

The pore formation and translocation machinery of Tc toxins is conserved; in contrast, the outer shell is structurally variable.
Abstract
Tc toxins use a syringe-like mechanism to penetrate the membrane and translocate toxic enzymes into the host cytosol. They are composed of three components: TcA, TcB, and TcC. Low-resolution structures of TcAs from different bacteria suggest a considerable difference in their architecture and possibly in their mechanism of action. Here, we present high-resolution structures of five TcAs from insect and human pathogens, which show a similar overall composition and domain organization. Essential structural features, including a trefoil protein knot, are present in all TcAs, suggesting a common mechanism of action. All TcAs form functional pores and can be combined with TcB-TcC subunits from other species to form active chimeric holotoxins. We identified a conserved ionic pair that stabilizes the shell, likely operating as a strong latch that only springs open after destabilization of other regions. Our results provide new insights into the architecture and mechanism of the Tc toxin family.
INTRODUCTION
Large multisubunit toxin complex (Tc) toxins are present in a variety of bacterial pathogens. They were first identified in the insect pathogenic bacterium Photorhabdus luminescens (1), and it was shown that these toxins have oral activity against insects (2). Consequently, the study of Tc toxins has become an important aspect in the search for novel biopesticides as an alternative to Bacillus thuringiensis (Bt) toxins. Bt toxins were the first biopesticides that were successfully used in transgenic plants. However, an increasing number of insects have developed resistance against Bt toxins (3), creating a crucial need for alternatives. Since Tc toxins are also encoded by human pathogenic bacteria like Yersinia pestis (4, 5), Yersinia pseudotuberculosis (4, 5), and Morganella morganii (6), the elucidation of the function and mechanism of action of these toxins is also of high medical relevance.
Tc toxins from P. luminescens are composed of three subunits: TcA, TcB, and TcC. The 1- to 1.4-MDa homopentameric TcA acts as an injecting device responsible for translocating the actual toxic component into host cells (7, 8). TcA consists of a preformed channel that is surrounded by a shell domain. Putative receptor-binding domains (RBDs) localized at the periphery of the shell possibly interact with receptors on the host cell membrane (9). pH-induced opening of the shell releases a putative entropic spring that drives the injection of the TcA channel into the membrane (9). TcB and TcC form a cocoon that harbors the actual toxic component, namely, the C-terminal hypervariable region (HVR) of TcC, which is autoproteolytically cleaved (9, 10). Binding of the TcB-TcC cocoon to the TcA channel via a six-bladed β-propeller triggers the opening of the cocoon and translocation of the toxic enzyme into the channel (9, 11). During this process, parts of the β-propeller completely unfold and refold into an alternative conformation upon binding (11). The enzyme passes through a narrow negatively charged constriction site inside the cocoon, most likely acting as an extruder that releases the unfolded protein with its C-terminus first into the translocation channel (11).
In our recent work, we demonstrated that the transmembrane helices of TcdA1 from P. luminescens rearrange once they enter the membrane to open the initially closed pore (12). The linker domain, which is stretched in the prepore state (9), is folded and tightly packed in a pocket that is formed between the shell and channel domains (12).
Besides the high-resolution structures of TcdA1 in its prepore and pore state (9, 11, 12), only low-resolution structures of two other TcAs have been determined. Whereas the TcA component of YenTCA1A2 from Yersinia entomophaga, like TcdA1 from P. luminescens, forms a pentameric bell-shaped complex with an inner channel and an outer shell (7), the structure of XptA1 from Xenorhabdus nematophila appears to have a tetrameric cage-like structure with a central cavity (13). This evidence would tend to suggest that Tc toxins might differ considerably in their architecture and, as a consequence, possibly in their mechanism of action.
To better understand the structural variety of Tc toxins and to obtain a holistic view on their architectural organization and function, we determined the structures of TcA components from different insect and human pathogenic bacteria using single-particle cryo–electron microscopy (cryo-EM). The study included TcdA1 and TcdA4 from P. luminescens (Pl-TcdA1 and Pl-TcdA4), XptA1 from X. nematophila (Xn-XptA1), TcdA4 from M. morganii (Mm-TcdA4), and TcaA-TcaB from Y. pseudotuberculosis (Yp-TcaATcaB). By using our single-particle processing software SPHIRE (14), we obtained resolutions of up to 2.8 Å.
Contrary to previous expectations, the structures revealed that the examined TcAs, including Xn-XptA1, share the same pentameric bottle-shaped structure and that the domain organization of these toxin components is almost identical. Our study demonstrates that functionally crucial, structural features—i.e., the linker domain, the translocation channel, and the TcB-binding domain—have high structural similarities across all analyzed TcAs.
The main structural differences between the TcAs are found at the periphery of the shell where the RBDs and the neuraminidase-like domain are located. Unexpectedly, the bottom of the shell, formed by the neuraminidase-like domain, is not completely closed in Xn-XptA1, Mm-TcdA4, and Yp-TcaATcaB. We identified a previously unknown coiled-coil domain in Yp-TcaATcaB that is not found in any of the other studied TcAs. It reaches out from the shell and interacts with the funnel-shaped TcB-binding domain of TcA. Inversed charges close to the tip of the translocation channel in Xn-XptA1, Mm-TcdA4, and Yp-TcaATcaB indicate a possible difference in protein translocation. Overall, our results suggest that Tc toxins from different organisms share a common central architecture, resulting in a conserved assembly and pore formation mechanism, while the variability in the RBDs enables the targeting of different hosts.
RESULTS
TcA toxin subunits share a common overall architecture
To exclude that the crystallization buffer or crystal contacts had an influence on our previous crystal structure of Pl-TcdA1, we first determined a 2.8-Å resolution structure of Pl-TcdA1 from P. luminescens using cryo-EM and single-particle analysis in SPHIRE (14). The overall structure of Pl-TcdA1 was the same, but the higher resolution, 2.8 Å in comparison to 4 Å, enabled us to improve the atomic model of the complex (Fig. 1A; figs. S1A and S2, A to E; and movie S1).
Fig. 1. Structures of five TcAs.
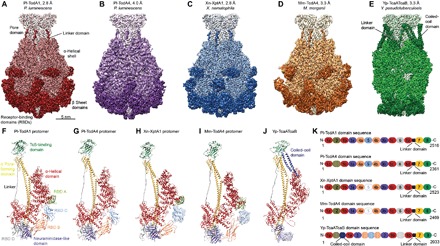
(A to E) Cryo-EM density maps of Pl-TcdA1, Pl-TcdA4, Xn-XptA1, Mm-TcdA4, and Yp-TcaATcaB, respectively, with the average resolutions according to 0.143 Fourier shell correlation. The color gradient from light to dark represents the pore domain with the TcB-binding domain, the α-helical shell, the β sheet domains, and the linker. (F to J) Structures of the TcA protomers. Pl-TcdA4 does not contain RBD A. RBD C was not well resolved in Xn-XptA1 and Mm-TcdA4 and is therefore not included in the models. Yp-TcaATcaB does not contain any RBD. The unique coiled-coil domain of Yp-TcaATcaB is highlighted in dark blue. Ninety-nine residues (amino acids 1140 to 1239) of the neuraminidase-like domain and the first 57 residues at the N-terminus (dotted line) Yp-TcaATcaB could not be built. The densities of the domains that could not be built are shown to indicate their location (H to K). The N-terminus Yp-TcaATcaB (residues 1 to 57) is depicted as red dotted line (J). (K) Domain organization of Pl-TcdA1, Pl-TcdA4, Xn-XptA1, Mm-TcdA4, and Yp-TcaATcaB. 1 = helical shell, 2 = RBD A, 3 = neuraminidase-like domain, 4 = RBD B, 5 = RBD C, 6 = RBD D, 7 = channel, and 8 = TcB-binding domain and the linker domain in black. Domains that could not be built are shown as solid circles, and domains that are missing in the sequence are hatched.
To compare the structure of Pl-TcdA1 with the structure of other TcA complexes, we heterologously expressed Pl-TcdA4, Xn-XptA1, Mm-TcdA4, and Yp-TcaATcaB in Escherichia coli and purified the proteins. While the TcAs from P. luminescens, X. nematophila, and M. morganii are single proteins, Yp-TcaATcaB is composed of two subunits (TcaA and TcaB). TcaA forms the α-helical shell, and TcaB comprises the pore-forming domain and the bottom part of the toxin (fig. S1F). To obtain complete complexes, Yp-TcaATcaB was expressed as a fusion construct (Materials and Methods).
We then determined the near-atomic structures of the different TcA complexes using cryo-EM and single-particle analysis yielding resolutions of 4.0 Å (Pl-TcdA4), 2.8 Å (Xn-XptA1), and 3.3 Å (Mm-TcdA4 and Yp-TcaATcaB) (Fig. 1, B to E; figs. S1, B to E, S2, and S3; and movie S1). The cryo-EM structures allowed us to build atomic models of the complexes, revealing that all five studied TcAs share the same overall architecture. They all have a pentameric bell-shaped structure consisting of a central pore-forming domain surrounded by an outer shell, which is made up of an α-helical part and β sheet domains. The linker domain, which connects the pore-forming domain with the outer shell, is present in all examined TcAs (Fig. 1, A to J).
In contrast to the previously reported low-resolution structure of Xn-XptA1, which suggested a tetramer with a cage-like structure (13), our structure of Xn-XptA1 reveals that it is a pentameric complex with a bell-shaped appearance similar to Pl-TcdA1 from P. luminescens. The relatively high amino acid sequence identity of approximately 46.7% between Xn-XptA1 and Pl-TcdA1 supports our results.
Differences in the shell domain
In all TcAs, the inner scaffold of the shell is composed of an α-helical domain (Fig. 1K). It can be divided into a large and a small lobe that are arranged perpendicular to each other forming an L-shape (Fig. 2A). The small lobe can be further subdivided into two pseudo-twofold symmetrical subunits in an X-shaped structure (Fig. 2A). The large lobe contains two different pseudo repeats (pseudo repeat 1 and pseudo repeat 2; Fig. 2, B and C), which are both composed of several helix-loop-helix motifs. The pseudo repeat 1 contains three repeating domains and has the same fold and organization in all analyzed TcAs (Fig. 2B). However, the pseudo repeat 2, which is composed of two helix-loop-helix motifs, reveals two additional insertions in the case of Yp-TcaATcaB, namely, an enlarged loop and a coiled-coil domain (Figs. 1, E and J, and 2, C and D). The coiled-coil domain reaches out to the TcB-binding domain (fig. S1F), thus connecting the two subunits, TcaA and TcaB. The coiled-coil domain probably increases the stability of the complex, which, in contrast to the other TcAs, does not contain any RBDs (see below). To test this hypothesis, we removed the domain by site-directed mutagenesis and replaced it by a linker of six amino acids (GRPSSG) to generate a short loop between the two deletion sites (Yp-TcaATcaB–∆622–714). The fusion construct Yp-TcaATcaB–∆622–714 expressed less well in E. coli than Yp-TcaATcaB(WT) and had the tendency to disassemble and aggregate, as judged by SDS–polyacrylamide gel electrophoresis and gel filtration (fig. S3, K and M). However, in negative-stain EM, we observed next to the complete toxin complexes many smaller particles that are absent in the wild-type (WT) sample (fig. S3, L and N). These findings suggest that Yp-TcaATcaB is less stable and shows a higher tendency to disassemble when the coiled-coil domain is removed.
Fig. 2. Organization of the α-helical shell.
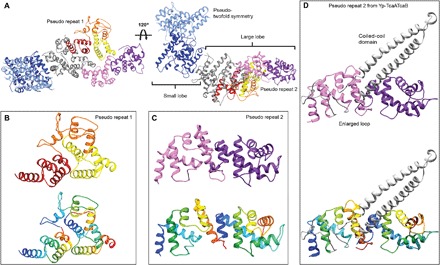
(A) The α-helical shell of a TcA protomer shown for Pl-TcdA1 can be divided into a small lobe (amino acids 1 to 160, 964 to 1090, 1608 to 1632, and 1762 to 1972) and a large lobe (amino acids 161 to 297 and 434 to 963). The small lobe has a pseudo-twofold symmetry (light and dark blue), resulting in an X-shaped structure. The large lobe contains two pseudo repeats; pseudo repeat 1 is shown in red, orange, and yellow, and pseudo repeat 2 is shown in rose and magenta. (B) The three repeating subdomains of pseudo repeat 1 are depicted in red, orange, and yellow in the upper panel and in rainbow colors (colored from blue to red from N- to C-terminus) in the lower panel. (C) The two repeating subdomains of pseudo repeat 2 are depicted in rose and magenta in the upper panel and in rainbow colors in the lower panel. (D) The pseudo repeat 2 in Yp-TcaATcaB shows the same overall fold, except for the insertion of the coiled-coil domain and an enlarged loop (both in gray).
The β sheet domains of the shell are less conserved than its α-helical domain (fig. S1G). The main differences are mostly located at the RBDs (Fig. 1, A to J). Pl-TcdA1 has four RBDs and a neuraminidase-like domain; RBD A is inserted in the large lobe, and RBD B, RBD C, and RBD D, as well as the neuraminidase-like domain, are inserted in the small lobe (Fig. 3D). Whereas Xn-XptA1 and Mm-TcdA4 have the same number of RBDs as Pl-TcdA1, Pl-TcdA4 misses RBD A, resulting in a slimmer shaped molecule (Fig. 1, A to D). Yp-TcaATcaB does not contain any RBD, only the neuraminidase-like domain (Fig. 1, E and J). This leads to a more distinct and slimmer shape of the shell domain of Yp-TcaATcaB. The N-terminal domain of TcaA (residues 1 to 57) and a larger peripheral domain of the neuraminidase-like domain (residues 1140 to 1239) were not resolved in Yp-TcaATcaB. Because of the missing RBDs, these regions are probably more flexible than in the other TcAs.
Fig. 3. A 31 trefoil protein knot is present in all TcAs.
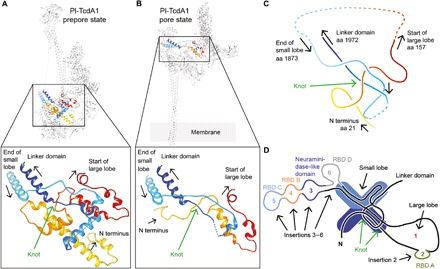
(A) A 31 trefoil protein knot is present in all five TcA prepores. The knot structure in a Pl-TcdA1 prepore protomer and a close-up view is presented. The polypeptide chain is highlighted in rainbow colors. The dashed line indicates missing residues (amino acids 1933 to 1938). (B) The protein knot is also present in the Pl-TcdA1 protomer in the pore state (Protein Data Bank IDs: 5LKH and 5LKI). The dotted line indicates missing residues (amino acids 191 to 1946). (C) Simplified structure of the protein knot of Pl-TcdA1. The arrows indicate the direction of sequence, and the amino acid (aa) numbering corresponds to Pl-TcdA1. (D) Scheme of the shell organization of a TcA protomer showing the location of the knot in the small lobe. RBD A to RBD D and the neuraminidase-like domain are inserted in the main sequence of the big and small lobes.
A conserved 31 trefoil protein knot stabilizes the base of the linker
The shell of all five TcAs contains a 31 trefoil (15) protein knot (Fig. 3A; fig. S4, A to J; and movie S2). The knot tightly connects the N-terminal part of the protein (residues 21 to 157 in Pl-TcdA1) with the last domain of the α-helical shell before the linker (residues 1873 to 1972 in Pl-TcdA1) (Fig. 3, C and D, and fig. S4, K and L). The protein knot is also present in the pore state of Pl-TcdA1 (12), indicating that it does not untangle during prepore-to-pore transition (Fig. 3B).
A 31 trefoil protein knot is the most simple and common knot type. It mostly occurs in smaller proteins or enzymes such as methyltransferases and carbonic anhydrases (16). With ~280 kDa (mass of Pl-TcdA1 monomer), TcA is, to our knowledge, by far the largest protein for which such a knot has been observed. The folding mechanism of a knotted protein has been shown to be quite complex, and the folding rate of knotted proteins is decreased (17). These findings suggest that it is rather surprising that the complex TcA structure contains a knot.
In many cases, the function of protein knots is unknown. Several studies, however, indicate that a protein knot has a stabilizing effect on proteins, particularly during mechanical stress and conformational changes (18). In the case of Tc toxins, the knot is indeed strengthening the base of the linker, which is especially put under strain during the prepore-to-pore transition (Fig. 3, A and B, fig. S4L, and movie S2). Consequently, the protein knot might also stabilize the stretched linker domain in the prepore state of TcA. Together, we conclude that the protein knot in TcA is an important conserved structural feature and propose that it is essential for the structural stability and function of all Tc toxins.
Differences in the pore domain
Between all TcAs, the pore domain is the part of the protein with the highest conservation (figs. S1G and S5A) and the highest structural similarity (fig. S1H). In addition, it is the among the best-resolved regions in all TcA reconstructions (figs. S2, E, J, and O, and S3, E and J). The differences between Pl-TcdA4 and Pl-TcdA1 are negligible in the pore domain. Hence, we will only compare Xn-XptA1, Mm-TcdA4, Yp-TcaATcaB, and Pl-TcdA1. The hydrophobic surface properties of the channel exterior are comparable in all TcAs (fig. S5B). The tip of their channels (5 nm) is highly hydrophobic, indicating that they have a similarly sized transmembrane domain (fig. S5B). Whereas the hydrophobic loops at the tip of the transmembrane domain are highly conserved, the residue residing in the center of each loop is variable, suggesting different interactions with the host membrane (fig. S5, A and D). The surface potentials are similar in Pl-TcdA1, Xn-XptA1, and Mm-TcdA4 with a patch of positive charges at the upper part and a patch of negative charges at the lower part of the α pore-forming domain. The channel of Yp-TcaATcaB, however, is more negatively charged at its outside and misses the prominent stretch of positive charges below the funnel (fig. S5C).
The lumen of the channel of Pl-TcdA1 contains several discrete bands of negative electrostatic potential (Fig. 4A) (9, 12). Therefore, Pl-TcdA1 is selective to cations if reconstituted in membranes without TcB-TcC (9, 19). The pattern of negative charges is also found in the lumen of the Xn-XptA1, Mm-TcdA4, and Yp-TcaATcaB. However, the lower one to two bands at their tip are positively charged instead (Fig. 4A). Despite distinct charges at the tip, the three TcAs are able to transport the HVR of TccC3 of P. luminescens (fig. S7). Further studies are needed to evaluate whether there is an ionic specific transport for the distinct TcAs and if the positive charged residues have an impact on the cargo transport.
Fig. 4. Comparison of the TcA channels.
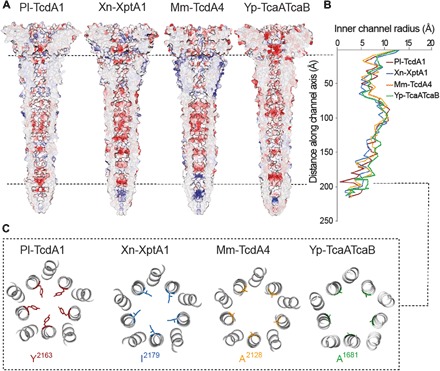
(A) Cross sections of the TcA channels demonstrating the electrostatic Coulomb potential in the channel lumen of Pl-TcdA1, Xn-XptA1, Mm-TcdA4, and Yp-TcaATcaB at pH 7. Positively charged (14 kcal/mol) and negatively charged (−14 kcal/mol) residues are colored in blue and red, respectively. (B) Graph depicting the inner channel radius of Pl-TcdA1 (red), Xn-XptA1 (blue), Mm-TcdA4 (orange), and Yp-TcaATcaB (green). At the narrowest position of the Pl-TcdA1 prepore (Y2163, indicated by the dashed line), the diameter of the channel is 3.9 Å. In Xn-XptA1, Mm-TcdA4, and Yp-TcaATcaB, the channel diameter reaches only 8.2, 7.8, and 8.4 Å at the narrowest part, respectively. (C) Slice through the channel at the position of the Pl-TcdA1 channel constriction (Y2163).
The channel lumen shows a comparable diameter profile for the four TcAs in the prepore state (Fig. 4B). Starting with a large diameter of approximately 26 Å for all TcAs at the TcB-interacting domain, the channel radius is decreased along the transport direction of the toxin. Between 40 and 65 Å from the channel entry, all four TcA channels are narrowed to a diameter of approximately 10 Å. Afterward, the channel widens up and reaches a diameter of up to 20 Å. At this position, there is an accumulation of hydrophobicity in the analyzed TcA channels (fig. S5E).
A major difference between the different TcAs can be found at the tip of the channel. Here, Y2163 in Pl-TcdA1 forms a constriction site with a diameter of 3.9 Å (Fig. 4C) (9) that opens after membrane insertion (12). Xn-XptA1, Mm-TcdA4, and Yp-TcaATcaB have smaller residues (I2179, A2128, and A1681, respectively) at this position, resulting in wider diameters with minima of 8.6, 8.9, and 8.4 Å, respectively (Fig. 4C). The tip is closed in all four TcAs in the prepore conformation (Fig. 4A).
To understand whether the minor differences in the channel properties have an influence on its function, we reconstituted TcA into black lipid bilayers and measured the conductance of the channel. All TcAs formed ion-permeable pores within a pH range of 4 to 11 (fig. S5, F to I). In accordance with our previous work, Pl-TcdA1 showed a higher pore-forming activity at extreme pH values compared to pH 6 (9), which goes along with the results obtained for Xn-XptA1 and Mm-TcdA4. Yp-TcaATcaB incorporated more readily into the membrane at pH 6, as judged by a higher rate of pore formation events in comparison to the other TcAs (fig. S5, G to I). In addition, Yp-TcaATcaB was, in general, less stable at more extreme pH values (fig. S6, A and B), which is also reflected in noisier single-channel currents at pH 4 and 11 (fig. S5, G and I, right).
While the conductance of Mm-TcdA4 is similar to Pl-TcdA1, the mean single-channel conductance values of Xn-XptA1 and Yp-TcaATcaB are 100 to 150 pS lower, independent of the pH. The charge distribution and the channel diameter influence the channel conductivity. Most likely, the varying charge distributions within the channel lumen (Fig. 4A) are responsible for the measured differences. However, although the channel diameter is comparable for the four TcAs in the prepore state (Fig. 4B), we cannot exclude more variable channel diameters for the distinct TcAs in the pore state, as it was shown that the pore diameter of Pl-TcdA1 is increased up to 42 Å after membrane insertion by cylindrical rearrangement of the 10 α helices (12).
The conservation of the TcB-binding domain allows the formation of chimeric holotoxins
The TcB-binding domain is crucial for the assembly of the holotoxin complex and is conserved among the four TcAs. Accordingly, the TcB-binding domains of TcAs from four different organisms (Pl-TcdA1, Xn-XptA1, Mm-TcdA4, and Yp-TcaATcaB) practically share identical folds (fig. S6C). The lowest variance is found in the central β sheet region, both with respect to amino acid conservation and root mean square deviation (RMSD) (fig. S6, C and D). Outward-facing loops are less conserved and differ slightly in their size. In the central β sheets, however, almost all residues are identical in the four TcAs (fig. S6, C and D).
The identical fold and high sequence conservation, together with the high conservation of the β-propeller of TcB (11), suggest that it is possible to form chimeric holotoxins by combining TcB-TcC complexes with TcA from different bacteria. When TcdB2 and TccC3 from P. luminescens is coexpressed with XptA2 from X. nematophila, a hybrid Tc is formed that is highly active against insects (20).
To examine whether TcB-TcC from P. luminescens (Pl-TcdB2-TccC3) can form holotoxins with TcAs from other organisms, we incubated it with all four TcAs. Using negative-stain EM, we observed holotoxin complexes for all combinations tested (Fig. 5, A to D). In the case of Mm-TcdA4 and Yp-TcaATcaB, however, some TcAs were still present that did not interact with TcB-TcC (Fig. 5, C and D). Expectedly, the measured affinities between Pl-TcdB2-TccC3 and Pl-TcdA1 (WT complex), Yp-TcaATcaB, and Xn-XptA1 were very high [dissociation constant (KD) = 0.63 ± 0.03 nM, KD = 0.95 ± 0.02 nM, and KD = 1.99 ± 0.15 nM, respectively] (Fig. 5E and fig. S6, E to H). In line with the EM results, the affinity between Pl-TcdB2-TccC3 and Mm-TcdA4 was one order of magnitude lower (KD = 11.3 ± 0.2 nM) (Fig. 5, C and E). All chimeric holotoxins were fully functional and active against human embryonic kidney (HEK) 293 T cells (fig. S7). However, the hybrid toxins were less effective than the native holotoxin complex formed by Pl-TcdA1 and Pl-TcdB2-TccC3, as 10 times higher concentrations of chimeric toxins are needed to achieve a comparable phenotype (fig. S7, C to E).
Fig. 5. Formation of chimeric holotoxins.
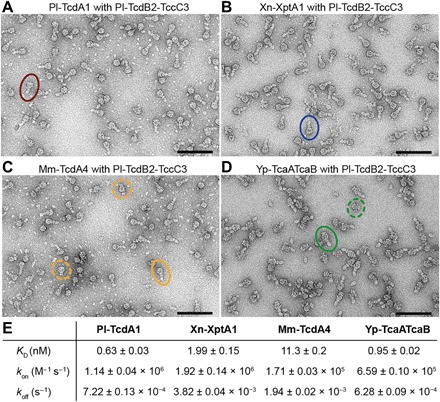
(A to D) Negative-stain electron micrographs after complex formation of different TcAs with TcdB2-TccC3 from P. luminescens and size exclusion chromatography. For each complex, a holotoxin particle is highlighted by circles. For Mm-TcdA4 and Yp-TcaATcaB, unbound TcAs are marked with dashed circles. Scale bars, 100 nm. (E) Table with the measured affinities for the chimeric complexes by biolayer interferometry including the dissociation constant (KD) and the on- and off-rate of complex formation (kon and koff, respectively). A global fit according to a 1:1 binding model was applied, including six to seven individual curves. The obtained parameters are the mean value ± the error of the fit. See also fig. S6.
Together, our results demonstrate that functional chimeric holotoxins can be assembled from different Tc subunits. This allows combining different host specificities with different enzymatic activities and therefore broadens the spectrum of the potential application of Tc toxins as biopesticides.
The electrostatic lock of the shell domain
The neuraminidase-like domain closes the shell at the bottom of Pl-TcdA1 (Fig. 6, A to C, and fig. S1A) (9). On the basis of the charge distribution in this region, we previously proposed that this domain functions as an electrostatic lock, which is closed at neutral pH and opens at high or low pH because of the repulsion of charged amino acids, triggering the prepore-to-pore transition of Pl-TcdA1 (9).
Fig. 6. Electrostatic potential of the neuraminidase-like domain and conserved ionic interactions in the shell of TcAs.
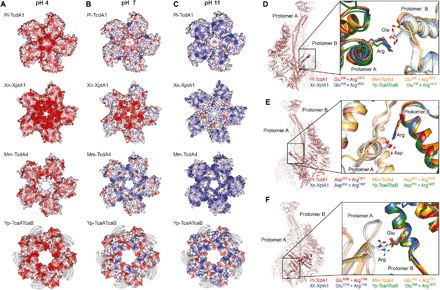
(A to C) Surface electrostatic Coulomb potential of the neuraminidase-like domain at different pH values, viewed from the bottom of TcA. Positively charged (14 kcal/mol) and negatively charged (−14 kcal/mol) residues are colored in blue and red, respectively. Surface electrostatic Coulomb potential at pH 4 (A), pH 7 (B), and pH 11 (C) are shown. The cryo-EM density map of the 99 residues, which could not be built in Yp-TcaATcaB, is depicted in gray. (D to F) Conserved ionic interactions between two protomers in Pl-TcdA1, Xn-XptA1, Mm-TcdA4, and Yp-TcaATcaB. The left panel shows two Pl-TcdA1 protomers indicating the different interaction sites, and the right panel presents close-up views of each interaction for Pl-TcdA1 (red), Xn-XptA1 (blue), Mm-TcdA4 (orange), and Yp-TcaATcaB (green). (D) Interaction of a glutamate (protomer A) with an arginine (protomer B). The residue distance is 3.8 Å in the prepore and 9.2 Å in the pore state of Pl-TcdA1. (E) Interaction of an aspartate (protomer A) with an arginine (protomer B). The residue distance is 3.5 Å in the prepore and 5.8 Å in the pore state of Pl-TcdA1. The interacting residues (D and E) belong to the α-helical shell domain. (F) Interaction of an arginine (neuraminidase-like domain of protomer A) with a glutamate (small lobe of protomer B). The residue distance is 3.9 Å in the prepore and 25 Å in the pore state of Pl-TcdA1.
To find out whether this is a general feature of TcAs, we compared their neuraminidase-like domains. The fold of the neuraminidase-like domain from different TcAs is almost identical (fig. S8, A, B, and F to I). Only the position of the second subdomain (residues 1140 to 1239) differs in Yp-TcaATcaB from that of the other TcAs (fig. S8C). Because of one loop per domain, which is five to eight residues shorter in Xn-XptA1, Mm-TcdA4, and Yp-TcaATcaB than in Pl-TcdA1 (fig. S8, D and E), the shells of these proteins are not closed at their bottom (Fig. 6, A to C). Therefore, this region cannot be responsible for holding together the shell domains in the prepore pentamer.
The surface electrostatic potentials at pH 4 and 11 of the neuraminidase-like domain of all TcAs show an accumulation of either positive or negative charges, leading to repulsions at these pH values (Fig. 6, A and C). At neutral pH, however, only Yp-TcaATcaB shares the equal distribution of opposite charges with Pl-TcdA1 (Fig. 6B). Xn-XptA1 and Mm-TcdA4 are more negatively or positively charged in this region, respectively (Fig. 6B), indicating that the shell is not stabilized by complimentarily charged residues in the neuraminidase-like domains. Nevertheless, these TcAs form stable prepore complexes at neutral pH and the prepore-to-pore transition could be induced by changing the pH, although at lower efficiency for Mm-TcdA4 and Yp-TcaATcaB (fig. S9). Thus, due to the differences in charge distribution among TcA neuraminidase-like domains and the relatively wide opening at the bottom of some of the TcA shells, we conclude that this domain is not responsible for the electrostatic lock mechanism that has been observed in all studied TcAs (fig. S9).
We therefore analyzed the interfaces between two shell domains to identify the conserved regions that could qualify as potential pH-sensitive elements. pH-dependent conformational changes have been shown to either implicate histidine-cation or anion-anion (glutamates and/or aspartates) interactions (21). The involved residue pairs switch from stabilizing to destabilizing conformation through pH shift–induced electrostatic repulsion.
We could not find any conserved anion-anion pair or histidine-cation pair in the examined TcA structures. We only identified a prominent cluster of three histidines in Pl-TcdA1 (H46 and H50 in protomer A and H1808 in protomer B) (fig. S8, J and K). However, the residues are not conserved among the TcAs, and the loop of protomer B containing H1808 is much shorter in Mm-TcdA4 and therefore out of range for an interaction with protomer A (fig. S8L). As a result, we exclude that this histidine cluster is important for the function of the TcAs in general.
Since we could not find a classical pH switch, we searched for other conserved ionic interactions in the shell and identified three buried pairs that are found at the interface between two protomers in all examined TcAs: E158 and R1873, D965 and R1971, and E1086 and R1166 (residue numbering of Pl-TcdA1) (Fig. 6, D to F). The interacting residues are in close spatial proximity in the prepore state (3.8, 3.5, and 3.9 Å, respectively) and are likely forming salt bridges. In the Pl-TcdA1 pore state, none of these interactions are possible because of the enlarged distance after the conformational change (9.8, 5.8, and 25 Å, respectively).
To analyze whether these interactions are involved in the pH shift–induced destabilization of the shell, we designed three mutants of Pl-TcdA1 in which the respective residues are mutated to alanine (E158A-R1873A, D965A-R1971A, and E1086A-R1166A). All mutants could be recombinantly expressed in E. coli. The purification of E158A-R1873A and D965A-R1971A yielded pure protein. Most of the E1086A-R1166A variant, however, aggregated during expression, and we could only partially purify small amounts of the protein (fig. S10, A to C). For E158A-R1873A and D965A-R1971A, we observed the characteristic bell-shaped particles, and the pore state could be induced by a pH shift to pH 11, indicating that these two variants behave like the wild type (fig. S10, A and B). Despite the impurity of Pl-TcdA1–E1086A-R1166A, we found TcAs in their pentameric prepore state in the sample (fig. S10C). Interestingly, the pore state of the protein was also present (fig. S10C), indicating that a part of the prepore complexes must have been transitioned to the pore state at neutral pH. This might also explain the aggregation during expression, since the pore state, where the hydrophobic transmembrane region is exposed, tends to aggregate in solution. Control mutants, where only one of the two residues was mutated to alanine, showed a similar expression pattern as the double mutant Pl-TcdA1–E1086A-R1166A (fig. S10E). This suggests that the E1086-R1166 residue pair is crucial for the stability of the shell domain at neutral pH.
On the basis of these results, one might be tempted to speculate that this residue pair acts as the electrostatic lock of TcA that opens at low and high pH, thereby destabilizing the shell. However, the pKa of the side chain groups of glutamate is 4.25 and that of arginine is 12.48. The pKa values are even further shifted for residues involved in salt bridges (22). It is therefore unlikely that the residues change their protonation state at the pH values that we used for inducing the pore state (pH 4.7 and 11, respectively).
Y1168, Y1205, and H1202 (numbering corresponding to Pl-TcdA1) are located in close vicinity to the E1086-R1166 residue pair (fig. S10F). The three residues are conserved in Pl-TcdA1, Xn-XptA1, and Mm-TcdA4, but not in Yp-TcaA-TcaB (fig. S10G). To examine whether they have an influence on the interaction between E1086 and R1166, we mutated the tyrosines to phenylalanines (Pl-TcdA1–Y1168F-Y1205F) and the histidine to alanine (Pl-TcdA1-H1202A) and analyzed the TcA variants (fig. S10, H and I). We observed that the characteristic bell-shaped particles and the pore state could be induced by a pH shift to pH 11, indicating that these two variants behave like the wild type (fig. S10, H and I). Therefore, these residues are obviously not influencing the E1086-R1166 residue pair.
We conclude that TcAs, in general, do not have a classic pH switch. The conserved residue pair E1086-R1166 is essential for the stability of the complex at neutral pH. However, it cannot act as a pH switch because of its composition. We propose instead that the destabilization of many electrostatic interactions at the interface between the shells of two protomers is responsible for the opening of the shell at low and high pH values. The E1086-R1166 pair probably operates as a latch that springs open only once the other regions are destabilized by electrostatic repulsions.
DISCUSSION
In our study, we investigated and compared five TcAs from four different organisms: P. luminescens, X. nematophila, M. morganii, and Y. pseudotuberculosis. The cryo-EM structures of the five TcAs combined with mutational and functional studies revealed that their overall architecture and mechanism of action is similar. Important structural features relevant for the mechanism of action—i.e., the linker, the toxin translocation channel, and the TcB-binding domain—are present in all analyzed structures. These parts are the most conserved, with respect to both structural organization and residue conservation. All TcAs have a 31 trefoil knot that is strengthening the base of the linker, which is especially put under strain during the prepore-to-pore transition. Therefore, the protein knot in TcA seems to be an important conserved structural feature of Tc toxins. In contrast, the β sheet domains of the TcA shell, comprising the RBDs, are less conserved, and parts of them are missing in some TcAs and show a higher structural flexibility than the rest of the protein.
The TcA from the opportunistic human pathogenic bacterium M. morganii, which can cause various infections, such as abscess, sepsis, and nosocomial infections following surgery, resulting in a high mortality rate (23), and the TcA from Y. pseudotuberculosis, the causative agent of Far-East Scarlet-like fever (24), are both functional Tc toxin components, suggesting that Tc toxins of these bacteria may contribute as active toxins to the pathogenic effect. The structural difference at their periphery, i.e., the RBDs, indicates that these toxins adapted to the interaction with different host cells during their evolution, which might result in the host specificity observed for TcAs from different species (4). In the extreme case of TcA from Y. pseudotuberculosis, which does not contain any RBD structurally similar to the RBDs of the other TcAs, an additional coiled-coil domain at the top of the shell compensates for the likely reduced stability of the complex due to the absence of RBDs.
TcAs are not only present in insects and human pathogenic bacteria but also have been identified in plant pathogenic bacteria such as Pseudomonas syringae and Pseudomonas fluorescens (25). Although we have not studied TcA from these bacteria, on the basis of our study, we propose that all TcAs, including those of plant pathogenic bacteria, share the same architecture and mechanism of action. The high structural conservation of Tc toxins allows the formation of functional chimeric holotoxins, opening up new avenues for the design of potential biopesticides.
An important step in the mechanism of action of Tc toxins is the destabilization of a pH-sensitive electrostatic lock in the shell that releases the channel at high or low pH values (9). Since we could not identify a classic pH switch, we propose that many electrostatic interactions at the shell-shell interfaces are responsible for the opening of the shell at low and high pH values instead. A conserved ionic pair that stabilizes the shell likely operates as a strong latch that only springs open after the electrostatic destabilization of other regions. Together, our study provides a holistic view on the architectural organization and function of Tc toxins that leads to new insights into the architecture and host specificity of the Tc toxin family.
MATERIALS AND METHODS
Protein expression and purification
The expression and purification of the five TcAs were performed in a similar manner. The sequences of tcdA1 (P. luminescens), tcdA4 (P. luminescens), xptA1 (X. nematophila), and tcdA4 (M. morganii) were cloned in pET19d (Novagen). The sequences of tcaa and tcab (Y. pseudotuberculosis) were cloned together in pET28a (Novagen) with a GS linker between the two fragments. All proteins were modified with an N-terminal hexahistidine tag. Expression was performed in E. coli BL21 (DE3) RIPL. Cells were transformed with the specific plasmid and grown in medium (Xn-XptA1, Pl-TcdA1, and Yp-TcaATcaB in LB medium and Mm-TcdA4 and Pl-TcdA4 in 2TY medium) until an optical density at 600 nm of 0.6 to 0.8 at 37°C. Protein expression was induced with 25 μM isopropyl-β-d-thiogalactopyranoside and performed at 18°C for 15 hours (Xn-XptA1 and Yp-TcaATcaB) or 20°C for 20 hours (Pl-TcdA1, Pl-TcdA4, and Mm-TcdA4). Cells were lysed in lysis buffer with 200 μM Pefabloc using a microfluidizer. Lysis buffers were either composed of 20 mM tris-HCl (pH 8), 150 mM NaCl, and 0.5% Triton X-100 for Xn-XptA1 and Yp-TcaATcaB or 50 mM tris-HCl (pH 8), 200 mM NaCl, and 0.05% Tween 20 for Pl-TcdA1, Mm-TcdA4, and Pl-TcdA4. Soluble proteins were separated from cell debris by centrifugation (38,000 rpm, 30 min, 4°C) and loaded on a 5-ml HisTrap FF column (GE Healthcare). The N-terminally His-tagged protein was eluted with 20 mM tris-HCl (pH 8), 150 mM NaCl, 0.05% Tween 20, and 500 mM imidazole. Protein-containing fractions were pooled and dialyzed against 20 mM tris-HCl (pH 8), 150 mM NaCl, and 0.05% Tween 20. Subsequently, the proteins were further purified by size exclusion chromatography using a Superose 6 10 to 300 column (GE Healthcare) for Xn-XptA1, Pl-TcdA4, and Yp-TcaATcaB or a Sephacryl S400 column (GE Healthcare) for Pl-TcdA1 and Mm-TcdA4. P. luminescens TcdB2-TccC3 was expressed and purified as described previously (8, 12). Site-directed mutagenesis of the WT Pl-TcdA1 and Yp-TcaATcaB was performed according to standard procedures to generate the following mutants: Pl-TcdA1–E158A-R1873A, Pl-TcdA1–D965A-R1971A, Pl-TcdA1–E1086A-R1166A, Pl-TcdA1-H1202A, Pl-TcdA1–Y1168F-Y1205F, and Yp-TcaATcaB–∆622–714. A linker of six amino acids (GRPSSG) was added in Yp-TcaATcaB–∆622–714 to generate a short loop between the two deletion sides. The Pl-TcdA1 and Yp-TcaATcaB mutants were expressed and purified like Pl-TcdA1(WT) and Yp-TcaATcaB(WT), respectively.
Negative-stain EM
Four microliters of the protein solutions (0.05 mg/ml) was applied on freshly glow-discharged copper grids (Agar Scientific, G400C), with an additional layer of 8-nm carbon film. After 40-s incubation time, the grids were blotted with Whatman No. 4 filter paper and stained with 0.75% uranyl formate. Images were recorded using a Tecnai G Spirit electron microscope (FEI) or a JEOL-JEM 1400 electron microscope operated at 120 kV and equipped with a TVIPS TemCam F416 detector.
Sample preparation for cryo-EM
QUANTIFOIL 2/1 grids with an additional 2-nm carbon layer were used for Pl-TcdA1, Xn-XptA1, and Mm-TcdA4. C-Flat 2/1 grids with a self-made additional carbon layer were used for Pl-TcdA4. QUANTIFOIL 2/1 grids without an additional layer were used for Yp-TcaATcaB. All samples were flash-frozen in liquid ethane using a Cryoplunger CP3 (Gatan) at 25°C and ~90% humidity. The grids were freshly glow-discharged before sample application. For Pl-TcdA1, 3 μl of protein solution (0.08 mg/ml) was incubated for 20 s and blotted for 1.8 s before plunging. For Pl-TcdA4, 4 μl of protein solution (0.1 mg/ml) was incubated for 40 s and blotted for 2 s before plunging. For Xn-XptA1, 4 μl of protein solution (0.1 mg/ml) was incubated for 40 s and blotted for 2.2 s before plunging. For Mm-TcdA4, 4 μl of protein solution (0.1 mg/ml) was incubated for 40 s and blotted for 2.5 s before plunging. For Yp-TcaATcaB, 4 μl of protein solution (3.5 mg/ml) was applied and directly blotted for 2 s.
Data acquisition
All datasets were collected on a Titan Krios transmission electron microscope (Thermo Fisher Scientific) equipped with a spherical-aberration corrector and an X-FEG (field emission gun). The dataset of Pl-TcdA4 was collected at the Netherlands Centre for Electron Nanoscopy (NeCEN) in Leiden. The other datasets were collected at the Max Planck Institute of Molecular Physiology, Dortmund. The images were recorded on a Falcon II direct electron detector for Pl-TcdA1, Pl-TcdA4, Xn-XptA1, and Mm-TcdA4 and on a Falcon III for Yp-TcaATcaB using the automated data collection software EPU (FEI). Four images were acquired per grid hole in all cases. Detailed parameters of data acquisition (total dose and number of frames) for each TcA are listed in fig. S1J.
Data processing
All images for each dataset were inspected manually, and micrographs with bad ice quality or high drift were discarded. Frame alignment was performed using MotionCorr v.2.1 (26) for Pl-TcdA4; MotionCor2 (27) for Xn-XptA1, Mm-TcdA4, and Yp-TcaATcaB; and unblur (28) for Pl-TcdA1. In total, unweighted full-dose images, dose-weighted images with full-dose and low-dose images (15 e−/Å2 for Pl-TcdA4 and 25 e−/Å2 for Xn-XptA1 and Mm-TcdA4) were generated. Data processing was performed with the software package SPHIRE (14). Contrast Transfer Function (CTF) estimation was performed with CTER (29) on the unweighted full-dose images. With the CTF assessment and the drift assessment tool in SPHIRE, micrographs with high defocus or drift were sorted out. The Pl-TcdA4 dataset was manually picked with EMAN2 boxer (30). For the Pl-TcdA1, Xn-XptA1, and Mm-TcdA4 datasets, we used the automated particle software Gautomatch (www.mrc-lmb.cam.ac.uk/kzhang/) for particle picking. Initially, ~2000 particles were manually picked with EMAN2 boxer and used for a first two-dimensional (2D) classification with ISAC to generate class averages as templates for Gautomatch. For Yp-TcaATcaB, crYOLO was used for automated particle picking (31). Fifteen images were manually picked and used as training data for crYOLO. Particles were extracted from the dose-weighted full-dose images. Initial and final particle numbers are listed in fig. S1J.
2D classification was performed using ISAC in SPHIRE (14). For all TcAs, 3D refinement (sxmeridien) and 3D sorting (sxsort3d, both implemented in SPHIRE) were performed with imposed C5 symmetry. We used the map of Pl-TcdA1 (EMD-2297) (8) and filtered it to 30 Å as an initial model. For the datasets of Pl-TcdA4, Xn-XptA1, and Mm-TcdA4, particles from the low-dose images were extracted, and the last iterations of the final refinement were performed in continuing mode with the low-dose particles. Sharpening was performed with a soft mask in SPHIRE, and the resolution was calculated between the two independently refined half maps at 0.143 Fourier shell correlation (FSC) criterion (figs. S2 and S3). The final densities were filtered to the estimated final average resolution. The local resolution of the obtained maps was determined using sxlocres of the SPHIRE software package, and the final density maps were colored accordingly to the local resolution in Chimera. Detailed parameters for each dataset are summarized in fig. S1J.
Model building
The sequences of the five TcAs were aligned using Clustal Omega. Homology models were generated for Xn-XptA1, Mm-TcdA4, Yp-TcaATcaB, and Pl-TcdA4 based on the Pl-TcdA1 crystal structure (Protein Data Bank ID: 1VW1) using MODELLER (32). The homology models were used as starting point for building the atomic models and were initially fitted in the EM density map using rigid body fitting in Chimera. Some regions showed already a good fit, especially the channel domain and the α-helical shell. Other parts, especially in the RBDs, showed no good agreement with the density maps. These domains were fitted separately into the corresponding density with flexible fitting using iMODFit (33). The single fitted domains were then merged together into a single model. Nevertheless, some regions still had to be built or remodeled de novo in Coot (34). For de novo model building, residues were only built when we could trace the backbone within the density; otherwise, we deleted these residues or complete domains (RBD C for Xn-XptA1 and Mm-TcdA4 and residues 1140 to 1239 for Yp-TcaATcaB). We used the real-space refinement tool of PHENIX (35) and Rosetta relaxation (36) to refine the models. The geometries of the final refined models were evaluated with MolProbity, and the data statistics are summarized in fig. S1J.
Structure analysis and visualization
UCSF Chimera was used for structure analysis, visualization, and figure preparation. Structure-based sequence alignments were generated using the T-COFFEE Expresso server. For surface representations, we used protonated proteins generated with the H++ server (37) according to pH 4, 7, and 11. For visualization of the protein surface electrostatics, the electrostatic Coulomb potential was calculated ranging from −14 to 14 kcal/mol in Chimera. The surface hydrophobicity of the TcA channels was colored according to the Chimera tool “define attribute,” with residue-specific scores as described previously (38). The conservation values were based on the sequence alignment and RMSD values on the structure-based sequence alignment. The pore diameters of Pl-TcdA1, Xn-XptA1, and Mm-TcdA4 were calculated with ChExVis (39) and MOLEOnline (40). The protein knot was identified and determined using the online server knotprot (15).
Black lipid membrane experiments
The single-channel conductivity was measured by black lipid membrane experiments. The membranes were formed using 1% solution of diphytanoyl phosphatidylcholine in n-decane (Avanti Polar Lipids, Alabaster, AL). The instrumentational setup consisted of a Teflon chamber with two compartments, which are connected by a small hole with a surface area of 0.4 mm2. The lipid solution was painted across the hole, resulting in membrane formation. After the membrane turned black, toxin was added to the cis side (the black side) of the chamber. The membrane current was measured with a pair of Ag/AgCl electrodes with salt bridges. The electrodes were switched in series with a voltage source and homemade current amplifier on the basis of a Burr-Brown operational amplifier as described previously (8). All measurements were performed with a membrane potential of 20 mV. For the measurements with different pH values, buffer containing 1 M KCl and 10 mM citric acid (pH 4), 10 mM MES (pH 6), or 10 mM CAPS (pH 11) was used. The average single-channel conductance was calculated from at least 70 pore insertion events. For the measurements at pH 4, 6, and 11, 3.7, 19.4, and 5.6 pmol of Pl-TcdA1, 0.28, 16.6, and 0.4 pmol of Xn-XptA1, 3, 28.1, and 4 pmol of Mm-TcdA4, and 29.3, 3.6, and 16.4 pmol of Yp-TcaATcaB were used, respectively.
Holotoxin formation
For the interaction studies with P. luminescens TcdB2-TccC3 and the different TcAs, the complex formation was induced by mixing 1 μM TcdB2-TccC3 and 0.5 μM of the different TcAs (pentamer concentration) and incubated for 1 hour at 4°C. To remove unbound TcdB2-TccC3, size exclusion chromatography was performed afterward using a Superose 6 increase 5/150 column (GE Healthcare). The retention fraction corresponding to the holotoxin with a total protein concentration of about 20 nM was then visualized by negative-stain EM.
Intoxication assay
HEK293T cells (Thermo Fisher Scientific) were intoxicated with preformed holotoxin composed of the different TcAs (Pl-TcdA1, Xn-XptA1, Mm-TcdA4, and Yp-TcaATcaB) and Pl-TcdB2-TccC3. Cells (2 × 104) in 400 μl of Dulbecco’s modified Eagle’s medium/F12 medium (Pan Biotech) were adherently grown overnight, and subsequently, 0.5, 5, or 10 nM chimeric holotoxin was added. Incubation was allowed to continue for 16 hours at 37°C before imaging. Experiments were performed in triplicate. Cells were not tested for mycoplasma contamination.
Biolayer interferometry
Affinities of Pl-TcdA1, Xn-XptA1, Mm-TcdA4, and Yp-TcaATcaB to Pl-TcdB2-TccC3 were determined by biolayer interferometry (BLI) using an Octet RED384 (FortéBio, Pall Life Sciences) and streptavidin (SA) biosensors. Pl-TcdB2-TccC3 was biotinylated in 20 mM Hepes-NaOH (pH 7.3), 200 mM NaCl, and 0.01% Tween 20 (labeling buffer) with EZ-Link Sulfo-NHS-LC-Biotin (Thermo Fisher Scientific) in a 1:3 molar ratio for 2 hours at room temperature, followed by 16 hours at 4°C. Unreacted biotin label was washed out using Amicon Ultra 100-kDa cutoff concentrators by diluting the sample two times with a 10-fold volume of measurement buffer [20 mM tris-HCl (pH 8.0), 200 mM NaCl, and 0.01% Tween 20] and reconcentrating back to the original volume.
Biotinylated Pl-TcdB2-TccC3 was immobilized on SA biosensors at a concentration of 40 μg/ml, followed by quenching with biotin (5 μg/ml). BLI sensorgrams were measured in three steps: baseline (300 s), association (20 s for Pl-TcdA1 and Xn-XptA1, 200 s for Mm-TcdA4, and 60 s for Yp-TcaATcaB, respectively), and dissociation (300 s for Yp-TcaATcaB and 200 s for the other TcAs). The sensorgrams were corrected for background association of the respective TcA on unloaded SA biosensors. On- and off-rates of binding were simultaneously determined by a global curve fit according to a 1:1 binding model. All BLI steps were performed in measurement buffer with additional bovine serum albumin (0.3 mg/ml).
Incubation of TcAs with nanodiscs at different pH values
The prepore-to-pore transition of Pl-TcdA1(WT) and the mutants was induced by pH shift from pH 8 to 11. For Pl-TcdA1(WT) and the three alanine mutants (Pl-TcdA1–E158A-R1873A, Pl-TcdA1–D965A-R1971A, and Pl-TcdA1–E1086A-R1166A), 50 nM toxin pentamer was mixed with 300 nM nanodiscs [MSP1D1-ΔH5-His with 1-palmitoyl-2-oleoyl-glycero-3-phosphocholine (POPC)] and dialyzed against 20 mM CAPS-NaOH (pH 11.0) and 150 mM NaCl over a period of 72 hours. For the mutants Pl-TcdA1-H1202A and Pl-TcdA1–Y1168F-Y1205F, 0.3 μM toxin and 2 μM nanodiscs (MSP2N2-His with POPC for Pl-TcdA1-H1202A and MSP1D1-ΔH5-His with POPC for Pl-TcdA1–Y1168F-Y1205F) were mixed and dialyzed for 72 hours against 20 mM CAPS-NaOH (pH 11) and 150 mM NaCl. For Xn-XptA1, 0.4 μM toxin and 9 μM nanodiscs [MSP1E3D1-His with brain polar lipids (Avanti Polar Lipids)] were mixed in the presence of 3 mM CaCl2 and dialyzed for 72 hours against 20 mM CAPS-NaOH (pH 11.0), 150 mM NaCl, and 3 mM CaCl2. For Mm-TcdA4, 0.8 μM toxin and 9 μM nanodiscs (MSP1D1-His with POPC) were mixed in the presence of 5 mM CaCl2 and dialyzed against 25 mM CAPS-NaOH (pH 11.2), 150 mM NaCl, and 5 mM CaCl2 for 48 hours. For Yp-TcaATcaB, 0.2 μM toxin and 2 μM nanodiscs (MSP2N2-His with POPC for low pH or MSP1D1-ΔH5-His with POPC for high pH) were mixed and dialyzed against 20 mM NaOAc (pH 4.7), 150 mM NaCl or 20 mM CAPS-NaOH (pH 10.5), and 150 mM NaCl. Pore formation was checked with negative-stain EM.
Nano-differential scanning fluorimetry measurements
The melting temperatures of the different TcAs were determined by nano-differential scanning fluorimetry in a Prometheus NT48 (NanoTemper). All measurements were performed in 20 mM tris-HCl (pH 8), 150 mm NaCl, and 0.05% Tween 20 using 100 nM TcA (monomer concentration) and a temperature gradient of 1°C/min over a temperature range of 20° to 90°C. For the pH stability test of Yp-TcaATcaB, a pH range from 4 up to 11 was used with 20 mM citric acid for pH 4, 4.5, and 5; 20 mM tris-HCl for pH 6, 7, and 8; 20 mM 2-(cyclohexylamino)ethanesulfonic acid (CHES) for pH 9 and 10; and 20 mM CAPS for pH 10.5 and 11 in addition to 150 mM NaCl and 0.05% Tween 20 in all buffers. Measurements were performed in triplicate.
Note added in proof: While this paper was under review, a high resolution structure of Y. entomophaga YenTcA1A2 became available (doi: 10.1038/s41467-019-09890-8).
Supplementary Material
Acknowledgments
We thank A. Elsner for purification of Mm-TcdA4 and Pl-TcdA1 and for assistance with site-directed mutagenesis. We thank K. Vogel-Bachmayr for the purification of Pl-TcB-TcC. We acknowledge the help of F. Merino during analysis of ionic interaction studies and B. Rath for the preliminary structure investigations of Yp-TcaATcaB. We gratefully thank O. Hofnagel and D. Prumbaum for their excellent assistance in EM. We gratefully acknowledge R. Matadeen and S. de Carlo (FEI Company) for image acquisition of the Pl-TcdA4 dataset at the NeCEN in Leiden. Funding: This work was supported by funds from the Max Planck Society (to S.R.) and the European Research Council under the European Union’s Seventh Framework Programme (FP7/2007-2013) (grant no. 615984; to S.R.). Author contributions: S.R. designed and supervised the project. F.L. processed and analyzed cryo-EM data and built atomic models of Xn-XptA1, Mm-TcdA4, and Yp-TcaATcaB. C.G. and D.R. processed Pl-TcdA4 and Pl-TcdA1 data, respectively, and D.R. built the atomic model of Pl-TcdA1. F.L. designed the proteins and performed structural analysis and mutational studies. R.B. and F.L. performed single-channel conductivity experiments. D.R. performed BLI and intoxication experiments. D.M. analyzed the organization of the α-helical shell. F.L. and D.R. prepared figures. F.L. and S.R. wrote the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. The cryo-EM densities of Pl-TcdA1, Xn-XptA1, Mm-TcdA4, Pl-TcdA4, and Yp-TcaATcaB have been deposited in the Electron Microscopy Data Bank under accession nos. 10033, 10034, 10035, 10036, and 10037, respectively. The corresponding coordinates have been deposited in the Protein Data Bank under accession nos. 6RW6, 6RW8, 6RW9, 6RWA, and 6RWB, respectively. Correspondence and requests for materials should be addressed to S.R. (stefan.raunser@mpi-dortmund.mpg.de).
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/10/eaax6497/DC1
Fig. S1. Structure and conservation of TcAs.
Fig. S2. Cryo-EM of Pl-TcdA1, Pl-TcdA4, and Xn-XptA1.
Fig. S3. Cryo-EM of Mm-TcdA4 and Yp-TcaATcaB and purification of Yp-TcaATcaB(WT) and Yp-TcaATcaB–∆622–714.
Fig. S4. A 31 trefoil protein knot is present in all five TcAs.
Fig. S5. Biophysical properties of the TcA channels.
Fig. S6. pH stability of Yp-TcaATcaB and characterization of chimeric holotoxin formation.
Fig. S7. Intoxication of HEK293T cells with chimeric holotoxins.
Fig. S8. Topology of neuraminidase-like domain and nonconserved cluster of three histidine residues.
Fig. S9. pH-induced pore formation of Xn-XptA1, Mm-TcdA4, and Yp-TcaATcaB.
Fig. S10. Mutational studies of Pl-TcdA1.
Movie S1. Cryo-EM density maps of Pl-TcdA1, Pl-TcdA4, Xn-XptA1, Mm-TcdA4, and Yp-TcaATcaB.
Movie S2. Molecular trefoil knot in Pl-TcdA1.
REFERENCES AND NOTES
- 1.Bowen D., Rocheleau T. A., Blackburn M., Andreev O., Golubeva E., Bhartia R., ffrench-Constant R., Insecticidal toxins from the bacterium Photorhabdus luminescens. Science 280, 2129–2132 (1998). [DOI] [PubMed] [Google Scholar]
- 2.Blackburn M., Golubeva E., Bowen D., Ffrench-Constant R., A novel insecticidal toxin from Photorhabdus luminescens, toxin complex a (Tca), and its histopathological effects on the midgut of manduca sexta. Appl. Environ. Microbiol. 64, 3036–3041 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tabashnik B. E., Brévault T., Carrière Y., Insect resistance to Bt crops: Lessons from the first billion acres. Nat. Biotechnol. 31, 510–521 (2013). [DOI] [PubMed] [Google Scholar]
- 4.Hares M. C., Hinchliffe S. J., Strong P. C. R., Eleftherianos I., Dowling A. J., ffrench-Constant R. H., Waterfield N., The Yersinia pseudotuberculosis and Yersinia pestis toxin complex is active against cultured mammalian cells. Microbiology 154, 3503–3517 (2008). [DOI] [PubMed] [Google Scholar]
- 5.Hinchliffe S. J., Isherwood K. E., Stabler R. A., Prentice M. B., Rakin A., Nichols R. A., Oyston P. C. F., Hinds J., Titball R. W., Wren B. W., Application of DNA microarrays to study the evolutionary genomics of Yersinia pestis and Yersinia pseudotuberculosis. Genome Res. 13, 2018–2029 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen Y.-T., Peng H.-L., Shia W.-C., Hsu F.-R., Ken C.-F., Tsao Y.-M., Chen C.-H., Liu C.-E., Hsieh M.-F., Chen H.-C., Tang C.-Y., Ku T.-H., Whole-genome sequencing and identification of Morganella morganii KT pathogenicity-related genes. BMC Genomics 13 (suppl. 7), S4 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Landsberg M. J., Jones S. A., Rothnagel R., Busby J. N., Marshall S. D. G., Simpson R. M., Lott J. S., Hankamer B., Hurst M. R. H., 3D structure of the Yersinia entomophaga toxin complex and implications for insecticidal activity. Proc. Natl. Acad. Sci. U.S.A. 108, 20544–20549 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gatsogiannis C., Lang A. E., Meusch D., Pfaumann V., Hofnagel O., Benz R., Aktories K., Raunser S., A syringe-like injection mechanism in Photorhabdus luminescens toxins. Nature 495, 520–523 (2013). [DOI] [PubMed] [Google Scholar]
- 9.Meusch D., Gatsogiannis C., Efremov R. G., Lang A. E., Hofnagel O., Vetter I. R., Aktories K., Raunser S., Mechanism of Tc toxin action revealed in molecular detail. Nature 508, 61–65 (2014). [DOI] [PubMed] [Google Scholar]
- 10.Busby J. N., Panjikar S., Landsberg M. J., Hurst M. R. H., Lott J. S., The BC component of ABC toxins is an RHS-repeat-containing protein encapsulation device. Nature 501, 547–550 (2013). [DOI] [PubMed] [Google Scholar]
- 11.Gatsogiannis C., Merino F., Roderer D., Balchin D., Schubert E., Kuhlee A., Hayer-Hartl M., Raunser S., Tc toxin activation requires unfolding and refolding of a β-propeller. Nature 563, 209–213 (2018). [DOI] [PubMed] [Google Scholar]
- 12.Gatsogiannis C., Merino F., Prumbaum D., Roderer D., Leidreiter F., Meusch D., Raunser S., Membrane insertion of a Tc toxin in near-atomic detail. Nat. Struct. Mol. Biol. 23, 884–890 (2016). [DOI] [PubMed] [Google Scholar]
- 13.Lee S. C., Stoilova-Mcphie S., Baxter L., Fülöp V., Henderson J., Rodger A., Roper D. I., Scott D. J., Smith C. J., Morgan J. A. W., Structural characterisation of the insecticidal toxin XptA1, reveals a 1.15 MDa tetramer with a cage-like structure. J. Mol. Biol. 366, 1558–1568 (2007). [DOI] [PubMed] [Google Scholar]
- 14.Moriya T., Saur M., Stabrin M., Merino F., Voicu H., Huang Z., Penczek P. A., Raunser S., Gatsogiannis C., High-resolution single particle analysis from electron cryo-microscopy images using SPHIRE. JoVE , e55448 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jamroz M., Niemyska W., Rawdon E. J., Stasiak A., Millett K. C., Sułkowski P., Sulkowska J. I., KnotProt: A database of proteins with knots and slipknots. Nucleic Acids Res. 43, D306–D314 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Virnau P., Mirny L. A., Kardar M., Intricate knots in proteins: Function and evolution. PLOS Comput. Biol. 2, e122 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dabrowski-Tumanski P., Stasiak A., Sulkowska J. I., In search of functional advantages of knots in proteins. PLOS ONE 11, e0165986 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Faísca P. F. N., Knotted proteins: A tangled tale of Structural Biology. Comput. Struct. Biotechnol. J. 13, 459–468 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lang A. E., Konukiewitz J., Aktories K., Benz R., TcdA1 of Photorhabdus luminescens: Electrophysiological analysis of pore formation and effector binding. Biophys. J. 105, 376–384 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sheets J. J., Hey T. D., Fencil K. J., Burton S. L., Ni W., Lang A. E., Benz R., Aktories K., Insecticidal toxin complex proteins from Xenorhabdus nematophilus: Structure and pore formation. J. Biol. Chem. 286, 22742–22749 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Harrison J. S., Higgins C. D., O’Meara M. J., Koellhoffer J. F., Kuhlman B. A., Lai J. R., Role of electrostatic repulsion in controlling pH-dependent conformational changes of viral fusion proteins. Structure 21, 1085–1096 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sundd M., Iverson N., Ibarra-Molero B., Sanchez-Ruiz J. M., Robertson A. D., Electrostatic interactions in ubiquitin: Stabilization of carboxylates by lysine amino groups. Biochemistry 41, 7586–7596 (2002). [DOI] [PubMed] [Google Scholar]
- 23.Lee I.-K., Liu J.-W., Clinical characteristics and risk factors for mortality in Morganella morganii bacteremia. J. Microbiol. Immunol. Infect. 39, 328–334 (2006). [PubMed] [Google Scholar]
- 24.Amphlett A., Far East Scarlet-Like Fever: A review of the epidemiology, symptomatology, and role of superantigenic toxin: Yersinia pseudotuberculosis-derived mitogen A. Open Forum Infect. Dis. 3, ofv202 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rangel L. I., Henkels M. D., Shaffer B. T., Walker F. L., Davis E. W. II, Stockwell V. O., Bruck D., Taylor B. J., Loper J. E., Characterization of toxin complex gene clusters and insect toxicity of bacteria representing four subgroups of Pseudomonas fluorescens. PLOS ONE 11, e0161120 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li X., Mooney P., Zheng S., Booth C. R., Braunfeld M. B., Gubbens S., Agard D. A., Cheng Y., Electron counting and beam-induced motion correction enable near-atomic-resolution single-particle cryo-EM. Nat. Methods 10, 584–590 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zheng S. Q., Palovcak E., Armache J.-P., Verba K. A., Cheng Y., Agard D. A., MotionCor2: Anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Grant T., Grigorieff N., Measuring the optimal exposure for single particle cryo-EM using a 2.6 Å reconstruction of rotavirus VP6. eLife 4, e06980 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Penczek P. A., Fang J., Li X., Cheng Y., Loerke J., Spahn C. M. T., CTER—Rapid estimation of CTF parameters with error assessment. Ultramicroscopy 140, 9–19 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tang G., Peng L., Baldwin P. R., Mann D. S., Jiang W., Rees I., Ludtke S. J., EMAN2: An extensible image processing suite for electron microscopy. J. Struct. Biol. 157, 38–46 (2007). [DOI] [PubMed] [Google Scholar]
- 31.Wagner T., Merino F., Stabrin M., Moriya T., Gatsogiannis C., Raunser S., SPHIRE-crYOLO: A fast and well-centering automated particle picker for cryo-EM. bioRxiv, 356584 (2018). [Google Scholar]
- 32.Šali A., Blundell T. L., Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 234, 779–815 (1993). [DOI] [PubMed] [Google Scholar]
- 33.Lopéz-Blanco J. R., Chacón P., iMODFIT: Efficient and robust flexible fitting based on vibrational analysis in internal coordinates. J. Struct. Biol. 184, 261–270 (2013). [DOI] [PubMed] [Google Scholar]
- 34.Emsley P., Lohkamp B., Scott W. G., Cowtan K., Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L.-W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H., PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang R. Y.-R., Kudryashev M., Li X., Egelman E. H., Basler M., Cheng Y., Baker D., DiMaio F., De novo protein structure determination from near-atomic-resolution cryo-EM maps. Nat. Methods 12, 335–338 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Anandakrishnan R., Aguilar B., Onufriev A. V., H++ 3.0: Automating pK prediction and the preparation of biomolecular structures for atomistic molecular modeling and simulations. Nucleic Acids Res. 40, W537–W541 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hessa T., Kim H., Bihlmaier K., Lundin C., Boekel J., Andersson H., Nilsson I., White S. H., von Heijne G., Recognition of transmembrane helices by the endoplasmic reticulum translocon. Nature 433, 377–381 (2005). [DOI] [PubMed] [Google Scholar]
- 39.Masood T. B., Sandhya S., Chandra N., Natarajan V., CHEXVIS: A tool for molecular channel extraction and visualization. BMC Bioinformatics 16, 119 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Berka K., Hanák O., Sehnal D., Banáš P., Navrátilová V., Jaiswal D., Ionescu C.-M., Svobodová Vařeková R., Koča J., Otyepka M., MOLEonline 2.0: Interactive web-based analysis of biomacromolecular channels. Nucleic Acids Res. 40, W222–W227 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/10/eaax6497/DC1
Fig. S1. Structure and conservation of TcAs.
Fig. S2. Cryo-EM of Pl-TcdA1, Pl-TcdA4, and Xn-XptA1.
Fig. S3. Cryo-EM of Mm-TcdA4 and Yp-TcaATcaB and purification of Yp-TcaATcaB(WT) and Yp-TcaATcaB–∆622–714.
Fig. S4. A 31 trefoil protein knot is present in all five TcAs.
Fig. S5. Biophysical properties of the TcA channels.
Fig. S6. pH stability of Yp-TcaATcaB and characterization of chimeric holotoxin formation.
Fig. S7. Intoxication of HEK293T cells with chimeric holotoxins.
Fig. S8. Topology of neuraminidase-like domain and nonconserved cluster of three histidine residues.
Fig. S9. pH-induced pore formation of Xn-XptA1, Mm-TcdA4, and Yp-TcaATcaB.
Fig. S10. Mutational studies of Pl-TcdA1.
Movie S1. Cryo-EM density maps of Pl-TcdA1, Pl-TcdA4, Xn-XptA1, Mm-TcdA4, and Yp-TcaATcaB.
Movie S2. Molecular trefoil knot in Pl-TcdA1.