

Abstract
β2- and β3-Amino acids are important chiral building blocks for the design of new pharmaceuticals and peptidomimetics. Here we report a straightforward regio- and enantiodivergent access to these compounds using a one-pot reaction composed of sparteine-mediated enantioselective lithiation of a Boc-1,3-oxazinane, transmetallation to zinc and direct or migratory Negishi coupling with an organic electrophile. The regioselectivity of the Negishi coupling was highly ligand-controlled and switchable to obtain the C4- or the C5-functionalized product exclusively. High enantioselectivities were achieved on a broad range of examples, and a catalytic version in chiral diamine was developed using the (+)-sparteine surrogate. Selected C4- and C5-functionalized Boc-1,3-oxazinanes were subsequently converted to highly enantio-enriched β2- and β3-amino acids with the (R) or (S) configuration, depending on the sparteine enantiomer employed in the lithiation step.
Introduction
β-Amino acids substituted at the 2- and 3-positions, named β2- and β3-amino acids, respectively, are very important chiral substructures found in natural products and active pharmaceutical ingredients (Fig. 1).1–3 In particular, their incorporation into peptides allows modulating the secondary structure of the latter and increasing their proteolytic stability, hence furnishing peptidomimetics with improved pharmacological value.4–5 Although much progress has been made in the enantioselective synthesis of β-amino acids, more direct and versatile methods are still highly sought after.3
Fig. 1.

Examples of natural products and active pharmaceutical ingredients containing β2- and β3-amino acids.
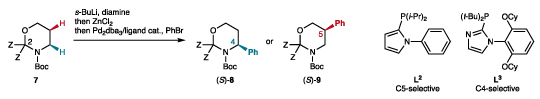
Migratory cross-couplings have emerged as interesting new methods to functionalize remote positions of alkyl chains and cyclic systems.6–8 In particular, our group showed that the use of appropriate ligands of palladium-based catalysts allows functionalization of various positions of the same reactant in a regiocontrolled fashion through a Pd migration mechanism.7 For instance, the Negishi coupling of racemic α-zincated Boc-piperidine, generated by Boc-directed α-lithiation of Boc-piperidine 1 with s-BuLi/TMEDA and transmetallation to zinc, leads to the C2 and C3-arylated racemic products 2-3 with good positional selectivity in the presence of appropriate phosphine ligands L1-L2 (Fig. 2a).9 In principle, enantioselective versions of these one-pot reactions may be developed by using a chiral base in the initial lithiation step.10–14 Indeed, in a seminal work, Campos and co-workers showed that the enantioselective lithiation of Boc-pyrrolidine 4 in the presence of (–)-sp (sp = sparteine),15 followed by one-pot Li-Zn transmetallation and Negishi coupling furnished C2-arylated products 5 efficiently and with high enantioselectivities, reflecting the enantiospecific nature of the Li-Zn transmetallation and cross-coupling steps (Fig. 2b).16 However, transposition to N-Bocpiperidine 1 was unsuccessful due to the lack of reactivity of the s-BuLi•sp complex towards this substrate.17 O’Brien and co-workers designed a less hindered surrogate of (+)-sp which provided enhanced reactivity in the lithiation step, but a modest yield and moderate enantioselectivity were observed in the Negishi arylation leading to product 6.18 These precedents, together with unsuccessful attempts employing other enantioselective lithiations, discouraged us from developing an enantioselective C3-selective arylation of Boc-piperidine 1. Alternatively, we turned to Boc-1,3-oxazinanes 7, which are protected forms of 3-aminopropanol, and hence very appealing substrates to develop a regiodivergent enantioselective functionalization strategy (Fig. 2c). The enantioselective lithiation of 7 with (–)-sp, which is currently unknown,19,20 would furnish α-lithiated intermediate A upon deprotonation of the pro-S hydrogen atom.15,17 Sequential transmetallations with ZnCl2 and the oxidative addition complex generated from Pd0/L and an electrophile R–X would afford complex B in a stereoretentive manner. In the presence of a bulky ligand such as L1 (see Fig. 2a), reductive elimination should be favored to give the C4-functionalized product 8. In the presence of a less bulky and more conformationally flexible ligand such as L2,7,9 stereospecific Pd migration should occur from B, based on previous calculations,9 via β-hydride elimination, providing complex C wherein Pd would remain bound to the same face of the molecule. π-Bond rotation and migratory insertion would deliver complex D, which would undergo reductive elimination to give rise to the enantioenriched C5-functionalized product 9. Isomers 8 and 9 would be simple precursors of β3-amino acid 10 and β2-amino acid 11, respectively, upon aminal cleavage and oxidation. Using (+)-sp21 instead of (–)-sp in the initial lithiation step would provide access to the enantiomeric end products ent-10 and ent-11 through the same sequence.
Fig. 2.

Lithiation/Negishi coupling of cyclic Boc-amines. a, The Negishi coupling of α-zincated Boc-piperidine 1 furnishes racemic C-2 (2) and C-3 (3) arylated products with good site-selectivity in the presence of appropriate phosphine ligands L1-L2. b, Enantioselective lithiation and direct Negishi coupling is effective for Boc-pyrrolidine, but not for Boc-piperidine. c, This work: design of a site- and enantioselective functionalization of Boc-1,3-oxazinanes and application to the synthesis of β-amino acids. Boc = tert-butyloxycarbonyl; TMEDA = N,N,N′,N′-tetramethylethylenediamine; dba = dibenzylideneacetone; NN* = chiral diamine; sp = sparteine.
Here we show that Boc-oxazinanes are lithiated efficiently and with high enantioselectivity using either stoichiometric or substoichiometric amounts of chiral base, and that the corresponding organozinc compounds obtained upon Li-Zn transmetallation undergo regiodivergent Negishi cross-coupling with nearly perfect ligand-controlled regioselectivity to give highly enantioenriched C4- and C5-functionalized products. The latter can be converted to valuable β2- and β3-amino acids upon aminal cleavage and oxidation.
Results
Optimization of the reaction conditions
We began our studies by investigating the one-pot arylation of Boc-1,3-oxazinanes 7a-d containing various C2 substituents, which were easily synthesized in two steps from 3-aminopropanol and various ketones (Table 1, see also Supplementary Table 1). The lithiation of compound 7a with the achiral s-BuLi•TMEDA complex, followed by transmetallation with ZnCl2 and cross-coupling with bromobenzene in the presence of the very bulky ligand L3, which we recently developed to avoid Pd migration in related Negishi couplings,22 furnished the C4-arylated product 8aa exclusively in moderate yield (entry 1). Using (+)-sp instead of TMEDA in the lithiation step furnished a promising e.r. of 75:25 (entry 2). Gratifyingly, replacing the methyl with ethyl groups on the oxazinane (7b) allowed increasing the e.r. to 95:5 (entry 3). The less hindered (+)-sp surrogate23 furnished a lower yield and enantioselectivity (entry 4). Further increasing the size of the Z groups was detrimental to the yield (entry 5). Finally, tuning the conditions by replacing ZnCl2 with Zn(OAc)2,24 phenyl bromide with phenyl nonaflate,22,25 and raising the cross-coupling temperature to 80 °C gave an improved efficiency (61%) and enantioselectivity (e.r. 97:3, entry 6). Then, we switched the ligand of the cross-coupling step to the less hindered and conformationally more flexible phosphine L2, which was previously designed to favor the migratory coupling of Boc-piperidines.9 Using substrate 7a and TMEDA in the lithiation step, the arylation site-selectivity was completely switched to the C5 position, with no trace of C4 isomer (entry 7). This ligand-controlled, total switch of selectivity in favor of the migratory arylation is remarkable, since we always obtained mixtures of isomers during previous studies on migratory couplings using an unbiased electrophile (e.g., Fig. 2a).7,9,26–28 This behavior might be related to the higher propensity of the 1,3-oxazinane, as compared to the piperidine ring, to reach the twist-boat conformation required to align the C–Pd and C–H bonds for the β-H elimination step initiating Pd migration.9 Moreover, the selectivity switch exerted by ligands L2-L3 can be explained by steric factors, i. e. the steric environment of the phosphorus atom and the rotation around the C–N axis, according to previous studies.9,22,27 Replacing TMEDA with (+)-sp provided 1,3-oxazinane 9aa with a similar e.r. (77.5:22.5) to the one observed for the C4-arylated product 8aa (75:25, entry 2), thus showing that the migration occurs with high enantiospecificity. In this case, increasing the bulk of the Z substituents (entries 9, 11, 12) furnished an optimal yield for n-propyl groups (entry 11), together with a high enantioselectivity. Similar to the C5-selective arylation, the (+)-sp surrogate gave a slightly lower e.r. than with (+)-sp (entry 10, compare with entry 9). Sparteine was kept for subsequent studies due to its lower price and higher availability of both enantiomers.
Table 1. Effect of selected parameters on the arylation of Boc-1,3-oxazinanes.
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Z | Reactant | Diamine | Ligand | 8/9a | Product | Yield (%)b | e.r.c |
| 1d | Me | 7a | TMEDA | L3 | >98:2 | 8aa | 54 | – |
| 2d | Me | 7a | (+)-sp | L3 | >98:2 | 8aa | 53 | 75:25 |
| 3d | Et | 7b | (+)-sp | L3 | >98:2 | 8ab | 51 | 95:5 |
| 4d | Et | 7b | (+)-sp surrogate | L3 | >98:2 | 8ab | 46 | 91:9 |
| 5d | n-Pr | 7c | (+)-sp | L3 | >98:2 | 8ac | 30 | 94:6 |
| 6e,f | Et | 7b | (+)-sp | L3 | >98:2 | 8ab | 61 | 97:3 |
| 7a | Me | 7a | TMEDA | L2 | <2:98 | 9aa | 65 | – |
| 8 | Me | 7a | (+)-sp | L2 | <2:98 | 9aa | 51 | 77.5:22.5 |
| 9 | Et | 7b | (+)-sp | L2 | <2:98 | 9ab | 48 | 97:3 |
| 10 | Et | 7b | (+)-sp surrogate | L2 | <2:98 | 9ab | 89 | 94:6 |
| 11f | n-Pr | 7c | (+)-sp | L2 | <2:98 | 9ac | 72 | 96.5:3.5 |
| 12 | –(CH2)4– | 7d | (+)-sp | L2 | <2:98 | 9ad | 23 | 85:15 |
Reaction conditions unless otherwise stated: 7 (1.0 equiv), s-BuLi (1.2 equiv), diamine (1.2 equiv), Et2O, –78 °C, 8 h, then ZnCl2/THF (1.2 equiv), –78→20 °C, 1 h, then removal of volatiles, then PhBr (0.7 equiv), Pd2dba3 (2.5 mol%), ligand (5 mol%), toluene, 80 °C, 17-24 h.
Measured by GCMS or 1H NMR analysis of the crude reaction mixture.
Yield of the isolated product.
Determined by HPLC on a chiral stationary phase.
Cross-coupling step performed at 60 °C instead of 80 °C.
Using Zn(OAc)2 instead of ZnCl2 and PhONf instead of PhBr.
Configuration and deuterium labelling
The absolute configuration of the arylated 1,3-oxazinanes 8ab and 9ac obtained using (+)-sp was determined to be (S) after cleavage of the aminal and comparison of the specific rotations of the corresponding Boc-aminoalcohols with literature data (see Supplementary References). In addition, quenching the organolithium intermediate obtained from 7c and (–)-sp with dimethyl sulfate followed by controlled aminal cleavage led to the known Boc-aminoalcohol 12 possessing the opposite orientation of the methyl group and the (S) configuration (Fig. 3a). This absolute configuration is expected from the sparteine-mediated enantioselective lithiations of other Boc-amines, which always give the same induction sense.15–18 In addition, the fact that the same sense of enantioselectivity is observed for compounds 12, 8ab and 9ac using a given enantiomer of sp confirms the expected stereoretentive nature of the various elementary steps depicted in Fig. 2c. Deuterium labelling experiments provided complementary insights (Fig. 3b). Performing the lithiation of 7b and 7c with the s-BuLi•(–)-sp complex/trapping with CD3OD twice, as reported by Hoppe,29 led to the deuterated 1,3-oxazinanes 7b-D and 7c-D with 96% and 98% deuterium incorporation at the C4 position, respectively. Then, lithiating compound 7b-D with the achiral s-BuLi•TMEDA complex and performing the C4-selective arylation under the same conditions as above furnished product 8ab-D with the same (S) absolute configuration as 8ab obtained using (+)-sp, with the same deuterium content as 7b-D and an e.r. of 95:5. The latter matches the theoretical value calculated with an e.r. of 97:3 for the sp-mediated lithiation step and the 96% deuterium incorporation (see Supplementary Figure 1). These values translate the fact that the TMEDA-mediated lithiation occurs with a very large kinetic isotope effect (KIE). Similarly large KIEs (kH/kD > 30) were already reported by Hoppe29 and Beak30 for the lithiation of O- and N-carbamates, respectively, and were ascribed to the tunnel effect.31 Performing the C5-selective arylation from 7c-D led to a similar outcome, with (S)-9ac-D being produced with a 97% deuterium content and an e.r. of 96:4. These results might be further exploited to synthesize isotopically-labelled β-amino acids.
Fig. 3.

Trapping experiments. TFA = trifluoroacetic acid. X = Cl or OAc, Y = Br or ONf (see Table 1). The deuterium contents were measured by 1H NMR. a Enantiomeric ratio of the mixtures of isotopomers, determined by HPLC on a chiral stationary phase.
Scope of the C4- and C5-functionalization using stoichiometric sparteine
Using the optimal conditions, the scope of the C4- and C5-functionalization was next examined using (+)-sp as the chiral diamine (Fig. 4, see also Supplementary Figure 2). The C5-functionalization (Fig. 4b) was found to be more general than the C4-functionalization (Fig. 4a), as the latter was mainly limited to aryl and heteroaryl nonaflates bearing substituents at the para or meta positions. This is probably due to the fact that the C4 position is more sterically hindered than the C5. Nevertheless, this C4-arylation performed satisfyingly with a range of aryl nonaflates containing electronically diverse substituents at the para (8b-f) and meta (8g-i) positions. More substituted aryl groups were also compatible (8j-k), as well as a naphthyl ring (8l) and diverse heteroaromatic systems (8m-p). In all cases, similar results were obtained, with yields in the range of 45-71% and excellent e.r. values (95:5-97:3) for the (S) enantiomers. As expected, simply using (–)-sp instead of (+)-sp in the lithiation step afforded the (R) enantiomers of 8c, 8f, 8j and 8l with similar levels of efficiency and enantioselectivity, hence demonstrating the enantiodivergent character of this method.
Fig. 4.

Scope of the C4- and C5-functionalization of Boc-1,3-oxazinanes. Reaction conditions: see Table 1, entries 6 (C4-functionalization) and 11 (C5-functionalization). E.r. values were determined by HPLC on a chiral stationary phase, either directly or on the Boc-protected aminoalcohol after cleavage of the aminal. The reference racemic products were synthesized using TMEDA instead of sp. a Using (–)-sp instead of (+)-sp. b Using L4 as the ligand. c Isolated as an inseparable 77:23 mixture of C5- and C6-isomers. d Performed on gram scale; 85% of (–)-sp was recovered.
As mentioned above, the scope of the C5 functionalization was found to be broader than the C4 functionalization (Fig. 4b). In addition to para (9b-h) and meta (9i-j)-substituted aryl bromides, ortho-substituted aryl bromides could be employed (9k-l). However, for product 9l bearing a strong electron-withdrawing CF3 group, ligand L4 (DavePhos)32 afforded an improved efficiency, as previously observed with Boc-piperidines.9 Disubstituted arenes (9m-n), a naphthalene (9o) and heteroarenes (9p-r) could be employed with similar levels of efficiency and enantioselectivity. Surprisingly, a ca. 3:1 mixture of the C5- and C6-isomers was isolated using 3-bromopyridine (see 9p). The C6-isomer was not detected in other cases, and the reason for this singularity is unclear at this point. In addition to (hetero)aryl bromides, alkenyl bromides also reacted successfully, as illustrated with products 9s-v. Once again, good yields and high enantioselectivities (e.r. 92:8-96:4) were achieved across all examples, and the corresponding products are potential precursors of β2-amino acids containing a range of useful functional groups upon further transformation of the alkene. In addition, similar to C4 functionalization, the use of (–)-sp afforded the (R) enantiomers of products 9ac, 9e, 9h, 9j, 9n, 9o, 9r and 9v with similarly good yields and enantioselectivities. Finally, the reaction leading to product (R)-9e could be conducted on gram scale with similar efficiency and enantioselectivity. Importantly, a simple aqueous extraction allowed to recover 85% of the engaged (–)-sp.
Development of a catalytic enantioselective version
Next, we turned to the development of a catalytic enantioselective version of this method via the diamine exchange method (Fig. 5). Indeed, O’Brien and co-workers reported the enantioselective deprotonation of Boc-pyrrolidine using a combination of substoichiometric (–)-sp or (+)-sp surrogate and stoichiometric diisopropylbispidine, an achiral diamine which reacts slowly in the lithiation with s-BuLi but is able to exchange with the chiral diamine on the lithiated intermediate.33–34 Different chiral diamines (0.3 equiv) were first tested in combination with diisopropylbispidine (1.3 equiv) in the C5-arylation providing (S)-9c, and consistent with O’Brien’s results the (+)-sp surrogate provided the best yield, together with a satisfying e.r. of 93:7. As a comparison, compound 9c was obtained in 18% yield, 95:5 e.r. using (+)-sp instead of the (+)-sp surrogate in combination with diisopropylbispidine, which probably indicates that (+)-sp is not easily displaced by diisopropylbispidine, unlike the (+)-sp surrogate. It should be noted that both enantiomers of the sparteine surrogate are in principle accessible, and hence this catalytic version can be also applied to synthesize both enantiomers of β-amino acids.35 These conditions were applied to both C4- and C5-selective arylations with a few aryl electrophiles. The yields and enantioselectivities were satisfying, although generally lower than those obtained with stoichiometric (+)-sp (compare to Fig. 4). Indeed, the enantioselectivity induced by the (+)-sp surrogate in the lithiation step is lower than (+)-sp (see Table 1). Nevertheless, these results represent a solid proof of concept showing the feasibility of catalytic enantioselective divergent functionalization.
Fig. 5.

Development of proof of concept catalytic enantioselective C4- and C5-arylations. Reaction conditions: 7 (1.0 equiv), s-BuLi (1.2 equiv), (+)-sp surrogate (0.3 equiv), diisopropylbispidine (1.3 equiv), Et2O, –78 °C, 8 h, then Zn(OAc)2 (C4-arylation) or ZnCl2 (C5-arylation), THF (1.2 equiv), –78→20 °C, 1 h, then removal of volatiles, then PhBr (0.7 equiv), Pd2dba3 (2.5 mol%), ligand (5 mol%), toluene, 80 °C, 17 h. a Using (+)-sp instead of the (+)-sp surrogate.
Application to the synthesis of β2- and β3-amino acids
To achieve our initial goal, we finally studied the transformation of selected C4- and C5-functionalized 1,3-oxazinanes into β2- and β3-amino acids (Fig. 6). A simple treatment with TFA in THF effected the cleavage of the aminal group to give the corresponding N-Boc-1,3-aminoalcohols, which are valuable chiral intermediates for asymmetric synthesis in their own right.36 Then, two one-step procedures were employed for their oxidation to the corresponding β-amino acids. Sharpless’ oxidation was employed for the less sensitive aminoalcohols (method A),37 whereas the method employing catalytic TEMPO and stoichiometric PIDA was preferred for more sensitive ones (method B).38 Selected functionalized oxazinanes 8-9 were hence converted to over twenty valuable β2- and β3-amino acids 10-11, most of them being new, bearing (hetero)aryl or alkenyl substituents in good yield and excellent enantiospecificity, thereby preserving the enantioselectivity achieved in the initial lithiation step. Notably, the racemization of the more sensitive β2-amino acids 11 was not observed under these conditions. Both (R) and (S) enantiomers of the β-amino acids are accessible through this method by simply changing the sparteine enantiomer in the lithiation step of the overall sequence. The X-ray diffraction analysis of product 11a obtained using (–)-sp confirmed the absolute configurations deduced from the comparison of specific rotations of the known β-amino alcohols (12, precursors of 10c and 10l) and acids (10a, 10e, 11a) synthesized in this study with the corresponding literature values. Of note, compound (R)-10c is a potential precursor of the β3-amino acid found in jasplakinolide (Fig. 1), upon cleavage of the methoxy group. Similarly, compound (S)-11e is a potential precursor of the β2-amino acid found in netarsudil upon reduction of the carboxylic ester. These two examples further illustrate the interest of the current methodology for the synthesis of bioactive molecules.
Fig. 6.

Application to the synthesis of β2- and β3-amino acids. Reaction conditions: 1. TFA, THF, 20 °C. 2. Method A: RuCl3 (5 mol%), NaIO4 (3 equiv), MeCN/H2O, 20 °C. Method B: TEMPO (20 mol%), PIDA (2 equiv), CH2Cl2/H2O, 20 °C. (S) enantiomers were obtained with (+)-sp and (R) enantiomers with (–)-sp. Yields in parentheses refer to the overall sequence from Boc-1,3-oxazinanes 7b-c. The e.r. were determined after derivatization to the corresponding methyl esters. a Obtained using method A. b Obtained using method B. c Thermal ellipsoids shown at 50 % probability. TEMPO = (2,2,6,6-tetramethylpiperidin-1-yl)oxyl; PIDA = (diacetoxyiodo)benzene.
Conclusion
Boc-1,3-oxazinanes are unique platforms for the selective functionalization at the C4- or C5-position using the one-pot directed lithiation, Li-Zn transmetallation and Negishi cross-coupling sequence. The regioselectivity of the cross-coupling step is highly ligand-controlled, and high enantioselectivities can be achieved for both C4- and C5-isomers using a chiral diamine in the lithiation step, by taking advantage of the enantiospecific character of the subsequent steps. A simple two-step transformation of the coupling products leads to enantioenriched β2- and β3-amino acids, which are important building blocks for the design of new pharmaceuticals and peptidomimetics.
Supplementary Material
Acknowledgements
This work was financially supported by the Swiss National Science Foundation (200021_165987) and the University of Basel. We thank Dr. A. Prescimone, University of Basel, for X-ray diffraction analysis, Dr. D. Häussinger, University of Basel, for NMR experiments, S. Mittelheisser and Dr. M. Pfeffer, University of Basel, for MS analysis, and Dr. J. Rotzler and Dr. F. Bächle (Solvias AG), for fruitful discussions.
Footnotes
Data availability. Data supporting the findings of this study are available in the Supplementary Information or from the corresponding author upon request. The Supplementary Information contains full details on the synthesis and characterization of compounds. CCDC 1913804 (compound (R)-11a) contains the supplementary crystallo-graphic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via https://www.ccdc.cam.ac.uk/structures/.
Author contributions
W.L. and K.-F.Z. designed and performed the experiments, analyzed experimental data and prepared the Supplementary Information. O.B. directed the investigations and prepared the manuscript.
Competing interests
The authors declare no competing interests.
References
- 1.Kudo F, Miyanaga A, Eguchi T. Biosynthesis of natural products containing β-amino acids. Nat Prod Rep. 2014;31:1056–1073. doi: 10.1039/c4np00007b. [DOI] [PubMed] [Google Scholar]
- 2.Ma JS. Unnatural amino acids in drug discovery. Chim Oggi. 2003;21:65–68. [Google Scholar]
- 3.Juaristi E, Soloshonok VA. Enantioselective synthesis of β-amino acids. Wiley-Interscience; Hoboken: 2005. [Google Scholar]
- 4.Steer DL, Lew RA, Perlmutter P, Smith AI, Aguilar M-I. β-Amino acids: versatile peptidomimetics. Curr Med Chem. 2002;9:811–822. doi: 10.2174/0929867024606759. [DOI] [PubMed] [Google Scholar]
- 5.Cabrele C, Martinek TA, Reiser O, Berlicki L. Peptides containing β-amino acid patterns: challenges and successes in medicinal chemistry. J Med Chem. 2014;57:9718–9739. doi: 10.1021/jm5010896. [DOI] [PubMed] [Google Scholar]
- 6.Vasseur A, Bruffaerts J, Marek I. Remote functionalization through alkene isomerization. Nat Chem. 2016;8:209–219. doi: 10.1038/nchem.2445. [DOI] [PubMed] [Google Scholar]
- 7.Baudoin O. Selectivity control in the palladium-catalyzed cross-coupling of alkyl nucleophiles. Chima. 2016;70:768–772. doi: 10.2533/chimia.2016.768. [DOI] [PubMed] [Google Scholar]
- 8.Sommer H, Juliá-Hernańdez F, Martin R, Marek I. Walking metals for remote functionalization. ACS Cent Sci. 2018;4:153–165. doi: 10.1021/acscentsci.8b00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Millet A, Larini P, Clot E, Baudoin O. Ligand-controlled β-selective C(sp3)–H arylation of N-Boc-piperidines. Chem Sci. 2013;4:2241–2247. [Google Scholar]
- 10.Selected enantioselective migratory cross-couplings involving carbo- or hydropalladation of olefins are given in references 11-14.
- 11.Werner EW, Mei T-S, Burckle AJ, Sigman MS. Enantioselective Heck arylations of acyclic alkenyl alcohols using a redox-relay strategy. Science. 2012;338:1455–1458. doi: 10.1126/science.1229208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Correia CRD, Oliveira CC, Salles AG, Jr, Santos EAF. The first examples of enantioselective Heck-Matsuda reaction: arylation of unactivated cyclic olefins using chiral bisoxazolines. Tetrahedron Lett. 2012;53:3325–3328. [Google Scholar]
- 13.Mei T-S, Patel HH, Sigman MS. Enantioselective construction of remote quaternary stereocenters. Nature. 2014;508:340–344. doi: 10.1038/nature13231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li L, Romano C, Mazet C. Palladium-catalyzed long-range deconjugative isomerization of highly substituted α,β-unsaturated carbonyl compounds. J Am Chem Soc. 2016;138:10344–10350. doi: 10.1021/jacs.6b06390. [DOI] [PubMed] [Google Scholar]
- 15.Beak P, Kerrick ST. Asymmetric deprotonations: enantioselective syntheses of 2-substituted (tert-butoxycarbonyl)pyrrolidines. J Am Chem Soc. 1991;113:9708–9710. [Google Scholar]
- 16.Campos KR, Klapars A, Waldman JH, Dormer PG, Chen C-y. Enantioselective, palladium-catalyzed α-arylation of N-Boc-pyrrolidine. J Am Chem Soc. 2006;128:3538–3539. doi: 10.1021/ja0605265. [DOI] [PubMed] [Google Scholar]
- 17.Bailey WF, Beak P, Kerrick ST, Ma S, Wiberg KB. An experimental and computational investigation of the enantioselective deprotonation of Boc-piperidine. J Am Chem Soc. 2002;124:1889–1896. doi: 10.1021/ja012169y. [DOI] [PubMed] [Google Scholar]
- 18.Stead D, Carbone G, O’Brien P, Campos KR, Coldham I, Sanderson A. Asymmetric deprotonation of N-Boc piperidine: React IR monitoring and mechanistic aspects. J Am Chem Soc. 2010;132:7260–7261. doi: 10.1021/ja102043e. [DOI] [PubMed] [Google Scholar]
- 19.The non-enantioselective α-lithiation of 2-methyl-Boc-oxazinane with s-BuLi/TMEDA was reported: Beak P, Yum EK. Lithiation of N-Boc-2-methyltetrahydro-1,3-oxazine: a synthetic equivalent for 1-lithio-3-hydroxy-1-propylamine. J Org Chem. 1993;58:823–824.
- 20.For the enantioselective lithiation of a Boc-piperazine: McDermott BP, Campbell AD, Ertan A. First example of s-BuLi/(–)-sparteine-mediated chiral deprotonation of a piperazine and proof of the sense of induction. Synlett. 2008:875–879.
- 21.Both enantiomers of sparteine are commercially available or readily prepared in multigram quantities from the seeds of Lupinus albus: Maulide N, Peng B, Mateus Afonso CA, Machado Frade RF. Process for converting lupanine into sparteine. WO 2014191261. 2014
- 22.Zhang K-F, Christoffel F, Baudoin O. Barbier-Negishi coupling of secondary alkyl bromides with triflates and nonaflates. Angew Chem Int Ed. 2018;57:1982–1986. doi: 10.1002/anie.201711990. [DOI] [PubMed] [Google Scholar]
- 23.Synthesized according to: Dixon AJ, McGrath MJ, O’Brien P. Synthesis of (+)-(1R,2S,9S)-11-methyl-7,11-diazatricyclo[7.3.1.02,7]tridecane, a (+)-sparteine surrogate. Org Synth. 2006;83:141–154.
- 24.Royal T, Baumgartner Y, Baudoin O. Enantioselective α-arylation of O-carbamates via sparteine-mediated lithiation and Negishi cross-coupling. Org Lett. 2017;19:166–169. doi: 10.1021/acs.orglett.6b03472. [DOI] [PubMed] [Google Scholar]
- 25.Rottlӓnder M, Knochel P. Palladium-catalyzed cross-coupling reactions with aryl nonaflates: a practical alternative to aryl triflates. J Org Chem. 1998;63:203–208. doi: 10.1021/jo971636k. [DOI] [PubMed] [Google Scholar]
- 26.Renaudat A, Jean-Gérard L, Jazzar R, Kefalidis CE, Clot E, Baudoin O. The palladium-catalyzed β arylation of carboxylic esters. Angew Chem Int Ed. 2010;49:7261–7265. doi: 10.1002/anie.201003544. [DOI] [PubMed] [Google Scholar]
- 27.Millet A, Dailler D, Larini P, Baudoin O. Ligand-controlled α- and β-arylation of acyclic N-Boc-amines. Angew Chem Int Ed. 2014;53:2678–2682. doi: 10.1002/anie.201310904. [DOI] [PubMed] [Google Scholar]
- 28.Dupuy S, Zhang K-F, Goutierre A-S, Baudoin O. Terminal-selective functionalization of alkyl chains by regioconvergent cross-coupling. Angew Chem Int Ed. 2016;55:14793–14797. doi: 10.1002/anie.201608535. [DOI] [PubMed] [Google Scholar]
- 29.Hoppe D, Paetow M, Hintze F. Stereodivergent enantioselective synthesis by exploiting unusually large kinetic H/D isotope effects on deprotonation. Angew Chem Int Ed Engl. 1993;32:394–396. [Google Scholar]
- 30.Gallagher DJ, Beak P. Complex-induced proximity effects: evidence for a prelithiation complex and a rate-determining deprotonation in the asymmetric lithiation of Boc-pyrrolidine by an i-PrLi/(–)-sparteine complex. J Org Chem. 1995;60:7092–7093. [Google Scholar]
- 31.Meisner J, Kästner J. Atom tunneling in chemistry. Angew Chem Int Ed. 2016;55:5400–5413. doi: 10.1002/anie.201511028. [DOI] [PubMed] [Google Scholar]
- 32.Old DW, Wolfe JP, Buchwald SL. A highly active catalyst for palladium-catalyzed cross-coupling reactions: room-temperature Suzuki couplings and amination of unactivated aryl chlorides. J Am Chem Soc. 1998;120:9722–9723. [Google Scholar]
- 33.McGrath MJ, O’Brien P. Catalytic asymmetric deprotonation using a ligand exchange approach. J Am Chem Soc. 2005;127:16378–16379. doi: 10.1021/ja056026d. [DOI] [PubMed] [Google Scholar]
- 34.Barker G, McGrath JL, Klapars A, Stead D, Zhou G, Campos KR, O’Brien P. Enantioselective, palladium-catalyzed α-arylation of N-Boc pyrrolidine: in situ React IR spectroscopic monitoring, scope, and synthetic applications. J Org Chem. 2011;76:5936–5953. doi: 10.1021/jo2011347. [DOI] [PubMed] [Google Scholar]
- 35.Firth JD, Canipa SJ, Ferris L, O'Brien P. Gram-scale synthesis of the (–)-sparteine surrogate and (–)-sparteine. Angew Chem Int Ed. 2018;57:223–226. doi: 10.1002/anie.201710261. [DOI] [PubMed] [Google Scholar]
- 36.Lait SM, Rankic DA, Keay BA. 1,3-Aminoalcohols and their derivatives in asymmetric organic synthesis. Chem Rev. 2007;107:767–796. doi: 10.1021/cr050065q. [DOI] [PubMed] [Google Scholar]
- 37.Carlsen PHJ, Katsuki T, Martin VS, Sharpless KB. A greatly improved procedure for ruthenium tetraoxide catalyzed oxidations of organic compounds. J Org Chem. 1981;46:3936–3938. [Google Scholar]
- 38.De Mico A, Margarita R, Parlanti L, Vescovi A, Piancatelli G. A versatile and highly selective hypervalent iodine (III)/2,2,6,6-tetramethyl-1-piperidinyloxyl-mediated oxidation of alcohols to carbonyl compounds. J Org Chem. 1997;62:6974–6977. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.