI scans and had complete blood sample measurements at baseline and 6 and 12 months after RYGB, a mixed-effects model analysis with Tukey’s post hoc test was used to assess differences between time points. For univariate linear regression analysis, nonnormally distributed data were first log-transformed, and Pearson correlation analysis was then performed. Statistical significance was determined by using 2-tailed Student’s t test and corrected for multiple comparisons using Bonferroni’s post hoc test. For multivariate linear regression analysis, the least-squares method with intercept and main effects models were used. All statistical analyses were performed on Graphpad Prism software (Version 8). Statistical significance was set at P < 0.05 for all statistical tests and adjusted to P < 0.0014 for univariate correlations after Bonferroni correction.
Study approval.
Patients gave written informed consent, and the original study was carried out in full accordance with the Declaration of Helsinki following approval by the Ethics Committee of the University of Leipzig (approval no. 132-11-18042011).
Author contributions
MKH and BP conceived the study, directed the analysis, and drafted the manuscript. MKH and MR performed the data analysis and produced the figures. All authors contributed to data generation. BP is the guarantor of this work.
Acknowledgments
The research in this manuscript was supported by the IFB AdiposityDiseases and the nutriCARD initiative, Federal Ministry of Education and Research (BMBF), Germany, project number FKZ 01E01001; the German Research Foundation (DFG) SFB 1052 in Obesity Mechanisms, project number 209933838 to BP and MR; and the SFB 874 in Integration and Representation of Sensory Processes, project number 122679504 to BP.
Version 1. 08/29/2019
In-Press Preview
Version 2. 10/03/2019
Electronic publication
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Copyright: © 2019, American Society for Clinical Investigation.
Reference information: JCI Insight. 2019;4(19):e131329.https://doi.org/10.1172/jci.insight.131329.
Contributor Information
Mohammed K. Hankir, Email: hankir_m@ukw.de.
Michael Rullmann, Email: michael.rullmann@medizin.uni-leipzig.de.
Florian Seyfried, Email: seyfried_f@ukw.de.
Sven Preusser, Email: sven.preusser@hof.uni-halle.de.
Sindy Poppitz, Email: poppitz@travdo-hotels.de.
Stefanie Heba, Email: stefanie.heba@rub.de.
Jana Hoyer, Email: Jana.Hoyer@tu-dresden.de.
Tatjana Schütz, Email: tatjana.schuetz@medizin.uni-leipzig.de.
Arne Dietrich, Email: arne.dietrich@medizin.uni-leipzig.de.
Karsten Müller, Email: karstenm@cbs.mpg.de.
Burkhard Pleger, Email: burkhard.pleger@bergmannsheil.de.
References
- 1.Mason EE, Ito C. Gastric bypass. Ann Surg. 1969;170(3):329–339. doi: 10.1097/00000658-196909010-00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Eickhoff H. Central Modulation of Energy Homeostasis and Cognitive Performance After Bariatric Surgery. Adv Neurobiol. 2017;19:213–236. doi: 10.1007/978-3-319-63260-5_9. [DOI] [PubMed] [Google Scholar]
- 3.Clemmensen C, Müller TD, Woods SC, Berthoud HR, Seeley RJ, Tschöp MH. Gut-Brain Cross-Talk in Metabolic Control. Cell. 2017;168(5):758–774. doi: 10.1016/j.cell.2017.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stefanidis A, Oldfield BJ. Neuroendocrine mechanisms underlying bariatric surgery: Insights from human studies and animal models. J Neuroendocrinol. 2017;29(10):e12534. doi: 10.1111/jne.12534. [DOI] [PubMed] [Google Scholar]
- 5.Zakeri R, Batterham RL. Potential mechanisms underlying the effect of bariatric surgery on eating behaviour. Curr Opin Endocrinol Diabetes Obes. 2018;25(1):3–11. doi: 10.1097/MED.0000000000000379. [DOI] [PubMed] [Google Scholar]
- 6.Hankir MK, Seyfried F, Miras AD, Cowley MA. Brain Feeding Circuits after Roux-en-Y Gastric Bypass. Trends Endocrinol Metab. 2018;29(4):218–237. doi: 10.1016/j.tem.2018.01.009. [DOI] [PubMed] [Google Scholar]
- 7.Kim KS, Seeley RJ, Sandoval DA. Signalling from the periphery to the brain that regulates energy homeostasis. Nat Rev Neurosci. 2018;19(4):185–196. doi: 10.1038/nrn.2018.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sinclair P, Brennan DJ, le Roux CW. Gut adaptation after metabolic surgery and its influences on the brain, liver and cancer. Nat Rev Gastroenterol Hepatol. 2018;15(10):606–624. doi: 10.1038/s41575-018-0057-y. [DOI] [PubMed] [Google Scholar]
- 9.Welbourn R, et al. Bariatric Surgery Worldwide: Baseline Demographic Description and One-Year Outcomes from the Fourth IFSO Global Registry Report 2018. Obes Surg. 2019;29(3):782–795. doi: 10.1007/s11695-018-3593-1. [DOI] [PubMed] [Google Scholar]
- 10.Larraufie P, et al. Important Role of the GLP-1 Axis for Glucose Homeostasis after Bariatric Surgery. Cell Rep. 2019;26(6):1399–1408.e6. doi: 10.1016/j.celrep.2019.01.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Flynn CR, et al. Bile diversion to the distal small intestine has comparable metabolic benefits to bariatric surgery. Nat Commun. 2015;6:7715. doi: 10.1038/ncomms8715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Albaugh VL, et al. Role of Bile Acids and GLP-1 in Mediating the Metabolic Improvements of Bariatric Surgery. Gastroenterology. 2019;156(4):1041–1051.e4. doi: 10.1053/j.gastro.2018.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Adams TD, et al. Weight and Metabolic Outcomes 12 Years after Gastric Bypass. N Engl J Med. 2017;377(12):1143–1155. doi: 10.1056/NEJMoa1700459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schauer PR, et al. Bariatric Surgery versus Intensive Medical Therapy for Diabetes - 5-Year Outcomes. N Engl J Med. 2017;376(7):641–651. doi: 10.1056/NEJMoa1600869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guarino D, et al. Postprandial hypoglycaemia after Roux-en-Y gastric bypass in individuals with type 2 diabetes. Diabetologia. 2019;62(1):178–186. doi: 10.1007/s00125-018-4737-5. [DOI] [PubMed] [Google Scholar]
- 16.Antoniewicz A, et al. Nutritional Deficiencies in Patients after Roux-en-Y Gastric Bypass and Sleeve Gastrectomy during 12-Month Follow-Up. Obes Surg. doi: 10.1007/s11695-019-03985-3. [published online ahead of print June 14, 2019]. [DOI] [PubMed] [Google Scholar]
- 17.Musella M, et al. Esophagogastric Neoplasms Following Bariatric Surgery: an Updated Systematic Review. Obes Surg. 2019;29(8):2660–2669. doi: 10.1007/s11695-019-03951-z. [DOI] [PubMed] [Google Scholar]
- 18.Yu EW. Fracture Risk After Roux-en-Y Gastric Bypass vs Adjustable Gastric Banding Among Medicare Beneficiaries. JAMA Surg. 2019;154(8):746–753. doi: 10.1001/jamasurg.2019.1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Blüher M. Obesity: global epidemiology and pathogenesis. Nat Rev Endocrinol. 2019;15(5):288–298. doi: 10.1038/s41574-019-0176-8. [DOI] [PubMed] [Google Scholar]
- 20.Cai D, Khor S. “Hypothalamic Microinflammation” Paradigm in Aging and Metabolic Diseases. Cell Metab. 2019;30(1):19–35. doi: 10.1016/j.cmet.2019.05.021. [DOI] [PubMed] [Google Scholar]
- 21.Zhang X, Zhang G, Zhang H, Karin M, Bai H, Cai D. Hypothalamic IKKbeta/NF-kappaB and ER stress link overnutrition to energy imbalance and obesity. Cell. 2008;135(1):61–73. doi: 10.1016/j.cell.2008.07.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kleinridders A, et al. MyD88 signaling in the CNS is required for development of fatty acid-induced leptin resistance and diet-induced obesity. Cell Metab. 2009;10(4):249–259. doi: 10.1016/j.cmet.2009.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gupta S, Knight AG, Gupta S, Keller JN, Bruce-Keller AJ. Saturated long-chain fatty acids activate inflammatory signaling in astrocytes. J Neurochem. 2012;120(6):1060–1071. doi: 10.1111/j.1471-4159.2012.07660.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Valdearcos M, Robblee MM, Benjamin DI, Nomura DK, Xu AW, Koliwad SK. Microglia dictate the impact of saturated fat consumption on hypothalamic inflammation and neuronal function. Cell Rep. 2014;9(6):2124–2138. doi: 10.1016/j.celrep.2014.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Valdearcos M, et al. Microglial Inflammatory Signaling Orchestrates the Hypothalamic Immune Response to Dietary Excess and Mediates Obesity Susceptibility. Cell Metab. 2018;27(6):1356. doi: 10.1016/j.cmet.2018.04.019. [DOI] [PubMed] [Google Scholar]
- 26.Douglass JD, Dorfman MD, Fasnacht R, Shaffer LD, Thaler JP. Astrocyte IKKβ/NF-κB signaling is required for diet-induced obesity and hypothalamic inflammation. Mol Metab. 2017;6(4):366–373. doi: 10.1016/j.molmet.2017.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang Y, Reichel JM, Han C, Zuniga-Hertz JP, Cai D. Astrocytic Process Plasticity and IKKβ/NF-κB in Central Control of Blood Glucose, Blood Pressure, and Body Weight. Cell Metab. 2017;25(5):1091–1102.e4. doi: 10.1016/j.cmet.2017.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thaler JP, et al. Obesity is associated with hypothalamic injury in rodents and humans. J Clin Invest. 2012;122(1):153–162. doi: 10.1172/JCI59660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schur EA, et al. Radiologic evidence that hypothalamic gliosis is associated with obesity and insulin resistance in humans. Obesity (Silver Spring) 2015;23(11):2142–2148. doi: 10.1002/oby.21248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kreutzer C, et al. Hypothalamic Inflammation in Human Obesity Is Mediated by Environmental and Genetic Factors. Diabetes. 2017;66(9):2407–2415. doi: 10.2337/db17-0067. [DOI] [PubMed] [Google Scholar]
- 31.Lee D, Thaler JP, Berkseth KE, Melhorn SJ, Schwartz MW, Schur EA. Longer T(2) relaxation time is a marker of hypothalamic gliosis in mice with diet-induced obesity. Am J Physiol Endocrinol Metab. 2013;304(11):E1245–E1250. doi: 10.1152/ajpendo.00020.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Puig J, et al. Hypothalamic damage is associated with inflammatory markers and worse cognitive performance in obese subjects. J Clin Endocrinol Metab. 2015;100(2):E276–E281. doi: 10.1210/jc.2014-2682. [DOI] [PubMed] [Google Scholar]
- 33.Jbabdi S, Sotiropoulos SN, Haber SN, Van Essen DC, Behrens TE. Measuring macroscopic brain connections in vivo. Nat Neurosci. 2015;18(11):1546–1555. doi: 10.1038/nn.4134. [DOI] [PubMed] [Google Scholar]
- 34.Baufeld C, Osterloh A, Prokop S, Miller KR, Heppner FL. High-fat diet-induced brain region-specific phenotypic spectrum of CNS resident microglia. Acta Neuropathol. 2016;132(3):361–375. doi: 10.1007/s00401-016-1595-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Seyfried F, Hankir MK. Could de-stressing the brain be the solution for long-term weight loss? Cell Stress. 2019;3(2):29–37. doi: 10.15698/cst2019.02.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Herrick MK, Favela KM, Simerly RB, Abumrad NN, Bingham NC. Attenuation of diet-induced hypothalamic inflammation following bariatric surgery in female mice. Mol Med. 2018;24(1):56. doi: 10.1186/s10020-018-0057-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Patkar PP, Hao Z, Mumphrey MB, Townsend RL, Berthoud HR, Shin AC. Unlike calorie restriction, Roux-en-Y gastric bypass surgery does not increase hypothalamic AgRP and NPY in mice on a high-fat diet. Int J Obes (Lond) doi: 10.1038/s41366.019.0328-x. doi: 10.1038/s41366.019.0328-x. [published online ahead of print February 4, 2019]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rullmann M, et al. Gastric-bypass surgery induced widespread neural plasticity of the obese human brain. Neuroimage. 2018;172:853–863. doi: 10.1016/j.neuroimage.2017.10.062. [DOI] [PubMed] [Google Scholar]
- 39.Glasser MF, Van Essen DC. Mapping human cortical areas in vivo based on myelin content as revealed by T1- and T2-weighted MRI. J Neurosci. 2011;31(32):11597–11616. doi: 10.1523/JNEUROSCI.2180-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bridge H, et al. Preserved extrastriate visual network in a monkey with substantial, naturally occurring damage to primary visual cortex. Elife. 2019;8:e42325. doi: 10.7554/eLife.42325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fasshauer M, Blüher M. Adipokines in health and disease. Trends Pharmacol Sci. 2015;36(7):461–470. doi: 10.1016/j.tips.2015.04.014. [DOI] [PubMed] [Google Scholar]
- 42.Berkseth KE, et al. Hypothalamic gliosis associated with high-fat diet feeding is reversible in mice: a combined immunohistochemical and magnetic resonance imaging study. Endocrinology. 2014;155(8):2858–2867. doi: 10.1210/en.2014-1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sewaybricker LE, et al. Salience network connectivity is reduced by a meal and influenced by genetic background and hypothalamic gliosis. Int J Obes (Lond) doi: 10.1038/s41366-019-0361-9. [published online ahead of print April 9, 2019]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Berkseth KE, et al. Hypothalamic Gliosis by MRI and Visceral Fat Mass Negatively Correlate with Plasma Testosterone Concentrations in Healthy Men. Obesity (Silver Spring) 2018;26(12):1898–1904. doi: 10.1002/oby.22324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tu TW, Williams RA, Lescher JD, Jikaria N, Turtzo LC, Frank JA. Radiological-pathological correlation of diffusion tensor and magnetization transfer imaging in a closed head traumatic brain injury model. Ann Neurol. 2016;79(6):907–920. doi: 10.1002/ana.24641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cazettes F, Cohen JI, Yau PL, Talbot H, Convit A. Obesity-mediated inflammation may damage the brain circuit that regulates food intake. Brain Res. 2011;1373:101–109. doi: 10.1016/j.brainres.2010.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Manley GT, et al. Aquaporin-4 deletion in mice reduces brain edema after acute water intoxication and ischemic stroke. Nat Med. 2000;6(2):159–163. doi: 10.1038/72256. [DOI] [PubMed] [Google Scholar]
- 48.Harrison L, Pfuhlmann K, Schriever SC, Pfluger PT. Profound weight loss induces reactive astrogliosis in the arcuate nucleus of obese mice. Mol Metab. 2019;24:149–155. doi: 10.1016/j.molmet.2019.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Frikke-Schmidt H, O’Rourke RW, Lumeng CN, Sandoval DA, Seeley RJ. Does bariatric surgery improve adipose tissue function? Obes Rev. 2016;17(9):795–809. doi: 10.1111/obr.12429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Douglass AM, et al. Central amygdala circuits modulate food consumption through a positive-valence mechanism. Nat Neurosci. 2017;20(10):1384–1394. doi: 10.1038/nn.4623. [DOI] [PubMed] [Google Scholar]
- 51.Land BB, et al. Medial prefrontal D1 dopamine neurons control food intake. Nat Neurosci. 2014;17(2):248–253. doi: 10.1038/nn.3625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ip CK, et al. Amygdala NPY Circuits Promote the Development of Accelerated Obesity under Chronic Stress Conditions. Cell Metab. 2019;30(1):111–128.e6. doi: 10.1016/j.cmet.2019.04.001. [DOI] [PubMed] [Google Scholar]
- 53.Cai H, Haubensak W, Anthony TE, Anderson DJ. Central amygdala PKC-δ(+) neurons mediate the influence of multiple anorexigenic signals. Nat Neurosci. 2014;17(9):1240–1248. doi: 10.1038/nn.3767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Almeida-Suhett CP, Graham A, Chen Y, Deuster P. Behavioral changes in male mice fed a high-fat diet are associated with IL-1β expression in specific brain regions. Physiol Behav. 2017;169:130–140. doi: 10.1016/j.physbeh.2016.11.016. [DOI] [PubMed] [Google Scholar]
- 55.Castro G, et al. Diet-induced obesity induces endoplasmic reticulum stress and insulin resistance in the amygdala of rats. FEBS Open Bio. 2013;3:443–449. doi: 10.1016/j.fob.2013.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Feng X, et al. IL1R1 is required for celastrol’s leptin-sensitization and antiobesity effects. Nat Med. 2019;25(4):575–582. doi: 10.1038/s41591-019-0358-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.García-Cáceres C, et al. Role of astrocytes, microglia, and tanycytes in brain control of systemic metabolism. Nat Neurosci. 2019;22(1):7–14. doi: 10.1038/s41593-018-0286-y. [DOI] [PubMed] [Google Scholar]
- 58.Hunt KF, et al. Differences in Regional Brain Responses to Food Ingestion After Roux-en-Y Gastric Bypass and the Role of Gut Peptides: A Neuroimaging Study. Diabetes Care. 2016;39(10):1787–1795. doi: 10.2337/dc15-2721. [DOI] [PubMed] [Google Scholar]
- 59.van de Sande-Lee S, et al. Partial reversibility of hypothalamic dysfunction and changes in brain activity after body mass reduction in obese subjects. Diabetes. 2011;60(6):1699–1704. doi: 10.2337/db10-1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Scholtz S, et al. Obese patients after gastric bypass surgery have lower brain-hedonic responses to food than after gastric banding. Gut. 2014;63(6):891–902. doi: 10.1136/gutjnl-2013-305008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Albaugh VL, Banan B, Ajouz H, Abumrad NN, Flynn CR. Bile acids and bariatric surgery. Mol Aspects Med. 2017;56:75–89. doi: 10.1016/j.mam.2017.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gao Y, et al. Hormones and diet, but not body weight, control hypothalamic microglial activity. Glia. 2014;62(1):17–25. doi: 10.1002/glia.22580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.le Roux CW, et al. Gut hormone profiles following bariatric surgery favor an anorectic state, facilitate weight loss, and improve metabolic parameters. Ann Surg. 2006;243(1):108–114. doi: 10.1097/01.sla.0000183349.16877.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhan J, et al. Diffusion Basis Spectrum and Diffusion Tensor Imaging Detect Hippocampal Inflammation and Dendritic Injury in a Virus-Induced Mouse Model of Epilepsy. Front Neurosci. 2018;12:77. doi: 10.3389/fnins.2018.00077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Logothetis NK. The neural basis of the blood-oxygen-level-dependent functional magnetic resonance imaging signal. Philos Trans R Soc Lond, B, Biol Sci. 2002;357(1424):1003–1037. doi: 10.1098/rstb.2002.1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sandiego CM, et al. Imaging robust microglial activation after lipopolysaccharide administration in humans with PET. Proc Natl Acad Sci USA. 2015;112(40):12468–12473. doi: 10.1073/pnas.1511003112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.van Haeften TW. Glucose tolerance, insulin sensitivity, and the homeostasis model assessment method. Diabetes Care. 1998;21(4):673–674. [PubMed] [Google Scholar]
- 68.Smith SM, et al. Advances in functional and structural MR image analysis and implementation as FSL. Neuroimage. 2004;23 Suppl 1:S208–S219. doi: 10.1016/j.neuroimage.2004.07.051. [DOI] [PubMed] [Google Scholar]
- 69.Oguz I, et al. DTIPrep: quality control of diffusion-weighted images. Front Neuroinform. 2014;8:4. doi: 10.3389/fninf.2014.00004. [DOI] [PMC free article] [PubMed] [Google Scholar]