

Abstract
Nitric oxide regulates BP by binding the reduced heme iron (Fe2+) in soluble guanylyl cyclase (sGC) and relaxing vascular smooth muscle cells (SMCs). We previously showed that sGC heme iron reduction (Fe3+ → Fe2+) is modulated by cytochrome b5 reductase 3 (CYB5R3). However, the in vivo role of SMC CYB5R3 in BP regulation remains elusive. Here, we generated conditional smooth muscle cell–specific Cyb5r3 KO mice (SMC CYB5R3–KO) to test if SMC CYB5R3 loss affects systemic BP in normotension and hypertension via regulation of the sGC redox state. SMC CYB5R3–KO mice exhibited a 5.84-mmHg increase in BP and impaired acetylcholine-induced vasodilation in mesenteric arteries compared with controls. To drive sGC oxidation and elevate BP, we infused mice with angiotensin II. We found that SMC CYB5R3–KO mice exhibited a 14.75-mmHg BP increase, and mesenteric arteries had diminished nitric oxide–dependent vasodilation but increased responsiveness to sGC heme-independent activator BAY 58-2667 over controls. Furthermore, acute injection of BAY 58-2667 in angiotensin II–treated SMC CYB5R3–KO mice showed greater BP reduction compared with controls. Together, these data provide the first in vivo evidence to our knowledge that SMC CYB5R3 is an sGC heme reductase in resistance arteries and provides resilience against systemic hypertension development.
Keywords: Vascular Biology
Keywords: Guanylate cyclase, Hypertension, Nitric oxide
Cytochrome b5 reductase 3 in smooth muscle cells impacts physiological blood pressure and confers resilience to systemic hypertension via modulating sGC redox state.
Introduction
In the vascular wall, nitric oxide (NO) produced by the endothelium binds soluble guanylyl cyclase (sGC) in vascular smooth muscle cells (SMCs) and converts guanosine-5′ triphosphate (GTP) to the second messenger molecule, cyclic guanosine 3′, 5′-monophosphate (cGMP) (1–3). Elevated cGMP, in turn, activates protein kinase G (PKG) leading to SMC relaxation, vasodilation, and a reduction in BP (4). A prerequisite for activation of NO-sensitive sGC is the presence of reduced heme iron (Fe2+) residing in the N-terminus of the heme-NO/O2-binding domain of the sGC-β1 subunit (1–3, 5–9). Binding of NO to reduced sGC heme elicits cleavage of the histidine-Fe2+ bond leading to subsequent activation and production of cGMP (10, 11). However, increased production of ROS such as hydrogen peroxide and peroxynitrite, as observed in cardiovascular disease, can lead to sGC heme oxidation (Fe2+ → Fe3+), rendering sGC insensitive or resistant to NO (10, 12, 13). Thus, preserving sGC heme in the reduced state, especially under oxidative stress conditions, is critical for NO-mediated regulation of vasomotor tone and BP control.
NADH cytochrome b5 reductase 3 (CYB5R3), also known as methemoglobin reductase, is a flavoprotein that has numerous enzymatic roles in cellular physiology. In RBCs, soluble CYB5R3 maintains hemoglobin in the reduced state permitting it to bind to oxygen (14, 15). Membrane-bound CYB5R3 has been shown to control elongation and desaturation of fatty acids (16, 17), cholesterol biosynthesis (18), drug metabolism (19, 20), and nutrient and oxidative stress responses (21). In endothelial cells, CYB5R3 has been found to reduce the heme of hemoglobin-α to mediate NO scavenging and impair NO diffusion from endothelial cells to SMCs at myoendothelial junctions (22, 23). Recently, we demonstrated for the first time that CYB5R3 is expressed in cultured SMCs and reduces the oxidized sGC heme (Fe3+→ Fe2+) (22, 24). Specifically, we showed that transient knockdown and pharmacological inhibition of CYB5R3 in SMCs impairs NO-mediated cGMP production by SMC in vitro and aortic relaxation via ex vivo myography (24). The in vivo role of CYB5R3 in SMCs, however, remains unknown.
Herein, we investigated whether SMC-derived CYB5R3 acts as an sGC heme reductase in vivo in the context of normotension and systemic hypertension. To test this, we generated tamoxifen-inducible, SMC-specific CYB5R3 KO mice (Cyb5r3Δ/ΔMyh11-CreERT2, hereafter referred to as SMC CYB5R3–KO). We found that SMC CYB5R3–KO mice had significantly higher mean arterial pressures (MAPs) as compared with WT controls (Cyb5r3wt/wtMyh11-CreERT2) that was further exacerbated with angiotensin II (Ang II)–induced hypertension. In addition, Ang II–treated SMC CYB5R3–KO mice show an enhanced vasodilation response to the sGC activator BAY 58-2667 compared with Ang II–treated WT mice. This finding suggests a loss of CYB5R3 in SMCs results in more oxidized or heme-deficient sGC. Combined, these data provide the first in vivo evidence that SMC CYB5R3 is an sGC heme reductase in resistance arteries and confers resilience to systemic hypertension.
Results
To determine the function of NADH CYB5R3 in SMCs in vivo, we generated Cyb5r3 floxed animals and crossed them with the previously characterized Myh11-CreERT2 mice (25) to generate tamoxifen-inducible SMC CYB5R3–KO animals (Cyb5r3Δ/ΔMyh11-CreERT2 or SMC CYB5R3–KO) (Supplemental Figure 1 and Table 1; supplemental material available online with this article; https://doi.org/10.1172/jci.insight.129183DS1). No gross morphological changes were observed as a consequence of SMC CYB5R3–KO, although they had significantly fewer circulating monocytes compared with WT mice (Table 2). We confirmed SMC-specific KO of Cyb5r3 via staining after tamoxifen-treated Cyb5r3wt/wtMyh11-CreERT2 (WT) and SMC CYB5R3–KO mice aorta and mesenteric arteries with CYB5R3. CYB5R3 levels were significantly reduced within the media areas of both the aorta and mesenteric arteries in SMC CYB5R3–KO (n = 5) compared with WT (n = 5) mice (Supplemental Figure 2, A–C). In order to determine if levels of sGC in SMCs were affected by the loss of SMC CYB5R3, the aorta and mesenteric arteries of WT and SMC CYB5R3–KO mice were stained for the beta subunit of sGC (sGC-β) (Supplemental Figure 3A). We observed no significant difference in sGC-β levels in the media of the aorta or mesenteric arteries between WT (n = 5) and SMC CYB5R3–KO (n = 4) animals (Supplemental Figure 3, B and C).
Table 1. Primers for genotyping Cyb5r3 mice.
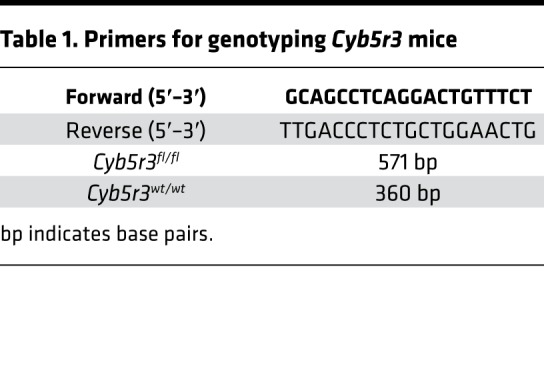
Table 2. Baseline hematology of WT and SMC CYB5R3–KO mice.

We hypothesized, given our previous work (24), that loss of SMC CYB5R3 would impair reduction of oxidized sGC (Fe2+ → Fe2+) consequently increasing vasoconstriction and BP. To test this idea, WT (n = 10) and SMC CYB5R3–KO (n = 10) mice were surgically implanted with telemetry units; BP and heart rate were continuously recorded for 24 hours (Figure 1A). We found that SMC CYB5R3–KO mice averaged a significant 5.84-mmHg higher mean arterial pressure (MAP) over the total 24-hour period (Figure 1B and Table 3). This baseline BP difference was driven by the fact that SMC CYB5R3–KO versus WT mice averaged significantly higher systolic and diastolic pressures over the total 24-hour period (Supplemental Figure 4, A and B). Consistent with this finding, SMC CYB5R3–KO mice averaged significantly lower heart rates over the total 24-hour period compared with WT mice (Figure 1C and Table 3).
Figure 1. SMC CYB5R3–KO mice have a significantly elevated MAP and reduced heart rate compared with WT mice under physiological conditions.

(A) The experimental design where WT (n = 10, gray) and SMC CYB5R3–KO (n = 10, red) mice after tamoxifen treatment were implanted with telemetry units and BP monitored continuously for 24 hours. (B) SMC CYB5R3–KO mice have a significantly increased MAP compared with WT mice. (C) SMC CYB5R3–KO mice have a significantly decreased heart rate compared with WT mice. (B and C) P values represent significant differences between WT and SMC CYB5R3–KO by 2-way repeated- measures ANOVA with *P < 0.05 by Sidak multiple comparison tests. Error bars represent ± SEM.
Table 3. Mean change between WT and SMC CYB5R3–KO telemetry measurements.

To determine the cause of the increased BP in SMC CYB5R3–KO mice, WT (n = 5–7) and SMC CYB5R3–KO (n = 7–8) mesenteric arteries were subjected to ex vivo wire myography to assess vessel reactivity (Figure 2A). Vasoconstriction with 10–7 M U46619, a thromboxane mimetic, showed no significant difference in vasoconstriction between WT and SMC CYB5R3–KO mice (Figure 2B). Next, we assessed WT versus SMC CYB5R3–KO mesenteric artery response to cumulative doses of 3 different vasodilators: acetylcholine, sodium nitroprusside (SNP), and BAY 58-2667 (sGC activator). Acetylcholine was used to assess whether the loss of SMC CYB5R3 affects endothelial cell-mediated vasodilation (26). We found that SMC CYB5R3–KO mice had a significantly impaired response to acetylcholine as compared with WT mice particularly at the 10–5 M dose (Figure 2C). To test whether NO-mediated vasodilation was disrupted in SMCs as a consequence of CYB5R3 loss, mesenteric arteries from WT and SMC CYB5R3–KO mice were given cumulative doses of SNP, a NO donor molecule that can deliver NO to reduced sGC in SMCs to induce vasodilation. We found no differences in SNP response between WT and SMC CYB5R3–KO mice (Figure 2D). In order to determine whether SMC CYB5R3 regulates the sGC state in these animals, we treated mesenteric arteries with the sGC activator BAY 58-2667. This compound activates oxidized sGC and heme-deficient sGC independent of NO in SMCs to induce cGMP production and vasodilation (4, 27). We found SMC CYB5R3–KO had a slight but significant impairment in vasodilation response to BAY 58-2667 compared with WT mice (Figure 2E). Combined, these data suggest that SMC CYB5R3 may lead to dysfunctional SMC and endothelial cross-talk under physiological conditions.
Figure 2. SMC CYB5R3–KO results in an acetylcholine-dependent impairment of vasodilation under physiological conditions.

(A) Experimental design showing that mesenteric arteries from WT (n = 5–7, gray) and SMC CYB5R3–KO (n = 7–8, red) mice were subjected to ex vivo wire myography to assess vasoreactivity. (B) There are no differences in WT and SMC CYB5R3–KO response to U46619-mediated vasoconstriction. P values determined by unpaired 2-tailed t test. Error bars are ± SEM. (C) WT mesenteric arteries had a significantly increased responsiveness to acetylcholine compared with SMC CYB5R3–KO mesenteric arteries. (D) No significant differences were seen between WT and SMC CYB5R3–KO responsiveness to the vasodilator SNP. (E) SMC CYB5R3–KO mice are less responsive to BAY 58-2667 compared with WT. (C–E) P values represent statistical differences between WT and SMC CYB5R3–KO by 2-way ANOVA with *P < 0.05 by post hoc Sidak multiple comparison tests. Error bars are ± SEM.
Next, we wanted to assess whether SMC CYB5R3 was important for BP regulation in the context of systemic hypertension. Hypertension and increased Ang II signaling has been shown to result in oxidation or loss of the sGC heme (28). WT (n = 8) and SMC CYB5R3–KO mice (n = 8) were implanted with osmotic mini-pumps delivering 750 ng kg–1 min–1 Ang II, and BP and heart rate changes were measured continuously for 14 days (Figure 3A). Ang II–induced hypertension resulted in a significant increase in mean arterial, systolic, and diastolic pressures and decreased heart rate in SMC CYB5R3–KO animals compared with WT animals during the first 7 days of Ang II treatment (Figure 3, B and C, and Supplemental Figure 5, A and B). Days 6–7 of Ang II treatment appeared to represent the culmination of the increased hypertensive state of SMC CYB5R3–KO mice compared with WT (Figure 3, D and E, Supplemental Figure 5, C and D). Days 6–7 of Ang II treatment further exacerbated the difference in the average MAP between WT and SMC CYB5R3–KO mice from the 5.84-mmHg difference in untreated animals to a peak 14.75-mmHg difference (Table 3). Notably, WT heart rate declined between days 8–9 of Ang II, while SMC CYB5R3–KO mice maintained a relatively consistent heart rate throughout the study (Figure 3C). Combined, the data indicate that SMC CYB5R3–KO mice, as compared with WT mice, are more susceptible to hypertensive agonists and likely have an impaired ability to mitigate Ang II effects.
Figure 3. SMC CYB5R3–KO mice have an exacerbated hypertensive response to Ang II compared with WT mice.

(A) Experimental design showing WT (n = 8, gray) and SMC CYB5R3–KO (n = 8, red) mice were implanted with telemetry units then subsequently with osmotic pumps delivering 750 ng kg–1 min–1 Ang II. BP was measured continuously 24 hours prior to and for the duration of Ang II treatment. (B) MAP (mmHg) and (C) heart rate (bpm) averaged and plotted per day for the course of 14 days shows SMC CYB5R3–KO mice have an increased MAP and decreased heart rate as compared with WT mice particularly during the first 7 days of Ang II treatment. Shaded regions highlight days 6–7 of Ang II where peak differences were observed between WT and SMC CYB5R3–KO. (D) The hourly MAP (mmHg) recording and (E) heart rate for the shaded regions in B and C for the 24 hours between days 6–7 of Ang II treatment. (A–E) The P value represents a difference across genotype by 2-way repeated-measures ANOVA with *P values < 0.05 as determined by post hoc Sidak multiple comparison tests. (B–E) Error bars are ± SEM.
After the 14 days of Ang II treatment, mesenteric arteries from WT (n = 9–12) and SMC CYB5R3–KO (n = 11–12) mice were subjected to ex vivo wire myography and treated with vasoconstrictor U46619 and cumulative doses of vasodilators (acetylcholine, SNP, and BAY 58-2667) to assess the impact of systemic hypertension on resistance artery function (Figure 4A). U46619-mediated vasoconstriction was significantly increased in Ang II–treated SMC CYB5R3–KO mice compared with WT animals (Figure 4B). Additionally, SMC CYB5R3–KO mesenteric arteries showed an impaired acetylcholine-mediated vasodilation compared with WT controls (Figure 4C). Cumulative treatment with NO-donor SNP resulted in a significant impairment of SMC CYB5R3–KO vasodilation compared with those in the WT group (Figure 4D). WT and SMC CYB5R3–KO mesenteric arteries were also treated with increasing concentrations of oxidized and heme-deficient sGC targeted vasodilator BAY 58-2667 where it was found that SMC CYB5R3–KO mice were significantly more responsive to BAY 58-2667– induced vasodilation than WT controls (Figure 4E). This finding indicates that loss of SMC CYB5R3 in Ang II–treated mesenteric arteries results in a greater shift in the sGC pool to a more oxidized or heme-deficient state as compared with WT mesenteric arteries. To further test this finding, we looked at cGMP pathway activation by quantifying downstream protein kinase G–specific (PKG-specific) phosphorylation of serine 239 on vasodilator-stimulator phosphoprotein (pVASP239). Mesenteric arteries isolated from 14-day Ang II–treated SMC CYB5R3–KO mice and given NO-donor diethylamine nonoate (DEA-NONOate) showed decreased levels of serine 239 phosphorylation of vasodilator-stimulated phosphoprotein (pVASP239) compared with WT mice (Supplemental Figure 6). To assess whether vasodilation in Ang II–treated conduit arteries was similarly affected by SMC CYB5R3 loss, we performed these same ex vivo myography experiments in aortas. We found no differences in phenylephrine-mediated vasoconstriction, or vasodilation via SNP or BAY 58-2667 between WT and SMC CYB5R3–KO aortas (Supplemental Figure 7, A, B, D, and E) However, WT hypertensive aorta and mesenteric arteries were found to be significantly more responsive to acetylcholine as compared with SMC CYB5R3–KO (Figure 4C and Supplemental Figure 7C). Taken together, these data provide evidence that SMC-derived CYB5R3 acts as a sGC reductase and affects NO-mediated vasodilation in hypertensive resistance arteries.
Figure 4. SMC CYB5R3 acts as an sGC heme reductase in the context of Ang II–induced systemic hypertension.

(A) Ex vivo wire myography experiments on mesenteric arteries from WT (n = 9–12, gray) and SMC CYB5R3–KO mice (n = 10–12, red) after 14 days Ang II (750 ng kg–1 min–1) treatment. (B) SMC CYB5R3–KO mice have increased U46619-mediated vasoconstriction than WT mice. P values determined by 2-tailed, unpaired t test. (C) SMC CYB5R3–KO mice are significantly less responsive to acetylcholine than WT mice. (D) WT mice are more responsive to vasodilator SNP than SMC CYB5R3–KO mice. (E) SMC CYB5R3–KO mice are more responsive than WT mice to BAY 58-2667. (C–E) P values represent statistical differences between WT and SMC CYB5R3–KO by 2-way ANOVA with *P < 0.05 representing significance by post hoc Sidak multiple comparison tests. Error bars are ± SEM.
To determine whether loss of SMC CYB5R3 had secondary effects on vessel and cardiac fibrosis or remodeling, aorta, mesenteric arteries, and heart tissue treated with Ang II for 7, 14, and 28 days were stained with Masson’s trichrome stain. We observed no gross differences in fibrosis of any of the tissues at any point of Ang II treatment (Supplemental Figure 8A, Supplemental Figure 9A, and Supplemental Figure 10A). We also found no significant differences in aorta or mesenteric artery medial area between WT and SMC CYB5R3–KO at any point of Ang II treatment (Supplemental Figure 8B and Supplemental Figure 9B). Similarly, no differences in cardiac hypertrophy or intracardiac fibrosis were observed between groups at any point of Ang II treatment (Supplemental Figure 10B).
To test whether loss of CYB5R3 in SMC can affect sGC heme redox state and consequently BP in vivo, hypertensive WT and SMC CYB5R3–KO mice were injected i.p. at 1 dose per day with increasing concentrations of 10–7–10–3 M BAY 58-2667 (Figure 5A). We reasoned these studies would extend our ex vivo myography vasodilation experiments in vivo for assessment of BAY 58-2667 influence on BP between WT and SMC CYB5R3–KO mice. Mice were anesthetized briefly via isoflurane when injected i.p. to minimize stress-induced BP changes. The first dose at 10–7 M was given to both WT and SMC CYB5R3–KO mice on day 20 of Ang II treatment. At the 10–4 M dose of BAY 58-2667 on day 23 of Ang II treatment (the equivalent of 0.04 mg/kg BAY 58-2667), we observed a decrease in MAP in both WT and SMC CYB5R3–KO mice between 15–30 minutes after injection, although no differences in response to BAY 58-2667 were observed between groups for up to 2 hours after injection at this dose (Figure 5B). Similarly, no significant differences in systolic or diastolic pressures were observed between WT and SMC CYB5R3–KO mice at this dose of BAY 58-2667 (Supplemental Figure 11, A and B). Finally, WT mice had an increased heart rate 15 minutes after injection of 0.04 mg/kg BAY58-2667 (Figure 5C).
Figure 5. Ang II–treated SMC CYB5R3–KO mice are more responsive to acute BAY 58-2667–induced vasodilation than WT controls.

(A) Experimental design with Ang II–treated WT (n = 4, gray) and SMC CYB5R3–KO mice (n = 4, red) receiving i.p. injections of BAY 58-2667 (1 dose/day) and changes in BP measured. (B) Acute injection (time: 0 minutes, purple arrow) with 0.04 mg/kg BAY 58-2667 resulted in no significant differences in change in MAP between WT and SMC CYB5R3–KO mice. (C) Injection of 0.04 mg/kg BAY 58-2667 (time: 0 minutes, purple arrow) results in a significant increase in change in heart rate in WT mice over SMC CYB5R3–KO at 15 minutes after injection. (D) The higher 0.4 mg/kg BAY 58-2667 dose (time: 0 minutes, purple arrow) resulted in a significant reduction in the change in MAP in SMC CYB5R3–KO mice compared with WT mice after injection, indicating SMC CYB5R3–KO mice are more sensitive to BAY 58-2667 BP-lowering effects. (E) Both WT and SMC CYB5R3–KO mice had an elevated change in heart rate after 0.4 mg/kg BAY 58-2667 injection (time: 0 minutes, purple arrow), although no differences in heart rate occurred between groups. (B–E) P value represents significant difference between WT and SMC CYB5R3–KO by 2-way repeated measures ANOVA with P < 0.05 by post hoc Sidak multiple comparison tests. Error bars are ± SEM.
Importantly, the subsequent higher 10–3 M dose, which is 0.4 mg/kg BAY 58-2667 given the following day resulted in a significant and drastic decrease in change in mean arterial BP in SMC CYB5R3–KO (n = 4) compared to WT mice (n = 4) (Figure 5D). SMC CYB5R3–KO mice also showed increased vasodilatory response to the 0.4 mg/kg BAY 58-2667 dose, as the change in systolic and diastolic pressures was significantly decreased after injection compared with WT controls (Supplemental Figure 11, C and D). Although the 0.4 mg/kg BAY 58-2667 dose increased the change in heart rate in both groups, no significant differences in change in heart rate were observed between WT and SMC CYB5R3–KO mice (Figure 5E). This finding suggests that the enhanced BAY 58-2667 blood pressure–lowering effects in Ang II–treated SMC CYB5R3–KO mice are the result of increased peripheral vascular resistance in these mice as compared with WT mice. Taken together, these data provide evidence that in Ang II–induced hypertensive conditions, CYB5R3 is the sGC reductase in SMC modulating the sGC heme state, thereby influencing BP and vessel function (Figure 6).
Figure 6. SMC CYB5R3 acts as an sGC reductase in systemic hypertension.

In normotension, NO binds to reduced heme sGC in SMC to induce vasodilation. Ang II causes systemic hypertension as well as oxidation and loss of the sGC heme rendering it insensitive to NO. CYB5R3 confers resilience to systemic hypertension by reducing the oxidized sGC heme back to its reduced state.
Discussion
The NO-sGC-cGMP pathway is well established in regulating systemic BP. We previously discovered in cultured SMCs that CYB5R3 restores NO-sGC-cGMP vasodilation signaling by reducing the oxidized sGC heme (Fe3+→Fe2+) (24, 29). However, the function of CYB5R3 in SMCs in vivo and its potential impact in BP regulation has yet to be explored. Using conditional, SMC-specific CYB5R3 KO mice, we provide the first evidence that SMC CYB5R3 is a sGC reductase and that SMC CYB5R3 confers protection against Ang II–induced systemic hypertension via regulation of the sGC redox state.
Under normal physiological conditions, we observed SMC CYB5R3–KO resulted in a significant elevation in physiological BP as compared with WT mice. Moreover, ex vivo myography studies on mesenteric arteries provide evidence that the elevation in physiological BP in SMC CYB5R3–KO mice involves dysfunction in endothelial cell acetylcholine-mediated signaling. Acetylcholine can vasodilate resistance arteries, in part, through the activation of endothelial nitric oxide synthase leading to NO synthesis, NO diffusion to SMCs, and then activation of reduced sGC-mediated signaling (26, 30). Independent of NO-sGC signaling, acetylcholine can induce vasodilation via activation of other endothelium-derived hyperpolarizing factor pathways involving lipooxygenases, cyclooxygenases, and cytochrome p450 enzymes (26, 31). In particular, cytochrome p450 enzymes can and may be regulated by CYB5R3 activity, adding additional complexity to the system (19, 32, 33). It is thus possible that SMC CYB5R3 may have an as yet unidentified role in one of these other mechanisms of acetylcholine action for vasodilation. Therefore, a further in-depth analysis of the complex and multifaceted acetylcholine-dependent pathway that is pertinent for SMC CYB5R3 regulation of physiological BP is warranted.
Most importantly, we provide evidence that CYB5R3 in SMCs acts as an sGC reductase in resistance arteries in the context of Ang II–induced systemic hypertension. We show Ang II–treated SMC CYB5R3–KO mice given sGC activator BAY 58-2667, which enhances cGMP production through acting on heme-deficient and oxidized sGC, resulted in a significant decrease in MAP in vivo and increased resistance artery vasodilation in ex vivo myography experiments compared with WT mice. In addition, NO-dependent cGMP production and vasodilation were blunted in Ang II SMC CYB5R3–KO mice compared with WT mice. Of interest, Ang II–treated WT and SMC CYB5R3–KO aortas did not differ in response to SNP or BAY 58-2667 but did show a blunted acetylcholine response in SMC CYB5R3–KO mice compared with WT controls. These data again highlight the importance of CYB5R3 in peripheral vascular resistance and a potential role for SMC CYB5R3 in acetylcholine-dependent vasodilation. Ang II–treated SMC CYB5R3–KO mice were also more sensitive to Ang II treatment with further enhanced elevation in MAP detected compared with WT mice for the first week of Ang II treatment. Combined, this data suggests that SMC CYB5R3, particularly in resistance arteries, confers protection from systemic hypertension. This finding is consistent with work published by Martin-Montalvo et al. showing that CYB5R3 overexpression confers protection from oxidative stress–induced lipid peroxidation and enhanced survival (17). During the second week of Ang II treatment, however, no differences in MAP or heart rate were detected between groups. Interestingly, at day 8 of Ang II treatment, the heart rate of the WT mice decreased to nearly the level of the SMC CYB5R3–KO group. We hypothesize these observations indicate a pressure natriuresis event and/or change in sympathetic drive that occurred in Ang II–treated WT mice that is absent in the SMC CYB5R3–KO mice.
We had previously published that spontaneously hypertensive rats treated with a CYB5R3 inhibitor acutely resulted in a decrease in MAP compared with vehicle-treated controls (29). It is worth considering these differences in observations are likely due to the net effect of pharmacological inhibition of CYB5R3 on both endothelial cells and SMCs. We previously found that pharmacological inhibition of CYB5R3 in aortas resulted in reduced acetylcholine and SNP-induced vasodilation providing evidence the CYB5R3 inhibitor likely has an impact on NO signaling in both SMCs and endothelial cells (24). In endothelial cells, CYB5R3 reduces ferric hemoglobin (methemoglobin)-α to its ferrous hemoglobin-α state (22, 23, 29). The oxidation of ferrous hemoglobin to ferric hemoglobin occurs very rapidly in the presence of oxygen, however, due to a NO dioxygenation reaction that occurs at a rate of 6–8 × 10–7 M–1 s–1 (22, 23, 29, 34). In comparison, the oxidation of ferrous sGC to ferric sGC, which does not involve oxygen or a NO dioxygenation reaction, likely occurs much more slowly. Taken together, it is plausible that the short-term, acute Ang II and CYB5R3 inhibitor treatment used in our previous studies in hypertensive rats was not given long enough to affect sGC, and therefore CYB5R3 action on hemoglobin-α in endothelial cells took precedence and caused the previously observed increased vasodilation response (9, 10).
In vitro, CYB5R3 has been shown to be the reductase for several hemoproteins including hemoglobin-α, cytoglobin, and as evidenced herein sGC (22–24, 29, 35, 36). Recently, Liu et al. showed that loss of CYB5R3 in cultured SMCs resulted in impaired NO scavenging (35, 36). This finding was attributed to CYB5R3 action on cytoglobin, which is found in SMCs and via NO dioxygenase reaction transitions from a ferrous to ferric heme state (35, 36). In addition, global genetic KO of cytoglobin was shown to result in systemic hypotension in untreated and Ang II–treated mice, the opposite of our observations in our SMC CYB5R3–KO mice (36). Combined with the fact that we observed no differences in SNP vasoreactivity between WT and SMC CYB5R3–KO mesenteric arteries under physiological conditions, our data suggest CYB5R3 likely does not act on cytoglobin in SMCs under physiological conditions in vivo.
Ang II treatment has been shown to increase NADPH oxidase-dependent production of superoxide at the plasma membrane and contributes to mitochondrial ROS generation (37). In SMCs specifically, numerous studies have provided evidence that Ang II superoxide production is NADPH oxidase 1 (NOX1) dependent and that NOX1 is localized to caveolae at the plasma membrane (38–45). Interestingly, work in cardiac hypertrophy models has shown that cytosolic sGC-β1 can be oxidized while sGC-β1 sequestered in caveolae microdomains at the plasma membrane is protected from oxidation (46, 47). Consistent with these data, Zabel et al. have shown that sGC-β1 localized to the membrane of the rat heart is more sensitive to NO signaling (48). We previously published that in sGC oxidant 1h-[1,2,4]oxadiazolo-[4,3-a]quinoxaline-1-one–treated SMCs, there are CYB5R3-sGC interactions occurring mostly within the cytoplasm (24). Given that CYB5R3 is present in the plasma, mitochondrial, and endoplasmic reticulum membranes, an intriguing future study would be to determine if SMC CYB5R3 subcellular localization is important for regulation of NO-sGC signaling during oxidative stress. Moreover, the fact that Ang II SMC CYB5R3–KO mice still responded to SNPs suggests it may be possible that an unidentified reductase may also contribute to activation of NO-sGC vasodilation in SMCs in the context of systemic hypertension.
Combined, our data provide substantive evidence that CYB5R3 in SMCs regulates the NO-sGC-cGMP pathway by acting as an sGC reductase during systemic hypertension. It is important to recognize, however, that CYB5R3 has been shown to have varied roles in cellular processes including cellular respiration and metabolism (17, 21, 32, 33, 49–51), oxygen delivery (14, 15), and NO-sGC-cGMP signaling (22–24, 29, 35, 36). Additionally, data from our lab and others have shown that CYB5R3 reductase activity in hepatocytes (17, 21, 49), RBCs (14, 15), endothelial cells (22, 23), and SMCs (24) have the capacity to target different hemoproteins and redox molecules (e.g., hemoglobin-α, sGC, and coenzyme Q) that then differentially modulate cell signaling and functional output. A crucial future direction will be to determine how the differential use of CYB5R3 reductase activity in various cells and tissues translates to that of human vascular physiology and cardiovascular diseases. This route will be of utmost importance when considering personalized treatment of patients with and without genetic variants for CYB5R3 and the possibility that they might have different responsiveness to NO, sGC stimulators, and sGC activators.
Methods
Generation of Cyb5r3fl/fl and SMC-specific CYB5R3 KO mice (Cyb5r3 Myh11-CreERT2).
A Cyb5r3 gene-targeting plasmid was generated by the University of California Davis Knockout Mouse Project Repository (Cyb5r3tm1a(KOMP)Wtsi, CSD29891, reproduced and simplified in Supplemental Figure 1A and refs. 52, 53). Exon 3 of Cyb5r3 was identified via computational model to be a “critical exon” where excision of exon 3 would result in a frame-shift mutation and nonsense–mediated decay of the Cyb5r3 transcript conferring loss of CYB5R3 (52). This gene-targeting vector was transduced into JM8.N4 embryonic stem cells (54), which are an agouti C57BL/6N line and selected for neomycin resistance before being implanted into pseudo-pregnant C57BL/6J mice at the University of Alabama. C57BL/6NJ chimeras were then crossed to C57BL/6J FLPase mice to target FRT sites and remove the embryonic stem cell selection cassette. The resulting Cyb5r3 floxed female mice were then crossed to male Myh11-CreERT2 mice (019079, C57BL/6, Jackson Laboratory) (25) to generate tamoxifen-inducible SMC-specific Cyb5r3 floxed mice: Cyb5r3fl/flMyh11-CreERT2 (Supplemental Figure 1, B and C). Mice were backcrossed for 10 generations. Given that the Myh11-CreERT2 is present on the Y chromosome, only male mice were used in our studies. Intraperitoneal injections of 1 mg per day per mouse of tamoxifen (MilliporeSigma, T-5648) for 10 days into Cyb5r3fl/flMyh11-CreERT2 mice results in activation of Cre-loxp– mediated excision of exon 3 and loss of CYB5R3 expression solely in Myh11–expressing cells (Supplemental Figure 1C). Tamoxifen treatment therefore generates SMC-specific Cyb5r3 KO mice (Cyb5r3Δ /ΔMyh11-CreERT2 or SMC CYB5R3–KO, Supplemental Figure 1C and Table 1). Cyb5r3wt/wtMyh11-CreERT2 or WT mice from this breeding scheme were used as control animals. Both WT and SMC CYB5R3–KO mice were subjected to the same tamoxifen dose regimen prior to any analysis described herein. Mice were between 12–24 weeks at the completion of experiments. The exception to this is 1 SMC CYB5R3–KO mouse in the 7- day Ang II trichrome analysis and 2 SMC CYB5R3–KO mice in the 28-day Ang II trichome analysis, which includes mice at 40 weeks of age at the completion of the experiment.
Animal harvesting for immunohistochemical analysis.
Mice were euthanized by CO2 asphyxiation and aorta, heart, and mesenteric arteries were removed. Tissues were post-fixed overnight in 4% paraformaldehyde (sc-281692, Santa Cruz Biotechnology Inc.), paraffin embedded, and sectioned at a 7-μM thickness. Artery sections were deparaffinized with xylene and rehydrated by decreasing 100%–70% ethanol followed by distilled H2O. Sections underwent heat-mediated citric acid–based antigen retrieval (H-3300, Vector Laboratories) for 20 minutes and then cooled for 30 minutes at 4°C. Subsequently, sections were blocked with PBS containing 10% horse serum (MilliporeSigma, H1270) for 1 hour at room temperature. Primary antibodies for CYB5R3 (10894-1-AP, Proteintech, 1:100) or sGC-β (160897, Cayman Chemicals, 1:100) in conjunction with platelet and endothelial cell adhesion molecule 1 (PECAM1, sc-1506, Santa Cruz Biotechnology Inc., 1:250) were diluted in PBS with 10% horse serum and incubated on sections overnight at 4°C in a humidity chamber. One section per slide was stained with both rabbit (I-1000, Vector Laboratories) and goat IgG controls (I-5000, Vector Laboratories) matching the concentration of primary antibodies. Sections were then washed in PBS and incubated with the preconjugated smooth muscle α-actin–FITC (ACTA2-FITC; F3777, MilliporeSigma, clone 1A4, 1:500), DAPI (D3571, Thermo Fisher Scientific, 1:100), and secondary antibodies donkey anti-rabbit Alexa Fluor 596 (A21207, Invitrogen, 1:250), and donkey anti-goat Alexa Fluor 647 (A21447, Invitrogen, 1:250) for 1 hour at room temperature in a humidity chamber. Sections were then washed with PBS and cover-slipped with Prolong Gold Antifade with DAPI reagent (P36931, Invitrogen). Arteries were imaged using a Nikon A1 Confocal Laser microscope at ×40 magnification with 1096 x 1096 resolution at the Center for Biological Imaging at the University of Pittsburgh. Z-stacks in 1-μM increments were taken for both stained and IgG control sections. Representative images are the maximum-intensity projections of the Z-stack images (Supplemental Figure 2A and Supplemental Figure 3A). In ImageJ, regions of interest were drawn for the maximum-intensity projection of ACTA2 (SMCs), which represents the vessel media and superimposed onto the maximum-intensity projection for CYB5R3 and sGC. The raw integrated intensity per area in ImageJ was then quantified to represent the amount of CYB5R3 or sGC staining in the media (Supplemental Figure 2, B and C, and Supplemental Figure 3, B and C).
Tissues were also stained with a Masson’s Trichrome Kit (Thermo Fisher Scientific, 87019) according to the manufacturer’s protocol to assess tissue fibrosis. For aorta and mesenteric arteries, images were taken on the Olympus Provis 1 microscope at ×10 and ×40 objectives, respectively, and the medial area was quantified using ImagePro Plus V9 software (Supplemental Figures 7 and 8). For heart tissues, stained sections were imaged using Tissuegnostics Microscope at ×20 objective and stitched using TissueQuest Analysis Software (Snake Stitch, 8% overlap). The total tissue area was determined by converting individual images into binary images and measuring the area using Fiji software (Supplemental Figure 10).
Telemetry.
Mice were anesthetized with isoflurane, and telemetry units (HDX-10, Data Sciences International [DSI]) were surgically implanted in the right common carotid artery. Mice were allowed to recover for 2 weeks from surgery before telemetry units were turned on. BP was then measured continuously between 24–48 hours to assess baseline physiological BP (Figure 1 and Supplemental Figure 4). Measurements of mean arterial BP, heart rate, systolic pressure, and diastolic pressure were recorded by Ponemah Software (DSI) and exported into Microsoft Excel (version 16.16.6) for analysis. For Ang II studies (Figures 3 and 5 and Supplemental Figures 5 and 6), mice were then anesthetized with isoflurane, and osmotic 28-day mini-pumps (Model 1004, Alzet) were inserted s.c. between mice shoulders. Ang II was delivered from the pumps at a rate of 750 ng kg–1 min–1. BP was monitored continuously immediately after osmotic mini-pump insertion and for the duration of the experiments.
Mesenteric artery and aorta ex vivo wire myography.
Myography experiments were conducted similarly to those previously published by our lab (24). In summary, mice were euthanized by CO2 asphyxiation and mesenteric arteries were cleaned, removed, and placed in a physiological salt solution (PSS) containing 0.026-mM EDTA, 119-mM NaCl, 5.5-mM d-glucose, 25-mM NaHCO3, 4.7-mM KCl, 1.17-mM MgSO4, 1.18-mM KH2PO4, and 2.5-mM CaCl2. PSS was then set to a of pH 7.4 and bubbled with 95% O2 5% CO2 at 37°C. Vessels were placed on the wire myograph (Multiple Myograph Model 620 M, Danish Myotechnology) using a 25-μm wire and allowed to rest for 30 minutes in PSS. After rest, fresh PSS was added for an additional 10 minutes. The mesenteric arteries were then stretched to a tension equivalent to a physiological 80 mmHg of pressure. For aortic rings, vessels were gradually stretched to a tension of 500 mg. Potassium (60 mM) in PSS was then added to vessels for 5 minutes to constrict the vessels and determine vessel viability. Vessels are then washed 3 times with PSS and allowed to rest for 30 minutes. To test for vessel viability, untreated mice mesenteric arteries and Ang II–treated aortas were treated for 5 minutes with 60-mM potassium in PSS, while Ang II–treated mesenteric arteries were given increasing doses of constrictor U46619 (10–7–10–6 M, 16450, Cayman Chemicals) at 5-minute intervals until vessels reached maximal constriction. The vessels were then washed 3 times with PSS and allowed to rest for an additional 30 minutes. Untreated and Ang II–treated mesenteric arteries were then constricted with 10–7–10–6 M U46619, and Ang II–treated aortas were given 10–6–10–5 M phenylephrine in 5-minute intervals to induce maximal constriction before vasodilator treatment. After vessels reached maximal constriction, aorta and mesenteric arteries were given cumulative increasing doses of the following vasodilator drugs in 5-minute increments: acetylcholine (10–8–10–4 M, A6625, Sigma-Aldrich), (SNP, 10–9–10–4 M, 71778, Sigma-Aldrich), or BAY 58-2667 (10–12–10–6 M). Vessels were then treated with Ca2+–free PSS containing 100-μM SNP to determine maximal dilation. Myography data were recorded on Lab Chart Software (AD Instruments). The data were normalized to the change in maximal constriction by U46619 or phenylephrine to maximal dilation in Ca2+–free PSS to determine the reported percentage relaxation of the vessels (Figures 2 and 4, and Supplemental Figure 7).
cGMP pathway activation quantification.
Intracellular cGMP production activates PKG which phosphorylates serine 239 on pVASP (pVASP239). pVASP239 can serve as a surrogate indicator of cGMP activation (55). Mesenteric artery cascades from 2 separate mice of the same genotype were isolated, pooled, and then pooled material was split equally into 2 separate tubes each containing 1 ml of a 1-part Lonza Smooth Muscle Growth Medium-2 with SmGM-2 SingleQuot kit containing growth factors and 5% FBS (Lonza, CC-3182) to 9 parts solely Lonza Smooth Muscle Growth Medium-2 and were put into a 5% CO2 incubator at 37°C. For Supplemental Figure 6, the n = 3 reported therefore represents the equivalent of 6 WT and 6 SMC CYB5R3–KO mice total used for the experiment. Vessels from both tubes were then treated with 100-μM 3-isobutyl-1 methylxanthine for 15 minutes to inhibit phosphodiesterase activity. Vessels are subsequently either left untreated or treated with 100-μM NO-donor diethylamine NONOate (DEA-NONOate) for 30 minutes. Vessels are then washed 3 times with PBS and then placed in 1 X Cell Lysis buffer (Cell Signaling Technology, 9803) containing protease (MilliporeSigma, P8340) and phosphatase (MilliporeSigma, P5726) inhibitors and lysed. Tissue protein levels were then quantified. For analysis of protein phosphorylation levels by Western blot, lysates were boiled and run on 4%–12% NuPAGE Bis-Tris gels (Invitrogen, NP0335BOX). Proteins were then transferred to a nitrocellulose membrane (LiCOR, 926-31092) and blocked for 1 hour in 1 part LiCOR Odyssey blocking buffer (LiCOR Biosciences, 927-40000) to 1 part PBS at room temperature. Membranes were then probed for pVASP239 (Cell Signaling Technology, 3114S) and β-actin (Santa Cruz Biotechnology Inc., sc-47778) overnight in 1 part LiCOR Odyssey blocking buffer to 1 part PBS with 0.0625% tween 20 at 4°C. Membranes were washed with PBS and 0.05% tween 20 and then incubated with secondary antibodies (1:15,000; LiCOR IRDye, 680RD, 926-68072; LiCOR IRDye 800CW, 926-32213) for 1 hour at room temperature. Membranes were washed and imaged using a LiCOR Odyssey with bands quantified using Image Studio Lite software.
Acute i.p. injections of BAY 58-2667 in vivo.
Following telemetry implantation and Ang II treatment (750 ng kg–1 min–1) via osmotic mini-pump, WT (n = 4) and SMC CYB5R3–KO mice (n = 4) were briefly anesthetized (< 2 min per mouse) with isoflurane and injected i.p. at 1 dose per day of increasing concentrations of 10–7–10–3 M BAY 58-2667. With this design, we rationalized we could mimic our ex vivo myography experiments in vivo. Mice were briefly anesthetized with isoflurane for each i.p. injection to minimize stress-induced variation in BP in the animals. Injections began at day 20 of Ang II treatment with the 10–7 M BAY 58-2667 dose given. As BP was measured continuously, we were able to assess the impact of BAY 58-2667 injection at each dose per day. We found that the 10–4 M and subsequent 10–3 M doses of BAY 58-2667 at days 23 and 24 of Ang II treatment, respectively led to a sufficient reduction in MAP and increase in heart rate at 30 minutes after injection in WT mice suggesting the drug was efficacious at these doses (Figure 5 and Supplemental Figure 11). We then extrapolated that these i.p. 10–4 M and 10–3 M doses of BAY 58-2667 were equivalent to 0.04 and 0.4 mg/kg BAY 58-2667 in our mice. The time to briefly anesthetize and inject the 8 animals i.p. (4 WT and 4 SMC CYB5R3–KO) used in this study averaged 15 minutes. To analyze if BP and heart rate changed as a consequence of acute doses of BAY 58-2667, BP and heart rate parameters were averaged across 15-minute intervals and normalized to the readings taken 15 minutes prior to the start of injection (Figure 5 and Supplemental Figure 11).
Statistics.
Statistical analyses were done using Graphpad Prism Software version 7.0d. For telemetry experiments, data normality was assessed and analyzed by 2-way repeated measures ANOVA with post hoc Sidak’s multiple comparisons test (Figure 1, B and C; Figure 3, B–E; Figure 5, B–E; Supplemental Figure 4, A and B; Supplemental Figure 5, A–D; and Supplemental Figure 6, A–D). For ex vivo myography, 2-way ANOVA analyses were conducted for vasodilator dose-response treatments with P values representing significance across genotype and *P < 0.05 representing significance by post hoc Sidak multiple comparison test (Figure 2, C–E; Figure 4, C–E; and Supplemental Figure 7, C–E). For the vasoconstrictor responses, immunohistochemical staining, and Western blot analyses, data normality was assessed by Shapiro-Wilk test and analyzed by unpaired 2-tailed t test, $unpaired 2-tailed t test with Welch’s correction, or ^Mann-Whitney U test based on normality results (Figure 2B; Figure 4B; Supplemental Figure 2, B and C; Supplemental Figure 3, B and C; Supplemental Figure 6B; Supplemental Figure 8B; Supplemental Figure 9B; and Supplemental Figure 10B).
Study approval.
All animal studies were approved by the University of Pittsburgh Institutional Animal Care and Use Committee (Protocol no. 17019505).
Author contributions
BGD, SAH, and ACS conceived and designed the experiments. BGD prepared tissues for histology and performed immunofluorescence staining and analysis. SAH performed ex vivo myography and telemetry experiments with both BGD and SAH analyzing data from these experiments. HMS and NH imaged and analyzed trichrome-stained arteries and heart, respectively under BGD’s direction. MPM, SAH, and BGD harvested mesenteric arteries and conducted pVASP239 analysis. BGD, SAH, IM, DF, PS, and ACS interpreted results of experiments. BGD and ACS prepared figures and wrote the manuscript. SAH and ACS edited the manuscript. BGD, SAH, HMS, MPM, NH, IM, DF, PS, and ACS approved the final version of the manuscript for submission.
Supplementary Material
Acknowledgments
These studies were supported, in whole or in part, by NIH grants R01 HL 133864, R01 HL 128304, AHA Grant-in-Aid 16GRNT27250146, AHA Established Investigator Award 34770095, Bayer AG Sponsored Research Funds, Vitalant and the Hemophilia Center of Western Pennsylvania (ACS), and NIH T32 DK007052 to BGD. We would like to thank Robert Kesterson for designing and generating Cyb5r3 floxed animals, Michele Mulkeen at the Rangos Histology Core Lab for processing our tissue samples, and Delphine Gomez for providing access to ImagePro Software.
Version 1. 09/05/2019
In-Press Preview
Version 2. 10/03/2019
Electronic publication
Funding Statement
no number associated with this grant.
no number associated with this grant
Footnotes
Conflict of interest: IM, DF, and PS are full-time employees of Bayer AG Pharmaceuticals. BGD, SAH, HMS, MPM, and ACS received research support for this work, in part, through a restricted research grant of Bayer AG Pharmaceuticals.
Copyright: © 2019, American Society for Clinical Investigation.
Reference information: JCI Insight. 2019;4(19):e129183.https://doi.org/10.1172/jci.insight.129183.
Contributor Information
Brittany G. Durgin, Email: durginb@pitt.edu.
Scott A. Hahn, Email: sch63@pitt.edu.
Heidi M. Schmidt, Email: HES81@pitt.edu.
Megan P. Miller, Email: mpl8@pitt.edu.
Neha Hafeez, Email: Hafeez.Neha@medstudent.pitt.edu.
Ilka Mathar, Email: ilka.mathar@bayer.com.
Daniel Freitag, Email: daniel.freitag@bayer.com.
Peter Sandner, Email: peter.sandner@bayer.com.
Adam C. Straub, Email: astraub@pitt.edu.
References
- 1.Lee YC, Martin E, Murad F. Human recombinant soluble guanylyl cyclase: expression, purification, and regulation. Proc Natl Acad Sci U S A. 2000;97(20):10763–10768. doi: 10.1073/pnas.190333697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhao Y, Marletta MA. Localization of the heme binding region in soluble guanylate cyclase. Biochemistry. 1997;36(50):15959–15964. doi: 10.1021/bi971825x. [DOI] [PubMed] [Google Scholar]
- 3.Karow DS, Pan D, Davis JH, Behrends S, Mathies RA, Marletta MA. Characterization of functional heme domains from soluble guanylate cyclase. Biochemistry. 2005;44(49):16266–16274. doi: 10.1021/bi051601b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tsai EJ, Kass DA. Cyclic GMP signaling in cardiovascular pathophysiology and therapeutics. Pharmacol Ther. 2009;122(3):216–238. doi: 10.1016/j.pharmthera.2009.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shah RC, Sanker S, Wood KC, Durgin BG, Straub AC. Redox regulation of soluble guanylyl cyclase. Nitric Oxide. 2018;76:97–104. doi: 10.1016/j.niox.2018.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Underbakke ES, Iavarone AT, Marletta MA. Higher-order interactions bridge the nitric oxide receptor and catalytic domains of soluble guanylate cyclase. Proc Natl Acad Sci U S A. 2013;110(17):6777–6782. doi: 10.1073/pnas.1301934110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ma X, Sayed N, Baskaran P, Beuve A, van den Akker F. PAS-mediated dimerization of soluble guanylyl cyclase revealed by signal transduction histidine kinase domain crystal structure. J Biol Chem. 2008;283(2):1167–1178. doi: 10.1074/jbc.M706218200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wedel B, Harteneck C, Foerster J, Friebe A, Schultz G, Koesling D. Functional domains of soluble guanylyl cyclase. J Biol Chem. 1995;270(42):24871–24875. doi: 10.1074/jbc.270.42.24871. [DOI] [PubMed] [Google Scholar]
- 9.Rothkegel C, et al. Dimerization region of soluble guanylate cyclase characterized by bimolecular fluorescence complementation in vivo. Mol Pharmacol. 2007;72(5):1181–1190. doi: 10.1124/mol.107.036368. [DOI] [PubMed] [Google Scholar]
- 10.Stasch JP, et al. Targeting the heme-oxidized nitric oxide receptor for selective vasodilatation of diseased blood vessels. J Clin Invest. 2006;116(9):2552–2561. doi: 10.1172/JCI28371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schrammel A, Behrends S, Schmidt K, Koesling D, Mayer B. Characterization of 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one as a heme-site inhibitor of nitric oxide-sensitive guanylyl cyclase. Mol Pharmacol. 1996;50(1):1–5. [PubMed] [Google Scholar]
- 12.Stasch JP, et al. NO- and haeme-independent activation of soluble guanylyl cyclase: molecular basis and cardiovascular implications of a new pharmacological principle. Br J Pharmacol. 2002;136(5):773–783. doi: 10.1038/sj.bjp.0704778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kalk P, et al. NO-independent activation of soluble guanylate cyclase prevents disease progression in rats with 5/6 nephrectomy. Br J Pharmacol. 2006;148(6):853–859. doi: 10.1038/sj.bjp.0706792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hultquist DE, Passon PG. Catalysis of methaemoglobin reduction by erythrocyte cytochrome B5 and cytochrome B5 reductase. Nature New Biol. 1971;229(8):252–254. doi: 10.1038/newbio229252a0. [DOI] [PubMed] [Google Scholar]
- 15.Passon PG, Hultsquist DE. Soluble cytochrome b5 reductase from human erythrocytes. Biochim Biophys Acta. 1972;275(1):62–73. doi: 10.1016/0005-2728(72)90024-2. [DOI] [PubMed] [Google Scholar]
- 16.Oshino N, Imai Y, Sato R. A function of cytochrome b5 in fatty acid desaturation by rat liver microsomes. J Biochem. 1971;69(1):155–167. doi: 10.1093/oxfordjournals.jbchem.a129444. [DOI] [PubMed] [Google Scholar]
- 17.Martin-Montalvo A, et al. Cytochrome b5 reductase and the control of lipid metabolism and healthspan. NPJ Aging Mech Dis. 2016;2:16006. doi: 10.1038/npjamd.2016.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Reddy VV, Kupfer D, Caspi E. Mechanism of C-5 double bond introduction in the biosynthesis of cholesterol by rat liver microsomes. J Biol Chem. 1977;252(9):2797–2801. [PubMed] [Google Scholar]
- 19.Hildebrandt A, Estabrook RW. Evidence for the participation of cytochrome b5 in hepatic microsomal mixed-function oxidation reactions. Arch Biochem Biophys. 1971;143(1):66–79. doi: 10.1016/0003-9861(71)90186-X. [DOI] [PubMed] [Google Scholar]
- 20.Sacco JC, Trepanier LA. Cytochrome b5 and NADH cytochrome b5 reductase: genotype-phenotype correlations for hydroxylamine reduction. Pharmacogenet Genomics. 2010;20(1):26–37. doi: 10.1097/FPC.0b013e3283343296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Siendones E, et al. Membrane-bound CYB5R3 is a common effector of nutritional and oxidative stress response through FOXO3a and Nrf2. Antioxid Redox Signal. 2014;21(12):1708–1725. doi: 10.1089/ars.2013.5479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Straub AC, et al. Hemoglobin α/eNOS coupling at myoendothelial junctions is required for nitric oxide scavenging during vasoconstriction. Arterioscler Thromb Vasc Biol. 2014;34(12):2594–2600. doi: 10.1161/ATVBAHA.114.303974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Straub AC, et al. Endothelial cell expression of haemoglobin α regulates nitric oxide signalling. Nature. 2012;491(7424):473–477. doi: 10.1038/nature11626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rahaman MM, et al. Cytochrome b5 reductase 3 modulates soluble guanylate cyclase redox state and cGMP signaling. Circ Res. 2017;121(2):137–148. doi: 10.1161/CIRCRESAHA.117.310705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wirth A, et al. G12-G13-LARG-mediated signaling in vascular smooth muscle is required for salt-induced hypertension. Nat Med. 2008;14(1):64–68. doi: 10.1038/nm1666. [DOI] [PubMed] [Google Scholar]
- 26.Félétou M, Köhler R, Vanhoutte PM. Endothelium-derived vasoactive factors and hypertension: possible roles in pathogenesis and as treatment targets. Curr Hypertens Rep. 2010;12(4):267–275. doi: 10.1007/s11906-010-0118-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kraehling JR, Sessa WC. Contemporary approaches to modulating the nitric oxide-cGMP pathway in cardiovascular disease. Circ Res. 2017;120(7):1174–1182. doi: 10.1161/CIRCRESAHA.117.303776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thoonen R, Sips PY, Bloch KD, Buys ES. Pathophysiology of hypertension in the absence of nitric oxide/cyclic GMP signaling. Curr Hypertens Rep. 2013;15(1):47–58. doi: 10.1007/s11906-012-0320-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rahaman MM, et al. Structure guided chemical modifications of propylthiouracil reveal novel small molecule inhibitors of cytochrome b5 reductase 3 that increase nitric oxide bioavailability. J Biol Chem. 2015;290(27):16861–16872. doi: 10.1074/jbc.M114.629964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ghimire K, Altmann HM, Straub AC, Isenberg JS. Nitric oxide: what’s new to NO? Am J Physiol Cell Physiol. 2017;312(3):C254–C262. doi: 10.1152/ajpcell.00315.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Félétou M, Vanhoutte PM. EDHF: an update. Clin Sci. 2009;117(4):139–155. doi: 10.1042/CS20090096. [DOI] [PubMed] [Google Scholar]
- 32.de Cabo R, Burgess JR, Navas P. Adaptations to oxidative stress induced by vitamin E deficiency in rat liver. J Bioenerg Biomembr. 2006;38(5–6):309–317. doi: 10.1007/s10863-006-9050-1. [DOI] [PubMed] [Google Scholar]
- 33.Henderson CJ, McLaughlin LA, Wolf CR. Evidence that cytochrome b5 and cytochrome b5 reductase can act as sole electron donors to the hepatic cytochrome P450 system. Mol Pharmacol. 2013;83(6):1209–1217. doi: 10.1124/mol.112.084616. [DOI] [PubMed] [Google Scholar]
- 34.Helms C, Kim-Shapiro DB. Hemoglobin-mediated nitric oxide signaling. Free Radic Biol Med. 2013;61:464–472. doi: 10.1016/j.freeradbiomed.2013.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Amdahl MB, Sparacino-Watkins CE, Corti P, Gladwin MT, Tejero J. Efficient reduction of vertebrate cytoglobins by the cytochrome b5/cytochrome b5 reductase/NADH system. Biochemistry. 2017;56(30):3993–4004. doi: 10.1021/acs.biochem.7b00224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu X, et al. Cytoglobin regulates blood pressure and vascular tone through nitric oxide metabolism in the vascular wall. Nat Commun. 2017;8:14807. doi: 10.1038/ncomms14807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dikalov SI, Nazarewicz RR. Angiotensin II-induced production of mitochondrial reactive oxygen species: potential mechanisms and relevance for cardiovascular disease. Antioxid Redox Signal. 2013;19(10):1085–1094. doi: 10.1089/ars.2012.4604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brown DI, Griendling KK. Nox proteins in signal transduction. Free Radic Biol Med. 2009;47(9):1239–1253. doi: 10.1016/j.freeradbiomed.2009.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee MY, et al. Mechanisms of vascular smooth muscle NADPH oxidase 1 (Nox1) contribution to injury-induced neointimal formation. Arterioscler Thromb Vasc Biol. 2009;29(4):480–487. doi: 10.1161/ATVBAHA.108.181925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Drummond GR, Selemidis S, Griendling KK, Sobey CG. Combating oxidative stress in vascular disease: NADPH oxidases as therapeutic targets. Nat Rev Drug Discov. 2011;10(6):453–471. doi: 10.1038/nrd3403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gavazzi G, et al. Decreased blood pressure in NOX1-deficient mice. FEBS Lett. 2006;580(2):497–504. doi: 10.1016/j.febslet.2005.12.049. [DOI] [PubMed] [Google Scholar]
- 42.Gavazzi G, Deffert C, Trocme C, Schäppi M, Herrmann FR, Krause KH. NOX1 deficiency protects from aortic dissection in response to angiotensin II. Hypertension. 2007;50(1):189–196. doi: 10.1161/HYPERTENSIONAHA.107.089706. [DOI] [PubMed] [Google Scholar]
- 43.Dikalova A, et al. Nox1 overexpression potentiates angiotensin II-induced hypertension and vascular smooth muscle hypertrophy in transgenic mice. Circulation. 2005;112(17):2668–2676. doi: 10.1161/CIRCULATIONAHA.105.538934. [DOI] [PubMed] [Google Scholar]
- 44.Griendling KK, Ushio-Fukai M, Lassègue B, Alexander RW. Angiotensin II signaling in vascular smooth muscle. New concepts. Hypertension. 1997;29(1 pt 2):366–373. doi: 10.1161/01.hyp.29.1.366. [DOI] [PubMed] [Google Scholar]
- 45.Lassègue B, Griendling KK. NADPH oxidases: functions and pathologies in the vasculature. Arterioscler Thromb Vasc Biol. 2010;30(4):653–661. doi: 10.1161/ATVBAHA.108.181610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu Y, et al. Volume overload induces differential spatiotemporal regulation of myocardial soluble guanylyl cyclase in eccentric hypertrophy and heart failure. J Mol Cell Cardiol. 2013;60:72–83. doi: 10.1016/j.yjmcc.2013.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tsai EJ, et al. Pressure-overload-induced subcellular relocalization/oxidation of soluble guanylyl cyclase in the heart modulates enzyme stimulation. Circ Res. 2012;110(2):295–303. doi: 10.1161/CIRCRESAHA.111.259242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zabel U, et al. Calcium-dependent membrane association sensitizes soluble guanylyl cyclase to nitric oxide. Nat Cell Biol. 2002;4(4):307–311. doi: 10.1038/ncb775. [DOI] [PubMed] [Google Scholar]
- 49.Diaz-Ruiz A, et al. Overexpression of CYB5R3 and NQO1, two NAD+ -producing enzymes, mimics aspects of caloric restriction. Aging Cell. 2018;17(4):e12767. doi: 10.1111/acel.12767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Oshino N, Imai Y, Sato R. A function of cytochrome b5 in fatty acid desaturation by rat liver microsomes. J Biochem. 1971;69(1):155–167. doi: 10.1093/oxfordjournals.jbchem.a129444. [DOI] [PubMed] [Google Scholar]
- 51.Reddy VV, Kupfer D, Caspi E. Mechanism of C-5 double bond introduction in the biosynthesis of cholesterol by rat liver microsomes. J Biol Chem. 1977;252(9):2797–2801. [PubMed] [Google Scholar]
- 52.Skarnes WC, et al. A conditional knockout resource for the genome-wide study of mouse gene function. Nature. 2011;474(7351):337–342. doi: 10.1038/nature10163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Osterwalder M, Galli A, Rosen B, Skarnes WC, Zeller R, Lopez-Rios J. Dual RMCE for efficient re-engineering of mouse mutant alleles. Nat Methods. 2010;7(11):893–895. doi: 10.1038/nmeth.1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pettitt SJ, et al. Agouti C57BL/6N embryonic stem cells for mouse genetic resources. Nat Methods. 2009;6(7):493–495. doi: 10.1038/nmeth.1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Butt E, et al. cAMP- and cGMP-dependent protein kinase phosphorylation sites of the focal adhesion vasodilator-stimulated phosphoprotein (VASP) in vitro and in intact human platelets. J Biol Chem. 1994;269(20):14509–14517. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.