

We present evidence for the dual role of epigenetics and transposable elements have a dual role in the reproductive isolation of nascent species.
Abstract
The genomic shock hypothesis stipulates that the stress associated with divergent genome admixture can cause transposable element (TE) derepression, which could act as a postzygotic isolation mechanism. TEs affect gene structure, expression patterns, and chromosome organization and may have deleterious consequences when released. For these reasons, they are silenced by heterochromatin formation, which includes DNA methylation. Here, we show that a significant proportion of TEs are differentially methylated between the “dwarf” (limnetic) and the “normal” (benthic) whitefish, two nascent species that diverged some 15,000 generations ago within the Coregonus clupeaformis species complex. Moreover, TEs are overrepresented among loci that were demethylated in hybrids, indicative of their transcriptional derepression. These results are consistent with earlier studies in this system that revealed TE transcriptional derepression causes abnormal embryonic development and death of hybrids. Hence, this supports a role of DNA methylation reprogramming and TE derepression in postzygotic isolation of nascent animal species.
INTRODUCTION
Divergence between evolutionary lineages generates genetic incompatibilities that may lead to hybrid sterility and/or inviability [i.e., Bateson-Dobzhansky-Muller (BDM) model of postzygotic reproductive isolation] (1). Besides well-studied animal model systems (e.g., Drosophila sp. and Mus sp.), little is known about the molecular mechanisms that can keep nascent species apart (1–3). Recent work investigating the role of transposable elements (TEs) and epigenetics in speciation extends the BDM model to a broader interpretation (4–6). This raised the hypothesis that TE derepression in hybrids is caused by a genomic shock produced by the admixture of two parental genomes and may act as a postzygotic barrier (7).
TEs affect gene structure, expression patterns, and chromosome organization, but their mobilization can have profoundly deleterious consequences (8, 9). Inactivation of TEs can be achieved by mutation accumulation over time, but transcriptional silencing of active TEs is achieved by piRNAs (small noncoding RNA that interact with PIWI proteins) that mediate heterochromatin formation, including DNA methylation (an epigenetic process by which methyl groups are added to DNA) (4, 10–12). Hence, piRNAs can play a role in establishing persistent DNA methylation patterns, as genes exhibiting persistent transgenerational DNA methylation patterns often contain a TE insertion nearby (10–12). In nascent species, mutation accumulation is not expected to contribute substantially to TE inactivation in comparison to piRNA pathway proteins that show rapid evolution, which suggests an “evolutionary arms race” with TEs (13). Therefore, incompatibilities may arise in hybrids between divergent lineages, which may explain DNA methylation pattern changes and transcriptional derepression of TEs (1). Interplay between DNA methylation and TEs has been documented as a key element of postzygotic isolation in plants (4, 12), but its potential role in the reproductive isolation of nascent animal species remains to be investigated. Documenting the interplay between DNA methylation and TEs in nascent animal species is, thus, a necessary step toward our global understanding of the role of TEs in postzygotic isolation.
The “normal” and “dwarf” lake whitefish species pairs are salmonids belonging to the Coregonus clupeaformis complex that have recently diverged during Pleistocene glaciation (~15,000 generations/60,000 years BP) (14, 15). Following independent secondary contact (~3000 generations ago/12,000 years BP) in several lakes of the St. John River basin (northeastern of North America), competitive interactions led to the parallel evolution of a dwarf species specializing to a limnetic niche, while the normal remained benthic specialist (14, 15). Adaptive phenotypic differentiation of behavioral, physiological, and morphological traits has been amply documented in wild and controlled environments (14, 16, 17). Transcriptomic studies revealed that overexpressed transcripts in the dwarf species are associated with “survival processes” including metabolic processes, immune system, and detoxification functions, whereas overexpressed transcripts in the normal species are associated with growth functions (14, 18). Moreover, the very low level of genomic differentiation in protein-coding genes supports the predominant role of regulatory regions in driving reproductive barrier (14, 19). Hybrids show reduced sperm performance, increased mortality during development, chromosomal instability, larval malformation, and fluctuating asymmetry (14, 20). These phenotypic effects are associated with a transcriptome-wide breakdown and an enrichment of TEs among overexpressed transcripts, which indicates TE derepression in hybrids (21, 22). Dwarf and normal whitefish are thus an excellent system to investigate the role of DNA methylation on TE derepression in hybrids to act as a postzygotic reproductive barrier between nascent animal species.
Here, we test for TE enrichment in DNA methylation differentiation, which would support a role for DNA methylation in TE regulation. First, we test for TE enrichment in DNA methylation differentiation between species to show that a distinct evolutionary path of TE silencing has occurred during the speciation of dwarf and normal whitefish. Then, we test for TE enrichment associated to a hypermethylation pattern in parental lineages in comparison to hybrids to support a role of DNA demethylation on TE derepression and associated postzygotic isolation.
RESULTS
Global patterns of epigenomic and genomic variation in lake whitefish
To control for environmental variation, we reared fish in a common environment and sampled the liver of 24 adult fish from four groups: six dwarfs, six normals, six dwarf ♀ × normal hybrids, and six normal ♀ × dwarf hybrids. Because the liver is involved in a broad diversity of biological functions, it expresses a broad array of genes. The liver is also a very homogeneous tissue in cell types, thus mitigating potential artefactual biases in methylation levels assessment caused by differential representation of various cell types. Moreover, there is a liver transcriptome available for whitefish, and previous studies also revealed differential gene expression and TE derepression in this tissue between dwarf and normal whitefish (18, 22). We constructed Illumina sequencing libraries and applied reduced-representation bisulfite sequencing and an average of read depth of 10× average per fish. CpGs were then confirmed after being check for C-T DNA polymorphism (see Materials and Methods for further precision). Following filtering, 10,374 of 530,710 single-nucleotide polymorphisms (SNPs) and 423,652 of 4,761,101 CpGs were kept for downstream analyses. We observed a clear group differentiation at both genomic and epigenomic levels [95% confidence interval (CI) ellipses; Fig. 1, A and B]. Parental groups show maximum differentiation at both genomic and epigenomic levels, whereas hybrids were intermediate (Fig. 1, A and B). Genomic and epigenomic variations were strongly correlated (Mantel test: r = 0.69; 95% CI, 0.48 to 0.90; P < 0.001), although the amount of variance explained by groups was three times higher at the genomic compared with the epigenomic level (27% versus 10%; Fig. 1, C and D). All four groups showed a similar level of DNA methylation, with almost 80% of CpGs being methylated (Fig. 1E). Last, pairwise individual correlations of DNA methylation at CpG sites varied between 0.93 and 0.96 (all P < 0.001), showing three main clusters based on UPGMA (Unweighted Pair Group Method with Arithmetic mean), which indicate higher correlation within (i) dwarf species, (ii) normal species, and (iii) all hybrids (Fig. 1F).
Fig. 1. Epigenomic and genomic variations between dwarf and normal whitefish species and their reciprocal hybrids.
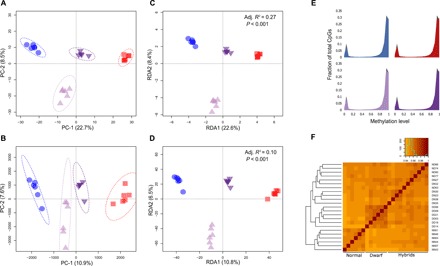
(A) Principal components analysis (PCA) on 10,374 SNPs. (B) PCA on 423,652 CpGs. (C) Redundancy analysis (RDA) on 10,374 SNPs where genetic variance is explained by the variable lineage. (D) RDA on 423,652 CpGs where epigenetic variance is explained by the variable lineage. Blue circles represent dwarf individuals (DD), red squares represent normal individuals (NN), dark purple down-triangles represent normal ♀-dwarf hybrids (ND), and pale purple up-triangles represent dwarf ♀-normal hybrids (DN). (E) Distribution of CpG according to their methylation level for pure and hybrid whitefish groups represented by the same color used above. (F) Pairwise individual correlation between CpG methylation levels. The tree was constructed using UPGMA methods, and the correlation matrix of level of methylation was observed among individuals.
Different DNA methylation of loci enriched in TEs between whitefish species
We tested for epigenetic differentiation between the two species and found 7272 CpGs distributed among 4188 loci with a methylation level of at least 20% (Fig. 2A). Among those 4188 loci, 1319 (31.5%) contain two or more CpGs with methylation differentiation in the same direction, which can modulate heterochromatin formation (23). To visualize epigenetic differentiation between the two species, we applied an unsupervised clustering on the 7272 CpGs in four k-means categories of CpGs (1 and 2: high and low methylation in normal species; 3 and 4: high and low methylation in dwarf species), which differentiated the two pure species (Fig. 2C). Mean variance among CpGs was significantly different (t test, all P < 0.001) between species within each k-means category of CpGs. In addition, dwarf whitefish shows an overall hypermethylation of the 7272 differently methylated CpGs (3859 hyper- versus 3413 hypomethylated CpGs; λ2 = 27.35, P < 0.001).
Fig. 2. DNA methylation differentiation between dwarf and normal whitefish species and their reciprocal hybrids.
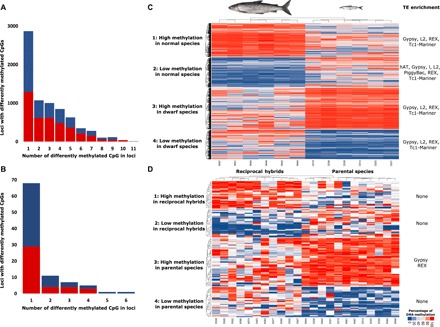
Methylation differentiation between pure dwarf and normal whitefish species (top panels) and between parental species and their hybrids (bottom panels). (A and B) Histograms representing the distribution of loci differentially methylated based on how many differently methylated CpGs (>20%) they harbor. Red and blue coloration represent hyper- and hypomethylation in dwarf in comparison to normal (A) and hybrids in comparison to parental lineages (B), respectively. (C and D) Heatmaps of differentially methylated CpGs (>20%) separated in four k-means groups that separated pure species (C) and pure species from hybrids (D). TE enrichment (FDR < 0.05) for each CpG category is provided at the right of each heatmap.
We annotated TEs based on a recent lake whitefish reference transcriptome (18), using the zebrafish (Danio rerio) TE database from Repbase (24) to annotate the 53,104 assembled transcripts. A total of 2643 transcripts (5.0% of the transcriptome) were annotated as TEs (tblastx; e-value < 1 × 10−6; Fig. 3A). To control for species-specific TEs in the transcriptome, we removed 1038 transcripts unique to one species (2.0%). We found TE superfamily proportions similar to our previous transcriptome study where we reported a significant overrepresentation of TEs among differentially expressed transcripts between dwarf and normal whitefish (22). These represent 5 superfamilies of long terminal repeat (LTR retrotransposons; 366 transcripts), 9 of non-LTR retrotransposons (658 transcripts), and 15 of DNA transposons (1599 transcripts; Fig. 3A). Here, we found an overrepresentation of TEs in loci harboring CpGs differently methylated between the two species (one-sided Fisher exact test, P < 0.001). In particular, we observed enrichment for Gypsy, REX, L2, and Tc1-Mariner superfamilies within each k-means CpG category [one-sided Fisher exact tests, false discovery rate (FDR) < 0.05; Fig. 2C]. In addition, a total of 38.5% of transcripts annotated as Gypsy harbor differentially methylated CpGs, 23.5% for REX, 14.0% for L2, and 11.1% for Tc1-Mariner (all supporting overrepresentation; one-sided Fisher exact tests, P < 0.001; Fig. 3B).
Fig. 3. Transposable element annotation and their proportions of DNA methylation differentiation between dwarf and normal whitefish species.
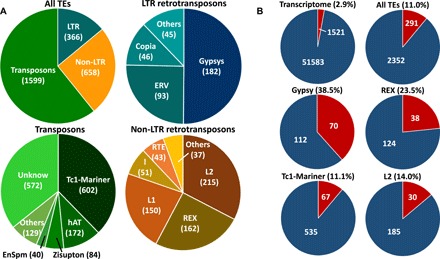
(A) Pie chart of annotated TEs in the transcriptome for all TEs, LTR retrotransposons, non-LTR retrotransposons, and transposons. (B) Proportion of transcripts harboring at least one CpG differently methylated in the entire transcriptome, in all transcripts annotated as TEs, in all transcripts annotated as a Gypsy TE superfamily, in addition to REX, Tc1-Mariner, and L2 superfamilies.
Hypomethylation of TEs in hybrids
To investigate the potential demethylation that may underlie transcriptional derepression of TEs in hybrid whitefish, we tested for methylation differentiation between parental species and hybrids. We found 142 CpGs distributed among 93 loci (Fig. 2B) with a methylation level higher than 20%. Among these, 25 (26.9%) contain at least two differentially methylated CpGs between parental species and hybrids, and none of them show methylation differentiation in the opposite direction. Loci harboring differentially methylated CpG between hybrids and pure species align to 56 transcripts (e-value < 1 × 10−6), of which 13 (23.2%) were TEs, which represents a highly significant overrepresentation of TEs present among differently methylated CpGs (one-sided Fisher exact test, P < 0.001). The heatmap distinguished hybrids from parents, but only the k-means CpG category 3 (high methylation in parental species) showed a difference in mean variance (t = 6.80, df = 99.43, P < 0.001; Fig. 2D), where hybrids are characterized by a higher variance at these CpGs in comparison to dwarf and normal individuals pooled together. Furthermore, this CpG category contains more transcripts harboring a TE than the others (8 of the 13; λ2 = 6.42, P = 0.011). This CpG category was also significantly enriched in Gypsy and REX TE superfamilies (one-sided Fisher exact tests, FDR with α = 0.05; Fig. 2D).
DISCUSSION
Global pattern of epigenomic and genomic variations in lake whitefish
We observed a strong association between genomic and epigenomic variation paired to a significantly higher genomic variation explained by groups, which was almost three times higher than epigenomic variation. Together, this provides support for a genetic basis of epigenomic variation between dwarf and normal whitefish. This is also supported by the intermediate position of hybrids in epigenomic variation, as would be expected under a Mendelian inheritance pattern. Unlike our study, a better relationship between lineages and epigenomic rather than genomic variation is generally observed in wild populations (25, 26). Thus, the higher association between lineages and genomic variation observed in our study suggests that the experimental conditions control for environmental effects on epigenomic variation. Furthermore, we observed a pattern of high methylation across the genome (around 80%), which is similar to previous reports in other vertebrates (27–29). We also observed a pronounced pairwise individual correlations at CpG sites. These two observations suggest that laboratory conditions did not cause an important stress generating stochastic variation in DNA methylation. Overall, this suggests that the variation we observed in whitefish methylome is reliable to address questions about the interplay between DNA methylation and TEs.
Different DNA methylation of loci enriched in TEs between whitefish species
We found a total of 7272 CpGs with methylation differentiation over 20% that differentiates the two whitefish species. In addition to global epigenomic variation pointed out above, this indicates an important methylation differentiation between dwarf and normal whitefish and supports the hypothesis of a distinct evolution of species methylome since their origin some 15,000 generations ago. In turn, this could contribute to phenotypic differentiation and postzygotic isolation based on the documented impact of DNA methylation on gene expression, DNA mutation rates, and TE silencing (10–12, 30). Furthermore, enrichment of Gypsy, REX, L2, and Tc1-Mariner TE superfamilies was present within each CpG category differentiating the two species. Moreover, our results show an overrepresentation of differently methylated CpGs in transcripts harboring these TE superfamilies, which may reflect both differentiation of their methylome and differentiation between dwarf and normal whitefish species associated to TE silencing during the speciation process. TE variation in activities, abundance, and diversity has often been associated with speciation events, and this is particularly true in fish evolution where their genomes show the highest diversity of TE superfamilies in vertebrates (31–34). Specifically, Gypsy, REX, L2, and Tc1-Mariner superfamilies show distinct activities throughout the evolutionary history of fish diversification, including salmonids (31–34). Given that regulation of DNA methylation is a major mechanism of TE silencing (10–12), specific pattern of DNA methylation on TEs suggests a first step than can potentially lead to TE derepression in hybrids and associated postzygotic isolation.
Hypomethylation of TEs in hybrids
We predicted that if TE derepression is caused by DNA methylation deregulation in hybrid, then we should observe an enrichment in TEs in a category of CpGs showing high methylation common to both parental species, but hypomethylation in hybrids. Here, we show that CpGs showing high methylation in both parental species and reduced methylation in hybrids are significantly enriched in Gypsy and REX TE superfamilies. This corroborates the observation of a transcriptional up-regulation of Gypsy and REX TE superfamilies that we previously reported in whitefish malformed hybrids (22). We propose that DNA hypomethylation is a likely molecular mechanism by which TEs are transcriptionally released, which may, in turn, contribute to postzygotic isolation by causing genomewide gene deregulation, as reported previously (14, 22).
Pronounced aneuploidy caused by mitotic and meiotic instability was also previously documented in whitefish hybrids (20). In marsupial hybrids from two different genera, global DNA hypomethylation patterns and TE derepression are associated with chromosomal remodeling (6). Wasp hybrids show TE derepression causing aneuploidy in the form of chromosome fragmentation (35). In addition, bursts of Gypsy TEs in supernumerary chromosome were observed in cyprinid speciation (36). In the light of those previous studies and considering the capacity of active TEs to affect chromosome structure (6, 8, 9), we propose that hybrid aneuploidy in lake whitefish may also be associated with TE hypomethylation.
CONCLUSION
Overall, our results point to (i) distinct evolution of TE silencing between dwarf and normal species and (ii) a role of DNA hypomethylation of TEs in hybrid dysfunction previously documented at the morphological, chromosomal, and transcriptional levels. Methylation patterns of the Gypsy and REX superfamilies were found to differ between whitefish species and be hypomethylated in hybrids relative to both parental forms. Thus, we propose that methylation of Gypsy and REX superfamilies contributes to the postzygotic isolation of these two nascent species. Next steps will be to (i) test for an amplification of DNA hypomethylation in Gypsy, REX, and other TEs in malformed backcross as expected by the BDM model, (ii) screen for mutations near TEs and inside the piRNA pathway proteins to identify sources of divergence underlying postzygotic isolation, and (iii) confirm that DNA hypomethylation affects TE activity in hybrids. Hence, this study supports a role of TE methylation reprogramming in hybrids as a mechanism of postzygotic barrier between nascent animal species. It also contributes to our global understanding on the role of the interplay between TEs and DNA methylation in speciation and adds to the growing evidence regarding their importance in evolutionary biology.
MATERIALS AND METHODS
Study design
This study aimed at testing the hypothesis of TE enrichment in DNA methylation differentiation between nascent animal species and its role as a postzygotic isolation mechanism associated with TE derepression in hybrids. To this end, we produced a Reduced Representation Bisulfite Sequencing library of two species of lake whitefish and their reciprocal hybrids, all raised in a common garden. We first tested for TE enrichment in DNA methylation differentiation between species to support a distinct evolution of TE silencing. Then, we tested for TE enrichment associated to a hypermethylation pattern in parental lineages in comparison to hybrids to support a role of DNA demethylation on TE derepression and associated postzygotic isolation. We annotated the lake whitefish transcriptome (18) with the zebrafish Repbase database (24) before testing for enrichment. All protocols were approved by Université Laval’s animal care committee (Protocol 82178).
Common garden and sample collection
Crosses were produced from parents originating from Témiscouata Lake (dwarf whitefish, Acadian lineage, 47°36′N, 68°45′W) and Aylmer Lake (normal whitefish, Atlantic lineage, 45°50′N, 71°26′W) in Québec, Canada, that were caught in November 2012. Artificial fertilization was conducted in the Laboratoire de Recherche en Sciences Aquatiques (Université Laval). Gametes from multiple females and males were mixed: The dwarf crosses were produced from 7 females and 12 males, the normal crosses from 5 females and 8 males, the ND crosses from 5 normal females and 12 dwarf males, and the DN crosses from 3 dwarf females and 2 normal males. The crosses were raised 5 years in a common garden, which consisted of freshwater tanks with a flow through system under identical temperatures, photoperiod, and diet. The livers of six individuals per group (total of 24) were sampled for subsequent analyses in spring 2017, a nonreproductive period of the species to mitigating putative epigenetic remodeling linked to sexual maturation.
DNA extraction and reduced representation bisulfite sequencing library preparation
The RRBS (Reduced Representation Bisulfite Sequencing) library preparation procedure was based on Gu et al. (37). DNeasy blood and tissue kit columns for the extraction of genomic DNA (gDNA) were used following the protocol guidelines (QIAGEN), and DNA quantification was calibrated with Quant-iT PicoGreen dsDNA (Fluoroskan Ascent FL; Thermo Fisher Scientific). Restriction enzyme Msp I was used to digest gDNA (600 ng), and magnetic beads (volume ratio, 1.8×) and 80% ethanol were used to clean and rinse twice the solution. Solution containing Klenow fragments (5 U; New England BioLabs Inc.) and dNTPs (0.5 mM dATP, 0.05 mM dGTP, 0.05 mM dCTP) were used for end-repair and A-tailing steps with an incubation step (30°C for 30 min and 37°C for 20 min). Magnetic beads (ratio 1.8×) and 80% ethanol were used to clean and rinse twice the solution. A master mix solution containing the T4 ligase buffer (1×; New England BioLabs Inc.) and a T4 ligase (2000 U) was used to ligate NEXTflex illumina DNA barcodes (Bio Scientific) during an overnight incubation at 16°C. Magnetic beads (ratio 1×) and 80% ethanol were used to clean and rinse the solution. Two rinses with magnetic beads (first wash ratio 0.7× and beads were discarded, second wash ratio 0.15×) were used for capturing specific fragments with lengths ranging from 200 to 400 base pairs (bp). A bisulfite conversion treatment was then conducted following EZ DNA Methylation-Gold Kit protocol recommendations (Zymo Research). Library quality and quantity were verified on a HiSense DNA Bioanalyzer 2100 chip (Agilent) and with Quant-iT PicoGreen assays (Fluoroskan Ascent FL; Thermo Labsystems), respectively. Libraries were sequenced on a HiSeq 2000 platform (five individuals by lane) at the McGill University and Génome Québec Innovation Centre (Montréal, QC) using a 100-bp single-end read approach.
Methylation and SNP calling
The software Cutadapt (38) implemented in Trim_Galore! v0.4.1 (www.bioinformatics.babraham.ac.uk/projects/trim_galore/) was first used to trim RRBS reads. After quality filtering and sample quality check, we removed one sample from the ND hybrid group that had low coverage (Methylkit “GetCoverageStats” function) and was identified as an outlier in a principal components analysis (PCA). Bismark v0.14.5 aligner (39) and its function “methyl extraction” were used on trimmed reads for a de novo alignment and extracting methylation information of cytosine in a CpG context. Only CpGs with a minimum coverage of 10× that do not show C-T DNA polymorphism in RAD sequencing (530,710 SNPs) were kept for subsequent analyses. In this way, variation in observed patterns of methylation cannot be confounded with C-T DNA polymorphism. Bisulfites sequencing estimates DNA methylation by converting nonmethylcytosine to uridine, which is then converted to thymine through sequencing. Without controlling for C-T DNA polymorphism, different allelic frequencies can thus be confounded as methylation differentiation (27). Methylation levels on normalized count across individuals were compiled with the MethylKit R package (40). Global DNA methylation patterns were explored by calculating the percentage of CpGs methylated with a minimum coverage of 10× for all individuals, as well as for pure normal and dwarf whitefish and their reciprocal hybrids using the function “getData” from the same R package. In addition, the fraction of total CpGs for each bin of 5% of methylation level (number of reads methylated divided by the total number of reads for a given CpG) was calculated for all four groups. Last, average Pearson correlation between levels of methylation at the CpG sites was calculated for each pairwise individual comparison.
For the RAD sequencing data, the software Cutadapt (38) was first used to trim adaptor sequences, length (80 bp) with default parameters. Librairies were demultiplexed using the “process_radtags” module in Stacks v1.44 (41). SNP calling was conducted using the Stacks v1.44 de novo approach. A reference catalog of consensus loci was first built using the “ustacks” module fixing minimum depth of coverage at four and allowing for up to three mismatches and the “cstacks” module with default parameters. The “sstacks” module allowed the mapping of individual reads to the reference catalog, and raw vcf file was produced using the “population” module. SNP filtering was conducted with the VCFtools v0.1.14 software (42), and only biallelic markers with minimum and maximum depths of coverage between 5× and 100×, minor allele frequency >0.05, minimum quality of 5, maximum missing data of 20%, and in Hardy-Weinberg equilibrium (P > 0.05) were conserved. A total of 10,374 from the initial 530,710 SNPs were kept for subsequent genomic analysis.
Genetic and DNA methylation variation
To visualize DNA methylation and SNP variation among dwarf and normal whitefish species and their reciprocal hybrids, we performed PCAs on all individuals for both the 423,652 CpGs and the 10,374 filtered SNP databases. Redundancy analyses (RDAs) were produced to assess the extent of variance in both DNA methylation and SNP variation explained by “lineage group” with the function “rda” of the VEGAN R package (43). RDA is a variant of canonical correlation analysis that gives less weight to rare variables, such as scoring error (44). Analyses of variance (ANOVAs; 1000 permutations) were performed to assess the global significance of each model, and the function “RsquareAdj” was used to estimate the percentage of variance explained in each RDA. Last, a Mantel test on Euclidean distance was performed with the function “mantel” to estimate the correlation between the genetic and the epigenetic datasets.
DNA methylation differentiation between groups
The Methylkit R package was used to retain the CpGs showing at least 20% of difference between groups (A, normal versus dwarf species; B, pure species versus hybrid crosses), with a q value <0.001. For the comparisons between pure species and their hybrids, we also verified that at least 20% of difference was observed between all of the following comparisons: dwarf versus DN hybrid, dwarf versus ND hybrid, normal versus DN hybrid, and normal versus ND hybrid. These additional filtering steps were performed to avoid cases of CpG differentiation caused by (i) differentiation between dwarf and normal species, where hybrids show more similarities to one species, and (ii) differentiation between DN hybrids and all other individuals, as shown by overall genomic variation in Fig. 1A. When more than one CpG showing differentiation higher than 20% was present on a single locus, we verified the directionality of these differentiation and removed loci that showed incongruence among CpGs, for instance, if one CpG was hypomethylated and another was hypermethylated. We found that 1319 (98.7%) of the 1337 loci in the pure dwarf versus normal comparison with two CpGs or more varied in the same direction, whereas all of the 25 loci identified between pure parental species and hybrids were found (binomial tests; both P < 0.001), thus indicating high congruence of nearby DNA methylation. Heatmap of the percentage of DNA methylation at differentially methylated CpG sites for each individual was conducted with the function “HeatmapAnnotation” of the R package ComplexHeatmap (45). Clustering of CpGs was based on a k-means where the number of groups was selected after considering the within-group sum of squares statistics (44). We tested for significant difference in the level average variance of methylation between groups among CpGs of each of the four k-means categories. Because average near 0 and 1 of a frequency variable is likely to produce smaller variance, we corrected the variance of the methylation level of a CpG by dividing it with the maximum variance, which is “s × (1 − s),” where s is the mean methylation level. Last, TE annotation was performed by a blast search (90 bp) against the annotated lake whitefish transcriptome (18) (see below).
Reference transcriptome, annotations, alignments, and enrichment test
We used a previously built reference transcriptome (53,104 contigs with an N50 of 1.77 kbp) (18). Briefly, raw liver RNA sequencing data from six normal and six dwarf individuals from wild populations (Cliff Lake) were cleaned and trimmed using Trimmomatic v0.36 (46). Individuals from Cliff Lake sympatric species pair are representative of their ancestral allopatric background (15). We de novo assembled a composite reference transcriptome for the two species, using Trinity v2.2.0 (47). We kept only contigs with an open reading frame longer than 300 bp and only the longest isoforms. We applied a scaling factor of one transcript per million to normalized raw read counts for subsequent gene expression analysis.
We then used a blastx approach against the zebrafish (D. rerio) Repbase database (24) to annotate in TEs the lake whitefish reference transcriptome. Briefly, we used Repbase as reference database to identify repetitive DNA elements in the built composite transcriptome and transfer the annotation information based on sequence similarities. Matches against the databases with an e-value below 1 × 10−6 were discarded, allowing the identification of TEs in our transcriptome based on sequence similarities.
We thus aligned RADtags from methylation data against the annotated reference transcriptome using blast v.2.6.0 (48) to physically link and annotate studied loci. Matches with an e-value below 1 × 10−6 were discarded. Moreover, we applied a stringent filtering step by keeping only one transcript per differently methylated locus, allowing to directly associate CpGs to transcripts (i.e., gene sequence or TEs). Enrichment was conducted after removing any duplicated transcript using one-sided Fisher exact tests (FDR = 0.05). Tests were produced for all TEs, and all specific TE superfamilies were encountered among all differently methylated loci.
Statistical analyses
All statistical analyses were performed, and graphs were drawn using the R v3.4.1 software (R Core Team; www.r-project.org). Correction for multiple tests using FDR was applied for testing CpGs differently methylated and TE enrichment. P < 0.05 was considered statistically significant.
Acknowledgments
We thank É. Normandeau, G. Côté, A. Dalziel, and M. Sevellec for whitefish rearing, bioinformatics support, and sampling. Four anonymous reviewers helped improve the article. Computations were carried out on the supercomputer Colosse, Université Laval, managed by Calcul Québec and Compute Canada and on local servers (Katak). Funding: This research was carried out with the Canadian Research Chair in genomics and conservation of aquatic resources led by L.B. (#DG-180965-2015). M.L. was supported by a postdoctoral scholarship from the Natural Sciences and Engineering Research Council of Canada (NSERC #BP-471682-2015) and Ressources Aquatic Québec (RAQ) travel awards. A.-M.C.-D. was supported by a postdoctoral scholarship from the Fonds de Recherche de Santé du Québec–Santé (FRQ-S #33616), the Natural Sciences and Engineering Research Council of Canada (NSERC #PDF-516851-2018), and the Lawski Foundation. Author contributions: M.L., L.B., and A.-M.D.-C. designed the research. M.L. and J.L.L. performed the research. M.K. and A.-M.D.-C. conducted rearing and sampling. C.R. contributed to new analytic tools. M.L. and J.L.L. analyzed the data. All authors participated in the writing of manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: The sequences reported in this paper have been deposited in the National Center for Biotechnology Information Sequence Read Archive (www.ncbi.nlm.nih.gov/sra/; BioProject accession no. PRJNA501856). All data needed to evaluate the conclusions in the paper are present in the paper. Additional data related to this paper may be requested from the authors.
REFERENCES AND NOTES
- 1.Presgraves D. C., The molecular evolutionary basis of species formation. Nat. Rev. Genet. 11, 175–180 (2010). [DOI] [PubMed] [Google Scholar]
- 2.Dion-Côté A.-M., Barbash D. A., Beyond speciation genes: An overview of genome stability in evolution and speciation. Curr. Opin. Genet. Dev. 47, 17–23 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Maheshwari S., Barbash D. A., The genetics of hybrid incompatibilities. Annu. Rev. Genet. 45, 331–355 (2011). [DOI] [PubMed] [Google Scholar]
- 4.Lafon-Placette C., Köhler C., Epigenetic mechanisms of postzygotic reproductive isolation in plants. Curr. Opin. Plant Biol. 23, 39–44 (2015). [DOI] [PubMed] [Google Scholar]
- 5.Quadrana L., Silveira A. B., Mayhew G. F., LeBlanc C., Martienssen R. A., Jeddeloh J. A., Colot V., The Arabidopsis thaliana mobilome and its impact at the species level. eLife 5, e15716 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.O’Neill R. J. W., O’Neill M. J., Graves J. A. M., Undermethylation associated with retroelement activation and chromosome remodelling in an interspecific mammalian hybrid. Science 393, 68–72 (1998). [DOI] [PubMed] [Google Scholar]
- 7.McClintock B., The significance of responses of the genome to challenge. Science 226, 792–801 (1984). [DOI] [PubMed] [Google Scholar]
- 8.Dubin M. J., Mittelsten Scheid O., Becker C., Transposons: A blessing curse. Curr. Opin. Plant Biol. 42, 23–29 (2018). [DOI] [PubMed] [Google Scholar]
- 9.Goodier J. L., Restricting retrotransposons: A review. Mobile DNA 7, 16 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rey O., Danchin E., Mirouze M., Loot C., Blanchet S., Adaptation to global change: A transposable element–epigenetics perspective. Trends Ecol. Evol. 31, 514–526 (2016). [DOI] [PubMed] [Google Scholar]
- 11.Hosaka A., Kakutani T., Transposable elements, genome evolution and transgenerational epigenetic variation. Curr. Opin. Genet. Dev. 49, 43–48 (2018). [DOI] [PubMed] [Google Scholar]
- 12.Weil C., Martienssen R., Epigenetic interactions between transposons and genes: Lessons from plants. Curr. Opin. Genet. Dev. 18, 188–192 (2008). [DOI] [PubMed] [Google Scholar]
- 13.Simkin A., Wong A., Poh Y.-P., Theurkauf W. E., Jensen J. D., Recurrent and recent selective sweeps in the piRNA pathway. Evolution 67, 1081–1090 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bernatchez L., Renault S., Whiteley A. R., Derome N., Jeukens J., Landry L., Nolet A. W., Østbye K., Rogers S. M., St-Cyr J., On the origin of species: Insights from the ecological genomics of lake whitefish. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 365, 1783–1800 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rougeux C., Bernatchez L., Gagnaire P.-A., Modeling the multiple facets of speciation-with-gene-flow toward inferring the divergence history of Lake Whitefish species pairs (Coregonus clupeaformis). Genome Biol. Evol. 9, 2057–2074 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dalziel A. C., Martin N., Laporte M., Guderley H., Bernatchez L., Adaptation and acclimation of aerobic exercise physiology in Lake Whitefish ecotypes (Coregonus clupeaformis). Evolution 69, 2167–2186 (2015). [DOI] [PubMed] [Google Scholar]
- 17.Laporte M., Dalziel A. C., Martin N., Bernatchez L., Adaptation and acclimation of traits associated with swimming capacity in Lake Whitefish (coregonus clupeaformis) ecotypes. BMC Evol. Biol. 16, 160 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rougeux C., Gagnaire P.-A., Praebel K., Seehausen O., Bernatchez L., Convergent transcriptomic landscapes under polygenic selection accompany inter-continental parallel evolution within a Nearctic Coregonus (Salmonidae) sister-species complex. bioRxiv 2018, 311464 (2018). [Google Scholar]
- 19.Hebert F. O., Renaut S., Bernatchez L., Targeted sequence capture and resequencing implies a predominant role of regulatory regions in the divergence of a sympatric lake whitefish species pair (Coregonus clupeaformis). Mol. Ecol. 22, 4896–4914 (2013). [DOI] [PubMed] [Google Scholar]
- 20.Dion-Côté A.-M., Symonová R., Ráb P., Bernatchez L., Reproductive isolation in a nascent species pair is associated with aneuploidy in hybrid offspring. Proc. Biol. Sci. 282, 20142862 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Renaut S., Bernatchez L., Transcriptome-wide signature of hybrid breakdown associated with intrinsic reproductive isolation in lake whitefish species pairs (Coregonus spp. Salmonidae). Heredity 106, 1003–1011 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dion-Côté A.-M., Renaut S., Normandeau E., Bernatchez L., RNA-seq reveals transcriptomic shock involving transposable elements reactivation in hybrids of young Lake Whitefish species. Mol. Biol. Evol. 31, 1188–1199 (2014). [DOI] [PubMed] [Google Scholar]
- 23.Lee J. Y., Lee T.-H., Effect of DNA methylation on structure of nucleosomes. J. Am. Chem. Soc. 134, 173–175 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bao W., Kojima K. K., Kohany O., Repbase Update, a database of repetitive elements in eukaryotic genomes. Mobile DNA 6, 11 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Skinner M. K., Gurerrero-Bosagna C., Haque M. M., Nilsson E. E., Koop J. A., Knutie J. A., Clayton D. H., Epigenetics and the evolution of Darwin’s finches. Genome Biol. Evol. 6, 1972–1989 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Smith T. A., Martin M. D., Nguyen M., Mendelson T. C., Epigenetic divergence as a potential first step in darter speciation. Mol. Ecol. 25, 1883–1894 (2016). [DOI] [PubMed] [Google Scholar]
- 27.Le Luyer J., Laporte M., Beacham T. D., Kaukinen K. H., Withler R. E., Leong J. S., Rondeau E. B., Koop B. F., Bernatchez L., Parallel epigenetic modifications induced by hatchery rearing in a Pacific salmon. Proc. Natl. Acad. Sci. 114, 12964–12969 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Derks M. F. L., Schachtschneider K. M., Madsen O., Schijlen E., Verhoeven K. J. F., van Oers K., Gene and transposable element methylation in great tit (Parus major) brain and blood. BMC Genomics 17, 332 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lister R., Pelizzola M., Dowen R. H., Hawkins R. D., Hon G., Tonti-Filippini J., Nery J. R., Lee L., Ye Z., Ngo Q.-M., Edsall L., Antosiewicz-Bourget A., Stewart R., Ruotti V., Millar H., Thompson J. A., Ren B., Ecker J. R., Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 462, 315–322 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tomkova M., Schuster-Böckler B., DNA Modifications: Naturally more error prone? Trends Genet. 34, 627–638 (2018). [DOI] [PubMed] [Google Scholar]
- 31.Gao B., Shen D., Xue S., Chen C., Cui H., Song C., The contribution of transposable elements to size variations between four teleost genomes. Mobile DNA 7, 4 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Volff J.-N., Körting C., Meyer A., Schartl M., Evolution and discontinuous distribution of Rex3 retrotransposons in fish. Mol. Biol. Evol. 18, 427–431 (2001). [DOI] [PubMed] [Google Scholar]
- 33.Volff J.-N., Körting C., Meyer A., Schartl M., Evolution and discontinuous distribution of Rex3 retrotransposons in fish. Mol. Biol. Evol. 18, 427–431 (2001). [DOI] [PubMed] [Google Scholar]
- 34.Ferreira D. C., Porto-Foresti F., Oliveira C., Foresti F., Transposable elements as a potential source for understanding the fish genome. Mob. Genet. Elem. 1, 112–117 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McVean G. T., Fractious chromosomes: Hybrid disruption and the origin of selfish genetic elements. Bioessays 17, 579–582 (1995). [DOI] [PubMed] [Google Scholar]
- 36.Ziegler C. G., Lamatsch D. K., Steinlein C., Engel W., Schartl M., Schmid M., The giant B chromosome of the cyprinid fish Alburnus alburnus harbours a retrotransposon-derived repetitive DNA sequence. Chromosome Res. 11, 23–35 (2003). [DOI] [PubMed] [Google Scholar]
- 37.Gu H., Smith Z. D., Bock C., Boyle P., Gnirke A., Meissner A., Preparation of reduced representation bisulfite sequencing libraries for genome-scale DNA methylation profiling. Nat. Protoc. 6, 468–481 (2011). [DOI] [PubMed] [Google Scholar]
- 38.Martin M., Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal 17, 10–12 (2011). [Google Scholar]
- 39.Krueger F., Andrews S. R., Bismark: A flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 27, 1571–1572 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Akalin A., Kormaksson M., Li S., Garrett-Bakelman F. E., Figueroa M. E., Melnick A., Mason C. E., methylKit: A comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genome Biol. 13, R87 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Catchen J., Hohenlohe P. A., Bassham S., Amores A., Cresko W. A., Stacks: An analysis tool set for population genomics. Mol. Ecol. 22, 3124–3140 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Danecek P., Auton A., Abecasis G., Albers C. A., Banks E., DePristo M. A., Handsaker R. E., Lunter G., Marth G. T., Sherry S. T., McVean G., Durbin R., The variant call format and VCFtools. Bioinformatics 27, 2156–2158 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Oksanen J., Blanchet G., Friendly M., Kindt R., Legendre P., McGlinn D., Minchin P.R., O’Hara R.B., Simpson G.L., Solymos P., Stevens M.H.H., Szoecs E., Wagner H., Vegan: Community ecology package. R package. Version 2, 0–3 (2012). [Google Scholar]
- 44.P. Legendre, L. Legendre, Numerical Ecology (Elsevier) vol. 24, ed. 3, 2012. [Google Scholar]
- 45.Gu Z., Eils R., Schlesner M., Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 32, 2847–2849 (2016). [DOI] [PubMed] [Google Scholar]
- 46.Magoč T., Salzberg S. L., FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27, 2957–2963 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Haas B. J., Papanicolaou A., Yassour M., Grabherr M., Blood P. D., Bowden J., Couger M. B., Eccles D., Li B., Lieber M., MacManes M. D., Ott M., Orvis J., Pochet N., Strozzi F., Weeks N., Westerman R., William T., Dewey C. N., Henschel R., LeDuc R. D., Friedman N., Regev A., De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 8, 1494–1512 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Altschul S. F., Gish W., Miller W., Myers E. W., Lipman D. J., Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990). [DOI] [PubMed] [Google Scholar]