

The diversity of secretory routes enabled the identification of specific inhibitors of CCR5 transport and HIV-1 infection.
Abstract
Using a cell-based assay monitoring differential protein transport in the secretory pathway coupled to high-content screening, we have identified three molecules that specifically reduce the delivery of the major co-receptor for HIV-1, CCR5, to the plasma membrane. They have no effect on the closely related receptors CCR1 and CXCR4. These molecules are also potent in primary macrophages as they markedly decrease HIV entry. At the molecular level, two of these molecules inhibit the critical palmitoylation of CCR5 and thereby block CCR5 in the early secretory pathway. Our results open a clear therapeutics avenue based on trafficking control and demonstrate that preventing HIV infection can be performed at the level of its receptor delivery.
INTRODUCTION
A large number of pathologies, from infectious and developmental diseases to cancers, depend on the activity of plasma membrane receptors, adhesion proteins, channels, etc. that are delivered from their site of synthesis in the endoplasmic reticulum (ER) to the plasma membrane through the secretory pathway. To perturb these protein functions, several tracks have been followed such as the development of agonists and antagonists, inhibitors of signaling, or enzymatic activity. Nevertheless, inhibition of the intracellular transport of these proteins has not been considered because intracellular routes were considered as too generic to represent a therapeutic target. However, a clear diversity of secretion routes for many different cargos has recently been unambiguously revealed (1). Taking advantage of this diversity, we set out to identify small molecules specifically inhibiting the transport of virus receptor, focusing on HIV-1 entry.
HIV-1 infects immune cells, in particular CD4+ T lymphocytes and macrophages, leading to AIDS. The cell entry of HIV-1 is initiated by the interaction of its surface envelope glycoprotein, gp120, with two host cell surface receptors: CD4 and a co-receptor. CC chemokine receptor 5 (CCR5) is the principal co-receptor for R5-tropic strains, responsible for the transmission and establishment of HIV-1 infection (2–6). Genetic polymorphism in the CCR5 gene has been correlated with HIV resistance. Individuals homozygous for the CCR5 delta32 allele do not express CCR5 at the cell surface and are resistant to HIV-1 infection (7, 8). An additional demonstration of the crucial role CCR5 plays in HIV-1 infection came from the long-term control of infection in a patient transplanted with stem cells from a delta32/delta32 individual (9). CCR5 delta32 individuals do not show major deficiencies due to the absence of cell surface CCR5, and as such, the therapy shows great promise. Consequently, several anti-HIV therapies targeting CCR5 have been developed, such as the drug maraviroc (10, 11), CCR5-blocking antibodies (12, 13), and CCR5 gene editing (14, 15). Of these, maraviroc is the only anti-HIV therapy targeting CCR5 currently used for the treatment of patients. By binding to CCR5, this small, nonpeptidic CCR5 ligand prevents the interaction of the HIV-1 gp120 to CCR5 via an allosteric mechanism.
CCR5 is a member of the class A G protein–coupled receptor (GPCR) family, containing seven transmembrane domains, which enters the secretory pathway at the level of the ER. It is exported from the ER to reach the Golgi complex and is then delivered to the plasma membrane. Little is known about the transport of CCR5 to the cell surface along the secretory pathway, and we tested whether specifically inhibiting delivery could provide an alternative therapeutic strategy. We used the retention using selective hooks (RUSH) assay (16) to quantitatively monitor the anterograde transport of CCR5. Furthermore, we coupled it to a high-content screening of chemical libraries to identify small molecules able to specifically inhibit CCR5 delivery to the plasma membrane. Of the three molecules identified, two inhibit the palmitoylation of the cysteine residues present in the cytoplasmic tail of CCR5, a molecular event that is critical for CCR5 to be transported to the plasma membrane. The incubation of the human primary target cells with either of the three molecules therefore resulted in a significant reduction in HIV-1 entry and de novo virus production.
Together, our data indicate that perturbation of CCR5 modification and more generally of its transport to the plasma membrane through the secretory pathway is a clear avenue for treatment. It also shows that the diversity of secretory routes represents an important and underexploited source for drug discovery.
RESULTS
Differential transport of CCR5 and TNF to the cell surface
The quantity of CCR5 present at the plasma membrane at steady state corresponds to a balance between the delivery of newly synthesized CCR5 and its endocytosis followed by recycling and degradation. To study the delivery of CCR5 to the plasma membrane and identify compounds that affect its anterograde transport, we synchronized its transport using the RUSH assay (16). Briefly, the cargo of interest is fused to a streptavidin binding peptide (SBP) and coexpressed with a resident protein of the ER, which is fused to streptavidin. The streptavidin “hook” retains the cargo upon synthesis due to the streptavidin-SBP interaction and prevents its export from the ER. The synchronized transport of the cargo is induced by the addition of biotin that rapidly enters cells, binds to streptavidin, and competes out SBP. We engineered a HeLa cell line stably expressing a version of CCR5 adapted to the RUSH assay (Str-KDEL_SBP-EGFP-CCR5). In the absence of biotin, CCR5 was localized in the ER (Fig. 1A, 0 min). As expected, addition of biotin enabled export of CCR5 from the ER toward the Golgi complex and its subsequent appearance at the cell surface (Fig. 1A, 30 to 120 min). Compared to another RUSH-adapted cargo, tumor necrosis factor (TNF), the transport kinetics were very different (Fig. 1A) (16, 17). For instance, while transport intermediates containing TNF were clearly visible from ER to Golgi and from Golgi to the plasma membrane, very few were detected for CCR5 (Fig. 1A and movies S1 and S2). To confirm this, we performed the visualization of simultaneously expressed CCR5 and TNF. The two cargos were segregated, especially at the level of the Golgi complex. First, CCR5 and TNF do not perfectly colocalize, and TNF is exported from the Golgi complex in tubular and vesicular transport carriers from which CCR5 was excluded (Fig. 1, B and C, and movie S3). Second, CCR5 appeared to reach the plasma membrane more slowly than TNF. To confirm this apparent kinetics difference, we quantified transport using flow cytometry. Enhanced green fluorescent protein (EGFP) is exposed to the extracellular face of the plasma membrane in both CCR5 and TNF constructs. This allows quantification of the kinetics of cell surface appearance using nonpermeabilized cells labeled with an anti-GFP antibody. This analysis confirmed that CCR5 is transported more slowly than TNF to the plasma membrane (Fig. 1D). Furthermore, it also revealed that once delivered at the plasma membrane, CCR5 is stable, while TNF rapidly disappears, consistent with our previous studies (16). Together, these results indicate that CCR5 and TNF have different transport characteristics that are likely sustained by distinct molecular machineries that could be selectively targeted.
Fig. 1. Differential anterograde transport of CCR5 and TNF.
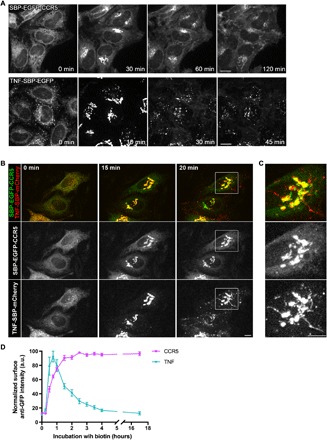
(A) Synchronized transport of CCR5 (top) and TNF (bottom) in HeLa cells stably expressing Str-KDEL_SBP-EGFP-CCR5 or Str-KDEL_TNF-SBP-EGFP. Trafficking was induced by addition of biotin at 0 min. Scale bar, 10 μm. (B) Dual-color imaging of the synchronized transport of SBP-EGFP-CCR5 and TNF-SBP-mCherry transiently coexpressed in HeLa cells. Streptavidin-KDEL was used as an ER hook. Release from the ER was induced by addition of biotin at 0 min. Scale bar, 10 μm. (C) Magnification (×2.8) of the Golgi complex region is displayed. Scale bar, 10 μm. (D) Kinetics of arrival of CCR5 (magenta) or TNF (cyan) to the cell surface after release from the ER measured by flow cytometry. Ratio of cell surface signal divided by GFP intensity was used for normalization. a.u., arbitrary units. The mean ± SEM of three experiments is shown. See also movies S1 to S3.
Identification of molecules inhibiting CCR5 secretion by high-content screening
To identify molecules that specifically inhibit the CCR5 plasma membrane delivery, we conducted high-content screenings of chemical libraries using HeLa cells stably expressing either RUSH-adapted CCR5 (Str-KDEL_SBP-EGFP-CCR5) or TNF (Str-KDEL_TNF-SBP-EGFP). They were plated in 384-well plates and incubated for 1.5 hours with small molecules (10 μM) from the following two chemical libraries: (i) an approved drug collection of 1200 drugs from Prestwick Chemicals and (ii) a drug collection of 2824 drugs obtained from the U.S. National Cancer Institute (NCI). As the molecules were dissolved in dimethyl sulfoxide (DMSO), an identical concentration of DMSO was used as negative control. Brefeldin A (BFA), which blocks secretion (18), and nocodazole, which disrupts microtubules and perturbs Golgi organization (19), were used as additional controls. In addition, biotin was omitted in some wells to validate the screening procedure and the analysis. Transport to the cell surface was induced by incubation with biotin for 2 hours (for CCR5) or 45 min (for TNF) according to the determined delivery kinetics (see Fig. 1). The localization of the cargos was determined using GFP fluorescence, while the fraction of the cargo present at the cell surface was quantified by immunolabeling using an anti-GFP antibody on fixed, but nonpermeabilized, cells. Nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI) for imaging and segmentation purposes (Fig. 2A).
Fig. 2. Identification of molecules inhibiting CCR5 transport by high-content differential screening.
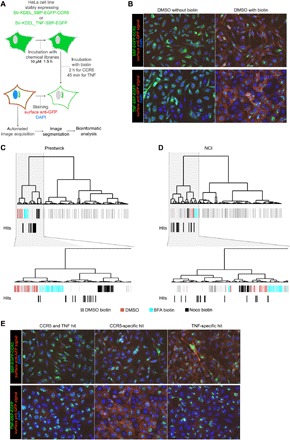
(A) Outline of the chemical screening strategy. (B) Micrographs from screening plates showing controls. DMSO without biotin and DMSO with biotin correspond to the conditions where no transport and normal transport occurred, respectively. HeLa cells stably expressing Str-KDEL_SBP-EGFP-CCR5 and Str-KDEL_TNF-SBP-EGFP are displayed in the top and bottom panels, respectively. Cargo at the plasma membrane was detected using anti-GFP antibodies on nonpermeabilized cells. (C and D) Clustering of molecules obtained after bioinformatics analysis of the Prestwick (C) and NCI (D) chemical library screening of CCR5 secretion. Below the dendrogram, each bar corresponds to a well of the plate. Molecules identified as hits were shifted one lane below for better visualization. BFA, brefeldin A; Noco, nocodazole. (E) Micrographs showing the three classes of hits detected. CCR5 and TNF hit corresponds to a molecule affecting transport of both CCR5 and TNF. CCR5-specific hit or TNF-specific hit corresponds to a molecule inhibiting only CCR5 or only TNF transport. HeLa cells stably expressing Str-KDEL_SBP-EGFP-CCR5 and Str-KDEL_TNF-SBP-EGFP are displayed in the top and bottom panels, respectively. Cargo at the plasma membrane was detected using anti-GFP antibodies on nonpermeabilized cells.
As expected, in the absence of biotin (DMSO without biotin), the GFP signal was restricted to the ER for both CCR5 and TNF, and almost no surface anti-GFP signal was visible (Fig. 2B). After incubation with biotin (DMSO with biotin), CCR5 and TNF reached the plasma membrane, as shown by a strong cell surface staining (see Fig. 2B). Using features obtained from image segmentation of these control conditions, a bioinformatics analysis was conducted to identify molecules from the Prestwick (Fig. 2C) and NCI libraries (Fig. 2D) that alter not only different parameters, such as CCR5 and TNF localization and plasma membrane delivery, but also cell organization or that induce cell death. Principal component analysis (PCA) and the resulting hierarchical clustering grouped conditions that altered secretion (i.e., DMSO without biotin, BFA with biotin, and nocodazole with biotin) and separated them from conditions inducing secretion (i.e., DMSO with biotin). The same approach was used to identify molecules from the libraries that altered transport and delivery of CCR5 or TNF. Several small molecules prevented secretion of both CCR5 and TNF (“CCR5 and TNF hit”). However, in addition to these generic inhibitors, several molecules specifically perturbed the secretion of either CCR5 (“CCR5-specific hit”) or TNF (“TNF-specific hit”) (Fig. 2E). This shows that the apparent qualitative and quantitative differences reported above that distinguish transport of CCR5 and TNF can be translated to specific inhibition.
Hit validation and specificity over two other chemokine receptors
The 15 strongest CCR5 hits (Table 1) were selected for further analysis. First, as a secondary screen, we evaluated their effects on the trafficking of two other chemokine receptors, CCR1 and CXCR4, closely related to CCR5. CCR5, CCR1, and CXCR4 belong to the class A subfamily (rhodopsin like) of GPCR, and CCR5 shares several ligands with CCR1 (namely, CCL3, CCL4, and CCL5). CCR1 and CXCR4 transport out of the ER was synchronized using the RUSH assay. The secretion of CCR1 and CXCR4 was slightly faster than that of CCR5, but like CCR5, they then remained stably expressed at the cell surface over several hours (Fig. 3A). To evaluate whether the 15 CCR5 hits also affect the delivery of CCR1 and CXCR4, we used an end-point analysis using the RUSH assay. After 2 hours of incubation with biotin, the presence of the cargo at the plasma membrane was quantitated using flow cytometry. CCR5 plasma delivery was monitored again, and the molecules were ranked according to their relative impact. Molecules 1 to 10 reduced the transport of CCR5 to the cell surface by less than 50%, while a stronger effect was observed for molecules 11 to 15. In particular, molecules 13, 14, and 15 reduced the cell surface delivery of CCR5 by more than 75% (Fig. 3B). These three molecules had only moderate effects on CCR1 and CXCR4 delivery (Fig. 3, C and D), demonstrating their specificity of action on the transport of CCR5 toward the cell surface. As an example, dual-color imaging of the synchronized transport of coexpressed CCR5 and CCR1 in cells treated or not with molecule 13 confirmed that it specifically inhibits CCR5 delivery (Fig. 3, E and F). Note that CCR5 was still observed in the ER and in the Golgi complex after more than 2 hours of biotin addition, indicating that molecule 13 inhibits the trafficking of CCR5 in the early secretory pathway. Together, these results demonstrate that molecules 13, 14, and 15 are not broad inhibitors of chemokine receptor transport but instead specifically target CCR5 transport.
Table 1. Names and molecular formulas of the molecules.
| # | Molecule | Molecular formula | Library |
| 1 | Etretinate | C23H30O3 | Prestwick |
| 2 | Pimethixene maleate | C23H23NO4S | Prestwick |
| 3 | NCI 169627 | C40H49NO14 | NCI |
| 4 | Clemastine fumarate | C25H30ClNO5 | Prestwick |
| 5 | Cilnidipine | C27H28N2O7 | Prestwick |
| 6 | Fluoxetine hydrochloride | C17H19ClF3NO | Prestwick |
| 7 | Deptropine citrate | C29H35NO8 | Prestwick |
| 8 | Parthenolide | C15H20O3 | Prestwick, NCI |
| 9 | NCI 1771 | C6H12N2S4 | NCI |
| 10 | Chlorprothixene hydrochloride | C18H19Cl2NS | Prestwick |
| 11 | NCI 111118 | C13H8Cl2S3 | NCI |
| 12 | NCI 228155 | C11H6N4O4S | NCI |
| 13 | Cadmium chloride | CdCl2 | NCI |
| 14 | NCI 68093, Zn pyrithione | C10H8N2O2S2Zn | NCI |
| 15 | NCI 333856, tetrocarcin A | C67H96N2O24.Na | NCI |
Fig. 3. Three small molecules inhibit the transport of CCR5 and do not affect transport of CCR1 or CXCR4.
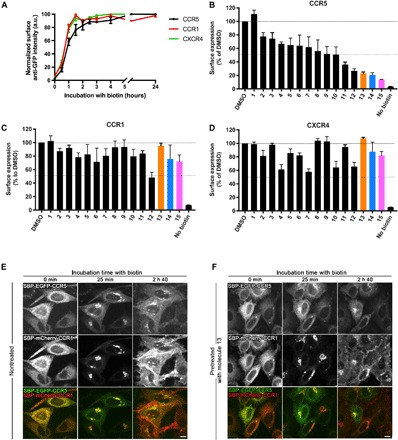
(A) Kinetics of synchronized transport of three chemokine receptors—CCR5 (black), CCR1 (red), and CXCR4 (green)—using the RUSH assay. Trafficking was induced by addition of biotin at time 0. Surface expression of CCR5, CCR1, and CXCR4 was measured by flow cytometry using an anti-GFP antibody. Ratio of cell surface signal to GFP intensity was used for normalization. The mean ± SEM of three experiments is shown. End-point measurement (2 hours) of the effects of the hit molecules on the trafficking of CCR5 (B), CCR1 (C), and CXCR4 (D) in HeLa cells. Cells were pretreated for 1.5 hours with molecules at 10 μM. Cargo present at the cell surface 2 hours after release from the ER was measured by flow cytometry using an anti-GFP antibody. Ratio of cell surface signal to GFP intensity was used for normalization. The mean ± SEM of three experiments is shown. Real-time synchronized secretion of CCR5 and CCR1 was monitored using dual-color imaging in nontreated HeLa cells (E) and in HeLa cells pretreated for 1.5 hours with 10 μM molecule 13 (F). Scale bars, 10 μm.
Molecules 13 and 14 inhibit the secretion of CCR5 via its cysteine-containing cytoplasmic tail and depend on its palmitoylation
Little is known about the key players controlling the CCR5 secretion and trafficking in the secretory pathway. CCR5 is a seven-transmembrane domain protein, with its N terminus facing the luminal/extracellular space and its C terminus in the cytoplasm (Fig. 4A). As the main differences between CCR1 and CCR5 are found in the C-terminal sequences, we examined the role of CCR5 cytoplasmic tail in mediating the effects of molecules 13, 14, and 15 on plasma membrane delivery. Chimeric receptors were constructed, whereby cytoplasmic tails of CCR5 and CCR1 were interchanged (Fig. 4B) and were tested in the RUSH assay in the presence of molecules 13, 14, and 15. First, all receptors either wild type (CCR5wt and CCR1wt) or chimeric (CCR5-CCR1tail and CCR1-CCR5tail) reach the cell surface with similar kinetics (Fig. 4C). However, incubation with molecules 13, 14, and 15 affected the secretion of the chimeras in different ways. The transport of both chimeras (CCR5-CCR1tail and CCR1-CCR5tail) was inhibited after exposure to molecule 15 by more than 40%, suggesting that the effect of this molecule is not targeted to the tail of CCR5. In contrast, molecules 13 and 14 inhibited the secretion of the constructs bearing the cytoplasmic tail of CCR5 (i.e., CCR5wt and CCR1-CCR5tail) by more than 40%. They were, however, quite inefficient against receptors bearing CCR1 tail (Fig. 4D). Although this inhibition was not as strong as the effect of these molecules on CCR5wt (40% inhibition for CCR1-CCR5 tail versus 90% for CCR5wt), this indicated that the presence of the cytoplasmic tail of CCR5 was necessary for the reduction of trafficking induced by molecules 13 and 14. The cytoplasmic tail of CCR5 contains three cysteine residues that require palmitoylation to ensure efficient secretion (20, 21). In contrast, the CCR1 cytoplasmic tail does not contain palmitoylated cysteine residues.
Fig. 4. Molecules 13 and 14 inhibit CCR5 secretion via its cytoplasmic tail and inhibit palmitoylation.
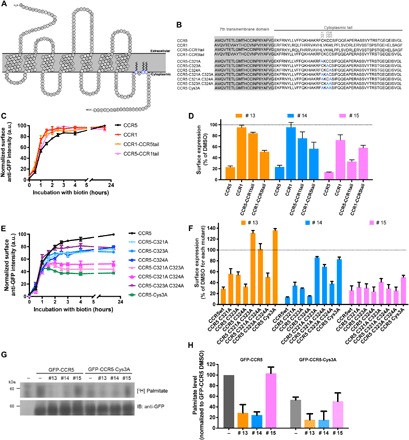
(A) Schematic representation of CCR5 with three palmitoylated cysteine residues indicated in blue. (B) Amino acid sequence of CCR5, CCR1, their chimeras, and the cysteine to alanine mutants used in this study. (C) Kinetics of the synchronized transport of CCR5/CCR1 chimeras to the cell surface measured by flow cytometry in HeLa cells transiently expressing the constructs. Release was induced by addition of biotin at time 0. The mean ± SEM of three experiments is shown. (D) End-point measurement (2 hours) by flow cytometry of the effects of molecules 13, 14, and 15 on the transport of the CCR5/CCR1 chimeras in HeLa cells after transient expression. Cells were pretreated for 1.5 hours with molecules at 10 μM. The mean ± SEM of three experiments is shown. (E) Kinetics of the synchronized transport of CCR5 cysteine mutants to the cell surface measured by flow cytometry in HeLa cells transiently expressing the corresponding constructs. Release was induced by addition of biotin at time 0. The mean ± SEM of three experiments is shown. (F) End-point measurement (2 hours) by flow cytometry of the effects of molecules 13, 14, and 15 on the transport of CCR5 cysteine mutants. HeLa cells transiently expressing the cysteine mutants were pretreated for 1.5 hours with molecules at 10 μM. The mean ± SEM of three experiments is shown. (G and H) Quantification of palmitoylation of either GFP-CCR5 or GFP-CCR5 Cys3A transiently expressed in HEK293T cells. [3H]Palmitate was incorporated for 4 hours in the presence of the compounds following a pretreatment with DMSO (−) and molecules 13, 14, and 15 for 30 min. A representative autoradiogram and immunoblot are shown (G), and the mean ± SEM of four experiments is shown.
To further study the role of the three cysteine residues in mediating the effect of molecules 13 and 14, a series of mutants were created with cysteine substituted by alanine, independently or in a combinatorial way (Fig. 4B). The kinetics of plasma membrane delivery of the CCR5 cysteine mutants were consistent with previous reports (20, 21). The secretion of the single cysteine mutants was decreased by 20%, while the secretion of the double and triple cysteine mutants was decreased by 50%, with the exception of the CCR5 C323A-C324A mutant that was decreased by only 20% (Fig. 4E).
We then tested the effect of molecules 13, 14, and 15 on the transport of the cysteine mutants. Molecule 13 did not reduce the secretion of CCR5 C321A-C323A, CCR5 C321A-C324A, and CCR5 Cys3A, while other mutants were affected (e.g., CCR5 C324A) (Fig. 4F). Similarly, although molecule 14 is more potent than molecule 13, it leads to the same relative inhibition. CCR5 C324A delivery was affected the strongest and to the same extent as CCR5wt. In contrast, in agreement with the results presented above, the inhibitory effect of molecule 15 was not affected by any of the cysteine mutation and relies on different CCR5 molecular features.
Because these cysteine residues were reported to be palmitoylated, we directly assessed the effects of molecules 13, 14, and 15 on CCR5 palmitoylation. In vivo metabolic labeling using radioactive palmitate of cells expressing either GFP-CCR5 or GFP-CCR5 Cys3A was performed (Fig. 4, G and H). Molecules 13 and 14 decreased the level of palmitoylated GFP-CCR5 by 70%. As expected, molecule 15 did not affect the palmitoylation level of GFP-CCR5.
As expected, CCR5 Cys3A showed a reduced level of palmitoylation (about 50%) compared to CCR5wt, although some palmitoylation signal was still observed. This may suggest that residual palmitoylation on cysteine other than Cys321, Cys323, and Cys324 may be occurring in the mutant. This residual signal was also decreased after incubation with molecules 13 and 14, while molecule 15 had no effect.
The palmitoyltransferase responsible for the palmitoylation of CCR5 is not known. We looked for pamitoyltransferases able to modify CCR5. DHHC3, DHHC7, and DHHC15 overexpression was found to increase palmitoylation of CCR5 (fig. S1A). Autopalmitoylation of DHHC3 and DHHC7 was inhibited to about 50% following incubation with molecules 13 and 14, whereas molecule 15 had no effect (fig. S1, B and C). These results suggest that molecules 13 and 14 may inhibit autopalmitoylation of DHHCs responsible for CCR5 palmitoylation and consequently palmitate transfer to CCR5.
Together, our results indicate that molecules 13 and 14 may share a similar mode of action inducing a strong reduction of CCR5 palmitoylation. In contrast, molecule 15 seems to affect the secretion of CCR5 by another, still elusive, mechanism.
Inhibition of HIV-1 infection in primary human macrophages
To validate the effect of molecules 13, 14, and 15 on the delivery of CCR5 to the cell surface, we monitored the expression of endogenous CCR5 in human monocyte–derived macrophages (hMDMs) after overnight treatment by flow cytometry.
These molecules, alone or in combination, after overnight incubation induced a small but significant decrease of CCR5 cell surface expression (19.1 to 23.8%) compared with DMSO treatment. The cell surface expression of CXCR4, however, was not significantly modified under the same conditions (P > 0.05; Fig. 5A and fig. S2A). These three molecules, alone or in combination, also did not induce major cytotoxicity (fig. S2B).
Fig. 5. Treatment with molecules 13, 14, and 15 decreases HIV-1 R5 infection in human macrophages.
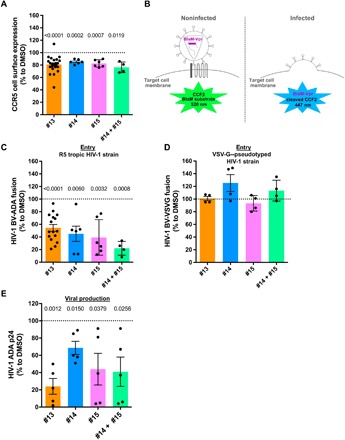
Primary human macrophages differentiated for 4 days with rhM-CSF were treated during 18 hours with molecule 13 at 10 μM, molecule 14 at 3 μM, molecule 15 at 1 μM, and molecules 14 and 15 at 1 μM (or DMSO at 0.1%). (A) Cell surface expression of CCR5 was measured by flow cytometry with specific antibodies. (B) Principles of the HIV-1 entry test used (22). Inhibition of fusion of HIV-1ADA (R5 tropic strain) (C) or HIV-1VSVG (VSVG pseudotyped) (D) containing BlaM-Vpr (BV) with CCF2/AM primary macrophages mediated by compounds. Total p24 amount (supernatants and lysates) of HIV-1ADA–infected macrophages was measured by ELISA (E). Each black point represents one donor analyzed independently. Statistical analyses were performed using Prism software. A one-sample t test was applied, and significant P values (<0.05) are indicated for each treatment compared to DMSO in (A), (C), and (E). The absence of a P value indicates that the results were not significantly different. Error bars correspond to SEM.
CCR5 is critical for HIV particles to bind target cells and mediate their entry by fusion. HIV entry was then investigated using the BlaM-Vpr fusion assay (22) after overnight treatment with molecules 13, 14, and 15 (Fig. 5B). The fusion of HIV-1 ADA (a strain that uses CCR5 as a co-receptor) with hMDMs was strongly decreased upon incubation with these molecules (by 45.7 to 78.0%) when compared with DMSO (Fig. 5C). Treatment with both molecules 14 and 15 led to further perturbation of viral entry. Under the same conditions, the entry of a vesicular stomatitis virus glycoprotein (VSVG)–pseudotyped virus (used as a CCR5-independent control) was not affected (Fig. 5D).
These data indicate that a small reduction of neosynthesized CCR5 expressed at the cell surface is sufficient to significantly affect R5-tropic HIV-1 entry into human macrophages. To further assess whether the perturbation of viral entry was sufficient to alter the viral cycle and production, the total amount of p24 capsid protein produced by macrophages was quantified. Viral production and secretion were both strongly reduced by 31.4 to 76.0% (Fig. 5E). Together, these results demonstrate that molecules specifically reducing CCR5 secretion at the cell surface impair HIV-1 infection of human macrophages.
DISCUSSION
The development of many pathologies relies on the efficient intracellular transport of proteins. Transport to the cell surface is particularly important, as adhesion proteins, channels, proteases, or receptors, for example, have to reach the plasma membrane to fulfill their functions. We thus reasoned that, instead of looking for molecules able to perturb their function when expressed at the plasma membrane, we may target their transport pathway to prevent their normal expression at the cell surface, hence exploring a novel therapeutic option. This option has been underexploited for at least two reasons.
On the one hand, it has long been thought that the diversity of secretory routes was low and that the bulk flow of membranes was responsible for most nonspecialized pathways. We now know that the diversity of pathways is high with several coats, adaptors, Golgi matrix proteins, or molecular motors active at the same transport stage. In addition, not only the molecular machinery of transport may be targeted but also the cargo itself may be targeted. Perturbation of its folding, modifications, or interaction with transport partners or membrane partitioning may perturb, or prevent, its transport.
On the other hand, as compared to the power and precision of the study of endocytosis and retrograde pathways, the diversity of the secretory pathway has long been difficult to study and target. Quantitative monitoring of the transport of proteins was only possible for selected proteins, and assays were hardly amenable to screening. The development of the RUSH assay (16) that allows us to cope with the diversity now allows us to overcome this limitation and specifically screen for inhibitory molecules.
To validate this new paradigm, we used the RUSH assay to screen for small molecules able to inhibit specifically the transport of CCR5 to the cell surface. CCR5 is essential for R5-tropic HIV-1 strain infections of human cells and represents a valuable therapeutic target. Individuals devoid of CCR5 expressed at the cell surface are resistant to HIV-1 infection, while people heterozygous for this deletion, who show a reduction of CCR5 cell surface expression, display slower progression of HIV infection (23). The absence of functional CCR5 seems not to be deleterious to these individuals, although increased susceptibility to infections, such as West Nile virus (24, 25) and tick-borne encephalitis (26), has been reported.
Several large-scale screens were conducted to identify protein regulators of HIV-1 infection, but none of them led to the identification of CCR5 secretion regulators (27–30). Little is known about the molecular players that regulate the secretion of CCR5. The involvement of the small guanosine triphosphatases (GTPases) Rab1, Rab8, and Rab11 has been proposed (31). Rab43 was recently reported to play a role in the export of several class A GPCRs from the ER (32), although its role in controlling the transport of CCR5 was not evaluated. The importance of palmitoylation of CCR5 was also revealed (20, 21). However, the palmitoyltransferase responsible for palmitoylation of CCR5 remains unknown.
RUSH-based differential screening, using TNF as a control reporter, allowed the identification of a set of inhibitory molecules. In particular, three molecules that strongly perturbed CCR5 transport were found to have no, or only moderate, effects on the secretion of the closely related CCR1 and CXCR4. Molecules 13, 14, and 15 were active on endogenous CCR5 secretion and reduced HIV-1 infection of human macrophages isolated from donors. Molecules 13, 14, and 15 are cadmium chloride (CdCl2), zinc pyrithione (C10H8N2O2S2Zn), and tetrocarcinA (C67H96N2O24), respectively. Cd and zinc pyrithione were found to share similar mechanisms of action because they both rely on the C-terminal cytoplasmic tail of CCR5 and, in particular, the cysteine residues in positions 321, 323, and 324 and inhibit CCR5 palmitoylation. At the molecular level, Cd affects protein function by binding to thiol groups. Direct binding of Cd to cysteine residues may prevent efficient palmitoylation of CCR5, hence affecting its transport, as previously reported (20, 21). Alternatively, palmitoyltransferase may represent the target of these molecules. Palmitoylation occurs in a two-step mechanism. First, palmitoyl–coenzyme A (CoA) is transferred to the DHHC cysteine-rich domain, leading to autoacylation of the palmitoyltransferase (33). In the second step, the palmitoyl group is transferred to the cysteine residues of the target protein. DHHC contains bound Zn (34). Cd is known to displace essential metals like Zn in metalloproteins [see (35) for review]. Zn pyrithione is an antifungal and antibacterial zinc chelator. As these two molecules may affect Zn-dependent enzymes, it is tempting to propose that Zn pyrithione and Cd bind to, and perturb, the palmitoyltransferase responsible for palmitoylation of CCR5.
Tetrocarcin A represents another class of CCR5 inhibitory molecule because it does not depend on the C-terminal tail and does not affect palmitoylation. It is an antibiotic (36), which antagonizes Bcl-2 anti-apoptotic function (37). The putative mode of action of tetrocarcin A on the trafficking of CCR5 remains unclear. Further studies are required to obtain a clearer view of tetrocarcin A’s mechanism of action, but it may represent an interesting unprecedently identified class of CCR5 inhibitory molecule.
The three molecules identified in our study exhibited inhibitory effects on HIV-1 infection for R5-tropic viruses at both the level of virus entry and viral particle production in human macrophages. Of major concern in anti-HIV therapies, particularly those targeting a receptor such as CCR5, is the emergence of escape viruses. To date, maraviroc is the only approved anti-HIV therapy targeting CCR5. Reports of the emergence of resistant viruses have since been published (38, 39). Targeting the host secretory pathway may avoid the emergence of such escape viruses. The molecules identified in this study induced a decrease in secretion of CCR5, resulting in a reduced expression at the cell surface. Reduction was small in primary cells but may target the conformations recognized by HIV-1. Because such molecules target the cellular machinery, viruses are less likely to escape. For example, if mutations were to arise in R5-tropic viruses, they would not be able to induce normal secretion of CCR5 and restore infection. This is a clear advantage over treatments based on competition or allosteric modifications. Combinatorial treatment may also reduce the amount of CCR5 at the cell surface and may therefore improve the efficacy of blocking antibodies or maraviroc.
In conclusion, our study confirms our model that proposes that the diversity of secretory routes can be exploited to identify molecules that specifically affect the transport of a given receptor. Targeting the transport and the function of target protein may thus represent a novel therapeutic paradigm.
MATERIALS AND METHODS
Cells
HeLa cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (Thermo Fisher Scientific) supplemented with 10% fetal calf serum (FCS; GE Healthcare), 1 mM sodium pyruvate, and penicillin and streptomycin (100 μg/ml) (Thermo Fisher Scientific). HeLa cells stably expressing Str-KDEL as a hook and either SBP-EGFP-CCR5 or TNF-SBP-EGFP as a reporter were obtained by transduction with lentiviral particles produced in human embryonic kidney (HEK) 293T. A clonal population was then selected using puromycin resistance and limiting dilution.
Human primary macrophages were isolated from the blood of healthy donors (Etablissement Français du Sang Ile-de-France, Site Trinité, #15/EFS/012) by density gradient sedimentation in Ficoll (GE Healthcare), followed by negative selection on magnetic beads (catalog no. 19059, Stem Cells) and adhesion on plastic at 37°C for 2 hours. Cells were then cultured in the presence of complete culture medium [RPMI 1640 supplemented with 10% FCS (Eurobio), streptomycin/penicillin (100 μg/ml), and 2 mM l-glutamine (Invitrogen/Gibco)] containing recombinant human macrophage colony-stimulating factor (rhM-CSF) (10 ng/ml) (R&D Systems) (40) for 4 to 5 days.
Plasmids and transfection
The DNA sequences corresponding to human CCR5 (P51681, UniProt), CCR1 (P322246, UniProt), and CXCR4 (P61073, UniProt) were purchased either as synthetic genes (GeneArt, Thermo Fisher Scientific) or as complementary DNA (cDNA) (Open Biosystems). They were cloned into RUSH plasmids downstream of Str-KDEL_IL2ss-SBP-EGFP or Str-KDEL_IL2ss-SBP-mCherry using Fse I and Pac I restriction enzymes (16). Str-KDEL_TNF-SBP-EGFP and Str-KDEL_TNF-SBP-mCherry plasmids have been described elsewhere (16). The CCR5-CCR1tail and CCR1-CCR5tail chimeras were generated from synthetic genes (GeneArt, Thermo Fisher Scientific) and cloned between Fse I and Pac I restriction sites. Mutations from cysteine to alanine in CCR5 tail were generated either by polymerase chain reaction assembly or by the insertion of small synthetic DNA fragments (gBlock from Integrated DNA Technologies). Protein sequences of chimeras and mutants are depicted in Fig. 4B. GFP-CCR5 and GFP-CCR5-Cys3A bear the interleukin-2 (IL-2) signal peptide upstream of GFP and either CCR5 wild-type or CCR5-Cys3A downstream of GFP. A modified version of pEGFP (Clontech) was used for their generation. All plasmids used in this study were verified by sequencing. HeLa cells were transfected using calcium phosphate as described previously (41).
The HIV-1ADA provirus plasmid (pHIV-1ADA) expressing the env gene of the HIV-1 R5-tropic strain ADA has been described elsewhere (42). pNL4.3Δ pNL4.3f was a gift from P. Benaroch (Institut Curie, Paris, France). The plasmid expressing the env gene of VSVG (pEnvVSVG) was a gift from S. Benichou (Institut Cochin, Paris, France).
High-content automated chemical screening
Chemical compounds were purchased from Prestwick Chemicals (Illkirch, France) corresponding to 1200 approved drugs [U.S. Food and Drug Administration (FDA), European Medicines Agency (EMA), and other agencies] dissolved in DMSO at 10 mM. A second library of 2824 compounds was provided by the NCI chemical libraries as follows: diversity set III, 1596 compounds; mechanistic set, 879 compounds; approved oncology drugs set II, 114 agents; and natural products set II, 235 agents. All NCI stock compounds were received in DMSO at a concentration of 10 mM except for mechanistic set (at 1 mM) (in a 96-well plate format). All libraries were reformatted in-house in 384-well plates. BFA and nocodazole were purchased from Sigma-Aldrich and used as control molecules.
For compound screening, cells (5.0 × 103 per well) were seeded on black clear-bottom 384-well plates (ViewPlate-384 Black, PerkinElmer) in 40 μl of complete medium. The screen was performed at the similar early cell passages (±2) for both replicates. Twenty-four hours after cell seeding, compounds were transferred robotically to plates containing cells using TeMO (MCA 384) (TECAN) to a final concentration of 10 μM and 0.5% of DMSO. Controls were added to columns 1, 2, 23, and 24 of each plate. After 90 min of compound incubation, cells were treated with 40 μM biotin for 45 min (for TNF) or 120 min (CCR5) at 37°C. Compound screens were performed in two independent replicate experiments at the BioPhenics Screening Laboratory (Institut Curie).
Cells were processed immediately after biotin treatment for immunofluorescence. Briefly, cells were fixed with 3% paraformaldehyde for 15 min and quenched with 50 mM NH4Cl in phosphate-buffered saline (PBS) solution for 10 min. For cell surface labeling, cells were incubated with anti-mouse GFP (1:800, Roche, catalog no. 814 460 001) diluted in 1% bovine serum albumin blocking solution for 45 min. Cells were then washed with PBS and incubated for 1 hour with Cy3-conjugated anti-mouse (1:600; catalog no. 715-165-151, Jackson ImmunoResearch). Nuclei were counterstained with DAPI (Life Technologies) for 45 min.
Image acquisition was performed using an INCell 2200 automated high-content screening fluorescence microscope (GE Healthcare) at a ×20 magnification (Nikon 20×/0.45). Four randomly selected image fields were acquired per wavelength, well, and replicate experiment. Image analysis to identify cells presenting predominantly cell surface or intracellular CCR5 and/or TNF localization was performed for each replicate experiment using the Multi Target analysis application module in the INCell analyzer Workstation 3.7 software (GE Healthcare). Results were reported as mean values from four image fields per well.
Bioinformatics analysis of the screens
Fields with less than 50 cells were filtered out after image segmentation. All cell features extracted from the image analysis step were normalized within each plate by subtracting the median value of control wells containing DMSO and biotin and dividing by the median absolute deviation of the same controls.
PCA was applied to normalized data for each dataset separately as a denoising method. From this PCA, the coordinates of wells in the subspace, defined by principal component with eigenvalue greater than one, were used to compute the Euclidean distance between wells. Hierarchical clustering with Ward’s agglomerative criterion was then applied on these distances. Two groups were identified from the clustering. The group with the positive controls was defined as the hits list. All analyses were performed using R statistical software.
Measurement of transport to the cell surface by flow cytometry
Cells (1 × 106) expressing RUSH constructs per condition were treated with 40 μM biotin to induce trafficking of the reporter and incubated at 37°C. At the desired time point, cells were washed once with PBS supplemented with 0.5 mM EDTA and incubated with PBS supplemented with 0.5 mM EDTA for 5 min at 37°C. Plates containing cells were then put on ice. Cells were resuspended and transferred to ice-cold tubes for centrifugation at 300g for 5 min at 4°C. Cell pellets were resuspended in cold PBS supplemented with 1% FCS for blocking and incubated for at least 10 min. After centrifugation, cell pellets were incubated in a solution of Alexa Fluor 647–coupled anti-GFP (catalog no. 565197, BD Pharmingen) prepared in PBS supplemented with 1% serum for 40 min on ice. Cells were washed three times in cold PBS and 1% serum and fixed with 2% paraformaldehyde (Electron Microscopy Sciences) for 15 min. Cells were washed twice with PBS before acquisition with an Accuri C6 flow cytometer. The intensity of the GFP signal (FL1) and the Alexa Fluor 647–conjugated antibody (FL4) was measured on GFP-positive cells. The FL4 signal was divided by the FL1 signal to normalize for the transfection level. The FL4/FL1 ratio for each condition was then normalized to the DMSO control.
Real-time imaging of the synchronized secretion
HeLa cells expressing either stably or transiently RUSH constructs were grown on 25-mm glass coverslips. Before imaging (after treatment), coverslips were transferred to an L-shape tubing–equipped Chamlide chamber (Live Cell Instrument). Trafficking was induced by exchanging Leibovitz medium (Life Technologies) with prewarmed Leibovitz medium supplemented with 40 μM biotin (Sigma-Aldrich). Imaging was performed at 37°C in a thermostat-controlled chamber using an Eclipse 80i microscope (Nikon) equipped with a spinning disc confocal head (Perkin) and an Ultra897 iXon camera (Andor). Image acquisition was performed using MetaMorph software (Molecular Devices). Maximum intensity projections of several z slices are shown (Figs. 1, A and C, and 3, E and F).
Measurement of CCR5 palmitoylation
HEK293T cells were transfected with GFP-CCR5 or GFP-CCR5 Cys3A mutant. Twenty-four hours after transfection, cells were labeled with [3H]palmitate (0.5 mCi/ml) for 4 hours. Cells were incubated with 10 μM of individual compounds for 30 min before labeling and for 4 hours together with [3H]palmitate. For compound (−), DMSO was added. For fluorography, after SDS–polyacrylamide gel electrophoresis of cell lysates, the gels were exposed for 11 to 16 days. n = 4 independent experiments. The mean ± SEM is shown.
Antibodies and reagents
The following antibodies were used: Alexa Fluor 647–coupled anti-GFP (catalog no. 565197, BD Pharmingen); Alexa Fluor 647 rat immunoglobulin G2a (IgG2a), κ isotype control (catalog no. 400526); Alexa Fluor 647 anti-human CD195 (catalog no. 313712) from BioLegend; phycoerythrin (PE) mouse IgG1, κ isotype control (catalog no. 550617); PE mouse IgG2a, κ isotype control (catalog no. 553457); fluorescein isothiocyanate (FITC) mouse IgG2a, κ isotype control (catalog no. 555573); FITC mouse IgG1 κ isotype control (catalog no. 555748); PE mouse anti-human CD184 (catalog no. 555974); FITC mouse anti-human CD4 (catalog no. 555346); FITC mouse anti-human CD3 (catalog no. 555332); and PE mouse anti-human CD11b/Mac1 (catalog no. 555388) from BD Biosciences.
The amount of lactate dehydrogenase (LDH) released into supernatants was quantified using a Pierce LDH Cytotoxicity Assay kit (catalog no. 88953, Thermo Fisher Scientific). The Alliance HIV-1 p24 ELISA (enzyme-linked immunosorbent assay) Kit (catalog no. NEK050, PerkinElmer) was used to determine the amount of capsid p24 protein in supernatants and cell lysates. LDH and p24 quantification were performed following the manufacturer’s instructions.
Flow cytometry analysis of receptor surface expression in macrophages
Cells were washed once with cold PBS and detached using 2 mM EDTA in PBS on ice. To analyze cell surface expression of receptors, cells were washed with PBS and stained with antibodies (see above) for 1 hour on ice in PBS and 2% FCS. Cells were then washed twice in cold PBS and analyzed by flow cytometry (Accuri C6, BD Biosciences). Propidium iodide (catalog no. P4864, Sigma-Aldrich), diluted in cold PBS at 0.1 μg/ml, was added to cells just before analysis.
Viral production and HIV-1 infection
Viral stocks
HIV-1 particles or pseudoparticles containing BlaM-Vpr were produced by cotransfection of HEK293T cells with proviral plasmids (pHIV-1ADA or pNL4.3ΔEnv and pEnvVSVG), pCMV-BlaM-Vpr encoding β-lactamase fused to the viral protein Vpr, and pAdvantage, as described elsewhere (22). After 48 hours of culture at 37°C, the virus-containing supernatant was filtered and stored at −80°C. Pseudoparticles containing BlaM-Vpr were ultracentrifuged at 60,000g for 90 min at 4°C on a sucrose cushion (20%). The virion-enriched pellet was resuspended in PBS and aliquoted for storage at −80°C. The amount of p24 antigen in the supernatants was quantified using an ELISA kit (PerkinElmer). HIV-1 infectious titers were also determined in HeLa TZM-bl cells (LTRlacZ, NIH reagent program) by scoring β-lactamase–positive cells 24 hours after infection, as described previously (43).
BlaM-Vpr viral fusion assay
After 18 hours of incubation with compounds, 1.5 × 105 primary macrophages were inoculated with the BlaM-Vpr–containing viruses (15 ng of p24 Gag) by 1-hour spinoculation at 4°C and incubated for 2.5 hours at 37°C. Cells were then loaded with CCF2/AM, the BlaM-Vpr substrate (2 hours at room temperature), and fixed. Enzymatic cleavage of CCF2/AM by β-lactamase (22) was measured by flow cytometry (LSR II, BD), and data were analyzed with FACSDiva software. The percentage of fusion corresponds to the percentage of cells displaying increased cleaved CCF2/AM fluorescence (447 nm).
HIV-1 infections
After treatment for 18 hours, human primary macrophages were infected with HIV-1ADA in six-well trays with a multiplicity of infection of 0.2, incubated for 24 hours at 37°C, and washed with culture medium without FCS. Cells were cultured for 24 hours at 37°C in complete culture medium supplemented with library compounds. After 24 hours, supernatant was harvested, and cells were lysed for 15 min at 4°C in lysis buffer [20 mM tris-HCl (pH 7.5), 150 mM NaCl, 0.5% NP-40, 50 mM NaF, and 1 mM sodium orthovanadate, supplemented with complete protease inhibitor cocktail; Roche Diagnostic]. Lysates were centrifuged at 10,000g for 10 min at 4°C, and the quantity of HIV-1 p24 in the postnuclear supernatants was determined by ELISA.
Supplementary Material
Acknowledgments
We thank A. Mularski and C. Rabouille for carefully reading and commenting on the manuscript. Funding: We acknowledge the Cell and Tissue Imaging Facility (PICT-IBiSA), Institut Curie, a member of the French National Research Infrastructure, France-BioImaging (ANR10-INBS-04). This work was supported by grants from CNRS, INSERM, Université Paris Descartes, Agence Nationale de la Recherche (2011 BSV3 025 02), and Agence Nationale de Recherches sur le Sida et les Hépatites (ANRS, AO2012-2) to A.B., F.P., and F.N., and Fondation pour la Recherche Médicale (FRM DEQ20130326518) to F.N. This work has received support under the “Investissements d’Avenir” program launched by the French government and implemented by ANR with the references ANR-10-LABX-62-IBEID and ANR-10-IDEX-0001-02 PSL. Author contributions: G.B., A.B., F.N., and F.P. designed the study and analyzed data. G.B., F.H., I.S., Y.F., and M.F. performed experiments and analyzed data. S.T., A.L., and E.D.N. set up, performed, and analyzed the chemical screening. P.G. conducted the bioinformatics analysis of the screens. G.B., F.H., A.B., F.N., F.H., and F.P. wrote the manuscript, which all coauthors commented on. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/10/eaax0821/DC1
Fig. S1. Molecules 13 and 14 inhibit autopalmitoylation of DHHC3 and DHHC7.
Fig. S2. Molecules 13, 14, and 15 do not alter CXCR4 surface expression or induce cytotoxicity in primary macrophages.
Movie S1. Synchronized transport of CCR5.
Movie S2. Synchronized transport of TNF.
Movie S3. Synchronized transport of CCR5 and TNF.
REFERENCES AND NOTES
- 1.Boncompain G., Perez F., The many routes of Golgi-dependent trafficking. Histochem. Cell Biol. 140, 251–260 (2013). [DOI] [PubMed] [Google Scholar]
- 2.Alkhatib G., Combadiere C., Broder C. C., Feng Y., Kennedy P. E., Murphy P. M., Berger E. A., CC CKR5: A RANTES, MIP-1α, MIP-1β receptor as a fusion cofactor for macrophage-tropic HIV-1. Science 272, 1955–1958 (1996). [DOI] [PubMed] [Google Scholar]
- 3.Choe H., Farzan M., Sun Y., Sullivan N., Rollins B., Ponath P. D., Wu L., Mackay C. R., LaRosa G., Newman W., Gerard N., Gerard C., Sodroski J., The β-chemokine receptors CCR3 and CCR5 facilitate infection by primary HIV-1 isolates. Cell 85, 1135–1148 (1996). [DOI] [PubMed] [Google Scholar]
- 4.Deng H., Liu R., Ellmeier W., Choe S., Unutmaz D., Burkhart M., Marzio P. D., Marmon S., Sutton R. E., Hill C. M., Davis C. B., Peiper S. C., Schall T. J., Littman D. R., Landau N. R., Identification of a major co-receptor for primary isolates of HIV-1. Nature 381, 661–666 (1996). [DOI] [PubMed] [Google Scholar]
- 5.Doranz B. J., Rucker J., Yi Y., Smyth R. J., Samson M., Peiper S. C., Parmentier M., Collman R. G., Doms R. W., A dual-tropic primary HIV-1 isolate that uses fusin and the β-chemokine receptors CKR-5, CKR-3, and CKR-2b as fusion cofactors. Cell 85, 1149–1158 (1996). [DOI] [PubMed] [Google Scholar]
- 6.Dragic T., Litwin V., Allaway G. P., Martin S. R., Huang Y., Nagashima K. A., Cayanan C., Maddon P. J., Koup R. A., Moore J. P., Paxton W. A., HIV-1 entry into CD4+ cells is mediated by the chemokine receptor CC-CKR-5. Nature 381, 667–673 (1996). [DOI] [PubMed] [Google Scholar]
- 7.Liu R., Paxton W. A., Choe S., Ceradini D., Martin S. R., Horuk R., MacDonald M. E., Stuhlmann H., Koup R. A., Landau N. R., Homozygous defect in HIV-1 coreceptor accounts for resistance of some multiply-exposed individuals to HIV-1 infection. Cell 86, 367–377 (1996). [DOI] [PubMed] [Google Scholar]
- 8.Samson M., Libert F., Doranz B. J., Rucker J., Liesnard C., Farber C.-M., Saragosti S., Lapouméroulie C., Cognaux J., Forceille C., Muyldermans G., Verhofstede C., Burtonboy G., Georges M., Imai T., Rana S., Yi Y., Smyth R. J., Collman R. G., Doms R. W., Vassart G., Parmentier M., Resistance to HIV-1 infection in Caucasian individuals bearing mutant alleles of the CCR-5 chemokine receptor gene. Nature 382, 722–725 (1996). [DOI] [PubMed] [Google Scholar]
- 9.Hütter G., Nowak D., Mossner M., Ganepola S., Müssig A., Allers K., Schneider T., Hofmann J., Kücherer C., Blau O., Blau I. W., Hofmann W. K., Thiel E., Long-term control of HIV by CCR5 Delta32/Delta32 stem cell transplantation. N. Engl. J. Med. 360, 692–698 (2009). [DOI] [PubMed] [Google Scholar]
- 10.Dorr P., Westby M., Dobbs S., Griffin P., Irvine B., Macartney M., Mori J., Rickett G., Smith-Burchnell C., Napier C., Webster R., Armour D., Price D., Stammen B., Wood A., Perros M., Maraviroc (UK-427,857), a potent, orally bioavailable, and selective small-molecule inhibitor of chemokine receptor CCR5 with broad-spectrum anti-human immunodeficiency virus type 1 activity. Antimicrob. Agents Chemother. 49, 4721–4732 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fätkenheuer G., Pozniak A. L., Johnson M. A., Plettenberg A., Staszewski S., Hoepelman A. I., Saag M. S., Goebel F. D., Rockstroh J. K., Dezube B. J., Jenkins T. M., Medhurst C., Sullivan J. F., Ridgway C., Abel S., James I. T., Youle M., van der Ryst E., Efficacy of short-term monotherapy with maraviroc, a new CCR5 antagonist, in patients infected with HIV-1. Nat. Med. 11, 1170–1172 (2005). [DOI] [PubMed] [Google Scholar]
- 12.Jacobson J. M., Saag M. S., Thompson M. A., Fischl M. A., Liporace R., Reichman R. C., Redfield R. R., Fichtenbaum C. J., Zingman B. S., Patel M. C., Murga J. D., Pemrick S. M., D’Ambrosio P., Michael M., Kroger H., Ly H., Rotshteyn Y., Buice R., Morris S. A., Stavola J. J., Maddon P. J., Kremer A. B., Olson W. C., Antiviral activity of single-dose PRO 140, a CCR5 monoclonal antibody, in HIV-infected adults. J. Infect. Dis. 198, 1345–1352 (2008). [DOI] [PubMed] [Google Scholar]
- 13.Trkola A., Ketas T. J., Nagashima K. A., Zhao L., Cilliers T., Morris L., Moore J. P., Maddon P. J., Olson W. C., Potent, broad-spectrum inhibition of human immunodeficiency virus type 1 by the CCR5 monoclonal antibody PRO 140. J. Virol. 75, 579–588 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Perez E. E., Wang J., Miller J. C., Jouvenot Y., Kim K. A., Liu O., Wang N., Lee G., Bartsevich V. V., Lee Y.-L., Guschin D. Y., Rupniewski I., Waite A. J., Carpenito C., Carroll R. G., S Orange J., Urnov F. D., Rebar E. J., Ando D., Gregory P. D., Riley J. L., Holmes M. C., June C. H., Establishment of HIV-1 resistance in CD4+ T cells by genome editing using zinc-finger nucleases. Nat. Biotechnol. 26, 808–816 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tebas P., Stein D., Tang W. W., Frank I., Wang S. Q., Lee G., Spratt S. K., Surosky R. T., Giedlin M. A., Nichol G., Holmes M. C., Gregory P. D., Ando D. G., Kalos M., Collman R. G., Binder-Scholl G., Plesa G., Hwang W.-T., Levine B. L., June C. H., Gene editing of CCR5 in autologous CD4 T cells of persons infected with HIV. N. Engl. J. Med. 370, 901–910 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Boncompain G., Divoux S., Gareil N., de Forges H., Lescure A., Latreche L., Mercanti V., Jollivet F., Raposo G., Perez F., Synchronization of secretory protein traffic in populations of cells. Nat. Methods 9, 493–498 (2012). [DOI] [PubMed] [Google Scholar]
- 17.Fourriere L., Divoux S., Roceri M., Perez F., Boncompain G., Microtubule-independent secretion requires functional maturation of Golgi elements. J. Cell Sci. 129, 3238–3250 (2016). [DOI] [PubMed] [Google Scholar]
- 18.Lippincott-Schwartz J., Yuan L. C., Bonifacino J. S., Klausner R. D., Rapid redistribution of Golgi proteins into the ER in cells treated with brefeldin A: Evidence for membrane cycling from Golgi to ER. Cell 56, 801–813 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cole N. B., Sciaky N., Marotta A., Song J., Lippincott-Schwartz J., Golgi dispersal during microtubule disruption: Regeneration of Golgi stacks at peripheral endoplasmic reticulum exit sites. Mol. Biol. Cell 7, 631–650 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Blanpain C., Wittamer V., Vanderwinden J.-M., Boom A., Renneboog B., Lee B., Le Poul E., El Asmar L., Govaerts C., Vassart G., Doms R. W., Parmentier M., Palmitoylation of CCR5 is critical for receptor trafficking and efficient activation of intracellular signaling pathways. J. Biol. Chem. 276, 23795–23804 (2001). [DOI] [PubMed] [Google Scholar]
- 21.Percherancier Y., Planchenault T., Valenzuela-Fernandez A., Virelizier J.-L., Arenzana-Seisdedos F., Bachelerie F., Palmitoylation-dependent control of degradation, life span, and membrane expression of the CCR5 receptor. J. Biol. Chem. 276, 31936–31944 (2001). [DOI] [PubMed] [Google Scholar]
- 22.Cavrois M., de Noronha C., Greene W. C., A sensitive and specific enzyme-based assay detecting HIV-1 virion fusion in primary T lymphocytes. Nat. Biotechnol. 20, 1151–1154 (2002). [DOI] [PubMed] [Google Scholar]
- 23.Benkirane M., Jin D. Y., Chun R. F., Koup R. A., Jeang K. T., Mechanism of transdominant inhibition of CCR5-mediated HIV-1 infection by ccr5Δ32. J. Biol. Chem. 272, 30603–30606 (1997). [DOI] [PubMed] [Google Scholar]
- 24.Glass W. G., McDermott D. H., Lim J. K., Lekhong S., Yu S. F., Frank W. A., Pape J., Cheshier R. C., Murphy P. M., CCR5 deficiency increases risk of symptomatic West Nile virus infection. J. Exp. Med. 203, 35–40 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lim J. K., Louie C. Y., Glaser C., Jean C., Johnson B., Johnson H., McDermott D. H., Murphy P. M., Genetic deficiency of chemokine receptor CCR5 is a strong risk factor for symptomatic West Nile virus infection: A meta-analysis of 4 cohorts in the US epidemic. J. Infect. Dis. 197, 262–265 (2008). [DOI] [PubMed] [Google Scholar]
- 26.Kindberg E., Mickienė A., Ax C., Åkerlind B., Vene S., Lindquist L., Lundkvist Å., Svensson L., A deletion in the chemokine receptor 5 (CCR5) gene is associated with tickborne encephalitis. J. Infect. Dis. 197, 266–269 (2008). [DOI] [PubMed] [Google Scholar]
- 27.Brass A. L., Dykxhoorn D. M., Benita Y., Yan N., Engelman A., Xavier R. J., Lieberman J., Elledge S. J., Identification of host proteins required for HIV infection through a functional genomic screen. Science 319, 921–926 (2008). [DOI] [PubMed] [Google Scholar]
- 28.König R., Zhou Y., Elleder D., Diamond T. L., Bonamy G. M., Irelan J. T., Chiang C. Y., Tu B. P., De Jesus P. D., Lilley C. E., Seidel S., Opaluch A. M., Caldwell J. S., Weitzman M. D., Kuhen K. L., Bandyopadhyay S., Ideker T., Orth A. P., Miraglia L. J., Bushman F. D., Young J. A., Chanda S. K., Global analysis of host-pathogen interactions that regulate early-stage HIV-1 replication. Cell 135, 49–60 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Park R. J., Wang T., Koundakjian D., Hultquist J. F., Lamothe-Molina P., Monel B., Schumann K., Yu H., Krupzcak K. M., Garcia-Beltran W., Piechocka-Trocha A., Krogan N. J., Marson A., Sabatini D. M., Lander E. S., Hacohen N., Walker B. D., A genome-wide CRISPR screen identifies a restricted set of HIV host dependency factors. Nat. Genet. 49, 193–203 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhou H., Xu M., Huang Q., Gates A. T., Zhang X. D., Castle J. C., Stec E., Ferrer M., Strulovici B., Hazuda D. J., Espeseth A. S., Genome-scale RNAi screen for host factors required for HIV replication. Cell Host Microbe 4, 495–504 (2008). [DOI] [PubMed] [Google Scholar]
- 31.Charette N., Holland P., Frazer J., Allen H., Dupré D. J., Dependence on different Rab GTPases for the trafficking of CXCR4 and CCR5 homo or heterodimers between the endoplasmic reticulum and plasma membrane in Jurkat cells. Cell. Signal. 23, 1738–1749 (2011). [DOI] [PubMed] [Google Scholar]
- 32.Li C., Wei Z., Fan Y., Huang W., Su Y., Li H., Dong Z., Fukuda M., Khater M., Wu G., The GTPase Rab43 controls the anterograde ER-Golgi trafficking and sorting of GPCRs. Cell Rep. 21, 1089–1101 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fukata M., Fukata Y., Adesnik H., Nicoll R. A., Bredt D. S., Identification of PSD-95 palmitoylating enzymes. Neuron 44, 987–996 (2004). [DOI] [PubMed] [Google Scholar]
- 34.Gottlieb C. D., Zhang S., Linder M. E., The cysteine-rich domain of the DHHC3 palmitoyltransferase is palmitoylated and contains tightly bound zinc. J. Biol. Chem. 290, 29259–29269 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tang L., Qiu R., Tang Y., Wang S., Cadmium-zinc exchange and their binary relationship in the structure of Zn-related proteins: A mini review. Metallomics 6, 1313–1323 (2014). [DOI] [PubMed] [Google Scholar]
- 36.Tomita F., Tamaoki T., Tetrocarcins, novel antitumor antibiotics. I. Producing organism, fermentation and antimicrobial activity. J. Antibiot. 33, 940–945 (1980). [DOI] [PubMed] [Google Scholar]
- 37.Nakashima T., Miura M., Hara M., Tetrocarcin A inhibits mitochondrial functions of Bcl-2 and suppresses its anti-apoptotic activity. Cancer Res. 60, 1229–1235 (2000). [PubMed] [Google Scholar]
- 38.Roche M., Jakobsen M. R., Sterjovski J., Ellett A., Posta F., Lee B., Jubb B., Westby M., Lewin S. R., Ramsland P. A., Churchill M. J., Gorry P. R., HIV-1 escape from the CCR5 antagonist maraviroc associated with an altered and less-efficient mechanism of gp120-CCR5 engagement that attenuates macrophage tropism. J. Virol. 85, 4330–4342 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tilton J. C., Wilen C. B., Didigu C. A., Sinha R., Harrison J. E., Agrawal-Gamse C., Henning E. A., Bushman F. D., Martin J. N., Deeks S. G., Doms R. W., A maraviroc-resistant HIV-1 with narrow cross-resistance to other CCR5 antagonists depends on both N-terminal and extracellular loop domains of drug-bound CCR5. J. Virol. 84, 10863–10876 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mazzolini J., Herit F., Bouchet J., Benmerah A., Benichou S., Niedergang F., Inhibition of phagocytosis in HIV-1-infected macrophages relies on Nef-dependent alteration of focal delivery of recycling compartments. Blood 115, 4226–4236 (2010). [DOI] [PubMed] [Google Scholar]
- 41.Jordan M., Schallhorn A., Wurm F. M., Transfecting mammalian cells: Optimization of critical parameters affecting calcium-phosphate precipitate formation. Nucleic Acids Res. 24, 596–601 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pleskoff O., Tréboute C., Brelot A., Heveker N., Seman M., Alizon M., Identification of a chemokine receptor encoded by human cytomegalovirus as a cofactor for HIV-1 entry. Science 276, 1874–1878 (1997). [DOI] [PubMed] [Google Scholar]
- 43.Clavel F., Charneau P., Fusion from without directed by human immunodeficiency virus particles. J. Virol. 68, 1179–1185 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/10/eaax0821/DC1
Fig. S1. Molecules 13 and 14 inhibit autopalmitoylation of DHHC3 and DHHC7.
Fig. S2. Molecules 13, 14, and 15 do not alter CXCR4 surface expression or induce cytotoxicity in primary macrophages.
Movie S1. Synchronized transport of CCR5.
Movie S2. Synchronized transport of TNF.
Movie S3. Synchronized transport of CCR5 and TNF.