

Abstract
The cardiac hormone atrial natriuretic peptide (ANP) is a central regulator of blood volume and a therapeutic target in hypertension and heart failure. Enhanced ANP activity in such conditions through inhibition of the degradative enzyme neprilysin has shown clinical efficacy but is complicated by consequences of simultaneous accumulation of a heterogeneous array of other hormones. Targets for specific ANP enhancement have not been available. Here, we describe a cis-acting antisense transcript (NPPA-AS1), which negatively regulates ANP expression in human cardiomyocytes. We show that NPPA-AS1 regulates ANP expression via facilitating NPPA repressor RE1-silencing transcription factor (REST) binding to its promoter, rather than forming an RNA duplex with ANP mRNA. Expression of ANP mRNA and NPPA-AS1 was increased and correlated in isolated strained human cardiomyocytes and in hearts from patients with advanced heart failure. Further, inhibition of NPPA-AS1 in vitro and in vivo resulted in increased myocardial expression of ANP, increased circulating ANP, increased renal cGMP, and lower blood pressure. The effects of NPPA-AS1 inhibition on NPPA expression in human cardiomyocytes were further marked under cell-strain conditions. Collectively, these results implicate the antisense transcript NPPA-AS1 as part of a physiologic self-regulatory ANP circuit and a viable target for specific ANP augmentation.
Keywords: Cardiology
Keywords: Heart failure, Noncoding RNAs, Transcription
A therapeutic strategy to enhance cardiac natriuretic peptide response by inhibition of a noncoding antisense RNA results in reduced blood pressure.
Introduction
The cardiac hormone atrial natriuretic peptide (ANP) is a central regulator of blood volume and blood pressure (BP) through its natriuretic, diuretic, and vasorelaxant properties (1, 2). ANP is produced and released from atrial cardiomyocytes (CMs) in response to increased cardiac filling pressures, is central in maintenance of cardiorenal homeostasis, and acts to reduce both cardiac preload and afterload in the context of hypertension and heart disease. Therapeutic augmentation of the natriuretic peptide system through inhibition of the inactivating enzyme neprilysin was recently demonstrated to confer beneficial effects on mortality and hospitalization in patients with heart failure (3). However, more targeted strategies for natriuretic peptide enhancement are desirable, to reduce the risk of adverse effects resulting from accumulation of the several other peptide targets of neprilysin, and to increase therapeutic efficacy. For example, the accumulation of bradykinin may increase risk of angioedema, while accumulation of amyloid β may at least in theory predispose to brain amyloid deposits, and the complex range of vasoactive hormones may predispose to both increased and decreased BP, depending on the patient (4). Therefore, the identification of pathways or molecules that regulate natriuretic peptide expression in CMs is of potential importance. Transcriptional regulation of the gene encoding ANP (NPPA) has been studied extensively (5), but a viable therapeutic target for the upregulation of NPPA expression remains elusive.
Natural antisense transcripts (NATs) are long noncoding RNAs (lncRNAs) that overlap protein-coding genes but are transcribed from the antisense strand (6). NATs may exert inhibitory effects on the transcription of their corresponding sense mRNA and are therefore highly specific therapeutic targets in contexts where augmentation of the coding gene is preferable (7). Several mechanisms whereby NATs regulate their coding counterpart have been reported, including duplex formation with complementary sequences in the sense transcript (8), interaction with regions of regulatory DNA (9), and chromatin-modifying enzymes (10). The NPPA locus contains several NATs with poorly defined function. A role in intron retention during splicing of NPPA mRNA has been proposed but the broader physiological role of these transcripts remains unclear (11). NATs can be targeted with high specificity by small synthetic oligonucleotide reagents, which inactivate and mark targets for degradation by nuclear ribonuclease H. Such reagents can be broadly internalized into cells through a range of mechanisms, including phagocytosis, pinocytosis, and clathrin- and caveolin-dependent endocytosis (12).
The aim of this study was to comprehensively characterize the function of the NPPA antisense transcript NPPA-AS1 in the human heart and assess the feasibility of this NAT as a target for specific natriuretic system augmentation based on GapmeRs in vitro and in vivo.
Results
NPPA-AS1 is located in nuclei of atrial CMs.
The NPPA locus contains a completely overlapping antisense RNA transcript, denoted NPPA-AS1 (Figure 1A) (11). To determine the function of NPPA-AS1, we first sought to comprehensively assess its tissue distribution. We obtained RNA sequencing (RNA-Seq) data for NPPA-AS1 and NPPA from GTEx v7 (https://gtexportal.org/home/), which includes 53 different tissues from 635 donors. Atrial appendage was the tissue with the second highest NPPA-AS1 expression overall (Figure 1B). Similarly to NPPA (Figure 1C), cardiac expression of NPPA-AS1 was restricted almost exclusively to atrial tissue (20-fold difference). We confirmed this expression pattern in RNA-Seq data from the Myocardial Applied Genomics Network (MAGNet) (13), including 22 ventricular and 101 atrial samples from unused donor hearts (Figure 1D). The 3 main cell types that comprise the myocardium are CMs, fibroblasts, and endothelial cells (2). We analyzed NPPA and NPPA-AS1 expression in primary human CMs, primary cardiac fibroblasts, and primary human cardiac microvascular endothelial cells by quantitative real-time PCR (qRT-PCR) (Figure 2A). NPPA expression was restricted exclusively to CMs, whereas NPPA-AS1 expression was detected in all cardiac cell types. To further determine the subcellular distribution of NPPA-AS1, cytoplasmic and nuclear RNA was extracted from human CMs derived from induced pluripotent stem cells (iPS-CMs). The expression of NPPA-AS1 was 5-fold higher in the nucleus than in the cytoplasm, whereas NPPA, expectedly, was present predominantly in the cytoplasm (Figure 2B). RNA fluorescent in situ hybridization (FISH) using an NPPA-AS1 probe in iPS-CMs confirmed that NPPA-AS1 was localized mainly to nuclei (Figure 2C). Approximately 60% of cells displayed a nuclear FISH signal, whereas only 4% of cells displayed a cytoplasmic signal. A 6-fold decrease in the proportion of cells with a nuclear FISH signal and a 2-fold increase in the number of unstained cells was observed in cells transfected with siRNA specific for NPPA-AS1, confirming specificity of the FISH probe (P < 0.001). To determine whether nuclear NPPA-AS1 was present as chromatin-enriched or soluble RNA (14), we performed nuclear fractionation followed by qRT-PCR (Figure 2D). Results revealed that the levels of NPPA-AS1 was 10-fold higher in the chromatin-enriched fraction, whereas the majority of NPPA was soluble (as expected for an mRNA). The observations that NPPA-AS1 is highly expressed in atrial tissue and enriched in CM chromatin suggest a potential role in regulation of its protein-coding counterpart NPPA.
Figure 1. Tissue distribution of NPPA-AS1.

(A) Schematic overview of the NPPA/NPPA-AS1 locus. Arrows indicate direction of transcription. Chromosomal position is indicated at the top (GRCh37/hg19 genome assembly). Expression of NPPA-AS1 (B) and NPPA (C) across 53 different human tissues based on RNA-Seq data from the GTEx database (v. 7). Cardiac tissues are highlighted. TPM, transcripts per million reads. (D) Expression of NPPA and NPPA-AS1 in left ventricular (LV, n = 22) and left atrial (LA, n = 101) tissue based on RNA-Seq data from the Myocardial Applied Genomics Network. FPKM, fragments per kilobase million.
Figure 2. Cellular and subcellular localization of NPPA-AS1.

(A) NPPA-AS1 and NPPA expression in cardiac cells assessed with qRT-PCR. Results are expressed relative to GAPDH. hCM, human primary cardiomyocytes, n = 3; hcFB, human cardiac fibroblasts, n = 3; hcMVEC, human cardiac microvascular endothelial cells, n = 2. N/D, not detected. (B) NPPA-AS1 and NPPA mRNA levels in nuclear (NUC) and cytoplasmic (CYT) RNA extracts from iPS-CMs measured by qRT-PCR. Results are expressed relative to GAPDH in each fraction. (C) Fluorescence in situ hybridization of NPPA-AS1 (red) in iPS-CMs. Cells were stained with Alexa Fluor 488–conjugated phalloidin (green) and nuclei were counterstained with DAPI (blue). Original magnification, ×20. The proportion of cells with nuclear and cytoplasmic FISH foci was quantified in cells transfected with siRNA specific for NPPA-AS1 or negative control siRNA. A total of 41 random cell-containing visual fields were analyzed. ***P < 0.01 by Mann-Whitney U test. (D) NPPA-AS1 and NPPA levels in nuclear fractions from iPS-CMs measured by qRT-PCR. CHE, chromatin-enriched; SOL, soluble. Results are expressed relative to 18S RNA.
NPPA-AS1 negatively regulates NPPA expression.
A common function of NATs is negative feedback regulation in cis of the expression of the corresponding protein-coding gene (15). We therefore assessed the expression of NPPA in human iPS-CMs following siRNA-mediated knockdown of NPPA-AS1. Successful knockdown of NPPA-AS1 was confirmed and a reciprocal increase in NPPA expression was observed (Figure 3). These observations suggest that NPPA-AS1 negatively regulates NPPA expression.
Figure 3. NPPA-AS1 inhibits NPPA expression.

iPS-CMs were transfected with scrambled negative control siRNA (siScr) or siRNA targeting NPPA-AS1 (siNPPA-AS1) for 48 hours. NPPA-AS1 and NPPA mRNA expression relative to GAPDH and normalized to the mean of the control cells in iPS-CMs was measured by qRT-PCR. Results are based on 3 separate experiments with 3–4 replicates per group. Mean and standard deviation are shown. ***P < 0.01 by Mann-Whitney U test.
NPPA-AS1 does not form an RNA-duplex with NPPA mRNA.
Next, we wanted to study the mechanism whereby NPPA-AS1 negatively influences NPPA expression. A previous study had suggested that NPPA-AS1 forms an RNA duplex with NPPA mRNA (11) and we sought to confirm this interaction using chromatin isolation by RNA purification (ChIRP) in human cardiac tissue with a probe set specific for NPPA-AS1, analyzing coprecipitated RNA with qRT-PCR (Figure 4). As expected, there was a substantial enrichment of precipitated NPPA-AS1 RNA. However, the amount of coprecipitated NPPA mRNA did not exceed that of an unrelated, nonoverlapping transcript (GAPDH). Thus, these results suggest that in human cardiac tissue, NPPA-AS1 exerts its effect on NPPA expression through a mechanism other than direct duplex formation.
Figure 4. NPPA-AS1 does not form a duplex with NPPA mRNA.
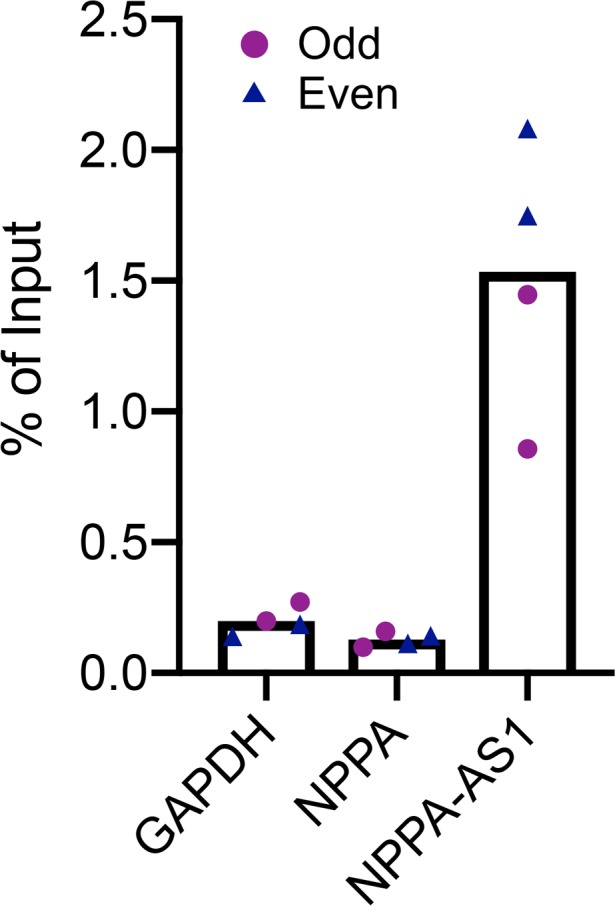
Chromatin isolation by RNA purification (ChIRP) in human cardiac tissue (n = 2). qRT-PCR quantification of NPPA in RNA coprecipitated with 2 independent ChIRP probe sets specific for NPPA-AS1 (“Even” and “Odd”). NPPA-AS1 and GAPDH mRNA was quantified as positive and negative controls, respectively. Results are expressed relative to input RNA.
NPPA-AS1 facilitates binding of the repressive transcription factor REST to the NPPA promoter.
Based on the observation that NPPA-AS1 is a chromatin-enriched transcript, we instead hypothesized that NPPA-AS1 regulates NPPA expression through binding to its promoter, as has been described for some NATs (16). To test this hypothesis, we performed ChIRP with the NPPA-AS1 probe set on human atrial tissue and quantified coprecipitated DNA using 6 primer pairs covering 1 kb upstream of the NPPA transcription start site (TSS) (Figure 5A). Primers specific for the GAPDH promoter were used as a negative control. As seen in Figure 5B, NPPA-AS1 coprecipitated with region E, approximately 560–700 bp upstream of the NPPA TSS. Interaction with region E was also seen in ventricular tissue (Supplemental Figure 1; supplemental material available online with this article; https://doi.org/10.1172/jci.insight.130978DS1).
Figure 5. NPPA-AS1 facilitates binding of the repressive transcription factor REST to the NPPA promoter.

(A) Schematic overview of the genomic region 1 kb upstream of the NPPA transcription start site (GRCh37/hg19 assembly). Indicated are the positions of the ChIRP primer pairs A–F. ENCODE ChIP-Seq transcription factor binding sites and DNase I hypersensitivity clusters are also indicated. The darkness of gray boxes is proportional to the maximum signal strength in the ENCODE v3 database. (B) qRT-PCR analysis of human atrial DNA coprecipitated with 2 independent ChIRP probe sets specific for NPPA-AS1 (“Even” and “Odd”). N/D, not detected. (C) Dot blots for protein coprecipitated with probes specific for NPPA-AS1. See complete unedited blots in the supplemental material. (D) Depiction of the noncanonical REST motif in the NPPA promoter and its overlap with region E. Indicated are the 2 half-sites and linker sequence as proposed by Johnson et al. (21). (E) REST ChIP-qPCR in iPS-CMs across the NPPA promoter. The GAPDH promoter was included as a negative control. The ChIP signal is expressed normalized to the negative control IgG ChIP for each region. The Kruskal-Wallis test was used to compare the ChIP signal for each individual region with the signal for the negative control (GAPDH) region. *P < 0.05, **P < 0.01 after adjustments for multiple comparisons using Dunn’s test. (F) REST ChIP-qPCR of iPS-CMs transfected with siRNA specific for REST, NPPA-AS1, or negative control siRNA, examining REST occupancy across regions C–F of the NPPA promoter. The ChIP signal is expressed relative to input DNA. *Two-way ANOVA was used to test the effect of each siRNA treatment on ChIP signal across all regions compared to negative control siRNA. REST occupancy was decreased with siREST and siNPPA-AS1 compared with negative control siRNA, P < 0.05 and P < 0.01, respectively. (G) qRT-PCR quantification of REST and NPPA expression in iPS-CMs transfected with siRNA specific for REST. Results are expressed relative to GAPDH and normalized to the mean of the control group. **P < 0.01, ***P < 0.001 using the Mann-Whitney U test. (H) Promoter reporter assay signals in cells cotransfected with pGLuc-NPPA or pGLuc-NPPAΔREST and siREST or siNPPA-AS1. Two-way ANOVA was used to test differences within and between groups. *P < 0.05, **P < 0.01, ***P < 0.001 after adjusting for multiple comparisons using Tukey’s test. For all graphs, mean and standard deviation are shown. All results are based on at least 2 separate experiments with 2–6 replicates for each condition or treatment group.
Analysis of this genomic region using ENCODE DNase I Hypersensitivity Peak Cluster (17) and Transcription Factor ChIP-Seq data (18–20) revealed that region E overlapped with a region characterized by DNase I hypersensitivity, indicative of accessible euchromatin, and binding sites for the transcription factors histone acetyltransferase (EP300), RE1-silencing transcription factor (REST), and upstream transcription factor 1 (USF1) (Figure 5A). We hypothesized that the interaction of NPPA-AS1 with the NPPA promoter modulates the binding of these transcription factors, either through competing for the binding sites or by facilitating recruitment. To assess whether NPPA-AS1 directly binds any of these 3 transcription factors, we performed ChIRP on human atrial tissue with the NPPA-AS1 probe set and analyzed coprecipitated proteins by dot blot. As seen in Figure 5C, REST, but not USF1 or EP300, was detected in the precipitated protein fraction. We confirmed this interaction with RNA immunoprecipitation (RIP) using an anti-REST antibody (Supplemental Figure 2).
Detailed sequence analysis of the NPPA promoter revealed that the 3′ end of region E overlapped with a noncanonical REST motif, as described by Johnson et al. (21) (Figure 5D). We sought to confirm that this motif was a de facto binding motif for REST in human CMs and test whether NPPA-AS1 was required for the recruitment of REST to this region. Binding of REST to regions A–F in iPS-CMs was assessed by chromatin immunoprecipitation (ChIP) followed by qRT-PCR. A region in the GAPDH promoter without a REST motif was included as a negative control. We observed enrichment of REST at regions C–F compared with an IgG control and with the negative-control region (Figure 5E). siRNA-mediated knockdown of REST in iPS-CMs significantly reduced REST occupancy across regions C–F (Figure 5F, P < 0.05), confirming the specificity of the ChIP signal. Interestingly, siRNA-mediated knockdown of NPPA-AS1 resulted in a similar decrease in the REST ChIP signal across regions C–F, suggesting that NPPA-AS1 is necessary for the recruitment of REST to the NPPA promoter (Figure 5F, P < 0.05). Importantly, knockdown of NPPA-AS1 did not affect REST gene expression (Supplemental Figure 3).
We next sought to confirm (a) that REST negatively regulates NPPA expression in human CMs; (b) that the binding of REST to the noncanonical motif overlapping region E was functional, negatively regulating NPPA expression; and (c) that the ability of NPPA-AS1 to regulate NPPA expression depends on this regulatory motif. To these ends, we cotransfected iPS-CMs with siRNA specific for either REST or NPPA-AS1 and a reporter vector expressing a luciferase gene under the control of the NPPA promoter (pGLuc-NPPA) or a vector where the REST motif had been deleted by site-directed mutagenesis (pGLuc-NPPAΔREST, Figure 5H). Transfection of iPS-CMs with siREST significantly increased NPPA expression and caused a significant increase in the reporter signal (Figure 5G), confirming that REST exerts a repressive effect on NPPA transcription and interacts with the NPPA promoter in human CMs. Deletion of the REST motif in and of itself caused a significant increase in reporter signal (Figure 5H, P < 0.001), and blunted the effect of REST knockdown, indicating that the REST motif is functional. Interestingly, siNPPA-AS1 also resulted in increased reporter signal from the intact vector (Figure 5H, P < 0.05), but this effect was abolished by deletion of the REST motif. Taken together, these results suggest that a possible mechanism whereby NPPA-AS1 inhibits NPPA transcription is by binding to REST and facilitating the interaction between this repressive transcription factor and a specific motif in the NPPA promoter. Interestingly, a residual effect of REST knockdown was observed even when the REST motif had been deleted (P < 0.05), suggesting that additional REST binding sites could be present in the NPPA promoter.
NPPA-AS1 is tethered to chromatin through RNA polymerase II.
We hypothesized that a cis-acting effect of NPPA-AS1 on the NPPA locus would be facilitated by tethering to local chromatin through RNA polymerase II (RNA Pol II) pausing. We assessed the amount of chromatin-enriched NPPA-AS1 in response to inhibition of RNA Pol II elongation by 5,6-dichloro-1-β-D-ribofuranosyl-1H-benzimidazole (DRB) treatment using nuclear fractioning and qRT-PCR. HOTTIP and XIST, 2 lncRNAs whose interaction with chromatin has previously been described to be polymerase dependent and independent, respectively, were included as controls. In cells treated with DRB, the proportion of chromatin-enriched NPPA-AS1 was significantly decreased (Figure 6A), indicating a role for RNA Pol II in facilitating the interaction between NPPA-AS1 and chromatin.
Figure 6. Tethering of NPPA-AS1 to chromatin is RNA polymerase II dependent.

(A) Assessment of the amount of chromatin-enriched NPPA-AS1 in response to DRB treatment. HEK293 cells were treated with 100 μM DRB or vehicle control for 2 hours and chromatin-enriched RNA was prepared using nuclear fractionation. Expression of NPPA-AS1, HOTTIP, and XIST was assessed using qRT-PCR. Data are expressed relative to 18S RNA and normalized to the mean of the control group. Results are based on 2 separate experiments with 3 replicates in each group. *P < 0.05, **P < 0.01 comparing expression between control and DRB-treated cells using the Mann-Whitney U test. Mean and standard deviation are shown. (B) Overview of the positions of each primer pair assessment of RNA polymerase II occupancy across the NPPA-AS1 promoter and gene body for ChIP-qPCR (GRCh37/hg19 assembly). (C) RNA polymerase II and negative control IgG ChIP signal for each genomic region in iPS-CMs. The signal is expressed relative to the DNA input sample. GB, gene body. Data are from 2 separate experiments with 2 technical replicates.
Given that the 3′ end of the NPPA-AS1 gene sits adjacent to the REST-binding region (region D and E in Figure 5A), we hypothesized that this region may be subject to transcriptional pausing, thereby tethering NPPA-AS1 and facilitating its interaction between REST and the NPPA promoter. To assess this, we performed ChIP with an anti–RNA Pol II antibody and quantified coprecipitated DNA across the NPPA-AS1 promoter, 5′ end, gene body, 3′ end, and in region E with qRT-PCR (Figure 6B). Results showed an enrichment of RNA Pol II in the 3′ end and region E compared with the rest of the NPPA-AS1 locus, consistent with polymerase pausing in the vicinity of the REST-binding region (Figure 6C).
NPPA and NPPA-AS1 are increased in stretched CMs and patients with heart failure.
We next aimed to determine whether the expression of NPPA-AS1 was altered during pathophysiological conditions. The increased mechanical stress on atrial CMs with increased ventricular filling pressures leads to a marked induction of NPPA gene expression (22), and we hypothesized that the expression of NPPA-AS1 would also be affected under these conditions. We subjected iPS-CMs to 10% cyclical strain for 48 hours, followed by 24 hours of normal culturing and continuously measured the expression of NPPA and NPPA-AS1 with qRT-PCR (Figure 7A). Importantly, expression of the reference gene GAPDH was not affected by the treatment (Supplemental Figure 4). The expression of NPPA at 48 hours was 2-fold higher than at baseline (Figure 7B, P < 0.05) and was accompanied by a similar increase in ANP protein levels (Figure 7C, P < 0.05). Notably, NPPA-AS1 expression showed an even more pronounced induction at the 48-hour time point (Figure 7B, P < 0.001). Interestingly, the transcriptional dynamics of NPPA and NPPA-AS1 differed in the response to cessation of the biomechanical stimulus. While NPPA expression continued to increase 6 hours after cessation of stretch, NPPA-AS1 expression decreased abruptly at this time point, suggesting that sustained transcription of the antisense transcript above baseline is dependent on continuous mechanical stimuli. We next investigated whether knockdown of NPPA-AS1 prior to stretch would affect the magnitude of the stretch-induced NPPA expression. iPS-CMs were transfected with siRNA against NPPA-AS1 or negative control siRNA before initiating stretch. Interestingly, stretch-induced NPPA expression was significantly higher in cells where NPPA-AS1 had been knocked down than in negative control cells (Figure 7D), suggesting that inhibition of this negative feedback mechanism can further enhance NPPA above normal strain-induced expression.
Figure 7. Mechanical strain increases NPPA and NPPA-AS1 expression in cardiomyocytes.

(A) Overview of the setup and design of the strain experiment. (B) NPPA and NPPA-AS1 expression during the time course of the experiment quantified by qRT-PCR. Expression is presented relative to GAPDH and normalized to the mean of the cells at time point 0 hours. Results are based on 3 separate experiments with 3 replicates in each group. Mean and standard deviation are shown. The Kruskal-Wallis test was used to test the difference in expression between baseline and each time point. *P < 0.05, ***P < 0.001 after adjustment for multiple comparisons using Dunn’s test. (C) Protein levels of ANP at baseline and after 48 hours of stretch. Results are expressed relative to GAPDH protein levels. Data are from 2 separate experiments with 3–4 replicates in each group. Shown are mean and standard deviation in each group. *P < 0.05 by Mann-Whitney U test. Shown below are representative blots for NPPA and GAPDH. US, unstretched; S, stretched. See complete unedited blots in the supplemental material. (D) NPPA expression in iPS-CMs first transfected with siRNA against NPPA-AS1 and then subjected to 48 hours of stretch. Two-way ANOVA with Tukey’s multiple-comparisons test was used to test differences within and between groups.*P < 0.05, **P < 0.01, ***P < 0.001. (E) RNA-Seq expression data for NPPA and NPPA-AS1 in ventricular tissue from heart failure patients (n = 42) and unused donor hearts (n = 22). ***P < 0.001 by Mann Whitney U test. (F) Correlation of NPPA and NPPA-AS1 in left ventricle and left atrium from heart failure and nonfailure donors. Pearson’s correlation coefficient and P value are shown.
To explore these findings in a clinical context, we assessed NPPA and NPPA-AS1 expression in a cohort of heart failure patients and nonfailure controls. RNA-Seq data from MAGNet (13), including left ventricular tissue from 42 heart failure patients and 22 nonfailure controls, as well as atrial tissue from 101 nonfailure donors, were analyzed. As seen in Figure 7E, both NPPA and NPPA-AS1 were significantly higher in ventricular tissue of heart failure patients compared with the control group. Moreover, we observed a statistically significant correlation between NPPA and NPPA-AS1 expression in both atrial and ventricular tissue (r = 0.43 and 0.58, respectively, Figure 7F).
NPPA expression is known to be induced during mammalian fetal heart development (23), with a peak during the second trimester in humans (24). To assess whether NPPA-AS1 is induced together with NPPA in the fetal human heart, we extracted data from a previously published data set containing RNA-Seq data from 35 human fetal samples from 6 tissues (heart, adrenal gland, intestine, kidney, lung, and stomach) collected between 10 and 20 weeks of gestational time (25). Cardiac expression of both NPPA and NPPA-AS1 followed the same temporal trend, with a sharp surge from week 10 to 11 followed by an approximately 50% decrease during weeks 17–20 (Supplemental Figure 5). Expression of NPPA and NPPA-AS1 was negligible in the other fetal tissues in the data set.
Taken together, these results indicate common transcriptional activation pathways for NPPA and NPPA-AS1, under physiological conditions as well as conditions of biomechanical stress and heart failure.
NPPA expression can be increased by therapeutic modulation of NPPA-AS1 in vivo.
Given that NPPA-AS1 is induced during pathophysiological conditions and acts as a negative feedback regulator of NPPA expression, we hypothesized that inhibition of NPPA-AS1 could be used as a therapeutic strategy for ANP augmentation. We wanted to evaluate the effects of Nppa antisense transcript knockdown on cardiac Nppa expression in mice using locked nucleic acid–based (LNA-based) antisense oligonucleotides (GapmeRs). The mouse Nppa locus contains a poorly conserved natural antisense transcript (Gm13054, here referred to as Nppa-as for clarity, Figure 8A). Cardiac expression of Nppa-as mirrored that of its human counterpart, with substantially higher expression in atria compared with ventricular tissue and general expression levels several orders of magnitude below that of the sense transcript (Supplemental Figure 6A). Moreover, ENCODE ChIP-Seq data showed that Rest binds to several regions across the Nppa locus, including in the promoter (Supplemental Figure 6B).
Figure 8. Knockdown of Nppa-as increases Nppa levels in vitro and in vivo.

(A) Schematic overview of the Nppa locus, including the natural antisense transcript Gm13054 on chromosome 4 (GRCm38/mm10 assembly). Arrows indicate direction of transcription. (B) Nppa-as and (C) Nppa expression in HL-1 cells transfected with different GapmeR designs and negative control, as measured by qRT-PCR. Expression is presented relative to Gapdh and normalized to the mean of the control group. Results are based on 3 separate experiments with 3 replicates in each group. Kruskal-Wallis was used to test the effect of each GapmeR design compared to the negative control. ***P < 0.001, **P < 0.01 after adjustment for multiple comparisons using Dunn’s test. (D) Nppa-as and (E) Nppa expression in atrial tissue of mice injected subcutaneously with 6.25, 12.5, or 25 mg/kg of GapmeR5 or 25 mg/kg of negative control GapmeR for 48 hours (n = 5–13 per group). Kruskal-Wallis was used to test the difference between animals treated with negative control and each of the G5 doses, as well as within G5 treatment groups. ***P < 0.001, *P < 0.05 after adjusting for multiple comparisons using Dunn’s test. (F) Plasma concentration of Anp in the saphenous vein of mice before and 48 hours after injection of the different Gapmer5 doses or 25 mg/kg of negative control GapmeR, as measured by ELISA (n = 5–13 per group). Repeated-measures 2-way ANOVA was used to test the difference before and after treatment in each group. *P < 0.05, **P < 0.01 after adjusting for multiple comparisons using Dunn’s test. (G) Systolic (SBP) and diastolic blood pressure (DBP) in mice before and 48 hours after GapmeR injections measured using a noninvasive tail cuff method (n = 5/group). Repeated-measures 2-way ANOVA was used to test for differences within groups. *P < 0.05 after adjusting for multiple comparisons using Dunn’s test. (H) Correlation of G5 dose with SBP and DBP in treated animals. Pearson’s correlation coefficient and P value are shown. (I) Kidney cGMP in each treatment group (n = 5/group) as measured by ELISA. Kruskal-Wallis was used to test the effect of each of the G5 doses compared to negative control GapmeR. *P < 0.05 after adjustment for multiple comparisons using Dunn’s test. For all graphs, mean and standard deviation are shown.
We evaluated the efficacy of 5 different GapmeR designs specific for Nppa-as in the murine CM cell line HL-1. GapmeR1 (G1) and Gapmer5 (G5) significantly inhibited Nppa-as expression in vitro compared with negative control GapmeR (Figure 8B). We next evaluated the ability of G1 and G5 to increase Nppa expression in dose-response experiments. G5 at 10 μM was sufficient to significantly increase Nppa expression, whereas 100 μM G1 was required to achieve the same effect (Figure 8C). Therefore, G5 was used for in vivo experiments.
First, the toxicity of GapmeR injections was assessed. Mice were injected subcutaneously with 25 mg/kg of G5, negative control, or PBS and the effect on body weight, liver- and kidney-to-body weight ratios, and the liver toxicity marker alanine aminotransferase were assessed after 48 hours. Both negative control and G5 were well tolerated (Supplemental Figure 7).
For evaluation of G5 efficacy, 3 doses of G5 (6.25, 12.5, and 25 mg/kg) or 25 mg/kg of negative control was injected subcutaneously in mice. G5 caused a dose-dependent knockdown of Nppa-as in atrial tissue and the 25 mg/kg dose resulted in a significant decrease as compared with mice injected with negative control (Figure 8D). A reciprocal, dose-dependent increase in Nppa expression was observed and the 25 mg/kg dose resulted in a statistically significant increase in Nppa expression compared with mice treated with negative control (Figure 8E, P < 0.01). Knockdown of Nppa-as was also observed in ventricular tissue, but did not result in increased Nppa expression (Supplemental Figure 8). Plasma Anp levels were significantly increased 48 hours after injection as compared with before injection in the 6.25 and 25 mg/kg treatment groups (Figure 8F). Furthermore, a significant decrease in systolic BP (SBP), and a trend toward decreased diastolic BP (DBP), was observed in the 25 mg/kg treatment group (Figure 8G). There was a significant negative correlation between dose and both SBP and DBP 48 hours after treatment (Figure 8H). Finally, kidney cGMP levels were numerically increased to a similar extent in all groups injected with G5 compared with negative control, but the effect was statistically significant only in the group that had received 6.25 mg/kg of G5 and marked in other groups (Figure 8I), indicating renal ANP receptor activation.
Discussion
Enhancing natriuretic peptide concentration in the circulation through inhibition of neprilysin has proven beneficial in the treatment of heart failure, but the broad substrate specificity of neprilysin confers a considerable risk of unspecific effects. In the present study, we have presented evidence that NPPA is regulated by a cis-acting antisense transcript, NPPA-AS1, that could constitute a potential target for specific natriuretic peptide augmentation.
Unlike its sense transcript, NPPA-AS1 is also expressed in noncardiac tissues. According to GTEx, testis is the tissue with the highest expression of NPPA-AS1. This is not surprising, since a general enrichment of noncoding RNA, including antisense transcripts, has been documented in this tissue before (23). This phenomenon has been described as a consequence of an overall permissive chromatin state in spermatocytes and spermatids and this “leaky” transcription pattern is likely functionally irrelevant (24). Noncardiac functions of NPPA-AS1 in the other 2 tissues with substantial NPPA-AS1 expression (pituitary and prostate), or effects in cardiac cell types other than CMs cannot be ruled out. However, considering that expression of the corresponding sense transcript (NPPA) in this case is extremely tissue and cell specific, these extracardiac or extracardiomyocytic actions would assume trans-acting effects of NPPA-AS1. Trans-acting antisense transcripts generally have at least partial sequence complementary to its target RNAs (25, 26). However, using a basic local alignment search, we could not identify a single mRNA transcript with sequence complementarity to NPPA-AS1 except NPPA. In addition, our data suggest that NPPA-AS1 is subject to transcriptional pausing at the 3′ end. This mechanism facilitates tethering of NPPA-AS1 to the NPPA locus and local effects. Thus, taking into account what is currently known about the regulation of gene expression by NAT, we consider the likelihood of NPPA-AS1 having additional trans-acting effects to be low. Previous work by Annilo et al. described a role for NPPA-AS1 in intron retention of NPPA mRNA through RNA duplex formation (11). The authors showed duplex formation by forced expression of NPPA and NPPA-AS1 in a mouse fibroblast cell line, which ignores the substantial differences in expression levels between the sense and antisense transcripts. These discrepancies might explain why we failed to detect any direct interaction between NPPA and NPPA-AS1 transcripts in a mechanistically relevant tissue, i.e., human myocardium.
Modulation of NPPA-AS1 in vitro and in vivo resulted in increased expression of NPPA. Intuitively, the positive correlation between the sense and antisense transcripts observed in human cardiac tissue might therefore seem contradictory. However, since the expression of both NPPA-AS1 and NPPA appears to be highly responsive to biomechanical stimulus, it rather appears that the 2 transcripts share common upstream regulatory pathways, and NPPA-AS1 acts as a natural negative feedback mechanism. Both NPPA and NPPA-AS1 transcription is induced in the failing heart, but it is impossible to gauge what cardiac NPPA expression and circulating ANP levels would be without the negative feedback loop provided by NPPA-AS1.
When considering NPPA-AS1 as a drug target, caution must also be given to the lack of sequence conservation between human and mouse NPPA antisense transcripts. It is well established that NATs generally exhibit poor sequence conservation (27, 28), but can still retain functional conservation (29). Indeed, it has been proposed that secondary and/or tertiary structures are more highly conserved in lncRNA transcripts (30, 31). Additionally, the human and mouse NPPA promoters show a high degree of conservation, indicating that mechanisms of transcriptional regulation are likely to be conserved (32). We show that inhibition of NPPA-AS1 and Nppa-as results in a similar upregulation of the sense transcript and although the molecular mechanism underlying these effects might differ in human and mouse CMs, our results provide proof of principle that pharmacological modulation of the Nppa antisense transcript with an oligonucleotide compound can be an effective and specific strategy for natriuretic peptide augmentation.
The phenotype of iPS-CMs that were used in the in vitro experiments is unclear with regard to atrial or ventricular representation. Based on previous work with the same cells (31, 33) and transcriptomic profiling performed by our group (data not shown), we conclude that these cells are a mix of atrial-, ventricular-, and nodal-like CMs. For instance, these cells express very high levels of atrial marker genes MYL7, NPPA, and NPPB, but similarly high levels of MYL2, MYH7, and KCNE1, markers of ventricular CMs. As such, these cells are not a perfect model for atrial CMs, and this limitation must be taken into account when interpreting the results of this study.
In human and murine atrial and ventricular tissues, we observed relatively low NPPA-AS1 expression in relation to NPPA. This difference in copy number in a sense/antisense pair is not uncommon, especially with regard to antisense transcripts with local effects on chromatin structure or as scaffolds for transcription factor complexes (34–36). Our data suggest that rather than binding to and forming a duplex with NPPA mRNA, NPPA-AS1 acts locally by binding to the NPPA promoter and facilitating recruitment of the transcription factor REST. It is likely that this mode of action in an overlapping DNA region only requires a few transcripts of RNA, considering that there are only 2 copies of a particular genomic region per cell.
We show that 25 mg/kg of G5 caused a significant decrease in atrial Nppa-as expression and a reciprocal increase in Nppa expression and plasma Anp levels. However, plasma Anp was also significantly increased in mice injected with the lowest dose of G5 even though these mice did not show an increase in atrial Nppa gene expression. This might reflect an unspecific effect of the GapmeR on other genes involved in Anp biogenesis and release. Future analysis of the cardiac transcriptome of injected mice must be performed in order to address this concern. Another possible explanation could be an unspecific effect of the negative control GapmeR on Nppa-as and Nppa expression, obscuring the magnitude of the effect of the active GapmeR. Again, further work, comparing atrial gene expression in mice injected with PBS to mice injected with negative control GapmeR is needed to address these issues. Regarding BP measurements, despite our efforts to train and adapt the animals to the measurement device, all mice were moderately hypertensive at baseline. Thus, the potential activation of homeostatic mechanisms at baseline might influence the outcomes of these experiments and represents a limitation of the current study.
Further work examining the effects of Nppa-as inhibition in models of conditions such as hypertension or heart failure will be necessary to further assess the relevance of this antisense RNA as a therapeutic target.
Methods
Cell culture.
Human iPS-CMs (Cellular Dynamics International), primary human cardiac fibroblasts (Cell Applications Inc.), human cardiac microvascular endothelial cells (Lonza), and primary human CMs (PromoCell) were grown in iCell Cardiomyocyte Plating or Maintenance Medium (Cellular Dynamics International), Fibroblast Growth Medium (Cell Applications Inc.), and Medium 200 with Low Serum Growth Supplement (Thermo Fisher Scientific), respectively, according to the manufacturers’ instructions. Cells were plated at a density of 1 × 106 cells/10-cm2 dish, 200,000 cells/chamber, or 200,000 cells/well in 12-well plates or 20,000 cells/well in 96-well plates.
siRNA-mediated knockdown of NPPA-AS1.
NPPA-AS1 in iPS-CMs was silenced by siRNA transfection (Thermo Fisher Scientific). In short, 200,000 CMs were seeded on 12-well plates and incubated at 37°C and 7% CO2. On day 5, cells were transfected with 40 nM siRNA specific for NPPA-AS1 (Thermo Fisher Scientific, n509550) or negative control (Thermo Fisher Scientific, AM4611) using Lipofectamine RNAiMAX transfection reagent (Life Technologies, catalog 13778-075) according to the manufacturer’s instructions. Cells were harvested after 48 hours for RNA extraction.
CM strain assay.
The stretch unit (STREX Cell Strain Instrument, B-Bridge International) was placed in an incubator. The silicon chambers (20 mm × 20 mm × 10 mm), including the plated iPS-CMs, were positioned in the unit with no tension on the membrane in the resting position. The cells were strained (60 cycles/min) in a uniaxial manner, with 10% elongation (2.0 mm) in one direction for 48 hours followed by 24 hours of culture without stretch. Cells were harvested for RNA and protein at 0, 48, 54, and 72 hours.
Gene expression.
Cells were lysed for RNA preparation by addition of QIAzol (Qiagen) for cells growing on regular culture plates, or with the addition of TRIzol (Thermo Fisher Scientific) with subsequent scraping with a rubber policeman for cells grown in silicon chambers. Total RNA was prepared using the miRNeasy Mini Kit (Qiagen) according to the manufacturer’s instructions. Cytoplasmic and nuclear RNA were extracted by using a Cytoplasmic & Nuclear RNA Purification Kit (Norgen Biotek) according to the manufacturer’s protocol.cDNA synthesis was performed using the RevertAid H Minus First-Strand cDNA Synthesis Kit (Thermo Fisher Scientific) with random hexamer primers according to the manufacturer’s instructions. Gene expression was assessed with qRT-PCR using TaqMan assays (Thermo Fisher Scientific) specific for NPPA (Hs00380230_g1), NPPA-AS1 (Hs01081097_m1), GAPDH (Hs02786624_g1), HOTTIP (Hs00955374_s1), RNA18s5 (Hs03928985_g1), XIST (Hs01079824_m1), Nppa (Mm01255747_g1), Gapdh (Mm99999915_g1), and 2× Universal TaqMan Master Mix (Thermo Fisher Scientific). A custom TaqMan assay was designed to measure the expression of murine Nppa-as (Gm13054) with the batch ID w1608882241000 (Thermo Fisher Scientific). qRT-PCR cycles were run on a StepOnePlus Real-Time PCR System (Thermo Fisher Scientific). Gene expression is presented either relative to that of the reference gene (2–dCt) or relative to the reference gene and normalized to the mean of the control samples (2–ddCt). For assessment of gene expression in cellular fractions (nuclei and cytoplasm), data were normalized to the expression of the reference gene in that fraction.
FISH.
A set of Quasar 670–conjugated RNA FISH probes were designed for NPPA-AS1 using the Stellaris Probe Designer v. 4.2 and acquired from LGC Biosearch. See Supplemental Table 1 for probe sequences. RNA FISH was performed according to the manufacturer’s protocol. iPS-CMs were seeded and cultured on glass coverslips in a 12-well plate. Five days after seeding, cells were transfected with siRNA specific for NPPA-AS1 or negative control siRNA as described above. Seventy-two hours after transfection, cells were fixed, permeabilized, and incubated with hybridization buffer containing 125 nM FISH probes overnight. The following day, cells were counterstained with Alexa Fluor 488–conjugated phalloidin (Thermo Fisher Scientific, A12379, diluted 1:40) for 30 minutes, washed, and mounted on microscopy slides with Fluoroshield with DAPI (Sigma-Aldrich, F6057) mounting medium and visualized with a Nikon LU4A Ti Microscope (Nikon Instruments). Positive FISH foci in the cytoplasm and nuclei of cells were counted in a total of 41 random cell-containing visual fields.
Western blot analysis.
Protein isolation was performed using the organic phase from the RNA extraction with TRIzol according to the manufacturer’s recommendations. Protein concentration was determined with the Pierce BCA Protein Assay Kit (Thermo Fisher Scientific) and 10 μg of protein was run in Bio-Rad 4%–10% Criterion gels for 90 minutes at 150 V. Proteins were transferred to 0.45-μm nitrocellulose membranes (Bio-Rad) for 90 minutes at 4°C at 100 V. The membrane was blocked in 5% BSA in TBS-T (20 mM Tris/HCl, 0.5 M NaCl, 0.05%, Tween-20; Bio-Rad) and incubated overnight with anti-ANP primary antibody (1:16,000, ab190001, Abcam). The next day, the membrane was washed with TBS-T and incubated with secondary antibody, washed with TBS-T, and developed with the SuperSignal West Femto Maximum Sensitivity Substrate (Thermo Fisher Scientific). The bands were visualized and analyzed using the LI-COR Odyssey (LI-COR Biosciences). ANP was normalized to GAPDH (1:1000, 2118, Cell Signaling Technology)
Nuclear fractionation.
Nuclear fractionation was performed on iPS-CMs and HEK293 cells (a gift from the Molecular Vascular Physiology group, Lund University) with the method described by Werner et al. (14). The levels of NPPA and NPPA-AS1 in the soluble and chromatin-enriched RNA fractions were analyzed with qRT-PCR as described above. In additional experiments, HEK293 cells were either treated with 100 μM DRB or vehicle control (DMSO) for 2 hours before nuclear fractionation.
Human cardiac gene expression.
RNA-Seq data from left atrial and ventricular tissue were acquired via the Myocardial Applied Genomics Network (MAGNet, https://www.med.upenn.edu/magnet/). Ventricular tissue was obtained from 42 explanted hearts (from heart failure patients, including 22 with dilated cardiomyopathy and 20 with ischemic heart disease) and 22 unused donor hearts. RNA was extracted from ventricular tissue using an RNeasy Lipid Tissue Total RNA Mini Kit (Qiagen). Poly-A libraries were prepared using the TruSeq RNA Sample Preparation Kit (Illumina) and sequenced using Illumina’s HiSeq 2000. Reads were aligned to the hg19 reference genome using TopHat with default options (37) and transcripts were assembled using Cufflinks (38). RNA-Seq data from left atrial tissue samples were obtained from 101 normal donors with no evidence of structural heart disease (39).
ChIRP.
ChIRP was performed on human atrial and ventricular tissue according to the method described by Chu et al. (40). Human cardiac tissue was obtained from explanted hearts after heart transplantation. The tissue was minced and homogenized using a dounce homogenizer. Cells were cross-linked with 1% glutaraldehyde at room temperature for 10 minutes on an end-to-end rotor and quenched with 1.25 M glycine. Cells were then lysed and sonicated (30 seconds on/45 seconds off pulse intervals) in a Bioruptor sonication device (Diagenode). A fragment size of 100 to 500 bp was verified by agarose gel electrophoresis. A ChIRP probe set specific for NPPA-AS1 was designed using the ChIRP Probe Designer with masking level set to 5, oligonucleotide length set to 20, and the minimum spacing length set to 80, and was acquired from LGC Biosearch Technologies (See Supplemental Table 1 for probe sequences). The obtained chromatin was hybridized with either the even- or odd-numbered probes (7 probes in each group) for 4 hours at 37°C. C-1 magnetic beads (Thermo Fisher Scientific) were used to capture the biotinylated chromatin, followed by 5 washes. For RNA elution, the beads were resuspended in RNA proteinase K buffer and proteinase K (Thermo Fisher Scientific) and incubated for 45 minutes at 50°C with shaking. Then, the samples were incubated for 10 minutes at 95°C and TRIzol was added, followed by 10 minutes of incubation, and RNA was extracted as described above. qRT-PCR was used to assess coprecipitation of NPPA-AS1, NPPA, and GAPDH as described above. Enrichment of coprecipitated RNA was assessed relative to input RNA. DNA was eluted from the beads by incubation with RNase A and RNase H twice for 30 minutes each at 37°C and with proteinase K treatment for 45 minutes at 50°C. Then, the DNA was purified using ChIP DNA Clean & Concentrator (ZymoResearch). DNA was analyzed by qRT-PCR using primers specific for 6 regions across the NPPA promoter (see Supplemental Table 1 for primer sequences). The enrichment of a specific region was assessed relative to input DNA. The detection of proteins was performed by dot blot, transferring 3 μL of the eluted protein from ChIRP to a nitrocellulose membrane (Bio-Rad). The membrane was blocked for 1 hour at room temperature, incubated with primary antibody against REST (Novusbio, NB100-756), USF1 (Abcam, 180717), and EP300 (Abcam, 14984), followed by secondary antibody incubation. The membrane was then developed using the SuperSignal West Femto Maximum Sensitivity Substrate (Thermo Fisher Scientific) and visualized using the LI-COR Odyssey.
RIP.
RIP was performed according to the manufacturer’s instructions (Sigma-Aldrich). Human atrial tissue was minced and lysed in harsh lysis buffer, including protease inhibitor cocktail, 1 mM DTT, and ribonuclease inhibitor (40 U/μL). REST antibody (5 μg, 17-641, Merck Millipore) or 5 μg IgG was incubated with magnetic beads for 30 minutes at room temperature with rotation for prebinding, followed by several wash steps. The antibody-coated beads were then resuspended in the IP reaction and incubated overnight at 4°C with rotation. After incubation, the bead-antibody-RNA complex was washed 5 times and resuspended in RIP wash buffer. RNA was purified using phenol-chloroform extraction. TRIzol and chloroform were added to the RIP reaction containing the beads, vortexed, and centrifuged for 10 minutes at 16,000 g at 4°C. Glycogen (5 mg/mL), 3 M sodium acetate, and 2-propanol were added to the aqueous phase and incubate for 6 hours at –80°C to precipitate the RNA. After centrifugation and washing with 80% ethanol, the RNA was resuspended in nuclease-free water. NPPA-AS1 was quantified in RNA immunoprecipitated with the REST antibody and a nonspecific IgG control antibody with qRT-PCR as described above. The qRT-PCR signal was normalized to that of an input-RNA control.
ChIP.
ChIP was performed using the SimpleChIP Plus Kit (Cell Signaling Technology) according to the manufacturer’s instructions. iPS-CMs (1 × 106) were seeded in 10-cm2 culture dishes and transfected on day 5 with 40 nM siRNA specific for REST (Thermo Fisher Scientific, s11933) or negative control (Thermo Fisher Scientific) using Lipofectamine RNAiMAX transfection reagent (Thermo Fisher Scientific). After 72 hours, cells were cross-linked in 1 % formaldehyde for 10 minutes at room temperature and quenched with glycine. Chromatin was digested by incubating with micrococcal nuclease (2000 gel units/μL) for 20 minutes at 37°C and nuclei were then lysed with 3 sets of 20-second pulses using an Ultrasonic Homogenizer (Sonics & Materials Inc.). A mean DNA fragment size of 100 to 500 bp was verified by agarose gel electrophoresis. Five micrograms of the digested chromatin was used per IP and incubated with 2 μg of normal rabbit IgG (Cell Signaling Technology, 2729) or 2 μg of antibody against REST (17-641, Merck Millipore) or anti–RNA Pol II (17-620, Merck Millipore) overnight at 4°C with rotation. Protein G Magnetic Beads were added to each IP and incubated for 2 hours at 4°C with rotation. Then, the chromatin was eluted from the antibody–magnetic beads at 65°C for 30 minutes and the crosslinks were reversed by heating at 65°C for 2 hours. The DNA was purified using ChIP DNA Clean & Concentrator (ZymoResearch). The DNA was analyzed by quantitative PCR using primers specific for distinct regions across the NPPA and NPPA-AS1 promoters and the NPPA-AS1 gene body (see Supplemental Table 1 for primer sequences). The enrichment of a specific region was assessed relative to input DNA.
In vivo Nppa-as inhibition.
Five LNA antisense oligonucleotide GapmeRs (G1–G5) against Nppa-as were custom designed using Exiqon’s Antisense LNA GapmeR Designer. Efficacy of Nppa-as knockdown was first evaluated in vitro. Briefly, 40,000 HL-1 cells were seeded in a 24-well plate and incubated at 37°C and 5% CO2. On day 5, cells were transfected with 10 nM, 50 nM, or 100 nM GapmeR (Exiqon) or Negative Control B (Exiqon) using RNAiMAX transfection reagent (Thermo Fisher Scientific). Cells were harvested after 48 hours for RNA extraction and qRT-PCR as described above. For in vivo knockdown of Nppa-as, 36 male C57BL/6JBomTac mice (Taconic Bioscience) were randomly selected for subcutaneous injection of 6.25, 12.5, or 25 mg/kg of G5 (n = 5–13) or negative control GapmeR (n = 13). After 48 hours, animals were sacrificed. A noninvasive tail cuff system (CODA for rodents, emka Technologies) was used for BP measurements before and 48 hours after injection of GapmeR. The mice were trained every day for 1 week before BP measurements to get accustomed to the tail cuff device and the experimenter. Mice were placed in the rodent restrainer with darkened chamber and heating and the tail cuff for recording SBP and DBP was placed close to the root of the tail. Fifteen minutes prior to measurements, mice were placed under a heating lamp to increase blood flow through the tail. Baseline BP measurements were sampled at the end of training, before injections, and by 48 hours after injection with GapmeR. BP values were averaged over more than 10 measurements. Peripheral venous blood was collected from the saphenous vein before and 48 hours after injection. Plasma was prepared by centrifugation at 1600 g for 15 minutes and stored at –80°C. Plasma Anp levels were assessed with the Atrial Natriuretic Peptide EIA Kit (Sigma-Aldrich) according to the manufacturer’s instructions. Atrial and ventricular tissue was dissected out for RNA extraction. One kidney per animal was dissected out for cGMP analysis. Tissue was homogenized using an Omni TH rotor-stator homogenizer (Omni International) and total RNA was prepared as described above. Kidney cGMP was measured with ELISA (R&D Systems) according to the manufacturer’s instructions. Kidneys were homogenized in the Cell Lysis Buffer provided in the kit and protein concentration was determined with the Pierce BCA Protein Assay Kit.
Promoter reporter Assay.
A GLuc-ON promoter reporter plasmid (GeneCopoeia) containing 1222 bp upstream and 93 bp downstream of the NPPA (accession number: NM_006172) promoter upstream of a Gaussia luciferase reporter was used for assessing NPPA promoter activity. The plasmid also contained a secondary reporter gene (secreted alkaline phosphatase, SEAP) under the control of the CMV promoter for normalization of the reporter signal for cell number and transfection efficiency. The noncanonical REST motif in the NPPA promoter was deleted using the Phusion Site-Directed Mutagenesis Kit (Thermo Fisher Scientific) according to the manufacturer’s instructions. Deletion of the sequence was confirmed with Sanger sequencing. See Supplemental Table 1 for primer sequences for site-directed mutagenesis and DNA sequencing. Plasmids were cotransfected with 40 nM siRNA specific for REST, NPPA-AS1, or negative control siRNA in iPS-CMs using Lipofectamine 3000 according to the manufacturer’s instructions. iPS-CMs were seeded at 20,000 per well in 96-well plates and transfected on day 5 after plating. Seventy-two hours after transfection, medium was aspirated. Gaussia luciferase and SEAP activity in the medium was measured using the Secrete-Pair Dual Luminescence Assay Kit (GeneCopoeia) according to the manufacturer’s instructions. Luminescence was measured on a ClarioStar plate reader (BMG Labtech) and the Gaussia luciferase reporter signal was normalized to the SEAP in each well.
Statistics.
All data are expressed as mean and standard deviation. Groups were compared using 2-tailed, nonparametric tests, including the Mann-Whitney U test for comparison of distributions between 2 groups and the Kruskal-Wallis test or 2-way ANOVA for multiple group comparisons. Results were considered statistically significant with P value < 0.05. Analyses were conducted in GraphPad Prism 7.
Study approval.
This study was conducted in accordance with the principles of the Declaration of Helsinki and was approved by the local ethics committee of Skane University Hospital in Lund. All patients provided written consent before participation. All animal procedures were performed according to protocols approved by the local ethics committee for animal research at Lund University.
Author contributions
SC conducted the majority of the experiments and analyzed and interpreted data. MKS designed and performed in vivo experiments and analyzed and interpreted results from these experiments. MM, CR, PTE, and TC contributed, analyzed, and interpreted data from heart failure patients and controls. JGS and OG interpreted results and wrote the manuscript. OG conceived the study. All authors provided input on the final version of the manuscript.
Supplementary Material
Acknowledgments
This work was supported by grants from the Swedish Heart and Lung Foundation (2016-0134 and 2016-0315), the Swedish Research Council (2017-02554), the European Research Council (ERC-STG-2015-679242), the Crafoord Foundation, the Elsa Golje Foundation, the Royal Physiographic Society, Lars Hiertas Minne, the Märta Winkler Foundation, governmental funding of clinical research within the Swedish National Health Service, a generous donation from the Knut and Alice Wallenberg foundation to the Wallenberg Center for Molecular Medicine in Lund, and funding from the Swedish Research Council (Linnaeus grant Dnr 349-2006-237, Strategic Research Area Exodiab Dnr 2009-1039) and Swedish Foundation for Strategic Research (Dnr IRC15-0067) to the Lund University Diabetes Center. Lund University Bioimaging Center (LBIC) is gratefully acknowledged for providing experimental resources.
Version 1. 09/10/2019
In-Press Preview
Version 2. 10/03/2019
Electronic publication
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Copyright: © 2019, American Society for Clinical Investigation.
Reference information: JCI Insight. 2019;4(19):e130978.https://doi.org/10.1172/jci.insight.130978.
Contributor Information
Selvi Celik, Email: selvi.celik@med.lu.se.
Mardjaneh Karbalaei Sadegh, Email: mardjaneh.karbalaei_sadegh@med.lu.se.
Michael Morley, Email: mmorley@mail.med.upenn.edu.
Carolina Roselli, Email: aroselli@broadinstitute.org.
Patrick T. Ellinor, Email: ellinor@mgh.harvard.edu.
Thomas Cappola, Email: thomas.cappola@uphs.upenn.edu.
J. Gustav Smith, Email: gustav.smith@med.lu.se.
References
- 1.de Bold AJ, Borenstein HB, Veress AT, Sonnenberg H. A rapid and potent natriuretic response to intravenous injection of atrial myocardial extract in rats. Life Sci. 1981;28(1):89–94. doi: 10.1016/0024-3205(81)90370-2. [DOI] [PubMed] [Google Scholar]
- 2.Potter LR, Yoder AR, Flora DR, Antos LK, Dickey DM. Natriuretic peptides: their structures, receptors, physiologic functions and therapeutic applications. Handb Exp Pharmacol. 2009;(191):341–366. doi: 10.1007/978-3-540-68964-5_15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McMurray JJ, et al. Angiotensin-neprilysin inhibition versus enalapril in heart failure. N Engl J Med. 2014;371(11):993–1004. doi: 10.1056/NEJMoa1409077. [DOI] [PubMed] [Google Scholar]
- 4.Chrysant SG. Benefits and pitfalls of sacubitril/valsartan treatment in patients with hypertension. J Clin Hypertens (Greenwich) 2018;20(2):351–355. doi: 10.1111/jch.13169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sergeeva IA, Christoffels VM. Regulation of expression of atrial and brain natriuretic peptide, biomarkers for heart development and disease. Biochim Biophys Acta. 2013;1832(12):2403–2413. doi: 10.1016/j.bbadis.2013.07.003. [DOI] [PubMed] [Google Scholar]
- 6.Khorkova O, Myers AJ, Hsiao J, Wahlestedt C. Natural antisense transcripts. Hum Mol Genet. 2014;23(R1):R54–R63. doi: 10.1093/hmg/ddu207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wahlestedt C. Targeting long non-coding RNA to therapeutically upregulate gene expression. Nat Rev Drug Discov. 2013;12(6):433–446. doi: 10.1038/nrd4018. [DOI] [PubMed] [Google Scholar]
- 8.Postepska-Igielska A, et al. LncRNA Khps1 regulates expression of the proto-oncogene SPHK1 via triplex-mediated changes in chromatin structure. Mol Cell. 2015;60(4):626–636. doi: 10.1016/j.molcel.2015.10.001. [DOI] [PubMed] [Google Scholar]
- 9.Faghihi MA, et al. Expression of a noncoding RNA is elevated in Alzheimer’s disease and drives rapid feed-forward regulation of beta-secretase. Nat Med. 2008;14(7):723–730. doi: 10.1038/nm1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nagano T, et al. The Air noncoding RNA epigenetically silences transcription by targeting G9a to chromatin. Science. 2008;322(5908):1717–1720. doi: 10.1126/science.1163802. [DOI] [PubMed] [Google Scholar]
- 11.Annilo T, Kepp K, Laan M. Natural antisense transcript of natriuretic peptide precursor A (NPPA): structural organization and modulation of NPPA expression. BMC Mol Biol. 2009;10:81. doi: 10.1186/1471-2199-10-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shen X, Corey DR. Chemistry, mechanism and clinical status of antisense oligonucleotides and duplex RNAs. Nucleic Acids Res. 2018;46(4):1584–1600. doi: 10.1093/nar/gkx1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu Y, et al. RNA-Seq identifies novel myocardial gene expression signatures of heart failure. Genomics. 2015;105(2):83–89. doi: 10.1016/j.ygeno.2014.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Werner MS, Ruthenburg AJ. Nuclear fractionation reveals thousands of chromatin-tethered noncoding RNAs adjacent to active genes. Cell Rep. 2015;12(7):1089–1098. doi: 10.1016/j.celrep.2015.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pelechano V, Steinmetz LM. Gene regulation by antisense transcription. Nat Rev Genet. 2013;14(12):880–893. doi: 10.1038/nrg3594. [DOI] [PubMed] [Google Scholar]
- 16.Kurokawa R. Promoter-associated long noncoding RNAs repress transcription through a RNA binding protein TLS. Adv Exp Med Biol. 2011;722:196–208. doi: 10.1007/978-1-4614-0332-6_12. [DOI] [PubMed] [Google Scholar]
- 17.Thurman RE, et al. The accessible chromatin landscape of the human genome. Nature. 2012;489(7414):75–82. doi: 10.1038/nature11232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gerstein MB, et al. Architecture of the human regulatory network derived from ENCODE data. Nature. 2012;489(7414):91–100. doi: 10.1038/nature11245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang J, et al. Sequence features and chromatin structure around the genomic regions bound by 119 human transcription factors. Genome Res. 2012;22(9):1798–1812. doi: 10.1101/gr.139105.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang J, et al. Factorbook.org: a Wiki-based database for transcription factor-binding data generated by the ENCODE consortium. Nucleic Acids Res. 2013;41(Database issue):D171–D176. doi: 10.1093/nar/gks1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Johnson DS, Mortazavi A, Myers RM, Wold B. Genome-wide mapping of in vivo protein-DNA interactions. Science. 2007;316(5830):1497–1502. doi: 10.1126/science.1141319. [DOI] [PubMed] [Google Scholar]
- 22.Pikkarainen S, et al. GATA-4 is a nuclear mediator of mechanical stretch-activated hypertrophic program. J Biol Chem. 2003;278(26):23807–23816. doi: 10.1074/jbc.M302719200. [DOI] [PubMed] [Google Scholar]
- 23.Man J, Barnett P, Christoffels VM. Structure and function of the Nppa-Nppb cluster locus during heart development and disease. Cell Mol Life Sci. 2018;75(8):1435–1444. doi: 10.1007/s00018-017-2737-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mercadier JJ, et al. Atrial natriuretic factor messenger ribonucleic acid and peptide in the human heart during ontogenic development. Biochem Biophys Res Commun. 1989;159(2):777–782. doi: 10.1016/0006-291X(89)90062-4. [DOI] [PubMed] [Google Scholar]
- 25.Szabo L, et al. Statistically based splicing detection reveals neural enrichment and tissue-specific induction of circular RNA during human fetal development. Genome Biol. 2015;16:126. doi: 10.1186/s13059-015-0690-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li JT, Zhang Y, Kong L, Liu QR, Wei L. Trans-natural antisense transcripts including noncoding RNAs in 10 species: implications for expression regulation. Nucleic Acids Res. 2008;36(15):4833–4844. doi: 10.1093/nar/gkn470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Johnsson P, Lipovich L, Grandér D, Morris KV. Evolutionary conservation of long non-coding RNAs; sequence, structure, function. Biochim Biophys Acta. 2014;1840(3):1063–1071. doi: 10.1016/j.bbagen.2013.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Piatek MJ, Henderson V, Zynad HS, Werner A. Natural antisense transcription from a comparative perspective. Genomics. 2016;108(2):56–63. doi: 10.1016/j.ygeno.2016.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pang KC, Frith MC, Mattick JS. Rapid evolution of noncoding RNAs: lack of conservation does not mean lack of function. Trends Genet. 2006;22(1):1–5. doi: 10.1016/j.tig.2005.10.003. [DOI] [PubMed] [Google Scholar]
- 30.Khaitovich P, et al. Functionality of intergenic transcription: an evolutionary comparison. PLoS Genet. 2006;2(10):e171. doi: 10.1371/journal.pgen.0020171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mathews DH, Moss WN, Turner DH. Folding and finding RNA secondary structure. Cold Spring Harb Perspect Biol. 2010;2(12):a003665. doi: 10.1101/cshperspect.a003665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Houweling AC, van Borren MM, Moorman AF, Christoffels VM. Expression and regulation of the atrial natriuretic factor encoding gene Nppa during development and disease. Cardiovasc Res. 2005;67(4):583–593. doi: 10.1016/j.cardiores.2005.06.013. [DOI] [PubMed] [Google Scholar]
- 33.Huo J, et al. Evaluation of batch variations in induced pluripotent stem cell-derived human cardiomyocytes from 2 major suppliers. Toxicol Sci. 2017;156(1):25–38. doi: 10.1093/toxsci/kfw235. [DOI] [PubMed] [Google Scholar]
- 34.Bernstein E, Allis CD. RNA meets chromatin. Genes Dev. 2005;19(14):1635–1655. doi: 10.1101/gad.1324305. [DOI] [PubMed] [Google Scholar]
- 35.Morris KV. Long antisense non-coding RNAs function to direct epigenetic complexes that regulate transcription in human cells. Epigenetics. 2009;4(5):296–301. doi: 10.4161/epi.4.5.9282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Clark BS, Blackshaw S. Long non-coding RNA-dependent transcriptional regulation in neuronal development and disease. Front Genet. 2014;5:164. doi: 10.3389/fgene.2014.00164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Trapnell C, et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 2012;7(3):562–578. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Roberts A, Pimentel H, Trapnell C, Pachter L. Identification of novel transcripts in annotated genomes using RNA-Seq. Bioinformatics. 2011;27(17):2325–2329. doi: 10.1093/bioinformatics/btr355. [DOI] [PubMed] [Google Scholar]
- 39.Roselli C, et al. Multi-ethnic genome-wide association study for atrial fibrillation. Nat Genet. 2018;50(9):1225–1233. doi: 10.1038/s41588-018-0133-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chu C, Quinn J, Chang HY. Chromatin isolation by RNA purification (ChIRP) J Vis Exp. 2012;(61):3912. doi: 10.3791/3912. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.